

Abstract
Our recent findings demonstrated that the Epstein-Barr virus-encoding small nonpolyadenylated RNA (EBER) confers resistance to various apoptotic stimuli and contributes to the maintenance of malignant phenotypes in Burkitt's lymphoma. In this study we investigated the role of EBER in the human epithelial Intestine 407 cell line, which is known to be susceptible to Fas (Apo1/CD95)-mediated apoptosis. Fas, a member of the tumor necrosis factor receptor family, transduces extracellular signals to the apoptotic cellular machinery, leading to cell death. Transfection of the EBER gene into Intestine 407 cells significantly protected the cells from Fas-mediated apoptosis, whereas EBER-negative cell lines underwent apoptosis after Fas treatment. EBER bound double-stranded RNA-dependent protein kinase R (PKR), an interferon-inducible serine/threonine kinase, and abrogated its kinase activity. Moreover, expression of the catalytically inactive dominant-negative PKR provided resistance to Fas-induced apoptosis. Expression of EBER or dominant-negative PKR also inhibited the cleavage of poly(ADP-ribose) polymerase, a mediator of the cellular response to DNA damage, downstream of the Fas-mediated apoptotic pathway. These results in combination indicate that EBER confers resistance to Fas-mediated apoptosis by blocking PKR activity in Intestine 407 cells, consistent with the idea that EBER contributes to the maintenance of epithelioid malignancies.
Fas (Apo-1/CD95), the receptor for Fas ligand (FasL), is a member of tumor necrosis factor receptor family and is expressed in many cell types. Fas mediates apoptosis under at least three kinds of physiological responses (1, 25): (i) peripheral deletion of activated mature T cells at the ends of an immune response; (ii) killing of targets such as cancer cells or virus-infected cells by cytotoxic T lymphocytes and NK cells; and (iii) killing of inflammatory cells at “immune-privileged” sites such as the eye. The ligation of FasL to Fas triggers the recruitment and activation of an adapter protein, FADD (Fas-associated death domain-containing protein) (4) and subsequent activation of caspase-8/FLICE (24), a family of cysteine proteases. Activated caspase-8 targets Bid, a proapoptotic member of the Bcl-2 family that can trigger a loss in mitochondrial transmembrane potential and cause an efflux of cytochrome c into the cytoplasm (23). Cytochrome c on translocating to the cytoplasm complexes with Apaf-1 to initiate formation of the apoptosome and activates procaspase-9. The initiation of both caspase-8 and -9, in turn, targets and activates terminator caspases such as caspase-3, -6, and -7 (22, 40, 45). These caspases target various proteins whose activities are regulated by proteolytic cleavage. For example, caspase-3 is responsible for cleavage of ICAD (30), resulting in oligonucleosome DNA fragmentation (7). α-Fodrin, which is important for the maintenance of membrane structure and cell surface protein function, is also an important target cleaved by caspases during apoptosis (43). Cleavage of α-fodrin leads to membrane malfunctions and cell shrinkage. Poly(ADP-ribose) polymerase (PARP), which is known as a mediator of the cellular response to DNA damage, (6) is a target of caspase-3 and -7. During apoptosis, PARP is cleaved from its intact form (110 kDa) into 85-kDa and 25-kDa fragments (37). This process separates the amino-terminal DNA-binding domain of the enzyme from the carboxyl-terminal catalytic domain, resulting in the loss of function of PARP. Other members of the bcl-2 family, including Bcl-2 (antiapoptotic) and Bax (proapoptotic) can also influence cell death, possibly by acting as outer mitochondrial membrane channel proteins that regulate cytochrome release into the cytosol (19). Numerous viruses have evolved a variety of strategies in order to antagonize Fas-induced apoptosis via their viral components. For example, adenovirus E1B protein, a Bcl-2 homologue, protects cells from Fas-mediated apoptosis (5), and viral FLIPs encoded by several gammaherpesviruses as well as the tumorigenic molluscipoxvirus interfere with apoptosis triggered by death receptors by sequestering FADD (39, 42). Other examples of viral inhibitors include the p35 protein of baculovirus (3) and CrmA of cowpox virus (38), which bind and inhibit multiple members of the caspase family.
Epstein-Barr virus (EBV), a member of the family of the human gammaherpesvirus, infects the majority of the human population and establishes a life-long latent infection (16, 28). EBV is associated with various human malignancies such as Burkitt's lymphoma (BL), AIDS-associated lymphoma, nasopharyngeal carcinoma and gastric carcinoma (GC). Two independent studies have shown that the EBV early lytic protein BHRF1, which has sequence homology to Bcl-2, confers resistance to Fas-mediated apoptosis, suggesting that BHRF1 plays a role in maximizing viral production during EBV's lytic cycle (8, 15).
Our previous findings demonstrated that the EBV-encoded small nonpolyadenylated RNAs (EBER1 and EBER2) contribute to the maintenance of malignant phenotypes in BL cells. EBER expression enables clonal growth in soft agarose, tumorigenicity in immunodeficient mice, resistance to a variety of apoptotic stimuli (18), and induction of human interleukin-10, an autocrine growth factor for BL cells (17). More recently, we found that EBER contributes to the protection from alpha-interferon (IFN-α)-induced apoptosis by inhibition of the PKR pathway in EBV-positive BL cells (26). However, any effect of EBER on Fas-induced apoptosis remains unclear.
Here we have investigated the role of EBER in human epithelial Intestine 407 cells, which are susceptible to Fas-induced apoptosis. We have found that (i) transfection of the EBER gene into Intestine 407 cells protected the cells from Fas-mediated apoptosis; (ii) EBER bound PKR and inhibited its kinase activity; (iii) expression of catalytically inactive dominant-negative PKR inhibited Fas-induced apoptosis; and (iv) expression of EBER or catalytically inactive dominant-negative PKR inhibited the cleavage of PARP. All these results indicate that EBER can play a role in the resistance to Fas-mediated apoptosis by inhibiting PKR activity and contributes to the maintenance of EBV-positive epithelioid malignancies.
MATERIALS AND METHODS
Cell line and culture.
The Intestine 407 epithelial cell line (kindly provided by Tsutomu Chiba, Kyoto University), derived from human ileum (13), was maintained in Dulbecco's modified Eagle's medium (Sigma) supplemented with 10% newborn calf serum (Invitrogen), penicillin (40 U/ml), and streptomycin (50 μg/ml) at 37°C in a 5% CO2 incubator. The EBV-positive BL Akata cell line (32, 33) was maintained in RPMI 1640 medium (Sigma) supplemented with 10% fetal bovine serum (Sigma), penicillin (40 U/ml), and streptomycin (50 μg/ml) at 37°C in a 5% CO2 incubator.
Plasmid, transfection, and cell cloning.
The EBER expression plasmid contains 10 tandem repeats of the EBER1 and EBER2 subfragment (bp 6288 to 7325) from the EcoRI K fragment of Akata EBV DNA and the neomycin resistance (Neor) gene driven by the simian virus 40 (SV40) promoter (18). Plasmids encoding the SV40-driven influenza virus-encoded hemagglutinin (HA)-tagged, catalytically inactive protein kinase R (PKR) K269A mutant (mPKR) and the SV40 promoter-driven FLAG-tagged PKR mutant that lacks a double-stranded RNA (dsRNA)-binding domain were described previously (26). Each of the plasmids was transfected into cells by Lipofectamine (Invitrogen) according to the manufacturer's instructions. For the cloning of stable transfectants, transfected cells were cultured for 2 days and transferred to 96-well, flat-bottom plates at 5,000 to 10,000 cells per well in complete culture medium containing 700 μg/ml G418 (Gibco-BRL) for selection.
Northern blot analysis.
Northern blot analysis was performed as described with some modifications (44). Briefly, total RNA was isolated using TRI reagent (Sigma). The RNAs (5 μg each) prepared from each cell clone were electrophoresed in 1.5% agarose gel containing 0.66 M formaldehyde and were transferred to a Hybond N+ nylon membrane (Amersham Pharmacia Biotec). Probes were prepared by PCR amplification using the primers 5′-AGGACCTACGCTGCCCTAGA-3′ (upstream) and 5′-AAAACATGCGGACCACCAGC-3′ (downstream) for EBER1, 5′-AGGACAG CCGTTGCCCTAGTGGTTTCG-3′ (upstream) and 5′-AAAAACAGCGGACAAGCCGA-3′ (downstream) for EBER2, and 5′-GCCTCCTGCACCACCAACTG-3′ (upstream) and 5′-CGACGCCTGCTTCACCACCTTCT-3′ (downstream) for GAPDH (glyceraldehyde-3-phosphate dehydrogenase). PCR was carried out for 30 cycles consisting of a denaturing step for 30 s at 94°C, annealing for 30 s at 53°C, and extension for 1 min at 72°C. Probe labeling was carried out using an AlkPhos direct labeling kit (Amersham Pharmacia Biotech), and signals were detected with a CDP-Star detection reagent (Amersham Pharmacia Biotech).
Apoptosis and caspase assay.
For induction of Fas-mediated apoptosis, cells were spread in six-well tissue culture plates at a density of 2.5 × 105 per well in Dulbecco's modified Eagle's medium containing 10% newborn calf serum. After 24 h, cells (5 × 105) were incubated in complete medium containing 15 ng/ml anti-Fas CH-11 monoclonal antibody (MBL) in the presence or absence of 2 μg/ml cycloheximide (CHX) for various times. To measure the induction of apoptosis, cells were harvested with phosphate-buffered saline (PBS) containing 50 mM EDTA and then subjected to a sub-G1 apoptosis assay using a FACScalibur (Becton Dickinson) as described previously (26, 27). Caspase-8 and -9 activities were measured using a fluorometric protease assay kit (MBL) according to the manufacturer's instructions. Briefly, cells treated with CH-11 antibody and CHX in six-well plates for various times were collected and lysed. Supernatants were incubated with Ile-Glu-Thr-Asp-AFC (where AFC is 7-amino-4-trifluoromethyl coumarin) and Leu-Glu-His-Asp-AFC as a substrate for caspase-8 and -9, respectively, for 1 h at 37°C. The amount of free AFC cleaved from substrate was determined using fluorescence detection (400 nm excitation and 505 nm emission).
UV cross-linking and immunoprecipitation.
The binding of EBER to PKR was examined by UV cross-linking and immunoprecipitation of PKR-RNA complexes. Cells (2 × 106) were transfected with FLAG-tagged PKR plasmids by Lipofectamine (Invitrogen) in 6-cm tissue culture dishes. At 48 h posttransfection, cells were washed with PBS and suspended in 2 ml of prechilled PBS. UV irradiation was performed for 5 min with an 8-W germicidal lamp at a distance of 4 cm (GC Gene Linker; Bio-Rad). N-laurylsarcosine was then added to the cell suspension at a final concentration of 0.5% (wt/vol). Cells were washed with PBS, harvested with PBS containing 0.5 mM EDTA, suspended in 200 μl of immunoprecipitation buffer (20 mM Tris-HCl, pH 7.4, 0.15 M NaCl, 5 mM EDTA, 1 μg/ml leupeptin, and 1 μg/ml pepstatin, and 1 mM phenylmethylsulfonyl fluoride [PMSF], 100-fold diluted phosphatase inhibitor cocktail I [Sigma]), and lysed by sonication for 15 s. After centrifugation at 15,000 rpm for 10 min at 4°C, the supernatant was preincubated with 20 μl of protein G-Sepharose (Amersham Pharmacia) in 1 ml of the same buffer for 1 h at 4°C. After centrifugation at 10,000 rpm for 1 s at 4°C, the supernatant was incubated with the anti-FLAG M2 antibody (Sigma) for 2 h at 4°C, followed by incubation with 20 μl of protein G-Sepharose for 1 h at 4°C. The bound complexes were precipitated at 10,000 rpm for 1 s at 4°C, and the immunoprecipitants were treated with proteinase K (final concentration, 10 μg/ml) and subjected to phenol extraction and ethanol precipitation for RNA isolation. Three-tenths of the RNA was used for cDNA synthesis. Thereafter, half of the synthesized cDNA was used for PCR for EBER1, and the remaining half of the cDNA was used for PCR for EBER2. One-fifth of the PCR product was subjected to agarose gel electrophoresis.
In vitro kinase assay.
The kinase activity of FLAG-PKR was examined by in vitro assay. The cells (5 × 106) were transfected with a FLAG-PKR expression plasmid by Lipofectamine in a 10-cm tissue culture dish. At 48 h posttransfection, the cells were washed with PBS, harvested with PBS containing 0.5 mM EDTA, and lysed in buffer I (20 mM Tris-HCl, pH 7.5, 50 mM KCl, 400 mM NaCl, 1 mM EDTA, 1 μg/ml leupeptin, 1 μg/ml pepstatin, 1 mM 2-mercaptoethanol, 1 mM PMSF, 100-fold diluted phosphatase inhibitor cocktail I, 0.1% Triton X-100 and 20% glycerol). Cell extracts corresponding to 3 × 106 cells were preincubated with 20 μl of protein G-Sepharose in 1 ml of buffer I for 1 h at 4°C. After centrifugation at 10,000 rpm for 1 s at 4°C, the supernatant was incubated with the anti-FLAG M2 antibody for 2 h at 4°C, followed by incubation with 20 μl of protein G-Sepharose for 1 h at 4°C. Immunoprecipitants were washed in buffer I three times, twice in buffer II (20 mM Tris-HCl, pH 7.5, 100 mM KCl, 0.1 mM EDTA, 1 μg/ml leupeptin, 1 μg/ml pepstatin, 1 mM PMSF, 400-fold diluted phosphates inhibitor cocktail I, and 20% glycerol), and then once in kinase reaction buffer (20 mM Tris-HCl, pH 7.5, 50 mM KCl, 0.01 mM EDTA, 2 mM MgCl2, 2 mM MnCl2, 0.1 mM PMSF, and 20% glycerol). Following the last wash, the immunoprecipitants were resuspended in 20 μl of kinase reaction buffer containing 2 μM [γ-32P]ATP (3,000 Ci/mmol) (NEN) and incubated for 15 min at 30°C. The reaction was stopped by the addition of 10 μl of fourfold-concentrated sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) sample buffer (500 mM Tris-HCl, pH 6.8, 10% SDS, 4% 2-mercaptoethanol, 0.4% bromophenol blue, and 40% glycerol). The samples were analyzed by 10% SDS-PAGE, and phosphorylation was visualized by autoradiography. The FLAG-PKR expression was determined by immunoblot analysis with anti-FLAG M2 antibody.
Immunoblot analysis.
Cells (1 × 105) were lysed in SDS-PAGE sample buffer, sonicated, and boiled for 5 min. The cell lysate was separated in 10% polyacrylamide gels and transferred to a nitrocellulose membrane (Schleicher and Schuell). After being blocked with Tris-buffered saline containing 2% nonfat dry milk (TBS-M; pH 7.6), the membrane was incubated for 2 h at room temperature with appropriate antibodies diluted in TBS-M. The antibodies used are monoclonal antibodies for FLAG-tagged protein (Sigma), β-actin (Sigma), PARP (BD Pharmingen), and ICAD (MBL) and polyclonal antibodies for PKR (Santa Cruz), HA-tagged protein (Santa Cruz), Bid (BD Pharmingen), and α-fodrin (BD Pharmingen). The membrane was washed three times in TBS-M containing 0.1% Tween 20 (TBS-T) and incubated with the secondary antibody, horseradish peroxidase-conjugated sheep antibody to immunoglobulin (Amersham Pharmacia) diluted 1:5,000 to 1:10,000 in TBS-M. After the second antibody reaction, the membranes were washed three times in TBS containing 0.1% Tween 20 and immersed in enhanced chemiluminescence solution (Amersham Pharmacia) as specified by the manufacturer, and subjected to autoradiography.
RESULTS
EBER confers resistance to Fas-mediated apoptosis in Intestine 407 cells.
To examine the effect of EBER on the Intestine 407 epithelial cell line, we transfected an EBER-expressing plasmid that contains 10 tandem repeats of the EBER1 and EBER2 subfragments of the Akata EBV genome into Intestine 407 cells and isolated clones expressing them stably. These clones showed similar levels of expression of EBER to that of EBV-infected Akata BL cell clones. The expression levels of EBER1 and EBER2 in the individual clones were determined by Northern blot analysis (Fig. 1). We then assessed the effect of EBER on Fas-induced apoptosis by comparison of three individual EBER-expressing clones with three cell clones expressing the neomycin resistance gene (Neor) as controls. The EBER- and Neor-expressing cell clones were incubated with and without 15 ng/ml anti-Fas CH-11 antibody in the presence of 2 μg/ml cycloheximide, which accelerates Fas-induced apoptosis, and flow cytometry was used to assess apoptosis by measuring the sub-G1 content of DNA in the cells. The Neor-expressing cell clones underwent apoptosis after treatment with anti-Fas antibody, while the EBER-expressing clones were relatively resistant to Fas-mediated apoptosis (Fig. 2).
FIG. 1.

EBER expression in Intestine 407 cell clones. EBER1, EBER2, and GAPDH expression were examined by Northern blot analysis in EBER- and Neor-expressing Intestine 407 cell clones. EBV-infected Akata BL cells were used as a positive control.
FIG. 2.

The effect of EBER on Fas-mediated apoptosis in Intestine 407 cells. Neor- and EBER-expressing Intestine 407 cell clones (2 × 105) were incubated in the medium with and without 15 ng/ml anti-Fas CH-11 antibody in the presence of 2 μg/ml CHX in six-well plates for 16 h, and the frequency of apoptotic cells was determined by a sub-G1 apoptosis assay using flow cytometry. Results are expressed as the means ± standard deviations of triplicate wells.
The effect of EBER on PKR autophosphorylation in Intestine 407 cells.
Our recent findings proved that EBER confers resistance to IFN-α-induced apoptosis by blocking PKR activity in several BL lines. We therefore examined whether EBER provides resistance to Fas-induced apoptosis by inhibiting PKR activity in Intestine 407 cells. First we examined the effect of EBER on PKR phosphorylation by an in vitro kinase assay. EBER- and Neor-expressing cell clones were transfected with a vector expressing FLAG-PKR and cultured for 48 h. FLAG-PKR was then immunoprecipitated with anti-FLAG M2 antibody followed by an in vitro assay in which poly(I) · poly(C) is added to activate the kinase. PKR was autophosphorylated in EBER-negative cell clones, while its autophosphorylation was significantly inhibited in EBER-expressing clones (Fig. 3).
FIG. 3.
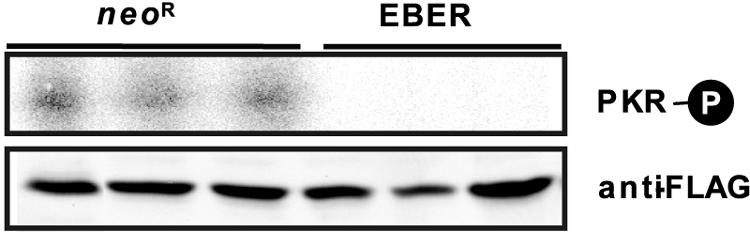
The effect of EBER on the phosphorylation of PKR in Intestine 407 cells. Neor- and EBER-expressing Intestine 407 cell clones (5 × 106) were transfected with a FLAG-tagged PKR-expressing plasmid by Lipofectamine. At 48 h posttransfection, FLAG-PKR was immunoprecipitated with anti-FLAG M2 antibody and subjected to an in vitro kinase assay in the presence of poly(I) · poly(C) (10 ng/ml). Phosphorylation of PKR was visualized by autoradiography.
The binding of EBER to PKR in Intestine 407 cells.
To clarify whether EBER binds to PKR in Intestine 407 cells directly, we used a UV cross-linking assay. EBER- and Neor-expressing cell clones were transfected with a FLAG-PKR expression plasmid. After UV irradiation, the complexes of FLAG-PKR and RNA were immunoprecipitated with anti-FLAG M2 antibody, RNA was extracted from the immunoprecipitants, and precipitated EBER was assayed by reverse transcription-PCR. Both EBER1 and EBER2 were found to be coprecipitated with FLAG-PKR in EBER-expressing cell clones but not in the control clones (Fig. 4). Neither EBER1 nor EBER2 was precipitated with the FLAG-tagged PKR mutant that lacks a dsRNA binding domain, indicating that the binding of EBER to PKR is specific (Fig. 4, EBER*). A parallel experiment was carried out using EBER-expressing cells transfected with a FLAG-PKR expression plasmid without UV cross-linking treatment (Fig. 4, −UV). The result of this parallel experiment indicated that EBER1 and EBER2 were also precipitated with anti-FLAG antibody, suggesting that the binding affinity of EBER to PKR is sufficient to detect binding even without UV cross-linking (21, 29, 41).
FIG. 4.

UV cross-linking assay for binding EBER to PKR in Intestine 407. Neor- and EBER-expressing Intestine 407 cell clones (5 × 106) were transfected with a FLAG-tagged PKR-expressing plasmid by Lipofectamine. As a control, an EBER-expressing cell clone was transfected with a FLAG-tagged mutant PKR plasmid, which lacks the dsRNA-binding domain (indicated as EBER*). At 48 h posttransfection, cells were treated with or without UV irradiation, and the bound complexes were immunoprecipitated with anti-FLAG antibody. The RNA fraction was extracted from the immunoprecipitants, and then EBER1 and EBER2 RNAs were amplified by reverse transcription-PCR.
EBER confers resistance to Fas-mediated apoptosis by blocking PKR activity.
To investigate further whether EBER provides resistance to Fas-induced apoptosis via blocking PKR's activity, Intestine 407 cells were transfected with a vector expressing HA-tagged, catalytically inactive PKR K269A mutant (mPKR) (26), and clones expressing it stably were isolated (Fig. 5A). These clones were treated with anti-Fas CH-11 antibody and CHX and then analyzed for apoptosis. The HA-mPKR-expressing clones showed significant resistance to Fas-induced apoptosis (Fig. 5B), indicating that EBER contributes to the protection of Intestine 407 cells from the Fas-mediated apoptotic pathway by blocking PKR activity.
FIG. 5.

The effect of dominant-negative PKR (mPKR) on phosphorylation of PKR and Fas-mediated apoptosis in Intestine 407 cells. A HA-tagged mPKR expression plasmid was transfected into Intestine 407 cells, and cell clones that stably expressed mPKR were selected with G418. (A) The effect of dominant-negative PKR (mPKR) on the phosphorylation of PKR. Expression levels of HA-tagged mPKR were determined by immunoblot analysis using anti-HA polyclonal antibody (top). Neor- and HA-tagged mPKR-expressing Intestine 407 cell clones (5 × 106) were transfected with a FLAG-tagged PKR-expressing plasmid by Lipofectamine. At 48 h posttransfection, FLAG-PKR was immunoprecipitated with anti-FLAG M2 antibody and subjected to an in vitro kinase assay in the presence of poly(I) · poly(C) (10 ng/ml). Phosphorylation of PKR was visualized by autoradiography. (B) The effect of mPKR on Fas-mediated apoptosis. Neor-, EBER- and HA-mPKR-expressing Intestine 407 cell clones (5 × 105) were incubated in the medium containing 15 ng/ml anti-Fas antibody and 2 μg/ml CHX for indicated times up to 36 h, and the frequency of apoptotic cells was determined by a sub-G1 apoptosis assay using flow cytometry. Results are expressed as the means ± standard deviations of triplicate wells.
Characterizing EBER's inhibition of Fas-mediated apoptosis.
In order to characterize the mechanism by which EBER down-regulates Fas-mediated apoptosis, several participants in Fas-mediated death signaling were analyzed in EBER-, mPKR- and Neor-expressing clones after treatment with CH-11 antibody and CHX. Caspase-8 and -9 activities were quantified by cleavage of their respective substrates, IETD-AFC and LEHD-AFC. A substrate of caspase-8, Bid, and substrates of caspase-3, PARP, ICAD, and α-fodrin were examined by immunoblot analysis to detect cleaved products. Fas treatment initiated cleavage of PARP in Neor-expressing clones but was inhibited in cells expressing EBER and mPKR (Fig. 6B). However, neither EBER nor mPKR expression inhibited the cleavages of the other tested caspase-3 targets, ICAD and α-fodrin, or of Bid (Fig. 6A). Also, no other significant differences were observed in other proapoptotic components such as caspase-8, caspase-9 (Fig. 6B), and Bid (Fig. 6A) in EBER-, mPKR- and Neor-expressing cells.
FIG. 6.

Characterizing EBER's inhibition of Fas-mediated apoptosis. Neor-, EBER- and mPKR-expressing cells (5 × 105) were incubated in the presence of 15 ng/ml anti-Fas antibody and 2 μg/ml CHX for the indicated times. (A) The effect of EBER and mPKR expression on cleavage of Bid, PARP, ICAD, and α-Fodrin. After Fas-treatment, cells were harvested and the cleavages of Bid, PARP, ICAD, and α-fodrin were analyzed by immunoblot analysis. (B) The effect of EBER and mPKR expression on the activities of caspase-8 and -9. At the indicated times, cells were collected and caspase activities were quantified by cleavage of IETD-AFC and LEHD-AFC as a substrate for caspase-8 and -9, respectively. The amount of free AFC cleaved from substrate was determined using fluorescence detection (400 nm excitation and 505 nm emission). The results are expressed as the means ± standard deviations of triplicate wells.
DISCUSSION
PKR is a key mediator of apoptotic signaling induced both by IFN-α and dsRNA (26, 35). It is well understood that PKR also simultaneously activates several different apoptotic pathways. Apoptosis induction by PKR can involve inhibiting translational initiation induced through phosphorylation of eIF2α (12, 31). PKR also can induce apoptosis by activating TRAF family proteins (9) and their NF-κB targets (12). The activation of NF-κB could contribute to the transcriptional upregulation of several genes involved in apoptosis, such as FasL (34), IRF-1 (36), c-Myc, (20), and p53 (46).
Several studies have also characterized the functional interaction between death receptor-induced apoptotic signals and the PKR pathway. These studies showed that PKR induces apoptosis through the activation of proapoptotic factors such as FADD (2), caspase-8 (10), and caspase-9 (11). PKR-induced apoptotic signaling through FADD has been shown to be independent of the aggregation of ligand-induced death receptors (2, 10). Moreover, expression of dominant-negative PKR mutants completely inhibited apoptosis induced by Fas (2, 35) (Fig. 5).
In this work we tested the effect of EBER expression on several proapoptotic factors associated with Fas-mediated apoptosis and found that cleavage of PARP was significantly inhibited in EBER-expressing cells (Fig. 6A). Furthermore, the expression of mPKR also inhibited Fas-induced cleavage of PARP. Interestingly, no other significant differences were observed in other targets for caspase-3 including ICAD and α-fodrin (Fig. 6A) or in other proapoptotic components such as caspase-8, caspase-9 (Fig. 6B), and Bid (Fig. 6A) in EBER-, mPKR- and Neor-expressing cells. Our findings strongly support EBER as being responsible for prevention of Fas-mediated apoptosis by inhibition of PKR, although the detailed mechanism remains to be clarified.
In a previous report we have shown that EBER plays a role in the resistance to IFN-α-induced apoptosis by blocking the PKR pathway in BL cells (26). IFN-α is an antiviral cytokine, which is essential for the host's innate immunity. We found that EBER expression also contributed to the resistance to IFN-α-induced apoptosis in Intestine 407 cells (data not shown). We suggest that EBER plays a crucial role in the survival of EBV-infected cells against host defense mechanisms by blocking both innate and acquired immune responses in epithelial cells.
In conclusion, our findings provide the first evidence that EBER confers resistance to Fas-mediated apoptosis. Recently, we have shown that EBER promotes insulin-like growth factor 1 expression in the GC-derived EBV-negative cell line NU-GC-3 and that the secreted insulin-like growth factor 1 acts as an autocrine growth factor (14). Taken together, these findings strongly suggest that EBER plays an important role in the development of not only EBV-associated lymphoid malignancies but also epithelioid malignancies.
Acknowledgments
This work was supported by grants-in-aid from the Ministry of Education, Science, Sports, Culture, and Technology, Japan.
REFERENCES
- 1.Ashkenazi, A., and V. M. Dixit. 1998. Death receptors: signaling and modulation. Science 281:1305-1308. [DOI] [PubMed] [Google Scholar]
- 2.Balachandran, S., C. N. Kim, W. C. Yeh, T. W. Mak, K. Bhalla, and G. N. Barber. 1998. Activation of the dsRNA-dependent protein kinase, PKR, induces apoptosis through FADD-mediated death signaling. EMBO J. 17:6888-6902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beidler, D. R., M. Tewari, P. D. Friesen, G. Poirier, and V. M. Dixit. 1995. The baculovirus p35 protein inhibits Fas- and tumor necrosis factor-induced apoptosis. J. Biol. Chem. 1995. 270:16526-16528. [DOI] [PubMed] [Google Scholar]
- 4.Chinnaiyan, A. M., K. O'Rourke, M. Tewari, and V. M. Dixit. 1995. FADD, a novel death domain-containing protein, interacts with the death domain of Fas and initiates apoptosis. Cell 81:505-512. [DOI] [PubMed] [Google Scholar]
- 5.Chiou, S. K., C. C. Tseng, L. Rao, and E. White. 1994. Functional complementation of the adenovirus E1B 19-kilodalton protein with Bcl-2 in the inhibition of apoptosis in infected cells. J. Virol. 68:6553-6566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Duriez, P. J., and G. M. Shah. 1997. Cleavage of poly(ADP-ribose) polymerase: a sensitive parameter to study cell death. Biochem. Cell Biol. 75:337-349. [PubMed] [Google Scholar]
- 7.Enari, M., H. Sakahira, H. Yokoyama, K. Okawa, A. Iwamatsu, and S. Nagata. 1998. A caspase-activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature 391:43-50. [DOI] [PubMed] [Google Scholar]
- 8.Foghsgaard, L., and M. Jaattela. 1997. The ability of BHRF1 to inhibit apoptosis is dependent on stimulus and cell type. J. Virol. 71:7509-7517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gil, J., M. A. Garcia, P. Gomez-Puertas, S. Guerra, J. Rullas, H. Nakano, J. Alcami, and M. Esteban. 2004. TRAF family proteins link PKR with NF-κB. Mol. Cell. Biol. 24:4502-4512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gil, J., and M. Esteban. 2000. The interferon-induced protein kinase (PKR), triggers apoptosis through FADD-mediated activation of Caspase 8 in a manner independent of Fas and TNF-alpha receptors. Oncogene 19:3665-3674. [DOI] [PubMed] [Google Scholar]
- 11.Gil, J., M. A. Garcia, and M. Esteban. 2002. Caspase 9 activation by the dsRNA-dependent protein kinase, PKR: molecular mechanism and relevance. FEBS Lett. 529:249-255. [DOI] [PubMed] [Google Scholar]
- 12.Gil, L., J. Alcami, and M. Esteban. 1999. Induction of apoptosis by double-stranded-RNA-dependent protein kinase (PKR) involves the α subunit of eukaryotic translation initiation factor 2 and NF-κB. Mol. Cell. Biol. 19:4653-4663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hazama, A., and Y. Okada. 1988. Ca2+ sensitivity of volume-regulatory K+ and Cl- channels in cultured human epithelial cells. J. Physiol. 402:687-702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Iwakiri, D., Y. Eizuru, M. Tokunaga, and K. Takada. 2003. Autocrine growth of Epstein-Barr virus-positive gastric carcinoma cells mediated by an Epstein-Barr virus-encoded small RNA. Cancer Res. 63:7062-7067. [PubMed] [Google Scholar]
- 15.Kawanishi, M. 1997. Epstein-Barr virus BHRF1 protein protects intestine 407 epithelial cells from apoptosis induced by tumor necrosis factor alpha and anti-Fas antibody. J. Virol. 71:3319-3322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kieff, E. 1996. Epstein-Barr virus and its replication. p. 2343-2396. In D. M. Knipe, B. Roizman, P. M. Howley, T. P. Monath, S. E. Straus, R. M. Chanock, J. L. Melnick, and B. N. Fields (ed.), Fields virology, 3rd ed. Lippincott Williams & Wilkins, Philadelphia, Pa.
- 17.Kitagaw, N., M. Goto, K. Kurozumi, S. Maruo, M. Fukayama, T. Naoe, M. Yasukawa, K. Hino, T. Suzuki, S. Todo, and K. Takada. 2000. Epstein-Barr virus-encoded poly(A)(-) RNA supports Burkitt's lymphoma growth through interleukin-10 induction. EMBO J. 19:6742-6750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Komano, J., S. Maruo, K. Kurozumi, T. Oda, and K. Takada. 1999. Oncogenic role of Epstein-Barr virus-encoded RNAs in Burkitt's lymphoma cell line Akata. J. Virol. 73:9827-9831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kroemer, G. 1997. The proto-oncogene Bcl-2 and its role in regulating apoptosis. Nat. Med. 3:614-620. [DOI] [PubMed] [Google Scholar]
- 20.La Rosa, F. A., J. W. Pierce, and G. E. Sonenshein. 1994. Differential regulation of the c-myc oncogene promoter by the NF-κB Rel family of transcription factors. Mol. Cell. Biol. 14:1039-1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lerner, M. R., N. C. Andrews, G. Miller, and J. A. Steitz. 1981. Two small RNAs encoded by Epstein-Barr virus and complexed with protein are precipitated by antibodies from patients with systemic lupus erythematosus. Proc. Natl. Acad. Sci. USA 78:805-809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li, P., D. Nijhawan, I. Budihardjo, S. M. Srinivasula, M. Ahmad, E. S. Alnemri, and X. Wang. 1997. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 91:479-489. [DOI] [PubMed] [Google Scholar]
- 23.Luo, X., I. Budihardjo, H. Zou, C. Slaughter, and X. Wang. 1998. Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell 94:481-490. [DOI] [PubMed] [Google Scholar]
- 24.Muzio, M., A. M. Chinnaiyan, F. C. Kischkel, K. O'Rourke, A. Shevchenko, J. Ni, C. Scaffidi, J. D. Bretz, M. Zhang, R. Gentz, M. Mann, P. H. Krammer, M. E. Peter, and V. M. Dixit. 1996. FLICE, a novel FADD-homologous ICE/CED-3-like protease, is recruited to the CD95 (Fas/APO-1) death-inducing signaling complex. Cell 85:817-827. [DOI] [PubMed] [Google Scholar]
- 25.Nagata, S. 1997. Apoptosis by death factor. Cell 88:355-365. [DOI] [PubMed] [Google Scholar]
- 26.Nanbo, A., K. Inoue, K. Adachi-Takasawa, and K. Takada. 2002. Epstein-Barr virus RNA confers resistance to interferon-alpha-induced apoptosis in Burkitt's lymphoma. EMBO J. 21:954-965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nicoletti, I., G. Migliorati, M. C. Pagliacci, F. Grignani, and C. Riccardi. 1991. A rapid and simple method for measuring thymocyte apoptosis by propidium iodide staining and flow cytometry. J. Immunol. Methods 139:271-279. [DOI] [PubMed] [Google Scholar]
- 28.Rickinson, A. B., and E. Kieff. 1996. Epstein-Barr virus, p. 2397-2446. In D. M. Knipe, B. Roizman, P. M. Howley, T. P. Monath, S. E. Straus, R. M. Chanock, J. L. Melnick, and B. N. Fields (ed.), Fields virology, 3rd ed. Lippincott Williams & Wilkins, Philadelphia, Pa.
- 29.Rosa, M. D., E. Gottlieb, M. R. Lerner, and J. A. Steitz. 1981. Striking similarities are exhibited by two small Epstein-Barr virus-encoded ribonucleic acids and the adenovirus-associated ribonucleic acids VAI and VAII. Mol. Cell. Biol. 1:785-796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sakahira, H., M. Enari, and S. Nagata. 1998. Cleavage of CAD inhibitor in CAD activation and DNA degradation during apoptosis. Nature 391:96-99. [DOI] [PubMed] [Google Scholar]
- 31.Srivastava, S. P., K. U. Kumar, and R. J. Kaufman. 1998. Phosphorylation of eukaryotic translation initiation factor 2 mediates apoptosis in response to activation of the double-stranded RNA-dependent protein kinase. J. Biol. Chem. 273:2416-2423. [DOI] [PubMed] [Google Scholar]
- 32.Takada, K. 1984. Cross-linking of cell surface immunoglobulins induces Epstein-Barr virus in Burkitt's lymphoma lines. Int. J. Cancer. 33:27-32. [DOI] [PubMed] [Google Scholar]
- 33.Takada, K., K. Horinouchi, Y. Ono, T. Aya, T. Osato, M. Takahashi, and S. Hayasaka. 1991. An Epstein-Barr virus-producer line Akata: establishment of the cell line and analysis of viral DNA. Virus Genes 5:147-156. [DOI] [PubMed] [Google Scholar]
- 34.Takahashi, T., M. Tanaka, T. Inazawa, T. Abe, T. Suda, and S. Nagata. 1994. Human Fas ligand: gene structure, chromosomal localization and species specificity. Int. Immunol. 6:1567-1574. [DOI] [PubMed] [Google Scholar]
- 35.Takizawa, T., K. Ohashi, and Y. Nakanishi. 1996. Possible Involvement of Double-Stranded RNA-Activated Protein Kinase in Cell Death by Influenza Virus Infection. 1996. J. Virol. 70:8128-8132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tanaka, N., M. Ishihara, M. Kitagawa, H. Harada, T. Kimura, T. Matsuyama, M. S. Lamphier, S. Aizawa, T. W. Mak, and T. Taniguchi. 1994. Cellular commitment to oncogene-induced transformation or apoptosis is dependent on the transcription factor IRF-1. Cell 77:829-839. [DOI] [PubMed] [Google Scholar]
- 37.Tewari, M., L. T. Quan, K. O'Rourke, S. Desnoyers, Z. Zeng, D. R. Beidler, G. G. Poirier, G. S. Salvesen, and V. M. Dixit. 1995. Yama/CPP32 beta, a mammalian homolog of CED-3, is a CrmA-inhibitable protease that cleaves the death substrate poly(ADP-ribose) polymerase. Cell 81:801-809. [DOI] [PubMed] [Google Scholar]
- 38.Tewari, M., and V. M. Dixit. 1995. Fas- and tumor necrosis factor-induced apoptosis is inhibited by the poxvirus CrmA gene product. J. Biol. Chem. 270:3255-3260. [DOI] [PubMed] [Google Scholar]
- 39.Thome, M., P. Schneider, K. Hofmann, H. Fickenscher, E. Meinl, F. Neipel, C. Mattmann, K. Burns, J. L. Bodmer, M. Schroter, C. Scaffidi, P. H. Krammer, M. E. Peter, and J. Tschopp. 1997. Viral FLICE-inhibitory proteins (FLIPs) prevent apoptosis induced by death receptors. Nature 386:517-521. [DOI] [PubMed] [Google Scholar]
- 40.Thornberry, N. A., and Y. Lazebnik. 1998. Caspases: enemies within. Science 281:1312-1316. [DOI] [PubMed] [Google Scholar]
- 41.Toczyski, D. P., A. G. Matera, D. C. Ward, and J. A. Steitz. 1994. The Epstein-Barr virus (EBV) small RNA EBER1 binds and relocalizes ribosomal protein L22 in EBV-infected human B lymphocytes. Proc. Natl. Acad. Sci. USA 91:3463-3467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tschopp, J., M. Irmler, and M. Thome. 1998. Inhibition of Fas death signals by FLIPs. Curr. Opin. Immunol. 10:552-558. [DOI] [PubMed] [Google Scholar]
- 43.Vanags, D. M., M. I. Porn-Ares, S. Coppola, D. H. Burgess, and S. Orrenius. 1996. Protease involvement in Fodrin cleavage and phosphatidylserine exposure in apoptosis. J. Biol. Chem. 271:31075-31085. [DOI] [PubMed] [Google Scholar]
- 44.Yajima, M., T. Kanda, and K. Takada. 2005. Critical role of Epstein-Barr Virus (EBV)-encoded RNA in efficient EBV-induced B-lymphocyte growth transformation. J. Virol. 79:4298-4307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang, J., X. Liu, K. Bhalla, C. N. Kim, A. M. Ibrado, J. Cai, T. I. Peng, D. P. Jones, and X. Wang. 1997. Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science 275:1129-1132. [DOI] [PubMed] [Google Scholar]
- 46.Yeung, M. C., and A. S. Lau. 1998. Tumor suppressor p53 as a component of the tumor necrosis factor-induced, protein kinase PKR-mediated apoptotic pathway in human promonocytic U937 cells. J. Biol. Chem. 273:25198-25202. [DOI] [PubMed] [Google Scholar]