

Abstract
New insights into the intra- and intercellular trafficking of drug delivery particles challenges the dogma of particles as static intracellular depots for sustained drug release. Recent discoveries in the cell-to-cell transfer of cellular constituents, including proteins, organelles, and microparticles sheds light on new ways to propagate signals and therapeutics. While beneficial for the dispersion of therapeutics at sites of pathologies, propagation of biological entities advancing disease states is less desirable. Mechanisms are presented for the transfer of porous silicon microparticles between cells. Direct cell-to-cell transfer of microparticles by means of membrane adhesion or using membrane extensions known as tunneling nanotubes is presented. Cellular relays, or shuttle cells, are also shown to mediate the transfer of microparticles between cells. These microparticle-transfer events appear to be stimulated by environmental cues, introducing a new paradigm of environmentally triggered propagation of cellular signals and rapid dispersion of particle-delivered therapeutics. The opportunity to use microparticles to study cellular transfer events and biological triggers that induce these events may aid in the discovery of therapeutics that limit the spread of disease.
1. Introduction
Cellular communication underlies the development of the organism, biological signaling, and a vast array of physiological processes that are essential for survival. Cells interact through: 1) direct contact, which allows for mutual recognition and activation of proteins and carbohydrates attached to the cell membrane;[1] 2) paracrine signaling via the release of short-lived signaling molecules into the extracellular fluid;[2,3] 3) endocrine signaling via the release of hormones, which have long half-lives and can interact with distant cells, and;[4,5] 4) synaptic signaling via the release of neurotransmitters.[6] Herein, we report an additional mechanism of cellular communication involving cell-to-cell transfer of membrane-bound micro-vesicles.
Biological vesicles are either released from the plasma membrane of the cell via fusion of an organelle’s outer membrane with the plasma membrane or via budding from the plasma membrane. The resulting membrane-bound vesicles are heterogeneous with respect to size (between 50 and 1000 nm[7,8]) and vary in composition depending on the donor cell type. These vesicles present different molecules on their surface (e.g. CD63, MHC[9,10]) and carry cargo unique in cytokines, mRNA and microRNA,[11] protein, lipids, and organelles,[12] thereby acting as biological vectors transporting information between cells. Circulating in the bloodstream, these biovesicles have been shown to mediate long-range cell activation, phenotypic changes, and inflammatory signals. We have demonstrated cellular release and re-uptake of membrane-bound microvesicles carrying cellular constituents and synthetic nanoparticles from both macrophages[8] and endothelial cells.[13] Upon introduction to donor cells, internalized nanoparticles are enclosed in endosomes, and based on transmission electron micrographs, the secretable microvesicles, with an average diameter of 1000 nm, contain nanoparticles and cellular constituents representative of various stages of endosomal maturation, including recycled membrane whirls, vesicular bodies, partially degraded rough ER, and ribosomes. The wealth of biological information contained in these vesicles represents a mode for mass communication of cellular information.
Recent discoveries have expanded the mechanisms of cell-to-cell communication to include the formation of membrane channels, known as tunneling nanotubes (TNT). TNT form through two mechanisms: 1) formation of actin-driven protrusions in one cell which then connect with acceptor cells, and; 2) cell-to-cell contact and formation of nano-tubular bridges between cells as they move apart.[14] These TNT introduce direct channeling between the cells, allowing for exchange of cellular constituents including cytoplasm, organelles (e.g. endosomes,[15] lysosomes, mitochondria),[16] nucleic acids,[17] membrane components,[18] and pathogens, such as bacteria[16] and viruses.[19] TNT were first characterized in rat pheochomocytoma cells[15] and have since been described in lymphocytes, monocytes,[20] dendritic cells,[21] T cells,[19] astrocytes, epithelial cells,[12] and endothelial progenitor cells.[22] With respect to synthetic nanoparticles, Mi and colleagues[23] have shown that nanoparticles, specifically quantum dots, are candidates for transport between cells through TNT.
In this study, we present evidence for TNT connecting human microvascular and umbilical vein endothelial cells. Using live confocal imaging, we observe the transfer of microparticles between cells via direct cell-to-cell contact, through microparticle release and reuptake by secondary cells, and via cells that appear to act as shuttles to carry the microparticles to recipient cells. We propose that these cellular ‘shuttles’ are highly dynamic cells that pick up cargo in one cell and deliver it to secondary cells, essentially acting as relays for signal propagation.
The release of microparticles in membrane-bound vesicles, as well as the trafficking of microparticles through TNT and cellular shuttles, highlights the potential for rapid dispersion of cargo across a population of cells and a potential route for long-range trafficking of complex signals. For therapeutic drug delivery vehicles, this translates into a mechanism for rapid, or potentially-triggered, transfer of therapeutics and/or molecular signals.
2. Results
2.1. Endothelial Cell Characterization and Function in the Presence of Silicon Microparticles
This study focused on microparticle transfer between endothelial cells using porous silicon microparticles. The quasi-hemispherical particles had diameters of 3.2 μm, with an aspect ratio of 1.3, and 47% porosity (Figure 1A). The average pore size was 6 nm. For all experiments, the microparticles were surface oxidized, presenting functional groups and a negative surface potential. APTES silylation of the microparticles was then performed for the purpose of conjugating chromophores and enhancing cellular uptake by endothelial cells. We previously demonstrated that microparticles with a negative surface potential have decreased cellular associations with endothelial cells following serum opsonization.[24,25]
Figure 1.
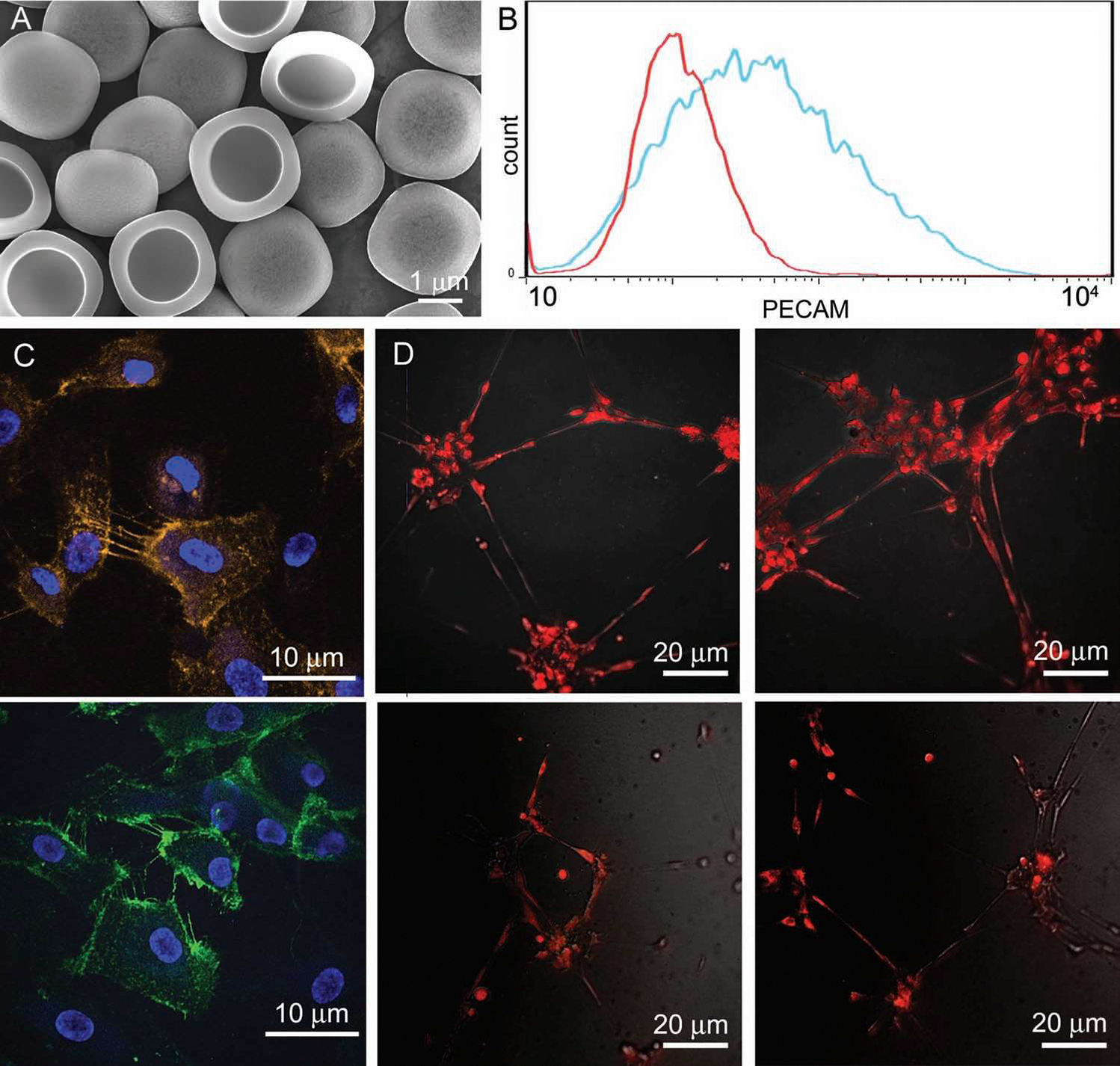
Phenotypic analysis of endothelial cells in culture. A) Scanning electron micrograph of porous silicon microparticles (mag. 10 000×; bar 1 μm). B) Flow cytometric analysis of PECAM expression by HMVEC (red–isotype control; blue–PECAM). C,D) Confocal micrographs showing PECAM expression on HMVEC (C; bars 10 μm) and endothelial tube formation by HMVEC cultured in a gel matrix (D; top row control, bottom row microparticle treated).
Human microvascular (HMVEC) and umbilical vein (HUVEC) endothelial cells were used as models to study microparticle transfer between endothelial cells. HMVEC were generated by Shao and Guo[26] by introducing the human telomerase catalytic protein into primary human microvascular endothelial cells.[26] These investigators demonstrated that the transformed cells retained endothelial cell-specific markers and exhibited angiogenic responses to appropriate stimuli. We confirmed that HMVEC are a homogeneous population expressing platelet endothelial cell adhesion molecule (PECAM) using flow cytometry (Figure 1B) and confocal microscopy (Figure 1C). Expression of PECAM was evident on all cells, with the highest levels of expression at sites of cell-to-cell contact, consistent with its role in cellular junctions, cell-cell communication and adhesion.[27]
We examined whether HMVEC would form endothelial tubes when cultured in matrigel, supporting normal physiological behavior (Figure 1D). Matrigel acts as a scaffold and source of growth and differentiation factors, promoting natural endothelial cell assembly into capillary-like structures. Both in the presence and absence of microparticles, cells spontaneously formed tube-like structures supporting their ability to properly differentiate, migrate, and form cell-to-cell adhesions.
2.2. Endothelial Tunneling Nanotubes (TNT)
We have previously demonstrated that porous silicon microparticles are internalized by HUVEC by a combination of phagocytosis and macropinocytosis.[24] Herein, the involvement of actin in microparticle uptake was re-confirmed by labeling cells with Alexa Fluor 555 phalloidin and blocking actin polymerization with cytochalasin B. In the presence of cytochalasin B, microparticles were seen surrounded by actin rings, supporting ‘frustrated phagocytosis’ of microparticles (Figure 2A). In the absence of actin polymerization, the presence of α-tubulin filaments in thick cellular extensions were easily detected (Figure 2B). Smaller actin-containing pseudopods, independent of tubulin, are also visible, as indicated by the arrow (as well as the inset) in Figure 2B.
Figure 2.

Endothelial TNT. A) Inhibition of phagocytosis of silicon microparticles (15 min incubation) by cytochalasin B (red: actin; green: microparticles; bar 5 μm). B) Actin (red) and tubulin (green) labeling of HUVEC treated with cytochalasin B and silicon microparticles (purple); bar 10 μm. C,D) Confocal micrographs show TNT connecting HUVEC in 2D cultures. Actin present in TNT is labeled with Alexa Fluor 555 phalloidin. Microparticles are shown in green, labeled with DyLight 488 and nuclei (left image) are labeled with DAPI (bar: left 20 μm, right 15 μm).
Confocal micrographs of phalloidin-labeled HUVECs following treatment with DyLight 488-labeled microparticles (Figure 2C,D) show an extended filopodium-like protrusion connecting two cells (Figure 2C), while smaller actin-driven pseudopods are seen between more proximal endothelial cells (Figure 2D). The presence of these cellular extensions provides support for the existence of TNT between endothelial cells.
Elongated cellular extensions connecting neighboring cells are also visible in electron micrographs. Transport of microparticles within the cellular extensions was seen in both scanning electron (SEM) (Figure 3A) and transmission electron (TEM) micrographs (Figure 3B). Microparticles on the cell surface in the SEM images are pseudo-colored in red to distinguish them from internalized microparticles. The intracellular location of the microparticles, as seen in the TEM images, are in the proximal ends of adjacent TNTs, supporting a role for TNT in microparticle exchange between cells. The TNTs form cellular connections at multiple points across the bordering membranes (Figure 3B, right image).
Figure 3.

Microparticle transfer via TNT. A) Pseudo-colored scanning electron micrographs show the presence of silicon microparticles in HUVEC TNT. Microparticles on the surface of the membrane are colored in red while internal microparticles are beneath the membrane [mag. 7.5 k and 2.5 k (cropped and enlarged); bars 1 and 5 μm]. B) Transmission electron micrographs of HMVEC show two connecting TNT with microparticles in the distal region of the membrane projection. Images are taken at 4000 and 8000 magnification, with the high magnification image highlighting connecting TNT (bars 10 and 3 μm).
2.3. Intercellular Transfer of Microparticles via Direct Cell-to-Cell Contact
Real-time imaging of direct cell-to-cell transfer of microparticles was captured using confocal microscopy. Images were captured every 5 min in 5 focal planes. In Field 1, we observed a single microparticle associating with 3 independent HMVEC over a span of 9.5 h. Pseudo-colored and unaltered confocal microscopy images of the three cells are shown Figure 4 and a movie spanning the course of 19 h is included in the supplemental data (SI, Movie S1). Selected time-lapse micrographs (Figure 4) show the microparticle as it begins and ends in association with the same cell, and intermittently associates with two other cells.
Figure 4.

Microparticle association with a population of neighboring endothelial cells. Select pseudo-colored (A) and unaltered (B) confocal images show the transfer of a single microparticle across 3 HMVEC over a period of 9.5 h. All tiffs are presented in SI, Movie S1.
We have previously shown that internalized silicon microparticles traffic to the peri-nuclear region of the cell in a microtubule-dependent manner.[13,24] Thus, we used nuclear-directed trafficking of the silicon microparticle as an indication that the particle was internalized, as opposed to interacting with the cell surface. Based on this metric, the microparticle (followed in Figure 4) transiently associates with ‘cell 1’ (red) and is then released from the cell surface into the surrounding media, where it then appears to “surf” across the membrane of ‘cell 2’ (blue) (Figure 4). Cell 2 makes direct contact with cell 3 (green), followed by microparticle engulfment and intracellular trafficking to the perinuclear region of cell 3. Cell 3 (green) retains the microparticle for 8 h, and then transfers the particle back to cell 1 (red) (Figure 4, Figure 5). Thus we observed that not all cell contact results in microparticle engulfment, and not all engulfment results in long-term retention of the microparticle. Particles can be internalized by an acceptor cell and move from the initial acceptor cell to surrounding cells.
Figure 5.

Microparticle transfer between HMVEC via direct cell-to-cell contact. A,B) Pseudo-colored and unaltered confocal micrographs show two adjacent HMVEC exchanging a silicon microparticle over a span of 90 min (see also SI, Movie S1). C) TEM micrographs support cell-to-cell transfer of microparticles at sites of direct cell-to-cell contact (4 k and 32 k magnification; bars 10 and 0.5 μm). D) Two silicon microparticles are shown in TEM micrographs (4 k and 32 k magnification; bars 10 and 0.5 μm) at a distance from the cell body surrounded by cell membranes (one microparticle is enlarged in the image to the right).
Step-by-step transfer of the microparticle from “cell 3” to “cell 1” (Figure 4) is shown in greater detail in Figure 5A,B. The process involves alignment of the two cells, with the acceptor cell expanding in size in the region of transfer to accommodate the microparticle. Events following the transfer, documented in supplemental Movie S1, include nuclear-directed trafficking of the microparticle following its return to “cell 1”. In a separate experiment, we captured TEM images of adjacent HMVEC, 4 hr after the introduction of microparticles, that lend further support for direct microparticle transfer between cells (Figure 5C). In the image in Figure 5C, the gap between the two adjacent cells is missing in the location directly adjacent to the microparticle, supporting localized fusion of the two cells. We also observed several microparticles distal from the cell enclosed in cellular membranes (Figure 5D). These membrane-enclosed microparticles may represent slices through TNTs or may be secreted membrane-bound microparticles.
2.4. Endothelial Shuttle Cells
In a separate field, during the same imaging session, we recorded microparticle transfer between two large endothelial cells mediated by a smaller, highly mobile endothelial cell (SI, Movie S2). Selected images show a highly dynamic ‘shuttle’ endothelial cell (green) consecutively mediating the transfer of two microparticles between two larger endothelial cells (red and blue). The ‘shuttle cell’ carrying the microparticle is initially attached to both of the larger cells. The microparticle traffics from one end of the shuttle cell to the opposite pole (Figure 6A). After the initial microparticle transfer, the shuttle cell returns to the initial donor cell and picks up a second microparticle (Figure 6B). Sequential images in the supplemental movie highlight the shuttle cell moving towards and attaching to the cell membrane at the precise location of the microparticle. The shuttle cell appears to wrap around the donor cell’s extended membrane section, extracting the microparticle.
Figure 6.

Endothelial-shuttles facilitate microparticle exchange. Pseudo-colored confocal micrographs show microparticle transfer between endothelial cells via mobile shuttle cells. HMVEC were imaged over a period of 19 h after the introduction of silicon microparticles. The transfer of two microparticles are shown in A and B as indicated by the arrows, with the order of events indicated with numbers. The corresponding unaltered tiffs are presented in SI, Movie S2.
2.5. Estimation of Microparticle Transfer Frequency
To assess the frequency of intercellular transfer of microparticles between cells, high throughput, imaging cytometry was used. As already shown, image-based analysis provides direct and unambiguous detection of particle transfer; the use of high throughput cytometry provides enough data to make statistically robust estimates. Two populations of endothelial cells, one containing red (Alexa Fluor 555)-labeled microparticles and the other containing green (Alexa Fluor 488)-labeled microparticles were co-cultured for 24 h. The imaging cytometer was then used to quantify particle transfer by identification of cells containing both red and green particles. Figure 7A shows typical images of an unlabeled control sample, the microparticle loaded pre-mixed samples and post-mixing cells which exhibit both colors of microparticle. Using the single-label populations, we set thresholds to discriminate spectral overlap from co-localization of signals from single cells; different color channel masks were also used to confirm spatial discrimination of the green and red particles. Under normal endothelial culture conditions (2% FCS), the dual-labeled cell percentage population after 24 h was 1%; however, when cells were cultured in serum-free media, the rate of microparticle exchange increased markedly to give a dual-labeled population of approximately 5% (Figure 7B).
Figure 7.

Estimation of silicon microparticle exchange between HMVEC. HMVEC were treated with single-label microparticles for 24 h followed by cell mixing for an additional 24 h. A) The Imagestream X flow cytometer and Ideas software were used to image cells and estimate the pixel-to-pixel intensity of red and green signals, providing a measure of spectral overlap and microparticle exchange between populations. B) Comparison of microparticle exchange between HMVEC in the presence and absence of serum.
Measurements of the individual, single color loaded, cell populations indicate that the initial mixed cell population is 40% green particle labeled, 35% red particle labeled, with the remaining 25% of cells unlabeled. There are, therefore, three routes for particle transfer: color stable, in which green or red particles move to similarly labeled cells; color exchange, in which cells acquire particles of different color; and color acquisition, when particles transfer to unlabeled cells. Thus our measurements of dual-labeled cells quantify the color exchange mechanism. Using the percentages of colored and unlabeled populations in the initial mix, the relative weights of stable, exchange and acquisition routes is predicted to be 1:1:2/3, assuming equal probability of exchange for all cells. From these considerations particle exchange is therefore estimated to have taken place in ~13% of the total cell population when using serum-free media. This estimate is obtained from a stationary measure at a fixed time point; however, the time lapse microscopy shows the particle transfer process to be highly dynamic with multiple exchange steps, in and out of cells. It is likely therefore that many more cells are involved in particle transfer than the snapshot population of 13%.
3. Discussion
Recent publicity on the cell to cell transfer of the protein Tau, contributing to the progression of Alzheimer’s disease, exemplifies exchange of cellular constituents traditionally thought to exist only in the cell of origin.[28] Aggregated intracellular Tau, which is responsible for the damaging neurofibrillary fibrils in the neurons of Alzheimer’s patients, was found to spread from cell to cell, mimicking the spread of the disease. The mechanism of transfer between cells was reported to be unknown,[28,29] potentially occurring through an exocytic process involving synaptic connections. Simultaneous uptake of dextran in cells in vitro suggested an endocytic route for internalization.[28] Our findings reported herein support the utility of using particles to study transfer events between cells and to aid in understanding biological triggers that induce these events.
The formation of pseudopods connecting cells is prevalent in antigen presenting cells, astrocytes, and endothelial progenitor cells, with lifetimes that vary depending on the type of cell.[14] In this study, we show that microvascular and umbilical vein endothelial cells communicate by means of TNT. Evidence of TNT was presented for both proximal and distal endothelial cells, with both actin and thicker, actin/tubulin filamentous links existing between cells. With regard to the transport of particles via TNT, Mi et al. reported[23] that Qdots, present in lysosomes, are transported along TNT and suggested that actin-mediated transport of lysosomes was unidirectional, while myosin-mediated transport was bidirectional, the latter requiring microtubules and interplay between dynein and kinesin.[30] Onfelt et al.[16] also reported that vesicular transport within TNT requires microtubules. The presence of microtubules in endothelial TNT supports a potential role for the transport of endosomes and other cellular vesicles within these structures.
We have previously documented that HMVEC and HUVEC are highly phagocytic with respect to the engulfment of silicon microparticles.[24,31] In this study, we present ultrastructural images supporting intracellular microparticle transport within TNT. The presence of microparticles at cell-to-cell adhesion sites within TNT supports a role for these pseudopods in microparticle transfer between endothelial cells. We have previously shown that microparticles in endothelial cells exist in endosomal membranes,[32] suggesting that microparticle transfer through TNT may be accompanied by transfer of the endosomal membrane and additional endocytic cargo. Rustom and colleagues[15] reported that organelles belonging to the endosomal/lysosomal system traffic unidirectionally through TNT to acceptor cells. They reported that the transferred endocytic vesicles were able to fuse with their counterparts in the new cell. Since early endosomes are a major site for integration of ligand activated membrane receptors and signaling complexes, this transfer of information constitutes an opportunity for rapid dissemination of signals across a community of cells. For drug delivery particles, this suggests a mechanism for rapid propagation of therapeutics across a population of neighboring cells.
We have observed alignment of neighboring cells that appears to enable direct cell-to-cell transfer of microparticles. We observed that cellular association with silicon microparticles did not always lead to internalization of the microparticles; in some cases the microparticle ‘surfed’ or glided across the cell membrane. The surfing of bacteria along TNT has been described as a mechanism for the transport of bacteria between macrophages. Onfelt et al.[16] reported that clusters of bacteria surfed along the surface of thin TNT and were internalized upon arrival at the cell body.
We have previously demonstrated that iron oxide nanoparticles are secreted from endothelial cells and macrophages in membrane-bound biovesicles,[8,13] with the potential for reuptake by secondary endothelial and tumor cells. The secreted nanoparticles were recovered in cell culture media by high speed centrifugation and magnetic capture, and were found to contain cellular constituents typically found in the endosomal/lysosomal pathway. Release of QD-containing vesicles into the extracellular medium has been reported in HeLa cells, with release based on vesicle shedding from the distal ends of filopodia.[33] Mesoporous silica particles have also been shown to be secreted by cells and taken up by recipient cells, transferring materials associated with membrane and vesicular trafficking, specifically α-actinin-4, cytoplasmic actin-1, and annexin A2.[34]
The presence of nano- and micro-particles in free vesicles in 2D endothelial cultures may reflect in vivo processes, such as transport of material in plasmalemmal vesicles.[35] The transportation of microparticles across the cell, as demonstrated in ‘shuttle’ endothelial cells in Movie S1, may mimic transcytosis, in which cargo is internalized by membrane invagination and resides in sealed vesicles that are transported across the cell and subsequently secreted from another plasmalemmal domain. In vivo, shuttle cells may also function to carry signals to more distal endothelial cells, initiating events such as neoangiogenesis.
4. Conclusion
In summary, endothelial cells are highly dynamic cells charged with rapid response to biological and chemical signals and events to maintain, strengthen, or reduce the endothelial barrier. The dynamic transfer of microparticles between cells, and the potential to manipulate the rate at which it occurs, may be a therapeutic advantage in that it permits rapid dissemination of therapeutics to a specific population of cells. The finding that serum-starvation increases the rate of microparticle transfer may indicate that biologically stressed microenvironments, such as the tumor microenvironment, may have innate signals that trigger these events, stimulating localized microparticle sharing between cells. In addition, based on the potential for propagation of disease based on material transport between cells, using particles to study such events would improve our understanding of these events and support the creation of therapeutics that limit the spread of disease.
5. Experimental Section
Cell Culture:
Human microvascular endothelial cells (HMVEC), received as a gift from Dr. Rong Shao (Biomedical Research Institute, Baystate Medical Center/University of Massachusetts at Amherst, Springfield, MA, USA), were grown in EBM medium(Lonza, Walkersville, MD) and Human Umbilical Vein Endothelial Cells (HUVEC), purchased from Lonza, were cultured in EBM-2 medium.
Microparticle Fabrication and Chemical Modification:
Mesoporous hemispherical silicon microparticles were manufactured employing semiconductor fabrication techniques in the Microelectronics Research Center at the University of Texas at Austin as previously described.[24,36] The microparticles were oxidized by incubation in piranha solution (1 volume H2O2 and 2 volumes of H2SO4) for 2 h at 100 °C. After the particles were washed with DI water, they were transferred to IPA and counted by means of Coulter Counter Multisizer 4 (Beckman Coulter, Brea CA). To introduce amine groups on the surface, microparticles were silane-modified with 2% (v/v) 3-aminopropyltriethoxysilane (APTES; Sigma, St Louis, MO) in IPA for 2 h at 35 °C. The microparticles were then washed with IPA and further modified with either DyLight 488 or 594 (Pierce, Rockford, IL) based on the manufacturer’s protocol.
Flow Cytometry:
HMVEC were collected by trypsinization, washed, and incubated in 1% BSA in PBS to reduce non-specific binding. Cells were labeled with either a non-specifc isotype control antibody or phycoerythrin-conjugated anti-PECAM antibody (BD Pharmigen, San Jose, CA) for 1 h at 4 °C. Cells were washed in PBS, fixed with 4% paraformaldehyde and analyzed by Flow Cytometry using a FACS Fortessa and FACSDiva software (BD Pharmigen, San Jose, CA).
Endothial Tube Formation:
3 × 104 HMVEC were labeled with Cell Tracker Orange (Invitrogen) in a 10 μm solution for 15 min at 37 °C. The cells were then washed twice with PBS and plated in a glass bottom chamber slide (4-well) on 80 μL of Geltrex (GIBCO), in the presence or absence of 488-labelled microparticles (1:15 cell: particle ratio). The next day cells were imaged with an A1 Nikon confocal microscope equipped with a 20x objective.
Confocal Microscopy:
HMVEC (1 × 105) were seeded in an 8-well glass bottom confocal chamber slide (BD Falcon, San Jose, CA) and the next day labeled with FITC-conjugated anti-PECAM antibody (BD Pharmingen, San Jose, CA), 1:25 dilution in a solution of 1% BSA in PBS, for 45 min at room temperature. Cells were then fixed with 4% paraformaldehyde, washed, and mounted in Vectashield mounting medium with DAPI (Vector Laboratories, Burlingame, CA) and imaged using an A1 Nikon confocal microscope. For live cell imaging, HMVEC (25 × 103 per well) were seeded into a 24 well-glass bottom dish (Mat Tek corporation, Ashland MA). Oxidized microparticles were added to the cells (1:5; cell to particles ratio). The samples were imaged in 5 focal planes every 5 min for 19 h using a 1 × 81 Olympus Micriscope equipped with a humidified, 37 °C incubator with 5% CO2, using a 20× objective.
HUVEC, grown on glass coverslips, were incubated with microparticles (1:10; cell:microparticle) for 60 min in serum-free media (containing 0.2% BSA) at 37 °C in the presence or absence of 10 mg/mL cytochalasin B (the inhibitor was introduced 60 min prior to and during incubation with the microparticles). Cells were then washed with PBS, fixed and permeabilized with 0.1% triton X-100. Non-specific binding was blocked by incubation in 1% BSA in PBS. Cells were labeled in blocking buffer containing 200 nM Alexa Fluor 555-conjugated phalloidin (Invitrogen) and anti-tubulin FITC-conjugated antibody (Abcam Inc., Cambridge, MA). Coverslips were washed with PBS and mounted on glass slides using Vectashield mounting media (Vector Laboratories, Burlingame, CA) containing a 1000-fold dilution DRAQ5 (Biostatus Limited, UK). Detection of silicon microparticles was based on autofluorescence or DyLight 488 labeling in the absence of tubulin labeling. Images were acquired using a Leica DM6000 upright confocal microscope equipped with a 63x oil immersion objective.
Transmission Electron Microscopy:
HMVEC were plated at 1 × 106 cells/well in a 6-well plate and the next day cells were incubated for 4 hr with silicon microparticles. Cells were subsequently fixed with a solution of 2% paraformaldehyde and 3% glutaraldehyde (Electron Microscopy Science, Hatfield, PA) in PBS. After washing, samples were then incubated with 0.1% cacodylate buffered tannic acid and subsequently with 1% cacodylate-buffered osmium tetroxide for 30 min, followed by staining with uranyl acetate.
The samples were then dehydrated in ethanol solutions, embedded in Epon resin and cured in an oven at 60 °C for 2 days to allow resin polymerization. Ultrathin sections were obtained with a Leica Ultracut microtome (Leica, Deerfield, IL) and further stained with uranyl acetate and lead citrate in a Leica EM Stainer. The samples were imaged with a JEM 1010 transmission electron microscope (JEOL, USA, Inc., Peabody, MA) with a voltage of 80 kV.
Scanning Electron Microscopy:
HUVEC (5 × 104) were seeded on silicon chip supports (Ted Pella, inc, Redding, CA) in a 24-well plate. Upon confluence, cell were incubated for 15 min with oxidized microparticles (ratio 10:1 particles per cell) at 37 °C in serum-free medium. Cells were then washed with PBS, fixed with 2.5% glutaraldehyde (Sigma-Aldrich, Saint Louis, MO) and washed in PBS. Samples were then dehydrated incubating in progressively more concentrated solutions of ethanol (30%, 50%, 70%, 90%, 95% and 100%) for 10 min each. As last step, cells were incubated 10 minutes with a solution of 50% (v/v) ethanol-hexamethyldisilazane (Sigma, Saint Luis, MO) followed by 5 min in 100% hexamethyldisilazane. Samples were dried overnight in a dessicator. Silicon chips were mounted on SEM stubs (Ted Pella, Inc. Redding, CA) with the help of conductive adhesive tape (12 mm OD Pelco, Ted Pella, Inc. Redding, CA), sputtered with a 10 nm film of gold by means of a Plasma Science CrC-150 Sputtering system (Torr International, Inc.) and imaged with an FEI Quanta 400 FEG-SEM (high vacuum, 20 kV, spot size 5).
Imagestream X Flow Cytometry:
1 × 105 HMVECs per well were seeded into a 6-well plate overnight, and then incubated for 24 h separately with either Dylight 594 or 488-labeled microparticles at a cell to particle ratio of 1:10. The two cell populations were then washed with PBS, trypsynized, and co-cultured for another 24 h in 2% FBS or serum-free media. Cell images were acquired with an Imagestream X flow cytometer (Amnis Corporation, Seattle, WA); bright field and fluorescence images were collected for each cell within sample sets of at least 10 000 cells. Untreated cells as well as the two single populations were imaged as controls.
All image analysis was done within the Ideas software environment (Amnis Corporation, Seattle, WA). Spectral compensation for cross-talk between fluorescence channels and correction for background auto fluorescence was implemented prior to image analysis. A two stage process was used to identify dual-labeled cells: 1) combinatorial gating based on fluorescence intensity was used to define a sub-population, positive for both red and green signals; 2) further discrimination was based on the use of an internalization parameter, defining the fraction of green fluorescence within a mask area corresponding to the red signal or a similarity coefficient, measuring the pixel by pixel correlation of green and red signal intensity. These internalization and similarity features were used to further refine the dual-labeled population by confirming that the red and green fluorescence were spatially distinct and so unambiguously attributable to differently labeled microparticles. The dual-labeled cell percentage was calculated as the average of the two values realized by the use of the internalization or similarity parameter while the difference between the values was taken as the measurement uncertainty.
Supplementary Material
Acknowledgements
The authors wish to thank Kenneth Dunner Jr. at the High Resolution Electron Microscopy Facility and Jared Burks at the Flow Cytometry and Cell Imaging Core Facility at the University of Texas MD Anderson Cancer Center for transmission electron and real-time confocal imaging. We also thank Matthew G. Landry, graphic designer in the Department of Nanomedicine at The Methodist Hospital Research Institute (TMHRI), for assistance with image preparation. The research was supported by funding provided by the DOD W81XWH-07-1-0596 (R.E.S.); TMHRI (R.E.S.), including the Ernest Cockrell Jr. Distinguished Endowed Chair (M.F.); the John S Dunn Distinguished Endowed Chair and 71 and WF Chao Foundation (S.T.C.W); NIH NIGMS RC2GM092599 (M.F., R.E.S.) and MH-58920 (A.J.B.); and the CPRIT Innovation for Cancer Prevention Research Training Program Pre-doctoral Fellowship (S.F.).
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
Silvia Ferrati, Department of Nanomedicine, The Methodist Hospital Research Institute, Houston, TX 77030, USA.
Sabeel Shamsudeen, Centre for Nanohealth, College of Engineering, Swansea University, Swansea, UK.
Huw D. Summers, Centre for Nanohealth, College of Engineering, Swansea University, Swansea, UK
Paul Rees, Centre for Nanohealth, College of Engineering, Swansea University, Swansea, UK.
James V. A. Abbey, Centre for Nanohealth, College of Engineering, Swansea University, Swansea, UK
Jeff Schmulen, Department of Nanomedicine, The Methodist Hospital Research Institute, Houston, TX 77030, USA.
Xuewu Liu, Department of Nanomedicine, The Methodist Hospital Research Institute, Houston, TX 77030, USA.
Stephen T. C. Wong, Department of Systems Medicine and Bioengineering, The Methodist Hospital Research Institute, Houston, TX 77030, USA
Andrew J. Bean, Department of Neurobiology and Anatomy, University of Texas Medical School, Houston, TX, USA Department of Pediatrics, M.D. Anderson Cancer Center, Houston, TX 77030, USA.
Mauro Ferrari, Department of Nanomedicine, The Methodist Hospital Research Institute, Houston, TX 77030, USA.
Rita E. Serda, Department of Nanomedicine, The Methodist Hospital Research Institute, Houston, TX 77030, USA.
References
- [1].Dainiak N, Blood 1991, 78, 264–276. [PubMed] [Google Scholar]
- [2].Denef C Clinics in endocrinology and metabolism 1986, 15, 1–32. [DOI] [PubMed] [Google Scholar]
- [3].Oka T, Yoshimura M, Clinics in Endocrinology and Metabolism 1986, 15, 79–97. [DOI] [PubMed] [Google Scholar]
- [4].Wiener H, New York State J. Medicine 1968, 68,1019–1038. [PubMed] [Google Scholar]
- [5].Scharrer B, J. Neurovisceral Relations 1969, 31, 1–20. [Google Scholar]
- [6].Badescu R, Balaceanu C, Nicolau E, Int. J. Biomedical Computing 1970, 1, 211–220. [DOI] [PubMed] [Google Scholar]
- [7].Couzin J, Science 2005, 308, 1862–1863. [DOI] [PubMed] [Google Scholar]
- [8].Serda RE, Mack A, van de Ven AL, Ferrati S, Dunner K Jr., Godin B, Chiappini C, Landry M, Brousseau L, Liu X, Bean AJ, Ferrari M, Small 2010, 6, 2691–2700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Iero M, Valenti R, Huber V, Filipazzi P, Parmiani G, Fais S, Rivoltini L, Cell Death Differ 2008, 15,80–88. [DOI] [PubMed] [Google Scholar]
- [10].Tan A, De La Pena H, Seifalian AM, Int. J. Nanomedicine 2010, 5, 889–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Valadi H, Ekstrom K, Bossios A, Sjostrand M, Lee JJ, Lotvall JO, Nat. Cell Biol. 2007, 9, 654–659. [DOI] [PubMed] [Google Scholar]
- [12].Gurke S, Barroso JF, Hodneland E, Bukoreshtliev NV, Schlicker O, Gerdes HH, Exp. Cell Res. 2008, 314, 3669–3683. [DOI] [PubMed] [Google Scholar]
- [13].Ferrati S, Mack A, Chiappini C, Liu X, Bean AJ, Ferrari M, Serda RE, Nanoscale 2010, 2, 1512–1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Gurke S, Barroso JF, Gerdes HH, Histochem. Cell Biol. 2008, 129, 539–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Rustom A, Saffrich R, Markovic I, Walther P, Gerdes HH, Science 2004, 303, 1007–1010. [DOI] [PubMed] [Google Scholar]
- [16].Onfelt B, Nedvetzki S, Benninger RK, Purbhoo MA, Sowinski S, Hume AN, Seabra MC, Neil MA, French PM, Davis DM, J. Immunol. 2006, 177, 8476–8483. [DOI] [PubMed] [Google Scholar]
- [17].Tavi P, Korhonen T, Hanninen SL, Bruton JD, Loof S, Simon A, Westerblad H, J. Cell Physiol. 2010, 223,376–383. [DOI] [PubMed] [Google Scholar]
- [18].Gerdes HH, Bukoreshtliev NV, Barroso JF, FEBS Lett. 2007, 581, 2194–2201. [DOI] [PubMed] [Google Scholar]
- [19].Sowinski S, Jolly C, Berninghausen O, Purbhoo MA, Chauveau A, Kohler K, Oddos S, Eissmann P, Brodsky FM, Hopkins C, Onfelt B, Sattentau Q, Davis DM, Nat. Cell Biol. 2008, 10, 211–219. [DOI] [PubMed] [Google Scholar]
- [20].Watkins SC, Salter RD, Immunity 2005, 23, 309–318. [DOI] [PubMed] [Google Scholar]
- [21].Rechavi O, Goldstein I, Kloog Y, FEBS Lett. 2009, 583, 1792–1799. [DOI] [PubMed] [Google Scholar]
- [22].Koyanagi M, Brandes RP, Haendeler J, Zeiher AM, Dimmeler S, Circ. Res. 2005, 96, 1039–1041. [DOI] [PubMed] [Google Scholar]
- [23].Mi L, Xiong R, Zhang Y, Yang W, Chen J-Y, Wang P-N, J. Biomat. Nanobio. 2011, 2, 172–179. [Google Scholar]
- [24].Serda RE, Gu J, Bhavane RC, Liu X, Chiappini C, Decuzzi P, Ferrari M, Biomaterials 2009, 30, 2440–2448. [DOI] [PubMed] [Google Scholar]
- [25].Serda RE, Blanco E, Mack A, Stafford SJ, Amra S, Li Q, van de Ven A, Tanaka T, Torchilin VP, Wiktorowicz JE, Ferrari M, Mol. Imaging 2011, 10, 43–55. [PMC free article] [PubMed] [Google Scholar]
- [26].Shao R, Guo X, Biochem. Biophys. Res. Commun. 2004, 321, 788–794. [DOI] [PubMed] [Google Scholar]
- [27].Newman PJ, Albelda SM, Nouvelle Revue Francaise d’Hematologie 1992, 34, S9–13. [PubMed] [Google Scholar]
- [28].Frost B, Jacks RL, Diamond MI, J. Biol. Chem. 2009, 284, 12845–12852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Liu L, Drouet V, Wu JW, Witter MP, Small SA, Clelland C, Duff K, PLoS One 2012, 7, e31302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Gross SP, Phys. Biol. 2004, 1, R1–11. [DOI] [PubMed] [Google Scholar]
- [31].Serda RE, Gu J, Burks JK, Ferrari K, Ferrari C, Ferrari M, Cytometry A 2009, 75, 752–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Serda RE, Ferrati S, Godin B, Tasciotti E, Liu X, Ferrari M, Nanoscale 2009, 1, 250–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Ruan G, Agrawal A, Marcus AI, Nie S, J. Am. Chem. Soc. 2007, 129, 14759–14766. [DOI] [PubMed] [Google Scholar]
- [34].Slowing II, Vivero-Escoto JL, Zhao Y, Kandel K, Peeraphatdit C, Trewyn BG, Lin VS-Y, Small, 7, 1526. [DOI] [PubMed] [Google Scholar]
- [35].Predescu SA, Predescu DN, Malik AB, Am. J. Physiol. Lung Cell Mol. Physiol. 2007, 293, L823–842. [DOI] [PubMed] [Google Scholar]
- [36].Chiappini C, Tasciotti E, Fakhoury JR, Fine D, Pullan L, Wang YC, Fu L, Liu X, Ferrari M, Chem Phys Chem 2010, 11, 1029–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.