

Abstract
This study aimed to evaluate the pharmacokinetics (PK), pharmacodynamics (PD), safety, and tolerability of SHR6508 injection, a new calcimimetic agent, in healthy Chinese subjects following single dose. This study utilized a placebo-controlled, single-dose, and dose-escalation design with four dose groups (0.5 mg, 2.5 mg, 5 mg, 10 mg). The trial started with a low dose and continuing to the next dose after completion of the out-of-group safety assessment of the previous dose group. Blood samples were collected at 15 time points to measure pharmacokinetic and pharmacodynamic parameters. Safety was assessed by therapeutic emergency adverse events (TEAEs), clinical laboratory tests, vital signs, electrocardiograms (ECGs), and physical examination. Of the 22 subjects who completed this study, 16 received SHR6508 Injection and 6 received placebo. In the 0.5–5 mg group, t1/2z was 8.8 h–28.3 h. Cmax and AUC increased proportionally with dose. PD results showed that SHR6508 dose dependently decreased iPTH and blood calcium levels in subjects in the 0.5–5-mg dose range; blood phosphorus levels in subjects in the 5 mg group tended to be elevated compared to those in the placebo group. Twenty-one TEAEs occurred in 12 subjects (54.5%), and no serious or severe TEAEs occurred. The overall safety and tolerability of a single intravenous dose of 0.5–5 mg SHR6508 in healthy subjects was favorable, exhibiting dose-dependent PK and PD properties.
Keywords: Chronic kidney disease, Calcimimetic agents, Secondary hyperparathyroidism, Safety
Introduction
Secondary hyperparathyroidism (SHPT) is a systemic disorder caused by parathyroid hormone secretion and is one of the common and serious complications of chronic kidney failure (Palmer et al. 2020). In patients with chronic kidney disease (CKD), the incidence of SHPT is 30–50%. SHPT accompanies almost the entire course of CKD, and the main pathophysiological changes include a persistent abnormal increase in parathyroid hormone secretion and abnormalities in blood calcium and blood phosphorus, which can cause multi-systemic damages to the blood system and cardiovascular and cerebral vascular systems, and is an independent risk factor for all-cause and independent risk factors for cardiovascular mortality. Therefore, effective treatment and standardized management of patients with SHPT is particularly important and is a clinical problem that needs to be solved urgently.
The current main therapeutic options for SHPT include phosphorus-lowering strategies, vitamin D analogs, and calcimimetics. Aluminum-containing and calcium- and phosphorus-containing binding agents commonly used in clinical practice carry risks of aluminum toxicity and hypercalcemia, respectively (Gokozan and Scognamiglio 2023). Vitamin D receptor agonists can inhibit the synthesis and secretion of parathormone (PTH) and promote the absorption of calcium in the small intestine, indirectly reducing the secretion of PTH, the efficacy of the treatment is accurate, but the clinical risk of triggering hypercalcemia. The mechanism of action of calcium-mimetic drugs is to activate the calcium-sensing receptor (CaSR) on the parathyroid master cells, and the activation of CaSR will inhibit the release and synthesis of PTH, thus lowering the level of PTH in the blood and at the same time lowering the level of blood calcium and blood phosphorus, which has a promising prospect for application (Palmer et al. 2020; Caligara et al. 1996). Elevated serum fibroblast growth factor 23 (FGF23) is associated with progression of CKD with complications such as heart failure, increased cardiovascular events and mortality. In this study, FGF23 showed a decreasing trend after administration, but no significant dose-dependent dependence was seen. Several possibilities have been reported regarding the mechanism by which calcimimetic reduce FGF23: one such possibility is an indirect pathway to reducing FGF23 by reducing serum calcium and p, and similar results were reported in a previous study on cinacalcet (Karaboyas et al. 2022).
Cinacalcet is the first marketed calcimimetic molecule to be administered orally; however, its dosage bioavailability is low, with clinical adverse effects such as upper gastrointestinal bleeding, nausea, vomiting, and diarrhea, and poor patient compliance (Gokozan and Scognamiglio 2023; Block et al. 2017). Parsabiv is a calcium-mimetic molecule, which is administered by intravenous infusion after the end of hemodialysis. Because the timing of administration coincides with the patient’s dialysis time, the patient’s compliance is better, and it is also helpful for the doctor to grasp the dosage of the patient's medication and make appropriate adjustments. However, there is a lack of long-term clinical application studies of this drug, and its effectiveness and safety need to be further explored. Gastrointestinal adverse reactions such as nausea and vomiting are the main factors limiting the long-term clinical application of the proposed calcium preparation, and other adverse reactions include hypocalcemia and myalgias (Zhang et al. 2022; Hai et al. 2017; Chandran et al. 2022).
The SHR6508 injection, developed by Shanghai Hengrui Pharmaceutical Co., LTD., a novel calcimimetic agent, which is intended to be used in patients with chronic kidney disease on maintenance hemodialysis combined with hyperparathyroidism. The aim of this study was to evaluate the pharmacokinetics, pharmacodynamics, and tolerability of SHR6508 injection in Chinese healthy subjects, and to provide a basis for the late clinical dosing regimen.
Methods
Ethics statement
This study was conducted in the Third Xiangya Hospital of Central South University, and has been approved by the Ethics Committee of the Third Xiangya Hospital of Central South University, Approval No. 21109. This clinical trial adheres to the Declaration of Helsinki (2013 edition), Good Clinical Practice (GCP) and related regulations. Before the start of the clinical trial, the subjects are informed of the details of the clinical trial, and all subjects have signed a written informed consent form. The trial was registered on Chictr.org.cn (ChiCTR2100048905).
Study participants
The participants of this trial were all healthy subjects. Eligible participants were female or male healthy volunteers 18–45 years old (including the boundary value) with a BMI between 19 and 26 kg/m2 (including the boundary value). At the same time, male participants are supposed to weigh at least 50.0 kg, female participants at least 45.0 kg. The participants had to commit that make no plans to get pregnant, donate sperms or ovums, and be willing to take physical contraceptive measures (including their partners) during the trial and for 3 months after the final dose. Those who have been judged by clinicians to be abnormal and clinically significant in the results of physical examination, vital signs examination, electrocardiogram, and laboratory tests (blood routine, urine routine, blood biochemistry, coagulation function, etc.) during the screening period will be considered unsuitable for participating in this study. All participants have been informed in detail about the nature of the trial, the potential benefits the trial brings them, and the probable inconveniences and underlying risk as well. Participants volunteered to join the trial and should be able to communicate with the investigators well, defer to the requirements of the trial and sign the written informed consent.
Study design
Thirty subjects were planned to be enrolled in this trial and randomized to receive SHR6508 or placebo. The trial flow chart is shown in Fig. 1. Based on the results of the preclinical repeated dose toxicity study, the no observed adverse effect level (NOAEL) of intravenous SHR6508 in Sprague–Dawley (SD) rats was 1 mg/kg with a mean plasma clearance of 15.4 mL/min/kg and a mean half-life (t1/2) of 4.12 h. The NOAEL dose in dogs was 0.20 mg/kg, the plasma clearance in vivo ranged from 4.47 to 4.69 mL/min/kg, and the half-life (t1/2) was 8.18 to 10.3 h. Based on the body surface area conversion, the rat NOAEL dose was converted to human equivalent dose (HED) 0.16 mg/kg, with average body weight 60 kg and safety factor of 10, calculating calculated maximum recommended starting dose (MRSD) 0.96 mg; the canine NOAEL dose was converted to human equivalent dose (HED) 0.12 mg/kg, with average body weight 60 kg and safety factor of 10, calculating MRSD of 0.72 mg.
Fig. 1.

Study Design. *Note: During the course of the study, this trial was terminated after completion of the study in the 5-mg group (group C) because the pharmacodynamic indicators analyzed in a blinded state met expectations
Considering the specifications of the preparation and the operability of the test, the lower dose of 0.5 mg was selected as the starting dose in this study. According to the preclinical findings, the maximum tolerated dose (MTD) > 20 mg/kg in body weight; the maximum tolerated dose (MTD) in dogs > 2.5 mg/kg for body weight, and the maximum tolerated dose of 10 mg was much lower than the human MTD calculated in rats and dogs, which was safe and controllable.
In addition, SHR6508 and the marketed drug etelcalcetide (Parsabiv) are both calcium-sensitive receptor agonists, with similar in vitro efficacy and similar PK characteristics in animals. According that the maximum single dose of Parsabiv in healthy humans was 10 mg for safe tolerance, 10 mg was selected as the highest climbing dose of single-dose tolerance test in this study. The trial follows a dose-escalation principle, starting with a low dose until completion of the safety assessment of the previous group before moving to the next dose group. Each subject received only one dose group, and the maximum climbing dose was initially set at 10 mg. The trial should be stopped at the earliest opportunity if the criteria for termination of the trial were met during the dose-escalation period, even if the maximum dose was not reached. During the course of the study, the trial was terminated at the completion of the 5-mg dose group (group C) because the pharmacodynamic parameters analyzed in a blinded fashion met expectations.
As a result, a total of 22 subjects completed the study, 16 of whom received SHR6508 and 6 received placebo. Subjects were admitted to the phase I ward before dinner on the day before each dose was administered in the study, had a uniformly light diet in the evening, and then fasted without water overnight for at least 10 h prior to dosing. Subjects were administered the drug by intravenous push on the morning of the day of administration for 30–60 s according to the randomization table.
Endpoints and assessments
The primary endpoint was the PK evaluation. The pharmacokinetic parameters of SHR6508 in plasma were calculated using a non-compartmental model (NCA). A total of 15 PK blood collection points were collected for each subject, 0 h (within 1 h before administration) and 2 min, 5 min, 15 min, and 30 min; 1 h, 2 h, 3 h, 4 h, 12 h, 24 h, 36 h, 48 h, and 72 h, about 3 mL of venous blood was collected after administration. The main pharmacokinetic parameters included time to peak (Tmax), peak concentration (Cmax), area under the curve from time zero to the last detectable blood concentration (AUC0-t), area under the curve extrapolated from time zero to time to infinity (AUC0-∞), terminal elimination half-life ( t1/2z), apparent clearance (CLz), apparent volume of distribution (Vz), mean residence time from time zero to the lowest detectable blood concentration (MRT0-t), and mean residence time extrapolated from time zero to time infinity MRT(0-∞). The main evaluation indexes included evaluating whether the different doses conformed to a linear kinetic process and evaluating the differences in the main pharmacokinetic parameters between the different administered dose groups.
PD parameters and safety profiles were assessed as the secondary endpoint. A total of 9 PD blood collection points for each subject were 0 h (within 1 h before administration), 10 min, 30 min, 1 h, 6 h, 10 h, 16 h, 24 h, and 48 h, about 7 mL of venous blood was collected after administration. The baseline value of each PD indicator was that measured from blood samples collected at 0 h (within 1 h before administration). Pharmacodynamic evaluation indexes included the percentage reduction of iPTH (intact parathyroid hormone) and serum-corrected calcium from baseline after administration of different dose groups, as well as the changes of FGF23 and phosphorus ions after administration of different dose groups. The calculation method of serum-corrected calcium is as follows: serum-corrected calcium [mg/dL] = serum calcium [mg/dL] + (4-albumin [g/dL]) 0.8. If not specifically specified, the last valid measurement before the first dose was taken as baseline. The blinding analysis revealed that the pharmacological effect of reducing iPTH and serum calcium in healthy subjects was dose-dependent.
Blood samples collected at the time specified in the protocol were retained as serum for anti-SHR6508 antibodies in the serum. The safety index included observing any adverse events, including abnormal clinical symptoms and vital signs, abnormalities in laboratory tests, occurring in all subjects during the clinical study period, recording their clinical presentation characteristics, severity, time of occurrence, duration, treatment and prognosis, and determining the correlation between them and the test drug. The safety examination items of this study include physical examination, vital signs, 12-lead electrocardiogram, blood routine, urine routine, blood chemistry, coagulation function, parathyroid hormone, blood transfusion, and blood pregnancy (women only).
The safety of the drugs was evaluated by the NCI-CTC AE (version 5.0) criteria. The early termination criteria for this study are as follows: according to the CTCAE 5.0 standard, more than 1/2 subjects in one dose group (2 subjects in group A, 4 in groups B, C, and D) had adverse events related to grade II and above, Or more than 1/4 of the subjects (1 patient in group A, groups B, C, D) with grade III to IV adverse events related to trial drug. It indicates that the subjects were obviously intolerant; the sponsor requests to terminate under the premise of fully protecting the rights and safety of the subjects; the drug administration regulatory department or the ethics committee ordered to terminate the trial due to major safety risks of the test drug; when the pharmacodynamic parameters are found to be reached or close to the expectation, the sponsor or the investigator may terminate the study prematurely if the higher dose group study is not required; other reasons for inappropriate to continue the trial as judged by the sponsor or investigator (Tables 1 and 2).
Table 1.
Table of the pharmacokinetic parameters
| Parameter | Unit | Instruction |
|---|---|---|
| Cmax | ng/mL | Peak concentration. It was obtained directly from the measured blood drug concentration–time data |
| Tmax | h | Peak time. It was obtained directly from the measured blood drug concentration–time data |
| AUC0-t | h*ng/mL | Area under the curve in the time from zero to the last detectable blood concentration. Calculated by the linear trapezoidal rule: AUC = (Ti + 1-Ti) (Ci + Ci + 1)/2, AUC 0-t is the sum of all AUC |
| AUC0-∞ | h*ng/mL | Area under the curve for extrapolation from zero to infinity time. AUC 0-∞ = AUC 0-t + Ct /λz (Ct is the last determinable blood concentration) |
| T1/2z | h | End-terminal elimination half-life. T1/2z = ln2/λz。 |
| Vz | L | Apparent volume of distribution. Vz = CLz/λz。 |
| CLz | L/h | Epigenetic clearance. CLz = dose of administration/AUC0-∞。 |
| λz | 1/h | Eliminate the rate constant, the negative number of the slope of the end of the curve for the semi-log drug, calculated using linear regression |
| MRT0-t | h | Average residence time in the time from 0 h to the lowest detectable plasma concentration. MRT0-t = AUMC0-t/AUC0-t。 |
| MRT0-∞ | h | Average dwell time for extrapolation from zero to infinity. MRT0-∞ = AUMC0-∞/AUC0-∞。 |
| AUC_%Extrap | / | Percentage of the residual area. AUC_%Extrap = [(AUC0-∞-AUC0-t)/AUC0-∞] × 100%。 |
Table 2.
Table of the PD parameters and the calculation method
| Parameter | Instruction |
|---|---|
| iPTH | Blood samples were collected at the defined time points to assess changes in iPTH levels relative to baseline |
| Serum corrected for calcium |
Blood samples were collected at the indicated time points to assess changes in serum-corrected calcium levels from baseline Serum-corrected calcium [mg/dL] = serum calcium [mg/dL] + (4-albumin [g/dL]) 0.8 |
| Phosphate ions | Blood samples were collected at the indicated time points to assess changes in phosphorus ion levels relative to baseline |
| FGF23 | Blood samples were collected at defined time points to assess changes in FGF23 levels from baseline |
Blinding
This trial is a randomized, double-blind clinical trial using stratified block randomization, stratified by dose group, randomization specialist using SAS statistics software (version 9.4 or above) to generate randomization tables and assign randomization numbers. The blinding base shall be made in two copies, sealed and stamped with a special seal for blindness by the random management unit, and kept in the clinical research unit and the sponsor respectively. The blinding or other relevant personnel produce the drug number and the corresponding treatment group by SAS software (version 9.4 or above), and perform the drug packaging according to the drug number. Blinding must be kept from subject enrollment, recording, and evaluation of trial results, trial process inspection to data management. Except for the non-blind personnel, the subjects, investigators, the medical staff involved in the trial or clinical evaluation, the sponsor participating in the clinical study evaluation, and the clinical research associate (CRA) involved in the trial are not blinded to the investigational drug the subject receives. The blinding process forms the blinding record preservation. In case of the following emergencies: (1) serious adverse events; (2) more than 1/2 subjects in a dose group had grade II or above trial drug-related adverse events, or more than 1/4 subjects had III to IV and drug-related adverse events; (3) the site principal investigator (PI) determines the subject for emergency rescue. If so, the PI shall urgently contact the sponsor project manager (PM) or CRA, and the PM or CRA notifies the randomization specialist to immediately perform the unblinding and save the time and reason of unblinding. Once unblinded, the subject was treated as a shedding case. After blinding verification, the data are locked, and the statisticians, the statistician, the principal investigator and the sponsor will inform the corresponding group of each subject code so as to conduct statistical analysis of all the data.
Statistics analysis
PK parameter calculations were done by Phoenix WinNonlin 8.0 (or above), and all other data were analyzed using SAS 9.4 (or above) software. The pharmacokinetic parameters of each subject were calculated by the non-atrial model. The arithmetic mean, standard deviation, coefficient of variation, median, maximum, minimum, and geometric mean of each parameter were also calculated. The pharmacokinetics concentration analysis set (PKPS) set was used to evaluate the linear kinetic characteristics of SHR6508 in the dose range of 0.5–10 mg in a single intravenous injection in healthy subjects, and to evaluate the differences in the main pharmacokinetic parameters between different administration dose groups. The geometric mean, geometric standard deviation, and percentage change in the mean, standard deviation, and change from baseline of the PD metrics at each measurement time point were summarized by dose group, and the trend plots of the subjects’ PD levels over time during the test period are given for each dose group.
Results
Study subjects
The study subjects of this trial were healthy subjects, 30 subjects were scheduled to be enrolled, 6 in group A (0.5 mg), 8 in group B (2.5 mg), group C (5 mg), and group D (10 mg). Of the 23 participants enrolled in this study, 22 completed all planned doses in accordance with the protocol. Twenty-two were randomly assigned to receive either SHR6508 or placebo. The number of subjects participating in this study was comparable between males and women, with subjects aged 20 to 28 years. The demographic characteristics of the subjects are shown in Table 3. The demographic parameters of the cohorts were generally balanced. There were no significant differences in demographic or baseline characteristics between the dose groups. This study lasted approximately 2 months, the first enrollment date was August 23, 2021, and the last completion date was October 8, 2021.
Table 3.
Demographic characteristics of subjects
| Placebo (N = 6) | SHR6508 0.5 mg (N = 5) |
SHR6508 2.5 mg (N = 6) |
SHR6508 5 mg (N = 6) |
Total (N = 23) | |
|---|---|---|---|---|---|
| Age (year) | |||||
| n | 6 | 5 | 6 | 6 | 23 |
| Mean (SD) | 28.3 (9.35) | 26.0 (5.48) | 22.2 (3.54) | 25.3 (9.46) | 25.4 (7.34) |
| Median | 25.0 | 26.0 | 22.5 | 21.5 | 23.0 |
| Q1, Q3 | 21.0, 39.0 | 22.0, 28.0 | 19.0, 23.0 | 20.0, 26.0 | 20.0, 28.0 |
| Min, Max | 19, 41 | 20, 34 | 18, 28 | 19, 44 | 18, 44 |
| Sex, n (%) | |||||
| Male | 3 (50.0) | 3 (60.0) | 3 (50.0) | 3 (50.0) | 12 (52.2) |
| Female | 3 (50.0) | 2 (40.0) | 3 (50.0) | 3 (50.0) | 11 (47.8) |
| Ethnic, n (%) | |||||
| Han | 5 (83.3) | 5 (100.0) | 6 (100.0) | 6 (100.0) | 22 (95.7) |
| Other | 1 (16.7) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (4.3) |
| Height (cm) | |||||
| n | 6 | 5 | 6 | 6 | 23 |
| Mean (SD) | 157.17 (8.262) | 163.60 (9.529) | 162.75 (9.032) | 163.08 (6.681) | 161.57 (8.242) |
| Median | 155.00 | 167.50 | 161.25 | 163.00 | 159.00 |
| Q1, Q3 | 150.50, 161.50 | 157.00, 169.50 | 156.50, 170.50 | 156.50, 169.00 | 156.00, 169.50 |
| Min, Max | 149.5, 171.5 | 150.5, 173.5 | 151.5, 175.5 | 156.0, 171.0 | 149.5, 175.5 |
| Weight (kg) | |||||
| n | 6 | 5 | 6 | 6 | 23 |
| Mean (SD) | 55.80 (8.326) | 59.26 (6.048) | 57.02 (9.505) | 58.42 (7.947) | 57.55 (7.686) |
| Median | 54.65 | 60.40 | 54.65 | 57.40 | 57.60 |
| Q1, Q3 | 47.90, 62.70 | 57.90, 60.90 | 48.90, 66.00 | 56.60, 59.20 | 50.20, 62.70 |
| Min, Max | 47.0, 67.9 | 50.2, 66.9 | 47.8, 70.1 | 47.6, 72.3 | 47.0, 72.3 |
| BMI (kg/m2) | |||||
| n | 6 | 5 | 6 | 6 | 23 |
| Mean (SD) | 22.48 (1.712) | 22.20 (2.075) | 21.40 (1.874) | 21.95 (2.462) | 22.00 (1.948) |
| Median | 22.00 | 22.20 | 20.65 | 22.05 | 21.80 |
| Q1, Q3 | 21.40, 23.10 | 20.20, 23.80 | 20.30, 22.80 | 19.40, 23.50 | 20.30, 23.50 |
| Min, Max | 20.8, 25.6 | 20.1, 24.7 | 19.5, 24.5 | 19.4, 25.3 | 19.4, 25.6 |
Efficacy
PK results
A total of 22 subjects completed the study, including 16 subjects who received SHR6508 injection and 6 subjects who received placebo. The main pharmacokinetic parameters of SHR6508 after single intravenous injection of SHR6508 in healthy subjects are shown in Table 4.
Table 4.
Plasma PK parameter table of SHR6508 in subjects after administration (PK parameter analysis set)
| PK parameter (units) | SHR6508 0.5 mg (N = 4) | SHR6508 2.5 mg (N = 6) | SHR6508 5 mg (N = 5) |
|---|---|---|---|
| Tmax (h)* | 0.058 (0.03–0.08) | 0.033 (0.03–0.08) | 0.033 (0.03–0.25) |
| Cmax (ng/mL) | 88.45 ± 49.87 (56.4) | 418.67 ± 122.16 (29.2) | 617.40 ± 312.58 (50.6) |
| AUC0-t (h*ng/mL) | 67.42 ± 16.94 (25.1) | 488.61 ± 65.78 (13.5) | 928.40 ± 158.99 (17.1) |
| AUC0-∞ (h*ng/mL) | 82.84 ± 18.67 (22.5) | 538.33 ± 79.25 (14.7) | 1041.99 ± 202.76 (19.5) |
| t1/2z (h) | 8.81 ± 1.97 (22.3) | 26.40 ± 4.24 (16.1) | 28.31 ± 3.88 (13.7) |
| CLz (L/h) | 6.27 ± 1.41 (22.5) | 4.73 ± 0.69 (14.6) | 4.94 ± 0.88 (17.9) |
| λz(h−1) | 0.082 ± 0.019 (23) | 0.027 ± 0.0042 (15.7) | 0.025 ± 0.0032 (13.1) |
| Vz (L) | 76.85 ± 6.10 (7.9) | 177.78 ± 22.75 (12.8) | 198.56 ± 24.16 (12.2) |
| AUC_%Extrap (%) | 18.94 ± 2.26 (12) | 9.08 ± 1.72 (18.9) | 10.60 ± 2.44 (23.1) |
*Tmax is calculated using the median (minimum, maximum), while all other PK parameters are calculated using mean ± SD (CV%)
The blood concentration peaked rapidly after a single intravenous injection of SHR6508 in healthy subjects (the median Tmax was 0.033–0.058 h), and no significant difference in median Tmax was observed between different dose groups. Except for the low-dose group, the pharmacokinetic parameters Vz, t1/2z, and CLz related to drug distribution and elimination were basically the same in other dose groups: the average half-life (t1/2z) of the 0.5 mg dose group was about 8.8 h; that of the 2.5 mg/5 mg dose group was about 26.4 h and 28.3 h, respectively (Table 3 and Figs. 2 and 3). Figures 2 and 3 indicate that the plasma exposure levels (Cmax and AUC) of SHR6508 increased proportionally with dose within the range of 0.5–5 mg, and showed linear pharmacokinetic characteristics. The results of variance analysis showed no significant differences in PK characteristics between different genders.
Fig. 2.

The average blood drug concentration–time curve (PK concentration analysis set, linear coordinates) of SHR6508 in healthy subjects
Fig. 3.

The average blood drug concentration–time curve (PK concentration analysis set, semi-logarithmic coordinates) of SHR6508 in healthy subjects
PD results
Upon completion of study in the 5 mg dose group (group C), a blinded analysis revealed a pharmacological signal of dose-dependent reduction in iPTH and blood calcium in healthy subjects following a single intravenous injection of the product. This trial was terminated early in that the pharmacodynamic profile met expectations. A total of 22 subjects were enrolled in the PDS. The iPTH concentration decreased rapidly in all dose groups and decreased to a minimum within 1 h after administration. iPTH levels continued to decline longer with increasing SHR6508 dose. In the 0.5-mg group, the mean percent change from baseline in iPTH was minimized to approximately − 35.5% at 10 min after administration; in the 2.5-mg group, the mean percent change from baseline in iPTH was minimized to approximately − 65.9% at 1 h after administration; and in the 5 mg group, the mean percent change from baseline in iPTH was minimized to approximately −66.9% at 30 min after administration. The iPTH concentrations in the test drug group essentially returned to placebo levels at 24–48 h (Fig. 4).
Fig. 4.
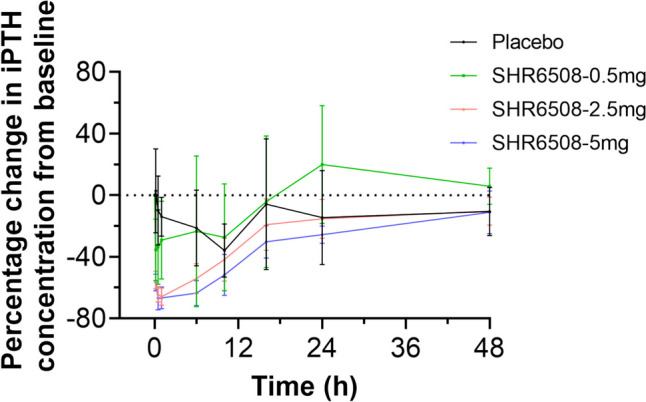
Graph of the percentage change in iPTH concentration relative to baseline over time in healthy subjects. Data are presented as mean ± standard deviation
Serum-corrected calcium concentrations showed a decreasing trend after dosing, and decreased with increasing dose administration. The mean percentage change in serum adjusted calcium concentration from baseline in the SHR6508 0.5, 2.5, and 5 mg groups decreased to a values of − 1.5%, − 7.1%, and − 7.5%, respectively, and then gradually returned to baseline after 24 h. The trend was similar in the 0.5-mg group and the placebo group (Fig. 5).
Fig. 5.

The percentage change in serum-corrected calcium concentration relative to baseline over time in healthy subjects
The FGF23 concentration decreased after administration in all dose groups, but no significant dose dependence was seen. In the 0.5-mg group, the mean percent change from baseline in FGF23 concentration decreased to a minimum of − 22.4% approximately at 10 min after administration; in the 2.5-mg group, the mean percent change from baseline in FGF23 concentration decreased to a minimum of − 28.0% approximately at 16 h after administration; in the 5-mg group, the mean percent change from baseline in FGF23 concentration at 24 h after administration decreased to a minimum of − 17.0% approximately. The FGF23 concentrations in all test drug groups were gradually recovered after 24 h (Fig. 6).
Fig. 6.
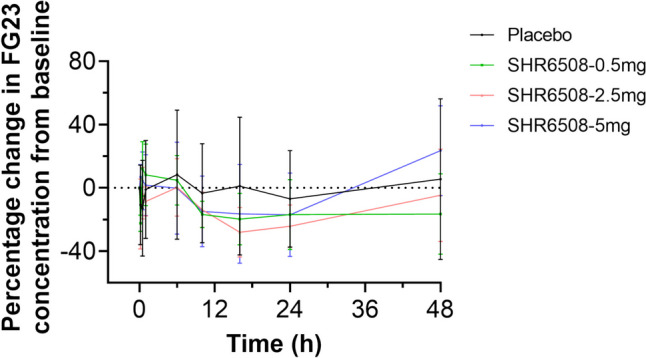
Percentage change in FGF23 concentration relative to baseline over time in healthy subjects
In the 0.5-mg and 2.5-mg groups, the blood phosphorus level trend was similar to that in the placebo group, with the phosphorus ion concentration rising to the maximum at 16 h after medication and gradually recovering after 24 h (Fig. 7). The phosphate ion concentration in the 5-mg group was the highest, about 28.9%, while the 0.5, 2.5 mg, and placebo groups was the highest at 16 h after dose, 10.0%, 14.8%, and 25.7%, respectively.
Fig. 7.
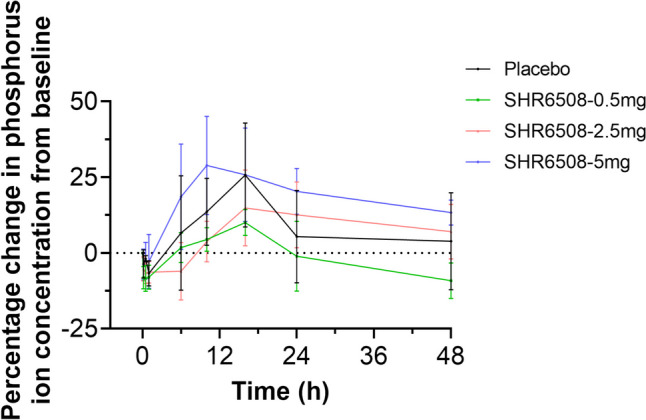
The percentage change in phosphorus ion concentration relative to baseline over time in healthy subjects
Assessment of safety
Of the 23 subjects enrolled in this study, 16 subjects received SHR6508 and 6 subjects received placebo, and only one of the remaining 22 subjects was included in the safety data set.
All safety analyses were based on safety data analysis from the 22 subjects in the safety analysis set. A total of 12 subjects (54.5%) experienced 21 TEAEs without drug-related adverse events. A total of 3 (50.0%) had TEAEs in the placebo group; 9 (56.3%) had TEAEs; TEAEs: 3 (75.0%) in the SHR6508 0.5-mg dose group; 5 (83.3%) had TEAEs in the SHR6508 2.5-mg dose group; and 1 (16.7%) had TEAEs in the SHR6508 5-mg dose group. No significant dose dependence was seen in the incidence of TEAEs. TEAEs in the SHR6508 group included positive bacterial test (3/16,18.8%), urinary protein (2/16, 12.5%), positive urinary leukocytes (2/16,12.5%), elevated blood triglycerides (2/16,12.5%), and other TEAEs were seen in single subjects. The TEAEs for 2 subjects in the placebo group were elevated blood triglycerides (2/6, 33.3%), and all other TEAEs were seen in a single subject.
All adverse events were Grade 1 in severity and were not possibly drug-related, leading to recovery/resolution or remission. No serious adverse events and adverse events attributable to death were reported during the trial, and no adverse events led to withdrawal from the trial. The subjects who completed the test had negative serum ADA test results and no anti-SHR6508 antibody was detected. Treatment-related adverse events by SOC and PT are listed in Table 5.
Table 5.
Treatment-related adverse events sorted by SOC and PT
| Placebo (N = 6) | SHR6508 0.5 mg (N = 4) |
SHR6508 2.5 mg (N = 6) |
SHR6508 5 mg (N = 6) |
Total (N = 16) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| SOC/PT | Events | n (%) | Events | n (%) | Events | n (%) | Events | n (%) | Events | n (%) |
| Number of subjects with at least 1 adverse event | 7 | 3 (50.0) | 3 | 3 (75.0) | 10 | 5 (83.3) | 1 | 1 (16.7) | 14 | 9 (56.3) |
| All kinds of inspections | 7 | 3 (50.0) | 3 | 3 (75.0) | 9 | 4 (66.7) | 1 | 1 (16.7) | 13 | 8 (50.0) |
| Positive urine leukocytes | 1 | 1 (16.7) | 0 | 0 (0.0) | 1 | 1 (16.7) | 1 | 1 (16.7) | 2 | 2 (12.5) |
| Positive urine occult blood | 0 | 0 (0.0) | 1 | 1 (25.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 1 | 1 (6.3) |
| Urine leukocyte esterase positive | 0 | 0 (0.0) | 0 | 0 (0.0) | 1 | 1 (16.7) | 0 | 0 (0.0) | 1 | 1 (6.3) |
| Positive urine red blood cells | 1 | 1 (16.7) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) |
| Protein in the urine is detected | 1 | 1 (16.7) | 0 | 0 (0.0) | 2 | 2 (33.3) | 0 | 0 (0.0) | 2 | 2 (12.5) |
| Increased heart rate | 0 | 0 (0.0) | 0 | 0 (0.0) | 1 | 1 (16.7) | 0 | 0 (0.0) | 1 | 1 (6.3) |
| Positive bacterial test | 1 | 1 (16.7) | 1 | 1 (25.0) | 2 | 2 (33.3) | 0 | 0 (0.0) | 3 | 3 (18.8) |
| Elevated vitamin C | 0 | 0 (0.0) | 0 | 0 (0.0) | 1 | 1 (16.7) | 0 | 0 (0.0) | 1 | 1 (6.3) |
| Elevated serum uric acid | 1 | 1 (16.7) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) |
| Elevated blood triglycerides | 2 | 2 (33.3) | 1 | 1 (25.0) | 1 | 1 (16.7) | 0 | 0 (0.0) | 2 | 2 (12.5) |
| Diseases of the heart organs | 0 | 0 (0.0) | 0 | 0 (0.0) | 1 | 1 (16.7) | 0 | 0 (0.0) | 1 | 1 (6.3) |
| First-degree atrioventricular block | 0 | 0 (0.0) | 0 | 0 (0.0) | 1 | 1 (16.7) | 0 | 0 (0.0) | 1 | 1 (6.3) |
Discussion
SHR6508 is a new calcimimetic molecule developed by Shanghai Hengrui Pharmaceutical Co., Ltd. This study is a safety, pharmacokinetics, and pharmacodynamic study in healthy adult subjects. Safety, pharmacokinetics, and pharmacodynamics of SHR6508 injections (0.5 mg, 2.5 mg, 5 mg, and placebo) investigated intravenous SHR6508 (0.5 mg, 2.5 mg, 5 mg, and placebo) in healthy adult subjects providing the development of underlying SHR6508 injections at a later stage. We found that a single-dose injection of SHR6508 in the range of 0.5–5 mg exhibited dose-dependent PK properties in healthy Chinese volunteers. PD analysis showed that SHR6508 reduced iPTH and serum calcium levels in a dose-dependent manner. Furthermore, all TEAEs were probably drug-unrelated, no drug-related adverse events occurred, and all TEAEs experienced recovery/resolution or remission. SHR6508 With controllable security.
We found that SHR6508 plasma drug concentration peaked rapidly at the end of administration, distributed rapidly and eliminated slowly after a single intravenous injection of 0.5–5 mg SHR6508 injection in healthy subjects. Among them, the 0.5-mg group had a faster clearance and shorter half-life, probably due to the lower dose administered, the blood concentration was below the lower limit of quantification after 24 h, which had some influence on the estimation of the terminal elimination term; the clearance and half-life results of the 2.5-mg group and the 5-mg group were closer to each other, and the average half-life of the two dose groups was about 26.4 h and 28.3 h, respectively. We performed the drug exposure of the different dose groups We analyzed the linear relationship between the different dose groups, and the results showed that the pharmacokinetics of SHR6508 injection in human beings in the dose range of 0.5–5 mg conformed to the linear kinetic characteristics.
PD result display that after a single injection of 0.5–5 mg of SHR6508 injection in healthy subjects, the body’s iPTH and blood calcium levels decreased, and this decrease was dose-dependent, which was in line with the expected effects of the product. The change in phosphorus levels is due to the blood biochemical change caused by the high dose of calmimetic, which is the extension and amplification effect of the pharmacological effect of calmimetic. Similar toxicity manifestations were seen in preclinical toxicological studies of similar drugs, which is related to the decrease of blood calcium after reducing PTH by calmimetic. An increase in phosphorus ion levels was observed in the SHR6508 5-mg group, which may be due to the normal physiological regulation caused by the decrease in iPTH levels. Overall, the results of this study demonstrated the safety and tolerability of SHR6508 in single injection doses of 0.5–5 mg, and SHR6508 raised no new safety concerns in healthy Chinese subjects.
Calcimimetics have been reported to be prone to gastrointestinal adverse effects. (Hai et al. 2017; Bover et al. 2016). In a phase 3 study comparing head-to-head with cinacalcet, SHPT patients were treated with either 2.5–15-mg evocalcet or 30–180-mg cinacalcet for 26 weeks, with comparable rates of gastrointestinal adverse reactions in both treatment groups (Chandran et al. 2022). However, evocalcet is a synthetic peptide, and therefore, potential issues regarding its efficacy and safety, such as allergy and infusion reactions, need to be addressed (Friedl and Zitt 2018; Cunningham et al. 2019). SHR6508 is also a kind of synthetic peptide, antibody testing is required to address these issues (Wolf et al. 2020), so perhaps longer term or larger studies are needed to assess the clinical relevance of anti-euthycal peptide antibodies (Block et al. 2017; Fukagawa et al. 2017; Martin et al. 2014). Our study demonstrated that SHR6508 was free of the known safety risks of similar species and the product, such as hypocalcemia, upper gastrointestinal bleeding and non-dynamic bone disease, and that no hemolysis-related adverse events occurred. The subjects who completed the trial had negative serum ADA tests and no anti-SHR6508 antibodies were detected. This suggests that SHR6508 has a manageable safety profile, particularly in terms of reducing gastrointestinal adverse events.
There are several limitations to this study. Firstly, although small sample sizes are generally acceptable for phase I trials, rare AEs may be difficult to detect. Therefore, further large-scale clinical trials are still needed to accurately prove the safety of SHR6508. Secondly, since healthy subjects are not representative of the patient population, the PK values of SHR6508 may vary when applied in clinical practice, and further data from long-term studies and clinical experience are needed in this regard.
Conclusion
In conclusion, our study evaluated the pharmacokinetic and pharmacodynamic profiles, as well as the safety and tolerability, of a single intravenous dose of SHR6508 in healthy subjects. The plasma drug exposure (Cmax and AUC) of SHR6508 increased proportionally with the administered dose, with essentially linear kinetics over the 0.5–5 mg dose range. Our results indicate that the overall safety and tolerability of a single intravenous injection of 0.5–5 mg of SHR6508 are favorable in healthy subjects, providing a new option for the treatment of SHPT.
Authors’ contributions
STZ is responsible for writing – original draft andwriting – review & editing. HYT IS responsible for validation. SY, XYX, CC, JH and GPY are responsible for Investigation, methodology, project administration and supervision. All authors made a significant contribution to the work reported, and have agreed on the journal to which the article has been submitted. The authors declare that all data were generated in-house and that no paper mill was used.
Funding
This study was sponsored by Shanghai Hengrui Biotech, Inc. and the Hunan Provincial Natural Science Foundation of China (No. 2023JJ60513).
Data availability
All source data for this work (or generated in this study) are available upon reasonable request.
Declarations
This study investigated the safety and tolerability of SHR6508 in healthy Chinese subjects and the pharmacokinetic characteristics and pharmacodynamics of SHR6508 in healthy subjects. This study provides evidence for subsequent clinical studies of SHR6508 and may provide a new option for the treatment of SHPT. This study is available as a preprint at 10.21203/rs.3.rs-4096983/v1.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Sheng-ting Zhang and Hong-yi Tan contributed equally to this work.
Contributor Information
Jie Huang, Email: cellahuang1988@163.com.
Guo-ping Yang, Email: ygp9880@126.com.
References
- Block GA, Bushinsky DA, Cheng S et al (2017) Effect of etelcalcetide vs cinacalcet on serum parathyroid hormone in patients receiving hemodialysis with secondary hyperparathyroidism: a randomized clinical trial. JAMA 317(2):156–164 [DOI] [PubMed] [Google Scholar]
- Bover J, Ureña P, Ruiz-García C et al (2016) Clinical and practical use of calcimimetics in dialysis patients with secondary hyperparathyroidism. Clin J Am Soc Nephrol 11(1):161–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caligara F, Giangrande A, Allaria P et al (1996) The PTH-calcium relationship curve in secondary hyperparathyroidism, an index of sensitivity and suppressibility of parathyroid glands. Nephrol Dial Transplant 11(Suppl 3):136–141 [DOI] [PubMed] [Google Scholar]
- Chandran M, Bilezikian JP, Lau J et al (2022) The efficacy and safety of cinacalcet in primary hyperparathyroidism: a systematic review and meta-analysis of randomized controlled trials and cohort studies. Rev Endocr Metab Disord 23(3):485–501 [DOI] [PubMed] [Google Scholar]
- Cunningham J, Block GA, Chertow GM et al (2019) Etelcalcetide is effective at all levels of severity of secondary hyperparathyroidism in hemodialysis patients. Kidney Int Rep 4(7):987–994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedl C, Zitt E (2018) Role of etelcalcetide in the management of secondary hyperparathyroidism in hemodialysis patients: a review on current data and place in therapy. Drug des Devel Ther 12:1589–1598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukagawa M, Yokoyama K, Shigematsu T et al (2017) A phase 3, multicentre, randomized, double-blind, placebo-controlled, parallel-group study to evaluate the efficacy and safety of etelcalcetide (ONO-5163/AMG 416), a novel intravenous calcimimetic, for secondary hyperparathyroidism in Japanese haemodialysis patients. Nephrol Dial Transplant 32(10):1723–1730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gokozan HN, Scognamiglio T (2023) Advances and updates in parathyroid pathology. Adv Anat Pathol 30(1):24–33 [DOI] [PubMed] [Google Scholar]
- Hai MTT, Guettier JM, Rosebraugh CJ (2017) Dosing of etelcalcetide and cinacalcet for secondary hyperparathyroidism. JAMA 317(20):2132 [DOI] [PubMed] [Google Scholar]
- Karaboyas A, Muenz D, Fuller DS et al (2022) Etelcalcetide utilization, dosing titration, and chronic kidney disease-mineral and bone disease (CKD-MBD) marker responses in US hemodialysis patients. Am J Kidney Dis 79(3):362–373 [DOI] [PubMed] [Google Scholar]
- Martin KJ, Bell G, Pickthorn K et al (2014) Velcalcetide (AMG 416), a novel peptide agonist of the calcium-sensing receptor, reduces serum parathyroid hormone and FGF23 levels in healthy male subjects. Nephrol Dial Transplant 29(2):385–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer SC, Mavridis D, Johnson DW et al (2020) Comparative effectiveness of calcimimetic agents for secondary hyperparathyroidism in adults: a systematic review and network meta-analysis. Am J Kidney Dis 76(3):321–330 [DOI] [PubMed] [Google Scholar]
- Wolf M, Block GA, Chertow GM et al (2020) Effects of etelcalcetide on fibroblast growth factor 23 in patients with secondary hyperparathyroidism receiving hemodialysis. Clin Kidney J 13(1):75–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang LX, Zhang B, Liu XY et al (2022) Advances in the treatment of secondary and tertiary hyperparathyroidism. Front Endocrinol (Lausanne) 13:1059828 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All source data for this work (or generated in this study) are available upon reasonable request.