

Abstract
This study reveals a novel role of water as a chiral inducer, demonstrating its ability to drive the asymmetric resolution of prochiral silver-nucleobase complexes. During crystallization, helical water columns spontaneously form, selectively recognizing one enantiomer of the silver complex. This enantiospecific interaction drives the separation of the P and M enantiomers, leading to the formation of enantiopure crystals, whose chirality was confirmed through X-ray crystallography.


Introduction
Water is the most abundant inorganic compound in nature, found both in the Earth’s crust and within living organisms. Hydrogen bonding, which is responsible for the primary structural cohesion between water dipoles, also drives the formation of water clusters. − Their arrangement has been elucidated in the gaseous, liquid, and solid phases, revealing intricate geometric configurations, including discrete and polymeric constructs such as linear chains, tapes, nanotubes, layers, or three-dimensional structures. ,
Water clusters typically build in the interstices of crystalline networks, filling the voids left by crystallizing molecules, thereby providing stability and, in many cases, modifying or even governing the molecular arrangement. More importantly, water clusters play a key role in many biological processes, such as protein folding, enzyme activity, or DNA stability, actively influencing their structure and properties. − In this regard, it is expected that a water cluster assembled around a DNA fragment will adopt its inherent chirality. , This concept can be extended to the environs of other chiral natural and artificial systems, in which chirality is transferred from an organic or metal–ligand template to a well-defined water network. − Conversely, water clusters can also induce the assembly of supramolecular nanostructures and tune their optical properties.
In recent years, the use of purine nucleobases as N-heterocyclic carbene (NHC) ligands has been increasingly explored. − Most reports have focused on the antiproliferative activity of caffeine- and theophylline-based metal complexes. − Additionally, the use of these bioinspired ligands has been expanded to catalytically − and optically active systems as a greener and more sustainable alternative to traditional NHCs.
Our interest in metal-nucleobase complexes, led us to design and investigate flexible nucleobase- and N-donor based ligands. − In this study, a bidentate ligand (2), in which two theophylline synthons are linked by a −CH2–CH2–O–CH2–CH2– ether group, assembles into a dinuclear silver complex (3) with intrinsic chirality. During the crystallization process of 3, spontaneous formation of helical water columns occurs, in which the water selectively recognizes a homoenantiomer of complex 3 over the other. This recognition promotes the growth of enantiopure crystals and enables the chiral resolution of 3.
Results and Discussion
Ligand bis[2-(7-theophyllinyl)ethyl]ether (1), was conveniently prepared in N,N-dimethylformamide (DMF) by reacting theophylline with bis(2-chloroethyl)ether (Scheme ). Addition of water to the DMF solution prompted the precipitation of 1, which was isolated by filtration in air. A further reaction of 1 with methyl triflate (MeOTf) allowed for the methylation of the N9 sites of the two purine bases, with the subsequent formation of the triflate salt, 2[OTf]2 (Scheme ). In order to introduce a more suitable anion (Br–) into the medium for the ensuing treatment with silver, NBu4Br was added to a solution of 2[OTf]2, facilitating the isolation of 2[Br]2. Finally, metalation of both C8 positions of theophylline fragments by Ag+ units was achieved upon treatment of 2[Br]2 with silver acetate, [Ag(OAc)], yielding [Ag2(OAc)2(2 –2H)] (3).
1. Formation of Compounds 1, 2[OTf]2, 2[Br]2, and 3 .

a (i) 0.5 eq bis(chloroethyl)ether, NaOH, DMF, 80 °C; (ii) 2 eq MeOTf, CH2Cl2, RT; (iii) [TBA]Br, THF, RT; (iv) 4 eq [Ag(OAc)], CH2Cl2, RT.
NMR Analysis
The 1H NMR spectrum of 1 (CDCl3, Supporting Information) is consistent with a symmetrical (theophylline-N7)–CH2–CH2–O–CH2–CH2–(theophylline-N7) array. N1-CH3 and N3-CH3 methyl groups are observed as singlets at 3.34 and 3.55 ppm, whereas the N7-CH2 and O-CH2 signals appear at 4.37 and 3.71 ppm, respectively. Besides, the H8 proton resonates as a singlet at 7.40 ppm. Further NMR characterization is provided in the Supporting Information: 1H–1H COSY, 13C{1H}-APT, 1H–13C HSQC, and 1H–13C HMBC spectra.
In 2[OTf]2, the presence of methyl groups at the N9 sites of theophylline and the resulting positive charge, delocalized in the heterocyclic rings, provoke the expected shift of the peaks in the 1H NMR spectrum. This is more pronounced in those attached to the imidazolium ring, N7-CH2 (4.67 ppm) and N9-CH3 (4.14 ppm). In addition, the shift of the H8 signal (8.79 ppm) confirms methylation at the neighboring N9 position. Moreover, 2[Br]2 displays a 1H NMR pattern similar to that of 2[OTf]2, and its solubility in water reveals the H ⇋ D isotopic exchange undergone at the H8 site (Supporting Information).
The synthesis of 3 entails 2-fold C8-deprotonation of cation 2 and further coordination of both Ag+ ions, which is unambiguously diagnosed by the absence of the H8 signals in its 1H NMR spectrum (Figure ). Further peak distribution is as follows: methyl groups from the theophylline scaffold appear as singlets at 4.27 (N9-CH3), 3.84 (N3-CH3) and 3.35 (N1-CH3) ppm, whereas those of the acetate anions (OAc-CH3) are observed at 1.90 ppm, confirming their coordination to the silver centers. Interestingly, N7-CH2 and O-CH2 resonate as broad pseudotriplets at 4.59 and 3.86 ppm. The free rotation around the sigma single bonds of the seven-membered chain, (theophylline-N7)–CH2–CH2–O–CH2–CH2–(theophylline-N7), would render each CH2 group chemically and magnetically equivalent, leading to pure triplets. However, the observed pseudotriplets, combined with the signal broadening, suggest hindered conformational rotation. This restriction increases the number of distinct energy states available for nuclear interactions, thus affecting the signal patterns.
1.

VT 1H NMR spectra (CD2Cl2, 400 MHz) of 3.
This interpretation is corroborated by temperature-dependent 1H NMR experiments, where cooling of the samples causes further signal broadening and noticeable chemical shift changes (Figure ). Such behavior reflects a dynamic system influenced by temperature, likely due to conformational constraints imposed by the molecular structure and/or interactions.
The 13C{1H} NMR spectrum of 3 provides additional evidence of fluxional behavior. The C8 carbenic atom resonates as a singlet at 187.06 ppm. Besides, peaks corresponding to the acetate moiety are observed at 177.57 ppm (C13) and 22.56 ppm (C14). Typically, the presence of 107Ag and 109Ag isotopes would produce observable carbon–silver coupling doublets. However, their absence might indicate a nonrigid carbene-silver bond. , This fluxional nature is further suggested by the broadening of the methyl acetate peak (1.90 ppm) at room temperature.
To better understand the dynamic processes, 1H DOSY NMR spectroscopy was employed, revealing a single species in solution with a hydrodynamic radius (r H) of 4.63 Å (D = 1.10 × 10–9 m2 s–1). This finding supports the hypothesis that any dissociation or reformation of the Ag–C bond could occur rapidly on the NMR time scale at room temperature, maintaining the observed monomeric species in solution. Together, these results emphasize the interplay between hindered rotations, fluxionality, and rapid equilibria in defining the solution behavior of the Ag-nucleobase complex 3.
Solid State Analysis
Compound 1 was crystallized by diffusion of diethyl ether into chloroform. After several days, single crystals of the hydrate 1·0.25H2O were obtained. The partial incorporation of water into the crystal lattice was attributed to the presence of moisture in the solvents. Its solid state structure (Figure ) is consistent with that determined by 1H NMR spectroscopy, where the −CH2–CH2–O–CH2–CH2– fragment is W-shaped and acts as a bridge between the two theophylline units, which are connected via their N7 sites. A notable feature of the geometry in compound 1 is that the C8–N7–C5 angles in both imidazole rings (105.86(15)°; 106.14(15)°) are larger than the corresponding C8–N9–C4 angles (103.22(15)°; 103.07(15)°), likely due to the coordination of the ether bridge at the latter positions. In addition, both purine rings are mutually tilted by 34.87(3)°.
2.

View of 1 with atom numbering scheme.
2-Fold methylation of compound 1 with methyl triflate afforded 2[OTf]2, whose structure was also determined by X-ray crystallography (Figure ). Intermolecular bond distances and angles are comparable to those of 1, except for the endocyclic C–N–C angles within the imidazole rings, which in 2 exhibit identical values due to methylation at N9: 107.77(19)° (C8a–N7a–C5a), 107.31(19)° (C8a–N9a–C4a), 107.45(19)° (C8b–N7b–C5b), and 107.55(19)° (C8b–N9b–C4b). Folding of cation 2 in 2[OTf]2 displays a slight deviation from that of 1, with both nucleobases tilted by 11.12(10)° in a nearly antiparallel arrangement, with their C8 positions oriented toward each other.
3.

Front view of cation 2 in 2[OTf]2.
Treatment of 2[OTf]2 with tetrabutylammonium bromide resulted in the formation of 2[Br]2, as described above. Interestingly, when 2[Br]2 was crystallized in methanolic media, crystals of the 2[Br]2·CH3OH adduct were obtained. Although interatomic distances and angles of cation 2 in 2[Br]2·CH3OH are almost identical to those in 2[OTf]2, its conformation differs. As depicted in Figure , cation 2 exhibits a U-conformation (C s symmetry), accommodating a Br– anion and a methanol molecule in its cavity, forming a host–guest system, in which Br– anion is connected by hydrogen bonding to methanol (MeOH···Br, 3.250(3) Å), and attached to the cationic host via electrostatics and anion−π interactions.
4.

Detail of the host–guest system {Br,MeOH ⊂ 2} in 2[Br]2·CH3OH. A Br– anion has been omitted for clarity.
To use cation 2 as an NHC ligand precursor, the C8 position of the nucleobase was deprotonated by adding four equivalents of silver acetate to a suspension of 2[Br]2 in dichloromethane. As anticipated above, this treatment resulted in the coordination of a silver ion to each nucleobase via its C8 site, with the silver linear coordination sphere completed by an acetate ligand, forming [Ag2(OAc)2(2 –2H)] (3). Compound 3 was initially isolated as a white, solvent-free powder. However, crystallization from a dimethyl sulfoxide (DMSO) solution yielded crystals that contained two solvent molecules in the asymmetric unit, identified as 3·2DMSO.
Figure provides a view of one enantiomer (see below) of 3 in 3·2DMSO. The C 2 symmetry observed in NMR measurements is disrupted in the crystal structure. As a result, while the coordination environments of silver are analogous, they are not identical in the solid state. Bond distances involving Ag+ ions are within the expected range: 2.083(3) Å (Ag1–C8a), 2.137(2) Å (Ag1–O11), 2.081(3) Å (Ag2–C8b), and 2.156(2) Å (Ag2–O21). In addition, separation between the two silver atoms is 3.9538(4) Å, which is probably too long to be considered a metallophilic interaction. Coordination angles involving silver atoms deviate slightly from linearity: C8a–Ag1–O11, 170.78(10)°; (C8b–Ag2–O21), 170.42(10)°. Regarding its spatial conformation, complex 3 adopts an antiparallel orientation, similar to that of 2[OTf]2, with the C8 positions facing each other and a mutual tilt angle of 20.00(6)°. The acetate ligands are twisted relative to the nucleobases attached to the same silver atom, with torsion angles of 23.10(13)° (Ag1) and 34.85(16)° (Ag2). The uncoordinated oxygen atoms (O12, O22) point toward the Hoogsteen edges of the respective nucleobases.
5.

Side view of the (P)-3 enantiomer in 3·2DMSO. For P- and M-chirality see Figure .
More importantly, 3 crystallizes in two chiral isomers (Figure ), P and M, both of which are present in the crystal packing of 3·2DMSO. Both forms exhibit nearly identical (enantiomeric) arrangements, with the chirality of 3 in the solid state arising from its three-dimensional folding. To describe the two enantiomers of 3, namely (P)-3 and (M)-3, a scheme analogous to that employed for helicenes is utilized. As depicted in Figure (left), a P-loop can be described starting at front-right acetate ligand, passing through its coordinated Ag and the adjoining nucleobase, then turning clockwise via the ether bridge to the other nucleobase, before reaching Ag′ and its attached back-left acetate ligand. A M-loop follows a counterclockwise rotation (Figure , right).
6.

Conceptual depiction of the helical chirality exhibited by the (P)-3 and (M)-3 enantiomers in 3·2DMSO.
Remarkably, the crystal packing of the racemic solvate 3·2DMSO exhibits a staggered arrangement, with π–π stacking interactions between symmetry-related nucleobases in an ···M···P··· pattern (M and P refer to the different enantiomers of 3), with stack distances of 3.4 Å (Figure ).
7.

Staggered arrangement of 3 in 3·2DMSO featuring π–π stacking. For P- and M-chirality see Figure .
Further crystallization assays were carried out by diffusing hexane into solutions of 3 in either dichloromethane or chloroform. Here, the use of undried solvents (taken directly from the bottle) resulted in the formation of crystalline hydrates with the formula 3·3H2O. Thus, during the crystallization process, trace amounts of water are extracted from solvents, resulting in the formation of stable, durable crystals. Once the crystals have formed and the solvents are removed, they remain stable without any water loss for weeks when stored in air at room temperature.
Under the microscope, the resulting crystals appeared indistinguishable, differing only slightly in size and other expected characteristics. To determine the chirality of the crystals, they were manually selected and mounted onto a diffractometer for characterization. The first crystal selected for X-ray diffraction analysis was assigned to the trigonal P3121 space group, and further identified as (M)-3·(P)-3H2O, where M indicates the helicity of 3 (Figure ) and P the helicity of the water chain (see below). Its absolute configuration was unambiguously established based on the Flack parameter: 0.029(8). In order to verify whether all the crystalline material exhibited the same chirality, several arbitrarily selected samples were analyzed. After multiple attempts, including X-ray measurements and refinements, we successfully identified a crystal that fitted within the P3221 space group. The Flack parameter of 0.017(9) confirmed it as (P)-3·(M)-3H2O, which is the enantiomer of (M)-3·(P)-3H2O. Subsequent measurements of other picked crystals showed an equal distribution of the two enantiomers.
Hereinafter, only the crystal structure of (M)-3·(P)-3H2O is described, as it is almost identical to that of (P)-3·(M)-3H2O. In (M)-3·(P)-3H2O, complex 3 exhibits a crystallographic C 2 axis that intersects the oxygen atom of the bridging ether, making the two halves of 3 symmetry-equivalent. Molecular arrangement of complex 3 in (M)-3·(P)-3H2O is similar to that exhibited in 3·2DMSO. However, while in the DMSO adduct the terminal oxygens of the acetate ligands are oriented toward the ether chain (Figure ), in (M)-3·(P)-3H2O and (P)-3·(M)-3H2O, they are rotated by 180°, pointing instead toward sugar edges of the theophylline fragments (Supporting Information).
The unit cell of (M)-3·(P)-3H2O contains complex 3 and two crystallographically different water molecules (O1w and O2w), both of which participate in hydrogen bonding, interacting with each other and/or with the acetate ligands. Hydrogen bonding distances are as follows: O1w···O1w′, 2.78(4) Å; O1w···O2w, 2.767(18) Å; O2w···O2w′, 2.878(16) Å; O1w···O12(acetate), 2.641(19) Å. In the crystal packing, the hydrogen bonding network between H2O molecules organizes into helical, enantiopure one-dimensional water chains. Specifically, symmetry related O2w water molecules form a P-helix that twists around the c axis (Figure ).
8.

Schematic representation of the P-helical backbone within the water cluster of (M)-3·(P)-3H2O. Red dot: oxygen atom.
The stability of this homochiral backbone is reinforced by envelope-like 5-membered water clusters, which surround the helix, and consist of three consecutive water molecules within the P-helix (O2w), along with two additional water molecules (O1w). The resulting fused pentagons form an intricate 1D array that preserves the P-helicity (Figure ).
In addition, the cisoid arrangement of both acetate terminal oxygens toward the sugar edges allows complex 3 to bind the water cluster via 2-fold O1w···O12 (acetate) hydrogen bonds (Figure ). These interactions are shorter than those involving water–water contacts (see above).
9.

Detail of the helical arrangement of 3 in (M)-3·(P)-3H2O around the respective chiral water chains.
Remarkably, the helical arrangement of the water array induces an enantiospecific hydrogen bonding interaction with complex 3, resulting in spontaneous chiral resolution. Such enantiomeric discrimination is not observed in the DMSO adduct. In particular, left-handed (M)-3 units are attached to the right-handed (P) water chains to form (M)-3·(P)-3H2O. In the case of the (P)-3·(M)-3H2O, the contrary occurs, and right-handed complex (P)-3 is recognized by the left-handed (M) water helix. Besides, positioning of silver complex 3 around the water chain results in a suprastructure that also maintains the helicity of the water cluster (Figure ).
10.
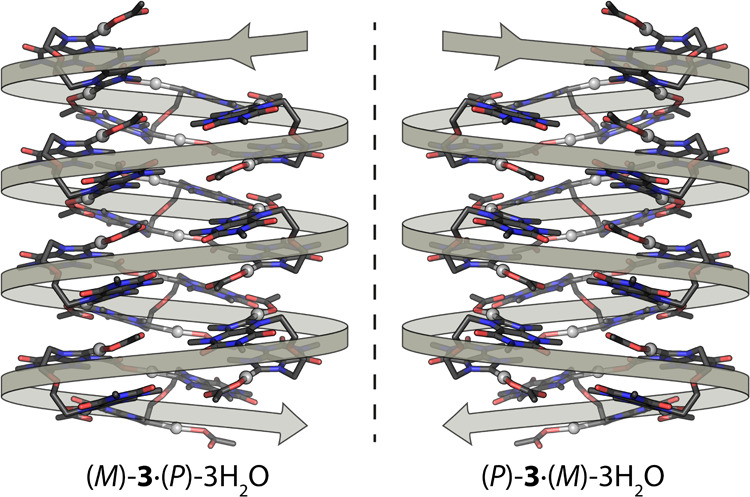
Two enantiomeric helical superstructures within the crystal packings of 3·3H2O.
To summarize, the transfer of chirality by a single achiral molecule is fundamentally limited. As demonstrated here, this situation changes when achiral molecules self-organize into supramolecular chiral assemblies. Such systems can generate chirality through interactions and molecular recognition, even in the absence of intrinsic chirality in the individual components.
Conclusions
The synthesis of the ether-nucleobase derivative 1 and the ditopic proligands 2[OTf]2 and 2[Br]2 facilitated the preparation of the silver-nucleobase complex 3, which was crystallized in DMSO and in chlorinated solvents.
In crystals grown from DMSO, complex 3 adopts two enantiomeric isomers, namely P and M, which arise from rotation around the ether bridge. Within the crystal lattice, both enantiomers alternate in intermolecular π–π interactions, stabilizing a racemic ladder-like structure. When crystallized from wet apolar solvents such as CHCl3 or CH2Cl2, crystalline prisms of 3·3H2O assemble to achieve 1D helical water columns. Moreover, (M)-3·(P)-3H2O crystallizes in the P3121 space group, where 3 adopts M-chirality, while the water array exhibits P-helicity. In contrast, the enantiomeric crystal (P)-3·(M)-3H2O is assigned to the P3221 space group.
The crystal lattice of 3·3H2O is primarily stabilized by hydrogen bonding interactions, with other packing and intermolecular forces contributing to a lesser extent. This suggests that the arrangement of water molecules and their interactions with the silver complex 3 are fundamental to its structural integrity.
Therefore, chiral resolution of enantiomers during crystallization occurs spontaneously, without chiral auxiliaries. In this process, water drives the formation of enantiopure crystals, underscoring its pivotal role as a natural inducer of chirality. These findings offer fresh insights into stereochemical organization and highlight the unique ability of water to influence chiral outcomes in supramolecular assemblies.
Experimental Section
General Procedures
All the reagents used in this work were purchased from commercial sources and used as received. Glassware was dried at 140 °C before use. Unless otherwise stated, all reactions were carried out under aerobic conditions. CH2Cl2 was obtained oxygen- and waterfree from a Solvent Purification System (Innovative Technologies). Crystallization procedures were performed using undried solvents. 1H and 13C{1H} NMR spectra were recorded on Bruker Avance 300 (300.13 and 75.48 MHz, respectively) and Bruker Avance 400 (400.16 and 100.61 MHz, respectively) spectrometers. Spectral assignments were achieved by combination of 1H–1H COSY, 13C{1H}-APT and 1H–13C HSQC/HMBC. NMR chemical shifts (expressed in parts per million) are referenced to residual solvent peaks (1H and 13C). Methanol was used as internal standard in 13C{1H} and 1H–13C HSQC/HMBC spectra carried out in D2O. Coupling constants, J, are given in hertz (Hz). High-resolution electrospray mass spectra (HRMS) were acquired using a MicroTOF-Q hybrid quadrupole time-of-flight spectrometer (Bruker Daltonics, Bremen, Germany). UV–visible spectra in solution were recorded on a JASCO V-670 UV–vis spectrophotometer.
X-ray diffraction data were collected on a Bruker D8 Venture diffractometer, using graphite-monochromated Mo Kα radiation (λ = 0.71073 Å). Diffracted intensities were integrated and corrected for absorption effects using the multiscan method. − Both procedures are included in the APEX4 package. All the structures were solved by direct methods with SHELXS and refined by full-matrix least-squares on F 2 with SHELXL.
Crystal Data for Compound 1·0.25H2O
C72H90N32O21, M r = 1739.75, colorless plate, monoclinic, P21/c, a = 7.2685(3) Å, b = 15.7196(6) Å, c = 17.4713(7) Å, β = 101.1997(13)°, V = 1958.22(14) Å3, Z = 1, T = 100(2) K, D calcd = 1.475 g cm–3, μ = 0.112 mm–1, absorption correction factors min. 0.090 max. 0.988, 69364 reflections, 4890 unique (R int = 0.1032), 3432 observed, R 1 = 0.0491 [I > 2σ(I)], wR 2(F 2) = 0.1322 (all data), GOF = 1.018. CCDC 2410723.
Crystal Data for Compound 2[OTf]2
C22H28F6N8O11S2, M r = 758.64, colorless prism, triclinic P–1, a = 9.4248(8) Å, b = 12.1373(11) Å, c = 13.4277(12) Å, α = 92.6242(12)°, β = 90.4004(12)°, γ = 103.6345(12)°, V = 1490.9(2) Å3, Z = 2, T = 100(2) K, D calcd = 1.690 g cm–3, μ = 0.289 mm–1, absorption correction factors min. 0.822 max. 0.918, 19195 reflections, 7261 unique (R int = 0.0408), 5185 observed, R 1 = 0.0484 [I > 2σ(I)], wR 2(F 2) = 0.1147 (all data), GOF = 1.041. CCDC 2410724.
Crystal Data for Compound 2[Br]2·CH3OH
C21H32Br2N8O6, M r = 652.36, colorless block, monoclinic P21/c, a = 8.4766(3) Å, b = 22.1517(6) Å, c = 14.5982(5) Å, β = 103.2819(13)°, V = 2667.80(15) Å3, Z = 4, T = 100(2) K, D calcd = 1.624 g cm–3, μ = 3.091 mm–1, absorption correction factors min. 0.739 max. 0.790, 109976 reflections, 6638 unique (R int = 0.0456), 6088 observed, R 1 = 0.0333 [I > 2σ(I)], wR 2(F 2) = 0.0938 (all data), GOF = 1.069. CCDC 2410725.
Crystal Data for Compound 3·2DMSO
C28H44Ag2N8O11S2, M r = 948.57, colorless block, monoclinic P21/c, a = 20.1200(11) Å, b = 8.5680(5) Å, c = 22.0957(12) Å, β = 106.777(2)°, V = 3646.9(4) Å3, Z = 4, T = 100(2) K, D calcd = 1.728 g cm–3, μ = 1.256 mm–1, absorption correction factors min. 0.727 max. 0.828, 107317 reflections, 9107 unique (R int = 0.0511), 7984 observed, R 1 = 0.0354 [I > 2σ(I)], wR 2(F 2) = 0.0899 (all data), GOF = 1.158. CCDC 2410726.
Crystal Data for Compound (M)-3·(P)-3H2O
C24H38Ag2N8O12, M r = 846.36, colorless prism, trigonal P3121, a = 19.5911(6) Å, c = 7.0016(3) Å, V = 2327.26(17)Å3, Z = 3, T = 100(2) K, D calcd = 1.812 g cm–3, μ = 1.336 mm–1, absorption correction factors min. 0.842 max. 0.899, 75560 reflections, 3880 unique (R int = 0.0545), 3763 observed, R 1 = 0.0938 [I > 2σ(I)], wR 2(F 2) = 0.1966 (all data), GOF = 1.092. CCDC 2410727.
Crystal Data for Compound (M)-3·(P)-3H2O
C24H38Ag2N8O12, M r = 846.36, colorless prism, trigonal P3221, a = 19.5955(4) Å, c = 7.0045(3) Å, V = 2329.27(14) Å3, Z = 3, T = 100(2) K, D calcd = 1.810 g cm–3, μ = 1.335 mm–1, absorption correction factors min. 0.805 max. 0.923, 90779 reflections, 3878 unique (R int = 0.0553), 3729 observed, R 1 = 0.0902 [I > 2σ(I)], wR 2(F 2) = 0.2029 (all data), GOF = 1.097. CCDC 2410728.
Synthesis of Bis(theophylline-N7-ethyl)ether, 1
Theophylline (3.30 g, 18.3 mmol) and NaOH (1.06 g, 26.5 mmol) were suspended in 20 mL of DMF and the mixture was heated to 50 °C. Then, bis(2-chloroethyl) ether (1.3 mL, 11.1 mmol) was added and the temperature was raised to 80 °C. After 40 h the mixture was cooled down to room temperature and 20 mL of water were added. The white suspension was filtered, washed with water (2 × 10 mL) and methanol (2 × 5 mL) and dried under vacuum to yield 1 as a white solid (3.00 g, 76%). 1H NMR (400 MHz, CDCl3, 298 K): δ 7.40 (s, 2H, H8), 4.37 (t, 3 J H–H = 4.5 Hz, 4H, N7-CH2), 3.71 (t, 3 J H–H = 4.5 Hz, 4H, O-CH2), 3.55 (s, 6H, N3-CH3), 3.34 (s, 6H, N1-CH3). 13C{1H} NMR (100 MHz, CDCl3, 298 K): δ 155.30 (C6), 151.63 (C2), 149.08 (C4), 141.93 (C8), 106.56 (C5), 69.38 (O-C), 46.89 (N7-C), 29.88 (N3-C), 28.02 (N1-C). HRMS (CHCl3/CH3CN) [M + Na]+ calcd. for C18H22N8O5Na: 453.1605; found: 453.1603.
Synthesis of Bis(theophylline-N9-methyl-N7-ethyl)ether Triflate, 2[OTf]2
Under argon atmosphere, methyl trifluoromethanesulfonate (0.75 mL, 6.65 mmol) was added to a suspension of 1 (1.00 g, 2.34 mmol) in 20 mL of dry dichloromethane and the mixture was stirred at room temperature. The suspension gradually disappeared upon 1 h of stirring, giving rise to a colorless solution. Shortly after, a white precipitate appeared, and the mixture was stirred at room temperature for 3 days. Filtration and washings with dichloromethane (2 × 10 mL) and diethyl ether (3 × 10 mL) afforded 2[OTf]2 as a white solid (1.56 g, 2.06 mmol, 88%). 1H NMR (400 MHz, CD3CN, 298 K): δ 8.79 (s, 2H, H8), 4.67 (t, 3 J H–H = 4.8 Hz, 4H, N7-CH2), 4.14 (s, 6H, N9-CH3), 3.81 (t, 3 J H–H = 4.8 Hz, 4H, O-CH2), 3.74 (s, 6H, N3-CH3), 3.31 (s, 6H, N1-CH3). 13C{1H} NMR (100 MHz, CD3CN, 298 K): δ 154.75 (C6), 151.45 (C2), 140.83 (C4), 140.11 (C8), 108.80 (C5), 68.63 (O-C), 49.83 (N7-C), 38.09 (N9-C), 32.22 (N3-C), 29.14 (N1-C). 19F NMR (282 MHz, CD3CN, 298 K): δ −79.40 (s, OTf). HRMS (CH3CN) [M – 2OTf]2+ calcd. for C20H28N8O5: 230.1086; found: 230.1093/[M – OTf]+ calcd. for C21H28N8O8SF3: 609.1697; found: 609.1727.
Synthesis of Bis(theophylline-N9-methyl-N7-ethyl)ether Bromide, 2[Br]2
2[OTf]2 (298 mg, 0.39 mmol) was suspended in 15 mL of THF and tetrabutylammonium bromide (326 mg, 1.01 mmol) was added. After stirring at room temperature for 15 h, the white precipitate was filtered off, washed with further THF and acetone (2 × 5 mL) and dried under vacuum to yield 2[Br]2 as a white solid (240 mg, 0.38 mmol, 98%). 1H NMR (300 MHz, D2O, 298 K): δ 4.75 (t, 3 J H–H = 4.9 Hz, 4H, N7-CH2), 4.24 (s, 6H, N9-CH3), 3.96 (t, 3 J H–H = 4.9 Hz, 4H, O-CH2), 3.83 (s, 6H, N3-CH3), 3.37 (s, 6H, N1-CH3). 13C{1H} NMR (100 MHz, D2O, 298 K): δ 155.12 (C6), 152.09 (C2), 140.46 (C4), 140.12 (t, 1 J C–D = 30.0 Hz, C8), 108.84 (C5), 68.64 (O-C), 49.25 (N7-C), 37.93 (N9-C), 32.38 (N3-C), 29.31 (N1-C). HRMS (CH3OH) [M – 2Br]2+ calcd. for C20H28N8O5: 230.1086; found: 230.1093/[M – Br]+ calcd. for C20H28N8O5Br: 541.1343; found: 541.1349.
Synthesis of 3
2[Br]2 (111 mg, 0.18 mmol) was suspended in 12 mL of dichloromethane and silver acetate (129 mg, 0.77 mmol) was added. The white precipitate gradually transformed into a yellowish suspension in the span of 15 min. After stirring overnight, the yellow solid was removed upon filtration through Celite and the colorless solution was concentrated to about 1 mL under reduced pressure. Addition of 10 mL of hexane afforded a white solid which was filtered, washed with further hexane (2 × 5 mL) and dried under vacuum (110 mg, 0.14 mmol, 77%). 1H NMR (400 MHz, CD2Cl2, 298 K): 4.59 (pt, 3 J H–H = 4.2 Hz, 4H, N7-CH2), 4.27 (s, 6H, N9-CH3), 3.86 (br-pt, 4H, O-CH2), 3.84 (s, 6H, N3-CH3), 3.35 (s, 6H, N1-CH3), 1.90 (s, 6H, OAc-CH3). 13C{1H} NMR (100 MHz, CD2Cl2, 298 K): 187.06 (C8), 177.57 (C13), 154.03 (C6), 151.32 (C2), 141.41 (C4), 109.46 (C5), 69.27 (O-C), 52.19 (N7-C), 40.47 (N9-C), 32.32 (N3-C), 28.99 (N1-C), 22.56 (C14). HRMS (CDCl3/CH3OH) [M – Ag – 2 OAc]+ calcd. for C20H26N8O5Ag: 565.1072; found: 565.1049.
Supplementary Material
Acknowledgments
Financial support from the University of Zaragoza, the Aragón Government (A.P. predoctoral fellow, E42_23R, E05_23R), and the MCIU/AEI/FEDER (PID2021-122406NB-I00 and PID2022-137208NB-I00) is kindly acknowledged.
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.inorgchem.4c05384.
1D and 2D NMR spectra for all compounds (PDF)
The authors declare no competing financial interest.
References
- Latimer W. M., Rodebush W. H.. Polarity and ionization from the standpoint of the Lewis Theory of Valence. J. Am. Chem. Soc. 1920;42(7):1419–1433. doi: 10.1021/ja01452a015. [DOI] [Google Scholar]
- Ludwig R.. Water: From Clusters to the Bulk. Angew. Chem., Int. Ed. 2001;40:1808–1827. doi: 10.1002/1521-3773(20010518)40:10<1808::AID-ANIE1808>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- Wang B., Jiang W., Dai X., Gao Y., Wang Z., Zhang R.-Q.. Molecular orbital analysis of the hydrogen bonded water dimer. Sci. Rep. 2016;6(1):22099. doi: 10.1038/srep22099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K., Cruzan J. D., Saykally R. J.. Water clusters. Science. 1996;271:929–933. doi: 10.1126/science.271.5251.929. [DOI] [Google Scholar]
- Keutsch F. N., Saykally R. J.. Water clusters: Untangling the mysteries of the liquid, one molecule at a time. Proc. Natl. Acad. Sci. U.S.A. 2001;98:10533–10540. doi: 10.1073/pnas.191266498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg, D. ; Kauzmann, W. . The Structure and Properties of Water; Oxford University Press, 2005. [Google Scholar]
- Infantes L., Motherwell S.. Water clusters in organic molecular crystals. CrystEngcomm. 2002;4:454–461. doi: 10.1039/b204934a. [DOI] [Google Scholar]
- Wei D. G., Wilson W. D., Neidle S.. Small-molecule Binding to the DNA Minor Groove Is Mediated by a Conserved Water Cluster. J. Am. Chem. Soc. 2013;135:1369–1377. doi: 10.1021/ja308952y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pullanchery S., Roke S.. Handy water: Chiral superstructures around peptide β-sheets. Proc. Natl. Acad. Sci. U.S.A. 2021;118(2):e2024376118. doi: 10.1073/pnas.2024376118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steber A. L., Temelso D., Kisiel Z., Schnell M., Pérez C.. Rotational dive into the water clusters on a simple sugar substrate. Proc. Natl. Acad. Sci. U.S.A. 2023;120:e2214970120. doi: 10.1073/pnas.2214970120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayaram B., Jain T.. The role of water in protein-DNA recognition. Annu. Rev. Biophys. Biomol. Struct. 2004;33:343–361. doi: 10.1146/annurev.biophys.33.110502.140414. [DOI] [PubMed] [Google Scholar]
- Ball P.. Water as an Active Constituent in Cell Biology. Chem. Rev. 2008;108:74–108. doi: 10.1021/cr068037a. [DOI] [PubMed] [Google Scholar]
- Zhong D., Pal S. K., Zewail A. H.. Biological water: A critique. Chem. Phys. Lett. 2011;503:1–11. doi: 10.1016/j.cplett.2010.12.077. [DOI] [Google Scholar]
- Duboué-Dijon E., Fogarty A. C., Hynes J. T., Laage D.. Dynamical Disorder in the DNA Hydration Shell. J. Am. Chem. Soc. 2016;138:7610–7620. doi: 10.1021/jacs.6b02715. [DOI] [PubMed] [Google Scholar]
- Kocsis I., Sorci M., Vanselous H., Murail S., Sanders S. E., Licsandru E., Legrand Y.-M., van der Lee A., Baaden M., Petersen P. B.. et al. Oriented chiral water wires in artificial transmembrane channels. Sci. Adv. 2018;4:eaao5603. doi: 10.1126/sciadv.aao5603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su D.-D., Barboiu M.. Hydrogen-bonded water-wires/clusters – Toward natural selectivity of artificial water channels. Coord. Chem. Rev. 2024;515:215973. doi: 10.1016/j.ccr.2024.215973. [DOI] [Google Scholar]
- Perets E. A., Yan E. C. Y.. The H2O Helix: The Chiral Water Superstructure Surrounding DNA. ACS Cent. Sci. 2017;3:683–685. doi: 10.1021/acscentsci.7b00229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDermott M. L., Vanselous H., Corcelli S. A., Petersen P. B.. DNA’s Chiral Spine of Hydration. ACS Cent. Sci. 2017;3:708–714. doi: 10.1021/acscentsci.7b00100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar N., Khullar S., Mandal S. K.. Encapsulation of a Water Octamer Chain in a Chiral 2D Sheetlike Supramolecular Coordination Network Composed of Dinickel-Dicarboxylate Subunits. ACS Omega. 2018;3:11062–11070. doi: 10.1021/acsomega.8b01355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu B., Wang S., Wang R., Xu J., Yuan D., Hou H.. Chiral Metallocycles Templated Novel Chiral Water Frameworks. Cryst. Growth Des. 2013;13:518–525. doi: 10.1021/cg300971r. [DOI] [Google Scholar]
- Han L.-L., Zhang X.-Y., Chen J.-S., Li Z.-H., Sun D.-F., Wang X.-P., Sun D.. Silver(I)/Bipyrazole/Dicarboxylate Interpenetrated Coordination Networks: Spontaneous Chiral Resolution, Modulation of Topologies, Water Clusters, and Photoluminescences. Cryst. Growth Des. 2014;14:2230–2239. doi: 10.1021/cg401805x. [DOI] [Google Scholar]
- Ganguly S., Mondal R.. Coordination Driven Self-Assembly in Co(II) Coordination Polymers Displaying Unprecedented Topology, Water Cluster, Chirality, and Spin-Canted Magnetic Behavior. Cryst. Growth Des. 2015;15:2211–2222. doi: 10.1021/cg5018592. [DOI] [Google Scholar]
- Otake K.-I., Otsubo K., Komatsu T., Dekura S., Taylor J. M., Ikeda R., Sugimoto K., Fujiwara A., Chou C.-P., Sakti A. W.. et al. Confined water-mediated high proton conduction in hydrophobic channel of a synthetic nanotube. Nat. Commun. 2020;11(1):843. doi: 10.1038/s41467-020-14627-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang X., Chu D., Jiang H., Gong W., Jiang C., Cui Y., Liu Y.. Supramolecular self-assembly of chiral helical tubular polymers with amplified circularly polarized luminescence. Mater. Chem. Front. 2020;4:2772–2781. doi: 10.1039/D0QM00416B. [DOI] [Google Scholar]
- Tan K. T., Tao S., Huang N., Jiang D.. Water cluster in hydrophobic crystalline porous covalent organic frameworks. Nat. Commun. 2021;12(1):6747. doi: 10.1038/s41467-021-27128-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan G., Zhu C., Liu Y., Fang Y., Cui Y.. Water clusters induced assembly of chiral organic microstructures showing reversible phase transformations and luminescence switching. Chem. Commun. 2010;46:2307–2309. doi: 10.1039/b923259a. [DOI] [PubMed] [Google Scholar]
- Brackemeyer D., Hervé A., To Brinke C. S., Jahnke M. C., Hahn F. E.. A Versatile Methodology for the Regioselective C8-Metalation of Purine Bases. J. Am. Chem. Soc. 2014;136:7841–7844. doi: 10.1021/ja5030904. [DOI] [PubMed] [Google Scholar]
- Leitao M. I. P. S., Gonzalez C., Francescato G., Filipiak Z., Petronilho A.. On the reactivity of mRNA Cap0: C–H oxidative addition of 7-methylguanosine to Pt0 and base pairing studies. Chem. Commun. 2020;56:13365–13368. doi: 10.1039/D0CC06075E. [DOI] [PubMed] [Google Scholar]
- Odena C., Santiago T. G., Linares M. L., Castellanos-Blanco N., McGuire R. T., Chaves-Arquero B., Alonso J. M., Diéguez-Vázquez A., Tan E., Alcázar J., Buijnsters P., Cañellas S., Martin R.. Late-Stage C(sp2)-C(sp3) Diversification via Nickel Oxidative Addition Complexes. J. Am. Chem. Soc. 2024;146:21264–21270. doi: 10.1021/jacs.4c08404. [DOI] [PubMed] [Google Scholar]
- Kascatan-Nebioglu A., Melaiye A., Hindi K., Durmus S., Panzner M. J., Hogue L. A., Mallett R. J., Hovis C. E., Coughenour M., Crosby S. D., Milsted A., Ely D. L., Tessier C. A., Cannon C. L., Youngs W. J.. Synthesis from Caffeine of a Mixed N-Heterocyclic Carbene-Silver Acetate Complex Active against Resistant Respiratory Pathogens. J. Med. Chem. 2006;49:6811–6818. doi: 10.1021/jm060711t. [DOI] [PubMed] [Google Scholar]
- Bertrand B., Stefan L., Pirrotta M., Monchaud D., Bodio E., Richard P., Le Gendre P., Warmerdam E., de Jager M. H., Groothuis G. M. M., Picquet M., Casini A.. Caffeine-Based Gold(I) N-Heterocyclic Carbenes as Possible Anticancer Agents: Synthesis and Biological Properties. Inorg. Chem. 2014;53:2296–2303. doi: 10.1021/ic403011h. [DOI] [PubMed] [Google Scholar]
- Meier-Menches S. M., Neuditschko B., Zappe K., Schaier M., Gerner M. C., Schmetterer K. G., Del Favero G., Bonsignore R., Cichna-Markl M., Koellensperger G.. et al. An Organometallic Gold(I) Bis-N-Heterocyclic Carbene Complex with Multimodal Activity in Ovarian Cancer Cells. Chem. - Eur. J. 2020;26:15528–15537. doi: 10.1002/chem.202003495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francescato G., Leitão M. I. P. S., Orsini G., Petronilho A.. Synthesis and Medicinal Applications of N-Heterocyclic Carbene Complexes Based on Caffeine and Other Xanthines. ChemMedchem. 2024;19:e202400118. doi: 10.1002/cmdc.202400118. [DOI] [PubMed] [Google Scholar]
- Zhang J.-J., Muenzner J. K., el Maaty M. A. A., Karge B., Schobert R., Wölfl S., Ott I.. A multi-target caffeine derived rhodium(I) N-heterocyclic carbene complex: Evaluation of the mechanism of action. Dalton Trans. 2016;45:13161–13168. doi: 10.1039/C6DT02025A. [DOI] [PubMed] [Google Scholar]
- Kaußler C., Wragg D., Schmidt C., Moreno-Alcántar G., Jandl C., Stephan J., Fischer R. A., Leoni S., Casini A., Bonsignore R.. Dynamical Docking” of Cyclic Dinuclear Au(I) Bis-N-heterocyclic Complexes Facilitates Their Binding to G-Quadruplexes. Inorg. Chem. 2022;61:20405–20423. doi: 10.1021/acs.inorgchem.2c03041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazars F., Delaude L.. Greening” Ruthenium-Arene Catalyst Precursors with N-Heterocyclic Carbene Ligands Derived from Caffeine and Theophylline. Organometallics. 2023;42:1589–1597. doi: 10.1021/acs.organomet.3c00166. [DOI] [Google Scholar]
- Zhang J., Rahman M. M., Zhao Q., Feliciano J., Bisz E., Dziuk B., Lalancette R., Szostak R., Szostak M.. N-Heterocyclic Carbene Complexes of Nickel(II) from Caffeine and Theophylline: Sustainable Alternative to Imidazol-2-ylidenes. Organometallics. 2022;41:1806–1815. doi: 10.1021/acs.organomet.2c00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szadkowska A., Staszko S., Zaorska E., Pawłowski R.. A theophylline based copper N-heterocyclic carbene complex: Synthesis and activity studies in green media. RSC Adv. 2016;6:44248–44253. doi: 10.1039/C6RA06682H. [DOI] [Google Scholar]
- Bysewski O., Klosterhalfen N., Jordan R., Kletsch L., Winter A., Klein A., Dietzek-Ivanšić B., Schubert U. S.. Luminescent Platinum(II) Complexes with a Tridentate Caffeine-Based NHC-Pincer Ligand: Synthesis, Electrochemistry and Photophysics. Eur. J. Inorg. Chem. 2024;27(5):e202300620. doi: 10.1002/ejic.202300620. [DOI] [Google Scholar]
- Chaudhary A., Mathur D., Gaba R., Pasricha R., Sharma K.. Greening up organic reactions with caffeine: Applications, recent developments, and future directions. RSC Adv. 2024;14:8932–8962. doi: 10.1039/D4RA00432A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vellé A., Cebollada A., Iglesias M., Sanz Miguel P. J.. Argentophilicity as Essential Driving Force for a Dynamic Cation-Cation Host-Guest System: [Ag(acetonitrile)2]+ ⊂ [Ag2(bis-NHC)2]2+ (NHC = N-heterocyclic carbene) Inorg. Chem. 2014;53:10654–10659. doi: 10.1021/ic501715h. [DOI] [PubMed] [Google Scholar]
- Cebollada A., Vellé A., Iglesias M., Fullmer L. B., Goberna-Ferrón S., Nyman M., Sanz Miguel P. J.. Direct X-Ray Scattering Evidence for Metal–Metal Interactions in Solution at the Molecular Level. Angew. Chem., Int. Ed. 2015;54:12762–12766. doi: 10.1002/anie.201505736. [DOI] [PubMed] [Google Scholar]
- Vellé A., Rodríguez-Santiago L., Sodupe M., Sanz Miguel P. J.. Enhanced Metallophilicity in Metal-Carbene Systems: Stronger Character of Aurophilic Interactions in Solution. Chem. - Eur. J. 2020;26:997–1002. doi: 10.1002/chem.201904507. [DOI] [PubMed] [Google Scholar]
- Quintana M., Rodríguez-Rius A., Vellé A., Vives S., Sanz Miguel P. J., Triola G.. Dinuclear silver and gold bisNHC complexes as drug candidates for cancer therapy. Bioorg. Med. Chem. 2022;67:116814. doi: 10.1016/j.bmc.2022.116814. [DOI] [PubMed] [Google Scholar]
- Polo A., Gutiérrez Merino L., Rodríguez R., Sanz Miguel P. J.. Chirality at Metal in a Linear [Ag(NHC)2]+ Complex: Stereogenic C–Ag–C Axis, Atropisomerism and Role of π-π Interactions. Chem. - Eur. J. 2024;30:e202403239. doi: 10.1002/chem.202403239. [DOI] [PubMed] [Google Scholar]
- Vellé A., Cebollada A., Macías R., Iglesias M., Gil-Moles M., Sanz Miguel P. J.. From Imidazole toward Imidazolium Salts and N-Heterocyclic Carbene Ligands: Electronic and Geometrical Redistribution. ACS Omega. 2017;2:1392–1399. doi: 10.1021/acsomega.7b00138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kascatan-Nebioglu A., Panzner M. J., Garrison J. C., Tessier C. A., Youngs W. J.. Synthesis and Structural Characterization of N-Heterocyclic Carbene Complexes of Silver(I) and Rhodium(I) from Caffeine. Organometallics. 2004;23:1928–1931. doi: 10.1021/om030689r. [DOI] [Google Scholar]
- Wang H. M. J., Lin I. J. B.. Facile Synthesis of Silver(I)-Carbene Complexes. Useful Carbene Transfer Agents. Organometallics. 1998;17:972–975. doi: 10.1021/om9709704. [DOI] [Google Scholar]
- Schmidbaur H., Schier A.. Argentophilic Interactions. Angew. Chem., Int. Ed. 2015;54:746–784. doi: 10.1002/anie.201405936. [DOI] [PubMed] [Google Scholar]
- Bruker. Area-Detector Integration Software, version 6.01; Bruker, Madison, 2001. [Google Scholar]
- Bruker. Area Detector Absorption Program, Bruker, Madison, WI, 1996. [Google Scholar]
- Krause L., Herbst-Irmer R., Sheldrick G. M., Stalke D.. Comparison of silver and molybdenum microfocus X-ray sources for single-crystal structure determination. J. Appl. Crystallogr. 2015;48:3–10. doi: 10.1107/S1600576714022985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheldrick G. M.. A short history of SHELX. Acta Crystallogr., Sect. A:Found. Crystallogr. 2008;A64:112–122. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- Sheldrick G. M.. Crystal structure refinement with SHELXL. Acta Crystallogr., Sect. C: Struct. Chem. 2015;71:3–8. doi: 10.1107/S2053229614024218. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.