

Abstract
We describe a new approach for the preparation of inactivated retroviruses for vaccine application. The lipid domain of the viral envelope was selectively targeted to inactivate proteins and lipids therein and block fusion of the virus with the target cell membrane. In this way, complete elimination of the infectivity of human immunodeficiency virus (HIV) and simian immunodeficiency virus (SIV) could be achieved with preservation of antigenic determinants on the surface of the viral envelope. Inactivation was accomplished by modification of proteins and lipids in the viral envelope using the hydrophobic photoinduced alkylating probe 1,5 iodonaphthylazide (INA). Treatment of HIV and SIV isolates with INA plus light completely blocked fusion of the viral envelope and abolished infectivity. The inactivated virus remained structurally unchanged, with no detectable loss of viral proteins. Modifications to envelope and nucleocapsid proteins were detected by changes in their elution pattern on reverse-phase high-performance liquid chromatography. These modifications had no effect on primary and secondary structure epitopes as determined by monoclonal antibodies. Likewise, the inactivated HIV reacted as well as the live virus with the conformation-sensitive and broadly neutralizing anti-HIV type 1 monoclonal antibodies 2G12, b12, and 4E10. Targeting the lipid domain of biological membranes with hydrophobic alkylating compounds could be used as a general approach for inactivation of enveloped viruses and other pathogenic microorganisms for vaccine application.
Intensive efforts have been under way in the last few years to develop an anti-human immunodeficiency virus (HIV) vaccine that can protect humans from AIDS. Many of these efforts employ DNA-encoding viral genes in combination with viral vectors that express HIV genes, as well as recombinant immunomodulatory elements that are all administered in complex immunization regimens (for reviews, see references 7 and 29). The results of these studies proved very promising, since they demonstrated that some of these vaccines could protect macaques from disease but not from infection when challenged with infectious chimeric HIV type 1 [HIV-1]-simian immunodeficiency virus [SIV], designated SHIV. A common feature of these vaccines is that they employ gene-altering elements that appear increasingly unappealing for use in healthy human individuals. An alternative strategy for the development of an AIDS vaccine is a formulation based on the whole virus or virus-like particles. Broadly neutralizing antibodies against HIV-1 when administered passively by infusion are the only elements that could protect primates from infection by live viruses (2, 9, 16, 17). These antibodies developed spontaneously in response to whole-virus challenge in HIV-infected individuals and recognize structural epitopes present only on the intact virus (6, 11, 30, 31). The successful development of a whole virus-based vaccine against HIV for humans will be dependent on the complete elimination of live virus from the vaccine preparation. Several approaches for inactivation of HIV have been reported. These include chemical inactivation using formalin (18, 28) and ethyleneimine (20), UV and X-ray inactivation (12), and photochemical and photodynamic inactivation (3, 10) using psoralens and fluorescent dyes, respectively. These studies provided useful information and laid the foundation for most of the viral inactivation methods in use today. Mechanistically, these methods enlarge upon nucleic acid modification. A few years ago, a new method was introduced that made use of sulfhydryl-oxidizing reagents to inactivate retroviruses (24, 25). These reagents preferentially modify cysteines in the highly conserved zinc finger motif on the nucleocapsid protein of retroviruses and block viral replication. In these studies, 2,2′-dithiodipyridine (aldrithiol-2) was identified as a compound that could render HIV and SIV completely noninfectious while preserving the structure of the virion and the conformation of immunogenic epitopes for neutralizing antibodies (1, 27). Since the viral envelope proteins do not contain free sulfhydryls, their function remains unaffected, as they can still facilitate fusion of the inactivated virus with the target cell. Recently, it has been shown that immunization of macaques with aldrithiol-2-inactivated SIV conferred homologous protection against the development of disease in these animals when challenged with infectious SIV (14).
In this study, we examined if hydrophobic alkylating compounds that partitioned into the lipid bilayer of biological membranes could be used to block fusion of viruses with their target cell without compromising the overall integrity of the virus or the conformation of antigenic epitopes on viral envelope proteins. The general concept behind this approach is that the lipid bilayer of the membrane can be used as a multicomponent common target for inactivation of envelope viruses (and possibly other pathogenic organisms) for vaccine application. The alkylating agent that was used is the photoactivatable membrane probe 1,5-iodonaphthylazide (INA). INA is a nontoxic hydrophobic compound that has been used to label membrane-embedded domains of membrane proteins (4). When added to biological membranes, it partitions into the membrane bilayer and accumulates selectively in this domain. Upon irradiation with far-UV light, INA binds to membrane proteins and lipids deep into the lipid bilayer. This process causes the specific inactivation of integral membrane proteins and lipids embedded in this domain, while maintaining the integrity and activity of proteins that protrude outside the membrane (21). In this work, we demonstrate that INA treatment of HIV and SIV produced noninfectious and nonfusogenic viruses that are structurally intact and fully maintain the conformation of the immunogenic epitopes recognized by anti-HIV broadly neutralizing antibodies.
MATERIALS AND METHODS
Reagents and cells.
125INA was prepared as previously described (4). 3,3-Dioctadecyloxacarbocyanine (DiO) was from Molecular Probes. Glutaraldehyde was from Tousimis, Rockville, MD; sodium cacodylate, osmium, and Embed-812 epoxy resin were from Electron Microscope Sciences, Fort Washington, PA; and uranyl acetate and lead citrate were from Leica, Bannockburn, IL. The SIV p28 and HIV-1 p24 antigen capture assay kits were from the AIDS Vaccine Program, SAIC. HIV MN and SIV Mne were prepared by the AIDS Vaccine Program, SAIC (8). The TZM-bl indicator cell line (32) was obtained through the National Institutes of Health AIDS Research and Reference Reagent Program (ARRRP) from J. C. Kappes. Cell lines for AA2 clones 1 and 2 were generously provided by Raoul Benveniste (National Cancer Institute). HOSCD4X4 R5 cells were obtained from the ARRRP through Vineet KewalRamani and Dan Litman. The anti-gp120 polyclonal antiserum and the anti-p28 and anti-gp32 monoclonal antibodies were prepared by the AIDS Vaccine Program, SAIC. The anti-HIV-1 broadly neutralizing antibodies 2G12 and 4E10 were obtained through ARRRP from Herman Katinger (29, 30); B12 was obtained from Dennis Burton (6).
Preparation of viruses.
Viruses were purified from cell-free culture fluids by continuous-flow ultracentrifugation in a sucrose density gradient, followed by direct sedimentation to remove the sucrose as previously described (8). The virus was resuspended at a final concentration of 1,000 relative to the cell culture fluid, and single-use aliquots were stored in a liquid N2 vapor-phase freezer.
Treatment of virus with INA.
The indicated HIV-1 or SIV isolates were diluted in phosphate-buffered saline (PBS) to a final concentration of 0.7 mg/ml. INA from a stock of 30 mM in dimethyl sulfoxide (DMSO) was added to the virus suspension under dim light in three to four installments (for uniform mixing) to final concentrations as indicated in the individual experiments (2 to 200 μM). The suspension was incubated for 30 min at room temperature, and the virus was washed once by centrifugation and resuspended in PBS to the original volume. Glutathione, reduced form, was added to the suspension to final concentrations between 20 to 30 mM, and the sample was irradiated with UV light. The light source was a 100-W ozone-free mercury arc lamp placed in a lamphouse with a collector lens (Olympus). Samples were irradiated through a 310-nm cutoff filter placed in front of the lens (to allow transmission of the 313-, 334-, and 365-nm mercury emission bands) and through a water filter (to prevent sample heating) at a distance of 5 cm from the light source. At that point, the light dose was 10 mW/cm2 · s. Irradiation times were 2 min for sample volumes of up to 1 ml in a clear microcentrifuge tube or 5 min with mixing for volumes of 20 ml in a clear 50-ml tube.
Virus infectivity assay.
The infectivity of HIV-1(MN) and SIV Mne samples were determined by using AA2 clone 1 and clone 5 cells, respectively, as previously described (22). Cells were cultured in RPMI 1640 supplemented with 10% heat-inactivated fetal bovine serum, 2 mM l-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 0.1 μg/ml gentamicin, and 20 mM HEPES buffer. To determine infectious titers of untreated virus, serial dilutions of the virus were prepared in cell culture medium. One hundred microliters of virus from each dilution was added to 2.5 × 106 cells in 0.9 ml of medium (resulting in an additional 1:10 dilution of the virus) and incubated at 37°C in a 15-ml tube for 18 h while being slowly rocked. After the addition of 9 ml of incomplete medium, the cells were washed to remove unbound virus. The cells were washed twice more, and the pellet was resuspended in 5 ml of complete medium. One milliliter (5 × 105 cells) was placed in each of 4 wells of a 24-well plate (Costar no. 3524). Mock-infected cells were included as negative controls for each set of titrations. Plates were incubated at 37°C in a humidified, 5% CO2 incubator. On day 3, 1 ml of complete medium was added to each well. These cultures were passaged by removing 50% of the culture and adding fresh medium on days 3, 7, 10, 14, 17, and 21 to maintain actively dividing cell populations. On days 7, 14, and 21, samples were analyzed for viral capsid protein concentration to determine if progeny virions could be detected. These samples were clarified from cells by centrifugation at 600 × g for 5 min, and the supernatant was lysed by the addition of a 1/10 volume of 10% Triton X-100.Capsid protein concentrations were determined using either an SIV p28 or HIV-1 p24 antigen capture assay (house assay kits; AIDS Vaccine Program, SAIC). Samples were considered positive if they produced capsid protein concentrations above the antigen capture assay cutoff value and these concentrations increased over time. Infectious titers were determined by the Reed and Muench method (28) using capsid concentration results from day 21.
Infectivity assay for INA-treated virus.
To determine if INA-treated virus preparations were free of detectable infectious virus, samples were analyzed at the lowest possible dilution and against a greater number of target cells: 0.5 ml of INA-treated virus was added to 1.25 × 107 cells in 2.5 ml of medium and incubated at 37°C in a 15-ml tube for 18 h while being slowly rocked. Due to the high concentration of virus in the inoculum, these samples were washed four times by centrifugation to remove unbound virus. The resulting cell pellets were resuspended in 25 ml of medium (final cell concentration was 5.0 × 105/ml), planted in T75 flasks, and incubated at 37°C in 5% CO2. These cultures were passaged and analyzed for capsid antigen concentration. All the other details were as described above for the untreated virus.
Virus-induced cell-cell fusion.
AA2 clone 5 cells were inoculated with virus as described above for the infectivity analysis of INA-treated virus and microscopically monitored at a magnification of ×100 for virus-induced cell-cell fusion during the first 2 days.
Infectivity by luciferase reporter assay.
The assay was carried out as previously described (26) with TZM-bl cells. These are HeLa cells that were modified to express high levels of CD4 and CCR5, along with endogenous CXCR4. TZM-bl cells also contain HIV LTR-driven β-galactosidase and luciferase reporter cassettes that are activated by HIV tat expression. Cells (2 × 104 per well) were added to a 96-well microtiter plate (Falcon, Lincon Park, NJ) in 100 μl of complete medium and allowed to adhere 15 to 18 h at 37°C. Serial dilutions of HIV and INA-treated HIV were added to the cell monolayers in the presence of 40 μg/ml DEAE-dextran in Dulbecco's modified Eagle medium in a final volume of 100 μl. Viral infection was allowed to proceed for 2 h at 37°C, following which 100 μl of complete Dulbecco's modified Eagle medium was added. Luciferase activity was measured after 15 to 18 h using a Promega (Madison, WI) luciferase assay system kit. Briefly, the supernatants were removed, and the cells were lysed with the Steady Glo luciferase assay system. The light intensity of each well was measured with a luminometer. All infectivity assays were performed in duplicate.
Fusion assay by photosensitized labeling.
Fusion was determined as previously described (30). Briefly, HOSCD4X4 R5 cells that do not express the HLA type II antigen (HLA-DR) were incubated with 125INA together with the fluorescent membrane probe DiO in petri dishes, 10 cm in diameter. SIV Mne virions that are propagated in lymphocytes (HUT78) cells and carry HLA-DR on their envelope were added in the amount of 8 × 105 50% tissue culture infective doses (TCID50)/ml to each experimental group. The mixture was incubated at 37°C for 40 min, and then the dishes were irradiated for 1 min with a 488-nm light at the intensity of 10 mW/cm2 · min using an argon laser as the light source. At this wavelength, DiO activates 125INA by photosensitization and thus confines the radioactive labeling of proteins to the membrane of the target cell. The cells were lysed, and the HLA-DR α-subunit was isolated by immunoprecipitation. The extent of penetration of viral HLA-DR into the target cell membrane was determined by the extent of 125INA incorporation into this protein as measured by phosphor storage autoradiography using a Typhoon phosphorimager (Amersham).
Reversed-phase high-performance liquid chromatography (RP-HPLC) separation and analysis of viral proteins.
Viral samples were disrupted in 8 M Guanidine-HCl (Pierce, Rockford, IL) with or without 50 mM dithiothreitol (Calbiochem, La Jolla, CA) and fractionated by HPLC to isolate viral proteins. HPLC was performed at a flow rate of 300 μl/min on a 2.1- by 100-mm Poros R2/H narrow-bore column (Applied Biosystems), using aqueous acetonitrile-trifluoroacetic acid solvents and a Shimadzu HPLC system equipped with LC-10AD pumps, SCL-10A system controller, CTO-10AC oven, FRC-10A fraction collector, and SPD-M10AV diode array detector. The gradient of buffer B (0.1% trifluoracetic acid in acetonitrile) was as follows: 10 to 36.5%, 12 min; 36.5 to 37%, 4 min; 37 to 41%, 7 min; 41 to 70%, 12 min; and 70%, 5 min. A temperature of 55°C was maintained during HPLC separation. Peaks were detected by UV absorption at 206 and 280 nm and analyzed by Edman sequencing using an automated 477 Protein Sequencer (Applied Biosystems, Inc.), by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and by immunoblot analysis using the Enhanced ChemiLuminescence (ECL) procedure (Amersham Life Science, Arlington Heights, IL).
Western blot analysis of SIV Mne/HuT78 clone E11S.
Aliquots of viral samples were loaded on adjacent gel lanes (4 to 20%). Gel electrophoresis was performed under nonreducing or reducing conditions. Proteins were then transferred onto an Immobilon-P membrane (Millipore, Bedford, Mass.), and SIV-1 gp120 was detected using a rabbit polyclonal antisera generated against gp120. p28 and gp32 were detected with mouse monoclonal antibodies against these proteins. The signal was produced using ECL (Amersham, Arlington Heights, IL). gp120 and p28 were detected simultaneously, whereas gp32 was detected by stripping and reprobing the immunoblot. Stripping was carried out by incubating the membrane in a buffer containing 100 mM β-mercaptoethanol, 2% SDS, 62.5 mM Tris-HCl (pH 6.7) at 60°C for 30 min, followed by two 10-min washes in TBS (20 mM Tris-HCl [pH 7.6] and 137 mM NaCl) with 0.1% Tween-10 (Sigma). The filters were blocked for 2 h and reacted with a monoclonal anti-gp32 antibody.
Neutralizing antibody capture assay.
The antibody capture assay was carried out on the whole virus essentially as previously described (19). In short, 10 μg (each) of antibodies 2G12, B12, and 4E10 was coated on enzyme-linked immunosorbent assay (ELISA) plates in triplicates and blocked with bovine serum albumin. HIV-1(MN) was added for binding for 1 h at 37°C in the different amounts as indicated in the individual experiments. Control samples were wells without antibody. The unbound virus was removed by washing, and the captured virus was measured by p24 determination. The samples were lysed with 1% Triton in PBS, and the lysate was transferred to 96-well plates. These plates contained immobilized anti-p24 monoclonal antibody as part of an ELISA kit for p24 determination that was prepared by the AIDS Vaccine Program, SAIC.
Preparation of HIV and SIV for EM.
The preparation of viruses for electron microscopy (EM) analysis was carried out by Kunio Nagashima from the EM facility at SAIC, essentially as previously described (15). Briefly, a sedimented virus pellet was fixed in 2% glutaraldehyde in 0.1 M sodium cacodylate buffer (pH 7.0) for 2 h, followed by a postfixation with 1% osmium tetroxide in the same buffer for 1 h. En bloc staining was carried out with 0.5% uranyl acetate in 0.1 M acetate buffer (pH 4.5) for 1 h. The pellet was dehydrated in a series of graded ethanol solutions (e.g., 35%, 50%, 75%, 95%, and 100%) and 100% propylene oxide. The sample was then incubated overnight for infiltration with a 1:1 mixture of 100% propylene oxide and epoxy resin, embedded in pure epoxy resin, and cured for an additional 48 h at 55°C. Thin sections (each, 60 to 70 nm) were mounted on a naked copper grid, stained in uranyl acetate and lead citrate solutions, and stabilized by carbon evaporation. The samples were examined in the EM (Hitachi, Tokyo, Japan) operated at 75 kV; digital images were obtained with a charge-coupled device camera.
RESULTS
To assess the efficacy of INA treatment in viral inactivation for vaccine use, we tested two major parameters. The first parameter is functional and is evaluated by measuring the residual infectivity of the virus after INA treatment. The second parameter is the structural and conformational integrity of the virus after inactivation. All experiments were repeated at least two times.
The effect of INA treatment on virus activity.
Virus fusion activity was tested on SIV by monitoring virus-induced syncytium formation in target cells 47 h after infection. It was found that INA reduced the ability of the virus to induce cell-cell fusion (fusion from without) in a dose-dependent manner with no detected cell fusion with 200 μM INA (Fig. 1). Similar results were obtained 6.5 h after infection, conditions that monitor the effect of the added virus without the contribution of progeny viruses (not shown). Likewise, further incubation of the SIV-infected cells for up to 11 days showed a dose-dependent decrease in the production of the viral protein, p28 with no detectable production with 200 μM INA (Fig. 2). Similar results were obtained when infectivity was measured on INA-treated HIV after 21 days (Table 1). The effect of INA on HIV infectivity was also measured by the luciferase reporter gene assay (Fig. 3). The results demonstrated that at the highest dose (200 μM), INA completely blocked HIV infection with no detectable infectivity, even at very high doses of virus. SIV was also used to determine the effect of INA treatment on fusion of the virus with target cells. In these experiments, the controls of 200 μM INA with no UV irradiation (Fig. 2) and UV irradiation with no INA (not shown) were found to have no effect on the virus. Fusion at the plasma membrane level was measured by the extent of 125INA incorporation into viral HLA-DR by photosensitized labeling (22) and as described in Materials and Methods. The results shown in Fig. 4 demonstrate that INA treatment blocked fusion of the viral envelope with the target cell membrane in a dose-dependent manner. The extent of fusion inactivation within the range of concentrations used reached a maximum of about 75% at 2 μM INA. The low INA concentration used in this experiment for inactivation (relative to the experiments shown in Fig. 2 and 3) was to prevent interference with 125INA incorporation into viral membrane proteins on which the fusion assay was based. These conditions were optimized by preliminary experiments.
FIG. 1.

The effect of INA on virus induced cell-cell fusion. AA2 cells were infected with SIV that was preinactivated by the indicated concentrations of INA, as described in Materials and Methods. At 47 h after infection, cells were tested for the presence of syncytia induced by the virus. (A) Untreated control; (B) 2 μM INA; (C) 200 μM INA; (D) 200 μM INA with no UV irradiation.
FIG.2.

Dose-dependent inactivation of SIV infectivity by INA. AA2 cells were infected with SIV and treated with INA at the concentrations indicated (0.0 μM, 2 μM, 20 μM, and 200 μM). Infectivity was tested 11 days after infection by p28 determination as described in Materials and Methods. The experiment was repeated three times with similar results.
TABLE 1.
Infectivity of control and INA-treated HIV and SIV established by capsid production after 21 days of culture
| Treatment | Infectivity assay
|
||||||
|---|---|---|---|---|---|---|---|
| 1/dil | Avg capsid protein concn (pg/ml)c
|
Culture resultd
|
1/TCID50/mle | ||||
| Day 7 | Day 14 | Day 21 | Pos | Neg | |||
| SP1061-INAa | |||||||
| SIV Mne CL.E11S | |||||||
| Controlf | 103 | 21,669 | 76,680 | 67,525 | 4 | 0 | 2.2 × 106 |
| 104 | 1,263 | 71,517 | 60,856 | 4 | 0 | ||
| 105 | 0 | 62,876 | 70,193 | 4 | 0 | ||
| 106 | 0 | 18,334 | 48,966 | 3 | 1 | ||
| 107 | 0 | 0 | 0 | 0 | 4 | ||
| 108 | 0 | 0 | 0 | 0 | 4 | ||
| INA | 101 | 0 | 0 | 0 | 0 | 0 | 0 |
| SP1122-INAb | |||||||
| HIV-1 (MN) | |||||||
| Controlf | 103 | 213,795 | 188,411 | 588,818 | 4 | 0 | 4.6 × 104 |
| 104 | 6,317 | 41,027 | 503,443 | 4 | 0 | ||
| 105 | 375 | 0 | 0 | 1 | 3 | ||
| 106 | 0 | 0 | 0 | 0 | 4 | ||
| 107 | 0 | 0 | 0 | 0 | 4 | ||
| 108 | 0 | 0 | 0 | 0 | 4 | ||
| INA | 101 | 9,201 | 0 | 0 | 0 | 4 | 0 |
| 102 | 0 | 0 | 0 | 0 | 4 | ||
| 103 | 0 | 0 | 0 | 0 | 4 | ||
| 104 | 0 | 0 | 0 | 0 | 4 | ||
| 105 | 0 | 0 | 0 | 0 | 4 | ||
| 106 | 0 | 0 | 0 | 0 | 4 | ||
Treatment of infection SIV Mne/HvT78 clone E115, lot P3932; assay results versus AA2 clone 5 cells.
Treatment of infectious HIV-1 (MN)/H9 CL.4, lot 3941; assays results versus AA2 clone 1 cells.
Average of capsid protein concentrations determined from quadruplicate cultures.
Culture result as assessed on day 21. Pos, the number of the quadruplicate cultures that became productively infected with virus; Neg, the number of cultures that remained uninfected.
Reciprocal TCID50 was determined from the culture result data using the method of Reed and Muench (23).
Negative control cultures were maintained and remained uninfected thoughout the 21-day test period.
FIG. 3.

Inactivation of HIV infectivity by INA. HIV treated with 200 μM INA and untreated control was tested for infectivity by the luciferase reporter gene assay. Serial dilutions of HIV were added to TZM-bl cells, and luminescence was plotted against the indicated amounts of virus.
FIG. 4.

INA inhibits fusion of SIV envelope with the target cell membrane. SIV Mne was treated with INA at the concentrations indicated. Photosensitized labeling was used to determine fusion activity at the plasma membrane level by measuring the extent of 125INA incorporation into viral HLA-DR (α-chain) upon fusion with the target cell membrane. The maximal INA concentration applied for inactivation was 2 μM, because higher concentrations interfered with the incorporation of 125INA in the assay. The experiment was repeated two times with similar results.
The effect of INA treatment on the structural integrity of the virus.
To examine possible changes in structure of viral proteins as a result of inactivation, the following experiments were carried out. Viruses were recovered by centrifugation after INA inactivation, and samples were run on SDS-PAGE. The results shown in Fig. 5 indicate that INA treatment had no significant effect on the amount of virus recovered by sedimentation, as can be empirically determined by the total amount of protein in each lane. Likewise, the electrophoretic mobility of major viral proteins did not change as a result of INA inactivation. However, when the viral proteins were subjected to RP-HPLC, significant changes in the elution pattern of the viral proteins were observed on samples from the INA-treated SIV (Fig. 6). To further evaluate the extent of modification on viral proteins, three major viral proteins were analyzed by Western blotting for their susceptibility to recognition by antibodies under reducing and nonreducing conditions. Western blot analyses were carried out using monoclonal antibodies against p28 and gp32 and a polyclonal antibody against gp120. As is demonstrated in Fig. 7, INA treatment did not affect the reactivity of monoclonal antibodies toward linear epitopes (reducing conditions) or secondary structure epitopes (nonreducing conditions) on the envelope proteins gp120 and gp32, as well as the capsid protein p28. It was also of interest to test if the inactivated virus interacted with monoclonal antibodies that recognized conformational epitopes on the viral envelope proteins. INA-inactivated HIV was subjected to antibody capture assay with three different broadly neutralizing antibodies, two antibodies that recognize gp120 (2G12 and B12) and one that recognizes gp41 (4E10). The results shown in Fig. 8 demonstrate that the inactivated HIV reacts as well as the live virus with all these antibodies at all virus concentrations tested. Finally, to obtain a visual observation of the effect of inactivation on viruses, INA-treated and control HIV and SIV were fixed in and subjected to electron microscopy by negative staining. Figure 9 shows that inactivated viruses preserve their structure and integrity and are virtually indistinguishable from the untreated control viruses.
FIG. 5.
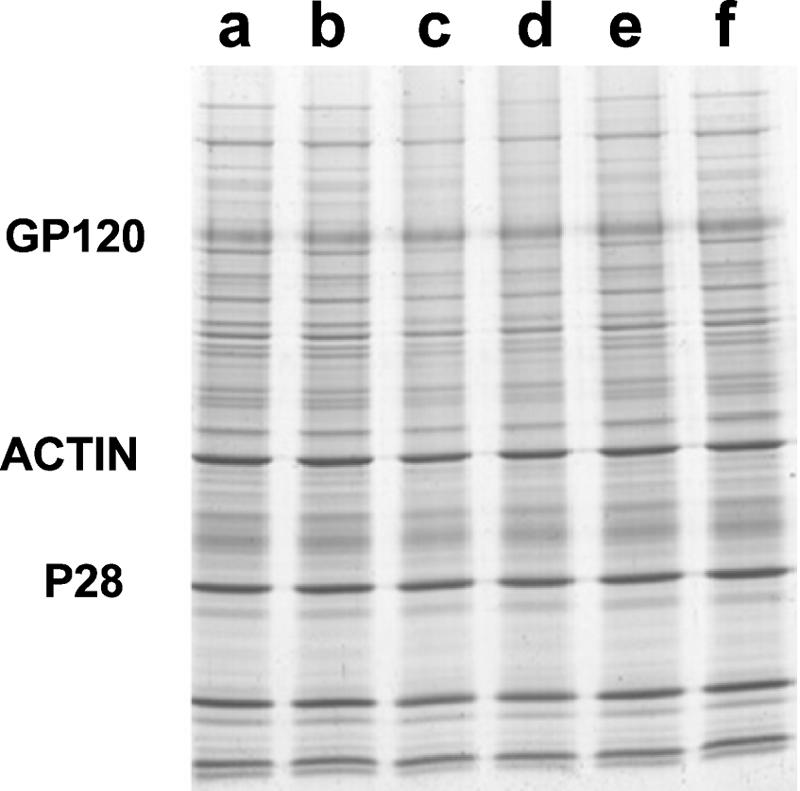
INA treatment does not affect the integrity of the virus. SIV isolate suspension was treated with INA as indicated, and the virus was recovered by centrifugation. The virus sample was then subjected to SDS-PAGE and Coomassie blue staining (25 μg/lane) to determine the total amount of protein recovered in each experimental group, as well as the recovery of the indicated major viral proteins. Lanes: a, control (buffer); b, 2% DMSO; c, 200 μM INA; d, 20 μM INA; e, 2 μM INA; f, 200 μM INA with no UV irradiation.
FIG. 6.

INA inactivation results in the modification of viral proteins. Samples of SIV (INA treated and nontreated control) were disrupted in 8 M guanidine hydrochloride, and proteins were subjected to separation by RP-HPLC. Individual proteins in the peaks were identified by Western blotting and sequencing. p8, p1/p2, p6, p17, and p28 are gag viral proteins, and gp120 and gp32 are viral envelope proteins. CL-II and actin are cellular proteins present in both microvesicles and virus.
FIG. 7.

INA inactivation does not affect the specific recognition of isolated viral proteins by antibodies. The indicated viral proteins from INA-treated SIV were analyzed by Western blot under reducing (R) and nonreducing (NR) conditions. Lanes: a nontreated virus; b, 1% DMSO; c, 200 μM INA; d, 20 μM INA; e, 2 μM INA; f, 200 μM INA (not photoactivated).
FIG. 8.

Inactivated HIV is recognized by conformational neutralizing antibodies, as well as the infectious virus. Antibody capture assay was carried out with increasing virus amounts with the indicated neutralizing monoclonal antibodies. 2G12 and B12 were used against epitopes on gp120 and 4E10 was used against gp41.
FIG. 9.

Electron microscopy of inactivated and control virions. Inactivated viruses and live viruses that were used in the experiments described above were fixed in glutaraldehyde, negatively stained, and visualized by electron microscopy as described in Materials and Methods. (A) HIV control; (B) inactivated HIV; (C) SIV control; (D) inactivated SIV. V, virion; Vsc, microvesicle.
DISCUSSION
In this study, we examined the concept that the lipid bilayer of the membrane can be used as a multicomponent common target for inactivation of enveloped viruses and possibly other pathogenic organisms for vaccine application. INA has been applied because this hydrophobic compound was originally developed to partition into the lipid domain of biological membranes and selectively bind upon irradiation to the transmembrane domains of integral membrane proteins.
Application of INA plus irradiation to SIV and HIV virions blocked the ability of the viral envelope to fuse with the target cell membrane. As expected, the elimination of the fusion ability resulted in a complete inactivation of both SIV and HIV with no detectable infectivity of the virus at 200 μM INA, as determined by two independent assays, the luciferase reporter gene assay and the assay for determination of p24-28 production as a result of viral infection. Inactivation of the virus with INA did not cause lysis of the virus, as evaluated by virus recovery by centrifugation; the sedimented virus appeared unchanged in the content and mass of major viral proteins. Analysis of the elution pattern of viral proteins with RP-HPLC showed significant alterations to the reverse-phase HPLC elution pattern of almost all viral proteins at 200 μM INA. This result is not surprising due to the high concentrations of INA relative to what has been used before for radioactive labeling (22) and signal transduction inactivation (21) in cells. These high concentrations were probably required to saturate the cellular microvesicles present in the viral isolates. However, these modifications were minor, since they were not manifested by any detectable change in the mass of these proteins, as determined by SDS-PAGE; they probably represent a small change in the hydrophobicity of these proteins conferred by INA binding. Likewise, there has been no effect on the recognition of linear and secondary structure epitopes of viral proteins by specific antibodies. It will be reasonable to assume that at high concentrations, a hydrophobic probe like INA will intercalate into the innermost hydrophobic domains of all proteins, including nonintegral membrane proteins like gp120 and nucleocapsid proteins. While modification of these domains by INA may further inactivate the virus, they are not likely to affect the overall three-dimensional structure of antigenic epitopes that are exposed to the aqueous environment. Consistent with this view is the observation that the three broadly neutralizing antibodies tested react with the INA-inactivated virus, as well as with the live infectious virus. These antibodies that originally developed spontaneously in HIV-infected individuals recognize conformational epitopes on the viral envelope that proved to be productive immunogens in humans. These epitopes remain unchanged on the envelope of INA-inactivated HIV. Support for the overall preservation of viral structure also comes from the electromicrographs of inactivated HIV and SIV that seem intact and structurally indistinguishable from nontreated virus. INA binds nonspecifically to envelope proteins (21) and lipids, thus targeting multiple inactivation sites that result in the loss of fusion activity. This mode of inactivation can be applied alone or possibly in conjunction with aldrithiol to create an orthogonal inactivation system that could produce an HIV particle that is both fusion impaired and reproduction incompetent. Recent emerging information suggests possible cytophatic effects of fusion-competent HIV envelope proteins on CD4+ T cells (5, 13). The elimination of fusion activity may be an important safety feature that will encourage application of vaccines based on whole inactivated HIV in humans. Obviously, more studies with animal models will be necessary to establish if this mode of inactivation can produce viruses that are safe and that elicit an effective immune response in vivo. A more general conclusion from this study is that the use of hydrophobic alkylating reagents that can target biological membranes could be applied to other envelope viruses or pathogens to produce inactivated microorganisms with preservation of surface protein conformation.
Acknowledgments
We thank Jeff Lifson from the AIDS Vaccine Program, SAIC, for his support in expert advice. We thank William Bohn, Jeremy Miller, Michael Poore, Robert Imming, Rodman Smith, and Terra Schaden-Ireland from the Biological Products Core Laboratory, as well as James D. Roser from the Protein Chemistry Laboratory AIDS Vaccine Program, SAIC, for their expert technical support. We thank Kunio Nagashima for his expert advice and execution of the EM analysis.
This work was funded in whole or in part with federal funds from the Intramural AIDS Targeted Antiviral Program and by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
REFERENCES
- 1.Arthur, O. L., J. W. Bess, Jr., E. N. Chertova, J. L. Rossio, M. T. Esser, R. E. Benveniste, L. E. Henderson, and J. D. Lifson. 1998. Chemical inactivation of retroviral infectivity by targeting nucleocapsid protein zinc fingers: a candidate SIV vaccine. AIDS Res. Hum. Retrovir. 14:S311-S319. [PubMed] [Google Scholar]
- 2.Baba, W. T., V. Liska, R. H. Lehman, J. Vlasak, W. Xu, S. Ayehunie, L. C. Cavacini, M. R. Posner, H. Kattinger, G. Stiegler, B. R. Bernacky, T. A. Rizvi, R. Schmidt, L. R. Hill, M. E. Keeling, Y. Lu, j. E. Wright, T. C. Chou, and R. M. Ruprecht. 2000. Human neutralizing monoclonal antibodies of the IgG1 subtype protect against mucosal simian-human immunodeficiency virus infection. Nat. Med. 6:200-206. [DOI] [PubMed] [Google Scholar]
- 3.Benade, E. L., Y. Shumaker, X. X. Chen, and Y. R. Dodd. 1994. Inactivation of free and cell-associated human immunodeficiency virus in platelet suspensions by aminomethyltrimethylpsoralen and ultraviolet light. Transfusion 34:680-684. [DOI] [PubMed] [Google Scholar]
- 4.Bercovici, T., and C. Gitler. 1978. 5-[125I]iodonaphthyl azide, a reagent to determine the penetration of proteins into the lipid bilayer of biological membranes. Biochemistry 17:1484-1489. [DOI] [PubMed] [Google Scholar]
- 5.Blanco, J., J. Barretina, B. Clotet, and A. J. Este. 2004. R5 HIV gp120-mediated cellular contacts induce the death of single CCR5-expressing CD4 T cells by a gp41-dependent mechanism. J. Leukoc. Biol. 76:804-881. [DOI] [PubMed] [Google Scholar]
- 6.Burton, R. D., J. Pyati, R. Koduri, S. J. Sharp, G. B. Thornton, P. W. H. I. Parren, L. S. W. Sawter, R. M. Hendry, N Dunlop, P. L. Nara, M. Lamancchia, E. Garatty, E. R. Stiehm, Y. J. Bryson, Y. Cao, J. P. Moore, D. D. Ho, and C. F. Barbas III. 1994. Efficient neutralization of primary isolates of HIV-1 by a recombinant human monoclonal antibody. Science 266:1024-1027. [DOI] [PubMed] [Google Scholar]
- 7.Calarota, A. S., and B. D. Weiner. 2003. Present status of human vaccine development. AIDS 17:S73-S84. [DOI] [PubMed] [Google Scholar]
- 8.Chertova, E., J. W. Bess, Jr., J. B. Crise, R. C. Sowder II, M. T. Schaden, J. M. Hilburn, J. A. Hoxie, R. E. Benveniste, J. D. Lifson, L. E. Henderson, and L. O. Arthur. 2002. Envelope glycoprotein incorporation, not shedding of surface envelope glycoprotein (gp120/SU), is the primary determinant of SU content of purified human immunodeficiency virus type 1 and simian immunodeficiency virus. J. Virol. 76:5315-5325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Conley, J. A., J. A. Kessler II, L. J. Boots, P. M. McKenna, W. A. Schleif, E. A. Emini, G. E. Mark III, H. Katinger, E. K. Kobb, S. T. Lunceford, S. R. Rouse, and K. K. Murthy. 1996. The consequence of passive administration of an anti-human immunodeficiency virus type 1 neutralizing monoclonal antibody before challenge of chimpanzees with a primary virus isolate. J. Virol. 70:6751-6758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dimitrov, S. D., and R. Blumenthal. 1994. Photoinactivation and kinetics of membrane fusion mediated by the human immunodeficiency virus type 1 envelope glycoprotein. J. Virol. 68:1956-1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.D'Souza, M. P., D. Livnat, J. A. Bradac, and S. H. Bridges. 1997. Evaluation of monoclonal antibodies to human immunodeficiency virus type 1 primary isolates by neutralization assays: performance criteria for selecting candidate antibodies for clinical trials. J. Infect. Dis. 175:1056-1062. [DOI] [PubMed] [Google Scholar]
- 12.Henderson, E. E., G. Tudor, and J. Yang. 1992. Inactivation of the human immunodeficiency virus type 1 (HIV-1) by ultraviolet and X irradiation. Radiat. Res. 131:169-176. [PubMed] [Google Scholar]
- 13.LaBonte, A. J., N. Madani, and J. Sodroski. 2003. Cytolysis by CCR5-using human immunodeficiency virus type 1 envelope glycoproteins is dependent on membrane fusion and can be inhibited by high levels of CD4 expression. J. Virol. 70:6645-6659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lifson, D. J., J. L. Rossio, M. Piatak, Jr., J. Bess, Jr., E. Chertova, D. K. Schneider, V. J. Coalter, B. Poore, R. F. Kiser, R. J. Imming, A. J. Scarzello, L. E. Henderson, W. G. Alvord, V. M. Hirsch, R. E. Benveniste, and L. O. Arthur. 2004. Evaluation of the safety, immunogenicity, and protective efficacy of whole inactivated simian immunodeficiency virus (SIV) vaccines with conformationally and functionally intact envelope glycoproteins. AIDS Res. Hum. Retrovir. 20:772-787. [DOI] [PubMed] [Google Scholar]
- 15.Mariner, J. M., J. B. McMahon, B. R. O'Keefer, K. Nagashima., and M. R. Boyd. 1998. The HIV-inactivating protein, cyanovirin-N, does not block gp120-mediated virus-to-cell binding. Biochem. Biophys. Res. Commun. 248:841-845. [DOI] [PubMed] [Google Scholar]
- 16.Mascola, R. J., G. Stiegler, T. C. VanCott, H. Kattinger, C. B. Carpenter, C. E. Hanson, H. Beary, D. Hayes, S. S. Frankel, D. L. Birx, and M. G. Lewis. 2000. Protection of macaques against vaginal transmission of a pathogenic HIV-1/SIV chimeric virus by passive infusion of neutralizing antibodies. Nat. Med. 6:207-210. [DOI] [PubMed] [Google Scholar]
- 17.Mascola, R. J., M. G. Lewis, G. Stiegler, D. Harris, T. C. VanCott, D. Hayes, M. K. Lauder, C. R. Brown, C. V. Sapan, S. S. Frankel, Y. Lu, M. L. Robb, H. Kattinger, and D. L. Birx. 1999. Protection of macaques against pathogenic simian/human immudodeficiency virus 89.6PD by passive transfer of neutralizing antibodies. J. Virol. 73:4009-4018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Murphey-Corb, M., L. N. Martin, M. Miller, M. West, S. Ohkawa, G. B. Baskin, J. Y. Zhang, S. D. Putney, A. C. Allison, D. A. Eppstein. 1989. A formalin-inactivated whole SIV vaccine confers protection in macaques. Science 246:1293-1297. [DOI] [PubMed] [Google Scholar]
- 19.Nyambi, N. P., S. Burda, L. Bastiani, and C. Williams. 2001. A virus binding assay for studying the antigenic landscape on intact, native, primary human immunodeficiency virus-type 1. J. Immunol. Methods 253:253-262. [DOI] [PubMed] [Google Scholar]
- 20.Race, E., C. A. Stein, M. D. Wigg, A. Baksh, M. Addawe, P. Frezza, and J. S. Oxford. 1995. A multistep procedure for the chemical inactivation of human immunodeficiency virus for use as an experimental vaccine. Vaccine 13:1567-1575. [DOI] [PubMed] [Google Scholar]
- 21.Raviv, Y., T. Bercovici, C. Gitler, and Y. Salomon. 1984. Selective photoinduced uncoupling of the response of adenylate cyclase to gonadotropins by 5-iodonaphtyl 1-azide. Biochemistry 23:503-508. [DOI] [PubMed] [Google Scholar]
- 22.Raviv, Y., M. Viard, J. W. Bess, Jr., and R. Blumenthal. 2002. Quantitative measurement of fusion of HIV-1 and SIV with cultured cells using photosensitized labeling. Virology 293:243-251. [DOI] [PubMed] [Google Scholar]
- 23.Reed, L. J., and H. Muench. 1938. A simple method of estimating 50 percent endpoints. Am. J. Hyg. 27:493-497. [Google Scholar]
- 24.Rein, A., D. E. Ott, J. Mirro, L. O. Arthur, W. Rice, and L. E. Henderson. 1996. Inactivation of murine leukemia virus by compounds that react with the zinc finger in the viral nucleocapsid protein. J. Virol. 70:4966-4972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rice, G. W., J. G. Supko, L. Malspeis, R. W. Buckheit, Jr., D. Clanton, M. Bu, L. Greham, C. A. Schaeffer, J. A. Turpin, J. Domagala, R. Gogliotti, J. P. Bader, S. M. Halliday, L. Cohen, R. C. Sowder II, L. O. Arthur, and L. E. Henderson. 1995. Inhibitors of HIV nucleocapsid protein zinc fingers as candidates for the treatment of AIDS. Science 270:1194-1197. [DOI] [PubMed] [Google Scholar]
- 26.Roos, W. J., F. M. Maughan, Z. Liao, K. E. J. Hildreth, and E. J. Clements. 2000. LuSIV cells: a reporter cell line for the detection and quantitation of a single cycle of HIV and SIV replication. Virology 273:307-315. [DOI] [PubMed] [Google Scholar]
- 27.Rossio, L. J., M. T. Esser, K. Suryanarayana, D. K. Schneider, J. W. Bess, Jr., G. M. Vasquez, T. A. Willtrout, E. Chertova, M. K. Grimes, Q. Sattentau, L. O. Arthur, L. E. Henederson, and J. D. Lifson. 1998. Inactivation of human immunodeficiency virus type 1 infectivity with preservation of conformational and functional integrity of virion surface proteins. J. Virol. 72:7992-8001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sattentau, O. J. 1995. Conservation of HIV-1 gp120 neutralizing epitopes after formalin inactivation. AIDS 9:1383-1385. [DOI] [PubMed] [Google Scholar]
- 29.Spearman, P. 2003. HIV vaccine development: Lessons from the past and promise for the future. Curr. HIV Res. 1:101-120. [DOI] [PubMed] [Google Scholar]
- 30.Stiegler, G., R. Kunert, M. Purtscher, S. Wolbank, R. Voglauer, F. Steidl, and H. Katinger. 2001. A potent cross-clade neutralizing human monoclonal antibody against a novel epitope on GP41 of human immunodeficiency virus type 1. AIDS Res. Hum. Retrovir. 17:1757-1765. [DOI] [PubMed] [Google Scholar]
- 31.Trkola, A., M. Purtscher, T. Muster, C. Ballaun, A. Buchacher, N. Sullivan, K. Srinivasan, J. Sodroski, J. P. Moore, and H. Katinger. 1996. Human monoclonal antibody 2G12 defines a distinctive neutralization epitope on the gp120 glycoprotein of human immunodeficiency virus type 1. J. Virol. 70:1100-1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wei, X., J. M. Decker, H. Liu, Z. Zhang, R. B. Arani, J. M. Kilby, M. S. Saag, X. Wu, G. M. Shaw, J. C. Kappes. 2002. Emergence of resistant human immunodeficiency virus type 1 in patients receiving fusion inhibitor (T-20) monotherapy. Antimicrob. Agents Chemother. 46:1896-1905. [DOI] [PMC free article] [PubMed] [Google Scholar]