

Abstract
The NTB-VPg protein of Tomato ringspot nepovirus is an integral membrane protein found in association with endoplasmic reticulum (ER)-derived membranes active in virus replication. A transmembrane helix present in a hydrophobic region at the C terminus of the NTB domain was previously shown to traverse the membranes, resulting in the translocation of the VPg domain in the lumen. We have now conducted an in planta analysis of membrane-targeting domains within NTB-VPg using in-frame fusions to the green fluorescent protein (GFP). As expected, the entire NTB-VPg protein directed the GFP fluorescence to ER membranes. GFP fusion proteins containing the C-terminal 86 amino acids of NTB-VPg also associated with ER membranes, resulting in ER-specific glycosylation at a naturally occurring glycosylation site in the VPg domain. Deletion of the hydrophobic region prevented the membrane association. The N-terminal 80 amino acids of NTB were also sufficient to direct the GFP fluorescence to intracellular membranes. A putative amphipathic helix in this region was necessary and sufficient to promote membrane association of the fusion proteins. Using in vitro membrane association assays and glycosylation site mapping, we show that the N terminus of NTB can be translocated in the lumen at least in vitro. This translocation was dependent on the presence of the putative amphipathic helix, suggesting that oligomeric forms of this helix traverse the membrane. Taken together, our results suggest that at least two distinct elements play a key role in the insertion of NTB-VPg in the membranes: a C-terminal transmembrane helix and an N-terminal amphipathic helix. An updated model of the topology of the protein in the membrane is presented.
Infection by positive-strand RNA viruses results in massive proliferation and modification of the structure of the host intracellular membranes. Viral replication occurs in association with intracellular membranes and is associated with various modified membrane structures, including membranous vesicles and membranous webs (8, 38, 44, 48). The specific nature of the membranes affected varies from one virus to another (e.g., endoplasmic reticulum [ER], tonoplast, mitochondrial membranes). Animal picornaviruses induce the formation of replication-competent membranous vesicles which are derived from the ER probably using mechanisms similar to those of the secretory pathway (17, 28, 43, 47, 51). Replication proteins and replication intermediates from plant viruses related to picornaviruses (e.g., potyviruses, comoviruses, and nepoviruses) are also found in association with ER-derived membranous vesicles (9, 10, 40, 46). It is thought that these vesicles constitute mini-virus factories which are physically separated from the cytoplasmic content of the cell and offer protective environments for viral RNA replication (44).
The initial targeting of viral replication proteins to the ER is a key step in the formation of replication-competent vesicles. One or several viral or host proteins act as membrane anchors for the replication complex. These membrane anchors have the ability to associate with the ER in the absence of other viral proteins. Other replication proteins are brought into the replication complex either as polyprotein precursors or through protein-protein interactions with the membrane anchor proteins (14, 44, 55). Viral membrane anchor proteins are usually integral membrane proteins and often have the ability to induce drastic modifications in membrane morphology (2, 4, 6, 11, 15, 19, 23, 24, 46, 53, 54). Various membrane association domains, including amphipathic helices and/or transmembrane helices, are involved in the association of the membrane anchors to the ER (6, 12, 16, 25, 36, 44, 60, 62). The association of the animal picornavirus membrane anchor proteins has been studied in detail; membrane association domains have been identified in the 3AB, 2B, and 2BC proteins, models for the topology of these proteins in the membrane have been established (1, 16, 25, 54), and the ability of these proteins to induce permeabilization of intracellular membranes has been extensively documented (1, 3, 35). Much less is known about membrane anchor proteins from plant viruses related to picornaviruses. Two comovirus replication proteins and one potyvirus replication protein have been shown to associate with ER membranes when expressed individually or in the context of a viral infection and to induce drastic modifications of the ER structure (11, 46). However, the mechanism of ER targeting and the topology of these proteins in the membrane are largely unknown. Several nepovirus proteins colocalize with membrane-bound replication complexes in infected cells (27, 30, 40). One of these proteins (NTB-VPg) is an integral membrane protein and can associate with microsomal membranes in vitro (30, 60). However, it is not known whether this protein can independently associate with ER membranes in planta and its possible role as a membrane anchor for the replication complex has not been confirmed.
The genome of Tomato ringspot nepovirus (ToRSV) consists of two molecules of RNA (45). RNA1 encodes a polyprotein (P1) containing the domains for the replication proteins, including the RNA-dependent RNA polymerase, a 3C-like proteinase, the genome-linked protein (VPg), a putative nucleoside triphosphate-binding protein (NTB), and two additional proteins (X1 and X2) of unknown function (42, 59, 61). The 3C-like proteinase is responsible for cleavage of P1 and of the RNA2-encoded polyprotein (13). The mature NTB protein along with several polyprotein precursors, notably NTB-VPg, are associated with ER-derived membranes active in virus replication (30). A hydrophobic domain at the C terminus of NTB contains a transmembrane helix that traverses the membranes in infected plants and in vitro, resulting in a luminal orientation of the VPg domain in at least a portion of the membrane-associated NTB-VPg protein (30, 60). In vitro, translocation of VPg in the lumen results in N-linked glycosylation at a glycosylation site within the VPg domain (60). In addition to the C-terminal hydrophobic domain, a putative amphipathic helix was identified at the N terminus of the protein that may also be involved in the interaction of the protein with the membranes, although this has not been confirmed experimentally (30).
In this study, we have analyzed membrane-targeting sequences within the NTB-VPg protein in planta using in-frame fusions to the green fluorescent protein (GFP) and have examined the topology of the N-terminal region of the NTB-VPg protein. We show that both the C-terminal hydrophobic domain and the N-terminal putative amphipathic helix can direct the GFP fluorescence to intracellular membranes. Our results also suggest that the putative amphipathic helix can promote the translocation of the N terminus of NTB into the lumen of the membrane at least in vitro.
MATERIALS AND METHODS
Plasmid constructions.
Plasmid pCD-327 (obtained from the Arabidopsis Biological Resource Center, Ohio State University, Columbus) contains the coding region for the red-shifted GFP (smRS-GFP) under the control of the Cauliflower mosaic virus 35S promoter (20). To introduce restriction sites allowing in-frame fusions to the C terminus of GFP, a fragment corresponding to the entire length of pCD-327 was amplified with primers 24 (Table 1) and 25 and Pfu polymerase (Stratagene). The PCR product was digested with KpnI and religated, resulting in plasmid psmRS-GFP(S-K) with unique SstI and KpnI sites. To construct pER-dsRed2, psmRS-GFP(S-K) was digested with EcoRI and treated with Klenow enzyme prior to religation, resulting in psmRS-GFP(S-KΔRI). The small HindIII-SstI fragment of pmGFP5-ER (63) was ligated into the corresponding sites of psmRS-GFP(S-KΔRI) to give psmRS-ER-GFP(S-KΔRI). A fragment containing the reading frame for dsRed2 was amplified from pDsRed2 (BD Biosciences Clontech) using primers 50 and 51, digested with EcoRI and SstI, and ligated into the corresponding sites of psmRS-ER-GFP(S-KΔRI) to give pER-dsRed2. pGFP-NV and its derivatives were constructed by amplifying the corresponding portion of the coding region of NTB-VPg using pMR10 (42) as a template and the following pairs of primers: 21 and 22 for pGFP-NV, 48 and 49 for pGFP-mN, 36 and 22 for pGFP-cNV3, 21 and 35 for pGFP-nN, 21 and 46 for pGFP-nN-TMD, and 47 and 35 for pGFP-nNΔTMD. The amplified fragments were digested with SstI and KpnI and inserted into the corresponding sites of psmRS-GFP(S-K). To construct pGFP-cNV3-T1229/A and pGFP-cNV3ΔTM1, fragments were amplified using primers 36 and 22 and pT7-cNV-T1229/A and pT7-cNVΔTM1 (60) as templates, respectively. To obtain a vector allowing fusions to the N terminus of GFP, a cDNA fragment containing the GFP coding region was amplified from pCD-327 using primers 39 and 2868, digested with BglII and SstI, and ligated into the BamHI and SstI sites of psmRS-GFP to give psmRS-GFPn(B-K) containing unique BamHI and KpnI sites. The internal AUG start codon at the beginning of the GFP open reading frame was then mutated to a GTG codon by site-directed mutagenesis to prevent the possibility of internal initiation at this site. This vector was used to insert cDNA fragments produced by amplification with the following pairs of primers: 40 and 53 for pNV-GFP, 40 and 35 for pnN-GFP, and 45 and 53 for pcNV3-GFP.
TABLE 1.
Primers used in plasmid constructions used in this study
| No. | Sequence (5′ to 3′)a | Commentsb |
|---|---|---|
| 21 | TATATGAGCTCGGTGGCGGATCAGGGCTCACTGACGTTTTTGG | 5′ NTB + SstI + GGGS (+) |
| 22 | GCGCGCGGTACCTTACTGTACAGATTGTGGGCGGA | 3′ VPg + KpnI + TAA (−) |
| 24 | GATCGAGGTACCTAAGAATTTCCCCGATCGTTCAAAC | pCD327 + KpnI (+) |
| 25 | TATAGGTACCTATGAGCTCTTTGTATAGTTCATCCATGCCATGTG | pCD327 + KpnI + SstI (−) |
| 30 | ACGCCCATGGTTCCTCTGAGTATCATGA | 5′ NTB + NcoI (+) |
| 31 | ACGCGTCGACCATTTTCCCGACAGCAGC | 3′ NTB + SalI (−) |
| 35 | GCGCGCCGGTACCAATTAACGTGGCAAGTTCACG | aa 870 NTB + KpnI (−) |
| 36 | TTATATGAGCTCGGTGGCGGAGGATTGTTCGTTGAAGCGTATGACTGG | aa 1153 NTB + SstI + GGGG (+) |
| 39 | TATAAGATCTAAGGAGATATAACAGGATCCCTTGGTACCGGAGGTGG AGGTATGAGTAAAGGAGAAGAACT | 5′ GFP + BglII + BamHI + KpnI + GGGG (+) |
| 40 | TTATATGGATCCATGGGGCTCACTGACGTTTTTGG | 5′ NTB + BamHI + ATG (+) |
| 45 | AGCGGATCCATGTTGTTCGTTGAAGCGTATGACTG | aa 1153 NTB + BamHI + ATG (+) |
| 46 | GCGCGCGGTACCAAGAGTATCAAGGGAATGGTG | aa 648 NTB + KpnI (−) |
| 47 | TTATATGAGCTCGGTGGCGGAGGAACGTTAATGGGGAAATTTGG | aa 649 NTB + SstI + GGGG (+) |
| 48 | TTATATGAGCTCGGTGGCGGAGGAAATTTTGATGTTGAAAAGTGGG | aa 702 NTB + SstI + GGGG (+) |
| 49 | GCGCGCGGTACCTTCTGTAAGTGAGCGCCCTG | aa 1152 NTB + KpnI (−) |
| 50 | TTATATGAATTCATGGCCTCCTCCGAGAACGTC | 5′ dsRed2 + EcoRI (+) |
| 51 | TTATTAGAGCTCTTAAAGCTCATCATGCAGGAACAGGTGGTGGCGGC | 3′ dsRed2 + SstI + HDEL + TAA (−) |
| 53 | GCGCGCGGTACCCTGTACACATTGTGGGCGGA | 3′ VPg + KpnI (−) |
| 54 | CTAGTACCATGGAACGAGCTATACAAGG | 5′ TBSV p19 + NcoI (+) |
| 55 | CTAGTAGGATCCTTACTCGCTTTCTTTTTCGAAG | 3′ TBSV p19 + BamHI + TAA (−) |
| 56 | ATAACGATGAGACGAACTCGAATCAAGATAATCC | Mut. 1st EcoRI site TBSV p19 (+) |
| 57 | GGATTATCTTGATTCGAGTTCGTCTCATCGTTAT | Mut. 1st EcoRI site TBSV p19 (−) |
| 58 | GACAGGACGGAAGCCTCACTGCACAGAGTC | Mut. 2nd EcoRI site TBSV p19 (+) |
| 59 | GACTCTGTGCAGTGAGGCTTCCGTCCTGTC | Mut. 2nd EcoRI site TBSV p19 (−) |
| 64 | TTATTATGGCCAACTCGACGTCACAAGGATCTCAGGCTCCAGTAGCACAG GGAGGTTCACAAGGAGAAGGGCTCACTGACGTTTTTTGGCG | 5′ NTB + MscI + N-glyc. site + ATG (+) |
| 65 | TTATTACTCGAGAATTAACGTGGCAAGTTCACG | aa 701 NTB + XhoI (−) |
| 71 | ACTCTTACGTTAATGGGGAAAACTGGCAAGCGCACTTCTT | Δ aa 654-686 NTB (+) |
| 72 | AAGAAGTGCGCTTGCCAGTTTTCCCCATTAACGTAAGAGT | Δ aa 654-686 NTB (−) |
| 73 | TTATTATGGCCAACTCGACGTCACAAGGATCTCAGGCTCCAGTAGCACAGG GAGGTTCACAAGGAGAAGATGGGCTAGTGCACCATTCCC | aa 649 NTB + MscI + N-glyc. site (+) |
| 75 | TATTACTCGAGATATCTCATTGTCGTTATCAATTGAATG | aa 870 NTB + XhoI (−) |
| 106 | TTATATGGATCCATGGCCAACTCGACGTCACAAGGATC | N-glyc. site + BamHI + ATG (+) |
| 109 | GCGCGCGGTACCTTACGCATAGTCAGGAACATCGTCTGGGTAAATTAAC GTGGCAAGTTCACG | aa 870 NTB + KpnI + HA tag + TAA (−) |
| 110 | CGACCAGGATCCATGGAACAAAAACTTATTTCTGAAGAAGATCTGTTGTTC GTTGAAGCGTATGACTGG | aa 1153 NTB + BamHI + myc + ATG (+) |
| 111 | GCGCGCGGTACCTTAGGCATAGTCAGGAACATCGTATGGGTACTGTACAG ATTGTGGGCGGA | 3′ VPg + KpnI + HA tag + TAA (−) |
| 2868 | CGCAAGACCGGCAACAGG | Within nos poly(A) signal (−) |
Introduced restriction sites are underlined. Point mutations or junction points in deletions are in bold and underlined. Insertions of amino acid spacers (GGGG or GGGS), of a consensus N-glycosylation site (N-glyc. site), and of epitope tags are in bold.
For each plasmid, the start point of the region homologous to the ToRSV sequence or other genes of interest is indicated (e.g., a specific amino acid of the ToRSV P1 sequence or that 5′ end or 3′ end of a specific protein domain). Additional features are also indicated, including introduced restriction sites, introduced start or stop codons (ATG or TAA), or linker amino acid sequences (in the one-letter code). Finally, the strand to which the primer corresponds (+ for coding strand and − for noncoding strand) is also indicated. In the case of primers used to introduce mutations, the nature of the mutations is indicated (i.e., deletion of specific amino acids or mutation [Mut.] of specific restriction sites).
To construct the agroinfiltration vectors, a HindIII-EcoRI fragment from pGFP-nN or pnN-GFP was inserted into the corresponding sites of binary vector pBIN19 (Clontech). The resulting plasmids, pBIN-GFP-nN and pBIN-nN-GFP, contained unique SstI/KpnI or BamHI/KpnI restriction sites bordering the NTB-derived sequence. These restriction sites were used to insert other fragments encoding regions of NTB-VPg fused to the C and N termini of GFP. Plasmid pBIN19-p19 containing the Tomato bushy stunt virus (TBSV) suppressor of gene silencing was constructed by amplifying a cDNA fragment corresponding to the TBSV p19 protein using an infectious TBSV cDNA clone as a template. The fragment was amplified using a previously described two-step PCR protocol (64), primers 54 and 55, and mutagenic primers 56, 57, 58, and 59 designed to abolish two internal EcoRI sites without affecting the amino acid sequence of the protein. The NcoI-BamHI-digested fragment was inserted into the corresponding sites of pBBI525 (52). A HindIII-EcoRI fragment from the resulting plasmid was then transferred into pBIN19.
To construct pT7-G-nN2, a fragment containing the N-terminal region of NTB was amplified using pMR10 as a template and primers 64 and 75, digested by MscI and XhoI, and introduced into the corresponding sites of pCITE-4a(+) (Novagen). Other plasmids were constructed in a similar manner using the following pairs of primers: 73 and 75 for pT7-G-nN2ΔTMD and 64 and 65 for pT7-G-nN. pT7-G-nN2ΔAH3 was produced using a two-step PCR protocol as above and mutagenic primers 71 and 72 in addition to primers 64 and 75. pT7-NV was described previously (60). pT7-N was constructed by amplifying a fragment containing the NTB coding region using primers 30 and 31 and inserting the NcoI-SalI-digested fragment into the corresponding sites of pCITE4a(+). pT7-NV-T1229/A was constructed by replacing the small XhoI fragment of pT7-NV with that of pT7-cNV-T1229/A (60). To obtain pT7-G-N, pT7-G-NV, and pT7-G-NV-T1229/A, the small BglII fragment of pT7-G-nN2 was replaced with a large BglII fragment from pT7-N, pT7-NV, or pT7-NV-T1229/A.
Plasmid pG-nN-HA was constructed by amplifying a fragment using primers 106 and 109 and pT7-G-nN as a template. The BamHI-KpnI-digested fragment was inserted into the corresponding sites of psmRS-GFP(S-K). The BamHI site in this plasmid is located upstream of the GFP coding region, which is therefore deleted from the resulting plasmid. Plasmids pNV-HA, pnN-HA, and pcNV3-HA were constructed in a similar manner using pGFP-NV as a template and primer pairs 40 and 111, 40 and 109, and 110 and 111, respectively. The BamHI-KpnI fragments of pG-nN-HA, pNV-HA, pnN-HA, and pcNV3-HA were ligated with the large BamHI-KpnI fragment of pBIN-GFP-NV, resulting in pBIN-G-nN-HA, pBIN-NV-HA, pBIN-nN-HA, and pBIN-cNV3-HA.
Biolistic delivery of plasmids into N. benthamiana plants and confocal microscopy.
Biolistic delivery of purified plasmids into N. benthamiana plant cells was done as previously described (64). A confocal microscope (Leica) was used to visualize the subcellular distribution patterns of the GFP and dsRed2 fusion proteins. In some cases, the fluorescence associated with GFP fusion proteins was weak; therefore, the green and red images were collected in a sequential manner to avoid cross talk between the channels. To minimize organelle movement between the collections of images, the channels were switched between lines with an average of four to eight data collections for each line. Green and red fluorescence images were merged using the Leica confocal software.
Agroinfiltration of N. benthamiana plants.
The binary vectors containing the plant expression cassettes of the targeting fusion proteins were transformed into Agrobacterium tumefaciens LBA4044 (Invitrogen) by electroporation. Colonies confirmed to contain the binary vector by PCR were used for agroinfiltration assays as previously described (57). To allow optimal expression of the fusion proteins, the TBSV p19 protein was coexpressed in the agroinfiltrated plant cells to prevent induction of posttranscriptional gene silencing (57). After infiltration, the plants were cultured in the greenhouse for 3 to 5 days. The infiltrated area of the inoculated leaves was collected for extraction.
Subcellular fractionation and deglycosylation assays.
Extraction of plant tissues and production of postnuclear (S3), soluble (S30), and membrane-enriched (P30) fractions were carried out as previously described (30, 46). The P30 fraction was resuspended in a volume of homogenization buffer corresponding to that of the S30 fraction to allow direct comparison of the protein concentration in each fraction. In some experiments, the P30 fraction was further concentrated by centrifugation at 100,000 × g for 1 h, followed by resuspension in 1/10 of the initial volume (P100 fraction). Membrane flotation assays were conducted essentially as previously described (7). Briefly, 800 μl of the S3, P30, or P100 fraction was adjusted to a final volume of 1.9 ml of 71.5% sucrose (wt/vol) in NTE buffer (7) and overlaid with 7 ml of 65% sucrose in NTE and then 3.1 ml of 10% sucrose in NTE. After centrifugation at 100,000 × g for 18 h, 12 1-ml fractions were collected from the bottom of the tube.
To purify hemagglutinin (HA) fusion proteins, 300 μl of the S3, S30, or P30 fraction was adjusted to a final concentration of 1% Triton X-100-0.1% sodium dodecyl sulfate (SDS)-0.05% sodium deoxycholate-150 mM NaCl, supplemented with 50 μl of resuspended anti-HA affinity matrix (Roche), and incubated overnight at 4°C with constant agitation. The matrix was pelleted and washed two times with homogenization buffer as described by the supplier. Captured proteins were released from the matrix by addition of 50 μl of protein loading buffer (34) to the pellet followed by boiling for 5 min.
For deglycosylation assays, protein loading buffer (34) was added to the P30 fractions (0.33 volume of a 4× stock solution). The samples were boiled for 5 min and centrifuged for 5 min, and the supernatant was collected. Nine volumes of the appropriate deglycosylation buffer was added to each sample along with either N-glycosidase F (PNGase F, 150 mU/50 μl of reaction mixture; Roche) or endoglycosidase H (Endo H, 1 U/50 μl of reaction mixture; Roche). The PNGase F buffer was 200 mM sodium phosphate buffer (pH 7)-25 mM EDTA-1% Triton X-100. The Endo H buffer was 100 mM sodium citrate buffer (pH 5)-100 mM β-mercaptoethanol-1% Triton X-100. The reaction mixtures were incubated at 37°C overnight. Protein loading buffer (0.33 volume of a 4× stock solution) was added, and the samples were boiled for 5 min.
Separation of proteins by SDS-polyacrylamide gel electrophoresis (PAGE) and immunodetection were conducted as previously described (30) using a mouse monoclonal anti-GFP antibody (BD Biosciences), a rabbit polyclonal anti-Bip antibody (donated by M. Chrispeels), a rabbit polyclonal anti-NTB antibody (59), or a mouse monoclonal anti-HA antibody (Bio/Can Scientific). The secondary antibodies were goat anti-mouse or goat anti-rabbit immunoglobulin G conjugated with horseradish peroxidase (Bio/Can).
In vitro translation assays.
Coupled in vitro transcription-translation reactions in the presence or the absence of canine microsomal membranes and deglycosylation assays of translation products were conducted as previously described (60).
Computer-assisted prediction of putative transmembrane helices and amphipathic helices.
Prediction of transmembrane helices in the N-terminal region of NTB was performed using the following programs: PHDhtm (41), Sosui (31), Tmpred (32), TopPred2 (58), TMHMM (50), HMMTOP (56), and MEMSAT2 (33). Prediction and projection of the putative amphipathic helix was conducted using the Antheprot program (22).
RESULTS
ER targeting of GFP fusion proteins containing the entire NTB-VPg protein.
We have previously shown that NTB-VPg is an integral membrane protein associated with ER-derived membranes in ToRSV-infected cells (30). To determine whether the NTB-VPg protein alone has the ability to associate with intracellular membranes, we examined the intracellular localization of NTB-VPg expressed independently of other viral proteins in planta. Both N-terminal and C-terminal fusions of NTB-VPg with GFP (GFP-NV and NV-GFP, Fig. 1A) were expressed in N. benthamiana plants (a natural host for ToRSV) using agroinfiltration. Five days after agroinfiltration, the leaves were collected and subcellular fractions enriched in soluble cytoplasmic proteins (S30 fraction) and in membrane-associated proteins (P30 fraction) were produced by differential centrifugation. Proteins were separated by SDS-PAGE and analyzed by immunoblot experiments using either an anti-GFP antibody (to detect GFP fusion proteins) or an antibody that recognizes an endogenous ER luminal protein (Bip) (30). As expected, Bip was detected predominantly in the membrane-enriched fraction while free GFP was detected predominantly in the S30 fraction (Fig. 1B, lanes 1 and 2 and lanes 5 and 6). The full-length 96-kDa GFP-NV and NV-GFP proteins were detected exclusively in the membrane-enriched fractions (P30) (Fig. 1B, lanes 11 to 14). The concentration of these proteins in the plant extracts was much lower than that of the free GFP, and extended exposure of the immunoblots was required to detect the proteins. Smaller proteins (of 28 to 32 kDa) were also detected in the S30 and P30 fractions derived from plants expressing GFP-NV and NV-GFP. One possibility is that they represent degradation products of the full-length proteins. The presence of the full-length GFP-NV and NV-GFP proteins in the P30 fraction suggested that they are membrane associated, although this experiment could not exclude the possibility that protein aggregation was responsible for the recovery of these proteins in the P30 fraction. The concentration of these proteins in the plant extracts was too low to allow membrane flotation experiments that would eliminate this possibility (see below).
FIG. 1.

Immunodetection of GFP fusion proteins containing the entire NTB-VPg protein or the central region of NTB. (A) Schematic representation of GFP fusion proteins. The hatched areas and the black squares represent the GFP and VPg domains, respectively. The black triangle indicates a putative amphipathic helix at the N terminus of NTB, and the asterisks represent stretches of hydrophobic amino acids. The expected molecular mass of each fusion protein is indicated in parentheses along with the amino acids of the P1 polyprotein included in each fusion protein (numbering according to Rott et al. [42]). (B) Immunodetection of GFP fusion proteins. N. benthamiana plants were agroinfiltrated with A. tumefaciens transformed with binary vectors allowing the expression of the various GFP fusion proteins as indicated above each lane. Plant extracts were fractionated into a soluble fraction (S) or a membrane-enriched fraction (P) by differential centrifugation as described in Materials and Methods (S30 and P30 fractions). Proteins were separated by SDS-PAGE (11% polyacrylamide) and immunodetected with anti-Bip (lanes 1 and 2) or anti-GFP (lanes 3 to 14) antibodies (Abs). The following amount of each fraction was loaded in the corresponding lanes: lanes 1 and 2, 5 μl; lanes 3 to 8, 2.5 μl; lanes 9 to 14, 10 μl. The film was exposed for 10 min (lanes 1 to 8) or 18 h (lanes 9 to 14). Migration of molecular mass standards is indicated on the left. ck, negative control agroinfiltrated with pBIN-p19 only. The arrow points to the band corresponding to the full-length GFP-NV and NV-GFP proteins.
The subcellular localization of the fusion proteins was examined in greater detail using confocal microscopy. To allow dual labeling of ER membranes and the various GFP fusion proteins, we engineered an ER-targeted red fluorescent protein (ER-dsRed2) using ER targeting and retention signals (including an N-terminal signal peptide and a C-terminal HDEL retention signal) previously described for an ER-targeted GFP reporter (63). The plasmids were delivered into N. benthamiana plant cells using biolistic bombardment. Twenty hours after transfection, the red fluorescence associated with ER-dsRed2 was clearly distinguished from the duller chloroplast autofluorescence (i.e., cells adjacent to those expressing the ER marker were not visible under the conditions used for the detection of the dsRed2 fluorescence; Fig. 2, panel 1, ER-dsRed2). The red fluorescence was concentrated in the perinuclear area and in the cortical network of the ER (Fig. 2, panels 1 to 3, ER-dsRed2), a pattern similar to that previously described for ER-GFP (11). In contrast, fluorescence associated with the free GFP was distributed throughout the cell and diffused through the nuclear pore (Fig. 2, panels 1 and 2, GFP). In some cases, the green fluorescence displayed a diffused network pattern that was in proximity to the ER network but did not coincide with the ER marker when examined at higher magnification (Fig. 2, panels 2 and 3). The subcellular localizations of free GFP and ER-dsRed2 did not change whether they were expressed individually or in combination (data not shown), suggesting that there was no interaction between the two proteins.
FIG. 2.

Subcellular localization of GFP fusion proteins containing the entire NTB-VPg protein or the central region of NTB. N. benthamiana leaf epidermal cells were transfected by biolistic delivery. Each transfection experiment included one plasmid allowing the ex pression of the GFP fusion protein as indicated on the left and one plasmid allowing the expression of the ER-dsRed2 marker. Individual transfected cells were examined by confocal microscopy 24 h after transfection, and a single slice is shown. The green channel shows the fluorescence associated with the GFP fusion proteins and the red channel shows the ER-dsRed2 fluorescence. The digitally superimposed images, where green and red signals that coincide produce a yellow signal, are also shown (merge). Panels 2, 5, 7, and 9 are close-up views of the region included in the white square in panels 1, 4, 6, and 8, respectively. A region of the cortical ER network is shown in panels 3 and 10. The bars on the merge panels represent 10 μm for panels 1, 4, 6, 8, and 11 and 2 μm for panels 3 and 10.
Fluorescence associated with GFP-NV and NV-GFP was concentrated around the nucleus or in a sharp web-like structure that coincided with the fluorescence associated with ER-dsRed2 (Fig. 2, panels 4 to 10). Aggregates of green and red fluorescence in the perinuclear area were occasionally observed in cells expressing GFP-NV or NV-GFP and ER-dsRed2 (panels 6 and 7 and data not shown) but not in cells expressing free GFP and ER-dsRed2. Taken together, these results suggested that GFP-NV and NV-GFP were directed to perinuclear and cortical ER membranes.
Absence of membrane-targeting domains in the central region of NTB.
We have previously suggested that sequences at the N and C termini of the NTB domain are involved in its association with the ER (30). To determine whether the central region of NTB contains unidentified membrane-targeting domains, a GFP fusion protein including only the central region of NTB (GFP-mN, Fig. 1A) was expressed in plants. After subcellular fractionation of plant extracts, GFP-mN was only detected in the S30 fraction (Fig. 1, lanes 7 and 8). The green fluorescence associated with GFP-mN (Fig. 2, panel 11) was similar to that observed for the free GFP and did not coincide with the ER-dsRed2 fluorescence. Taken together, these results indicated that GFP-mN does not associate with intracellular membranes and that the middle portion of NTB does not contain elements that could independently promote membrane association.
ER-specific glycosylation of GFP fusion proteins containing the hydrophobic domain in the C-terminal region of NTB-VPg.
We have previously shown that a truncated protein, cNV3, containing the hydrophobic domain located at the C terminus of NTB and the VPg domain could associate with canine microsomal membranes in vitro (60). Deletion of the hydrophobic region prevented in vitro membrane association (mutant ΔTM1) (60). To determine if cNV3 has the ability to associate with intracellular membranes in vivo, GFP was fused to the wild-type (WT) or ΔTM1 derivative of cNV3 (GFP-cNV3 and cNV3-GFP, Fig. 3A). Homogenates of leaves expressing these proteins were separated into S30 and P30 fractions. GFP-cNV3 (with an apparent molecular mass of 36 kDa) was present in both the S30 and P30 fractions (Fig. 3B, lanes 1 and 2). The full-length cNV3-GFP protein (with an apparent molecular mass of 38 kDa) was only detected in the P30 fraction, although smaller proteins were also detected in the S30 fraction (lanes 5 and 6). In contrast, both GFP-cNV3ΔTM1 and cNV3ΔTM1-GFP were present predominantly in the S30 fraction (lanes 3 and 4 and lanes 7 and 8). This result suggested that P30 fractionation of GFP-cNV3 and cNV3-GFP was dependent on the presence of the hydrophobic region.
FIG. 3.

Immunodetection of GFP fusion proteins containing the C-terminal region of NTB-VPg. (A) Schematic representation of GFP fusion proteins. The hatched box represents the GFP domain, the open box represents the NTB domain (with the hydrophobic region shown by the asterisk), and the black box represents the VPg domain. A conserved N-glycosylation site in the VPg domain is shown by the letter Y. The predicted molecular mass of each fusion protein is indicated in parentheses along with the amino acids of the P1 polyprotein included in the protein. The amino acids deleted in the ΔTM1 derivatives of GFP-cNV3 and cNV3-GFP are also indicated. (B) Immunodetection of GFP fusion proteins. Plant extracts from N. benthamiana plants agroinfiltrated with plasmids allowing the expression of WT or mutated versions of GFP-cNV3 and cNV3-GFP were subjected to differential centrifugation as indicated in Fig. 1. Proteins present in 5 μl of the soluble (S) or membrane-enriched (P) fraction were separated by SDS-PAGE (12% polyacrylamide) and immunodetected with the anti-GFP antibody. (C) Analysis of the glycosylation status of GFP-cNV3 and cNV3-GFP. Proteins present in membrane-enriched fractions (P30) derived from plants expressing the WT or mutated version (T/A mutant including a mutation of the VPg N-glycosylation site) of the fusion proteins were analyzed. In the case of the WT proteins, the P30 fractions were treated with two deglycosylation enzymes (PNGase F or Endo H) as indicated above each lane. The proteins were separated by SDS-PAGE as above and immunodetected with the anti-GFP antibody. The diamonds point to glycosylated forms of the proteins. Migration of molecular mass standards is indicated on the left.
Multiple forms of GFP-cNV3 and cNV3-GFP were detected in the membrane-enriched fractions, raising the possibility that membrane-dependent modification of the proteins, such as glycosylation, occurred in planta. We have previously shown that a naturally occurring N-glycosylation site (NST1229) in the VPg domain is glycosylated in vitro upon addition of canine microsomal membranes (60). To test for the presence of glycosylated forms of GFP-cNV3 and cNV3-GFP in the P30 fraction, we conducted two sets of experiments. In one set of experiments, we introduced a mutation in the glycosylation site (mutation T1229/A) in GFP-cNV3. This mutation was previously shown to prevent glycosylation in vitro (60). A protein with an apparent molecular mass of 42 kDa was detected in P30 fractions derived from plants expressing WT GFP-cNV3 but not in the P30 fraction derived from plants expressing the GFP-cNV3-T/A derivative (Fig. 3C, lanes 1 and 2) suggesting that this protein was a glycosylated form of GFP-cNV3. In a second set of experiments, P30 extracts derived from plants expressing WT GFP-cNV3 were treated with two deglycosylation enzymes specific for N-linked oligosaccharides. PNGase F releases all classes of N-linked oligosaccharides, including complex-type N-linked oligosaccharides produced in the Golgi, provided that they do not contain a fucose linked α(1-3) to Asn-GlcNAc, a structural motif present in many plant glycoproteins (37). Endo H digests the high-mannose carbohydrate side chains added in the ER but does not recognize oligosaccharides further modified in the Golgi (37). After treatment of the P30 extracts with either PNGase F or Endo H, the 42-kDa protein disappeared but was replaced by a new protein with an apparent molecular mass of 38 kDa (Fig. 3C, lanes 2 to 4). Similarly, in P30 fractions derived from plants expressing WT cNV3-GFP, a 42-kDa protein was eliminated after treatment with either deglycosylation enzyme (lanes 5 to 7). Possibly, the deglycosylated protein comigrated with the 38-kDa cNV3-GFP protein. These results confirmed the above suggestion that the 42-kDa protein is a glycosylated form of GFP-cNV3 and cNV3-GFP. The sensitivity of the 42-kDa glycosylated protein to Endo H also suggested that it was retained in the ER and did not translocate into the Golgi apparatus.
In addition to the 36- and 42-kDa proteins (detected for GFP-cNV3) and 38- and 42-kDa proteins (detected for cNV3-GFP), larger forms of the proteins (approximately 66 to 80 kDa) were observed, although the relative concentration of these proteins varied from one sample to another (Fig. 3C, lanes 2 and 5). Many membrane proteins can maintain their oligomeric structure in the presence of SDS (21). Therefore, the 66- to 80-kDa proteins may correspond to dimeric forms of the 36- to 42-kDa GFP-cNV3 and cNV3-GFP proteins. Interestingly, an 80-kDa protein present in the P30 fractions from plants expressing GFP-cNV3 (shown by the diamond in the upper region of the gel, lanes 2 to 4) was sensitive to the PNGase F and Endo H treatments, indicating that it was glycosylated. This result raised the possibility that dimerization of GFP-cNV3 occurred in the membrane environment with the VPg domain translocated in the lumen. Finally, in P30 extracts from plants expressing cNV3-GFP and cNV3-GFPΔTM1, smaller proteins (with apparent molecular masses of 31 to 33 kDa) were also detected predominantly in the S30 fractions (Fig. 3B, lanes 5 to 8). One possibility is that they are degradation products of the full-length proteins.
Partial ER association of GFP fusion proteins containing the hydrophobic domain in the C-terminal region of NTB-VPg.
In N. benthamiana plant cells, expression of GFP-cNV3 and cNV3-GFP proteins resulted in a fluorescence pattern that was suggestive of a partial association with ER membranes (Fig. 4, panels 1 to 4). Diffuse fluorescence was observed in the nucleus, suggesting the presence of soluble protein. However, there were also clear patterns of green fluorescence that coincided with the ER-dsRed2 fluorescence. The proportion of green fluorescence that coincided with the ER marker varied from cell to cell and with time but was in general higher in cells expressing cNV3-GFP than in cells expressing GFP-cNV3. The fluorescence in cells transfected with the GFP-cNV3 construct was predominantly diffuse at early time points (6 and 20 h posttransfection, data not shown), and it was not possible to conclusively determine whether fluorescence associated with membranes was also present at these time points. After 44 h, GFP-cNV3 was partially colocalized with the ER marker (panels 1 and 2). Taken together, these results suggested a partial association of GFP-cNV3 and cNV3-GFP with ER membranes. This result was consistent with the partial distribution of these proteins in the membrane-enriched fraction described above. Mutated derivatives of GFP-cNV3 and cNV3-GFP with a deletion of the entire hydrophobic region (ΔTM1 mutants, panels 5 to 8) did not localize to ER membranes, confirming the above suggestion that this region is responsible for the partial ER association of GFP-cNV3 and cNV3-GFP.
FIG. 4.

Subcellular localization of GFP fusion proteins containing the C-terminal region of NTB-VPg. N. benthamiana leaf epidermal cells were transfected by biolistic delivery with plasmids allowing the expression of the GFP fusion proteins indicated on the left and the ER-dsRed2 marker. Each panel represents a single slice of an individual transfected cell examined by confocal microscopy 44 h after transfection. As in Fig. 2, the green and red channels show the fluorescence associated with the GFP fusion proteins and the ER-dsRed2 protein, respectively. Panels 2, 4, 6, and 8 are close-up views of the region included in the white square in panels 1, 3, 5, and 7, respectively. The bars in the merge panels represent 10 μm.
Membrane association of GFP fusion proteins containing a putative amphipathic helix at the N terminus of NTB.
We have previously identified a putative amphipathic helix in the N-terminal region of NTB and suggested that this helix is involved in membrane association (predicted from amino acid [aa] 654 to aa 689, Fig. 5A) (30). A projection of the predicted amphipathic helix is shown in Fig. 5B. Examination of the N-terminal region of NTB with a number of transmembrane helix prediction programs (see Materials and Methods) also revealed the presence of a hydrophobic region located immediately upstream of the putative amphipathic helix which was predicted to form a transmembrane helix by some but not all prediction programs (aa 621 to aa 643, Fig. 5C). The degree of confidence in the Tmpred and Sosui predictions was not very strong (legend to Fig. 5C).
FIG. 5.

Computer-assisted analysis of putative membrane association domains in the N-terminal region of NTB. (A) Primary sequence of a putative amphipathic helix located in the N-terminal region of NTB. The dashed lines represent amino acids that were deleted by site-directed mutagenesis in mutant G-nN2ΔAH3. (B) Edmunson helical-wheel representation of the NTB N-terminal amphipathic helix. Hydrophobic and charged amino acids are shown in black and white, respectively. (C) Prediction of a putative transmembrane helix in the N-terminal region of NTB. The deduced amino acid sequence of the first 23 aa of the NTB domain are shown at the top. Below this sequence, the transmembrane helices predicted by each individual program are shown. Only three programs predicted a transmembrane helix at this position. The score for the Tmpred prediction for this transmembrane helix was 546, compared to a score of 2,698 for the previously identified transmembrane helix at the C terminus of NTB (only scores above 500 are considered significant). In the Sosui program, the N-terminal transmembrane helix was predicted to be a secondary transmembrane helix, while the transmembrane helix at the C terminus of NTB was predicted to be a primary transmembrane helix.
To evaluate the role of the N-terminal region of NTB in ER association, GFP was fused to the first 80 aa of NTB (GFP-nN and nN-GFP, Fig. 6A). In addition, fusion proteins GFP-nN-HR (containing only the upstream hydrophobic region) and GFP-nNΔHR (containing only the amphipathic helix) were also tested. Immunodetection of cell extracts derived from plants expressing the various GFP fusion proteins revealed that proteins containing the amphipathic helix (GFP-nN, GFP-nNΔHR, and nN-GFP) were present predominantly in the membrane-enriched fraction, while a protein that included only the upstream hydrophobic region (GFP-nN-HR) was almost exclusively in the soluble fraction (Fig. 6B). Larger forms of GFP-nN, GFP-nNΔHR, and nN-GFP were detected which may correspond to dimers and larger oligomers (lanes 1 and 2, lanes 5 and 6, and data not shown). These results suggested that the putative amphipathic helix is involved in membrane association or is responsible for protein aggregation. To distinguish between these possibilities, we conducted membrane flotation assays. Membrane-enriched fractions (P30) were adjusted to a concentration of 71.5% sucrose and deposited at the bottom of a sucrose step gradient. After high-speed centrifugation, soluble or aggregated proteins remain at the bottom of the gradient while membrane-associated proteins float at the interface between the 65% and 10% sucrose layers due to the buoyant density of lipids (7). We have shown that GFP-cNV3, present in P30 fractions, is glycosylated in an ER-specific manner in plants (Fig. 3C), demonstrating that it is membrane associated. Thus, this protein was used as a positive control for the membrane flotation assay. The collected fractions were probed with either anti-GFP antibody to detect the GFP fusion proteins or with anti-Bip antibody to detect the endogenous ER luminal protein. As expected, GFP-cNV3 and Bip partitioned predominantly at the interface between the 65% and 10% sucrose layers (fraction 9, Fig. 6C). In contrast, free GFP (present in a postnuclear fraction, S3) remained at the bottom of the gradient as previously described (7). GFP-nN partitioned into fraction 9, confirming that the protein is membrane associated. Similar results were obtained with GFP-nNΔHR (data not shown).
FIG. 6.

Immunodetection of GFP fusion proteins containing the N-terminal domain of NTB. (A) Schematic representation of GFP fusion proteins. As in Fig. 1, the hatched box represents the GFP domain and the open box represents the NTB domain (with the putative amphipathic helix shown by the triangle and a hydrophobic region shown by the asterisk). The predicted molecular mass of each fusion protein is indicated in parentheses along with the amino acids of the P1 polyprotein included in the proteins. (B) Immunodetection of GFP fusion proteins. Extracts from agroinfiltrated plants were fractionated into a soluble (S) and a membrane-enriched (P) fraction as described in Fig. 1. Proteins present in 2 μl of each fraction were separated by SDS-PAGE (12% polyacrylamide) and immunodetected with the anti-GFP antibody. Migration of molecular mass standards is shown on the left. (C) Membrane flotation assays. Subcellular fractions were deposited at the bottom of a sucrose step gradient as described in Materials and Methods. The starting material was a membrane-enriched fraction (P30) in the cases of GFP-cNV3 and GFP-nN and a postnuclear fraction (S3) in the case of the soluble GFP. GFP fusion proteins present in 25 μl of fractions 1 to 12 collected from the bottom of the gradient were separated by SDS-PAGE and detected by immunoblotting using anti-GFP antibodies. For detection of the endogenous Bip protein, fractions collected from the GFP-cNV3 gradient were probed with anti-Bip antibodies. Probing of fractions from the GFP and GFP-nN gradients with the anti-Bip antibodies resulted in similar results (data not shown). Only the portion of each gel displaying the monomeric form of the protein is shown, although larger forms of the proteins (probably corresponding to oligomers) were observed for GFP-cNV3 and GFP-nN. In the case of GFP-cNV3, the two bands shown correspond to the unmodified and glycosylated forms of the protein.
In N. benthamiana cells expressing GFP-nN, the green fluorescence coincided with the perinuclear ER-dsRed2 fluorescence and also partially with the ER-dsRed2 fluorescence associated with the cortical network (Fig. 7, panels 1 and 2). There were also aggregates of green fluorescence in the perinuclear area or throughout the cell, often in close proximity to the ER network (panel 2). In cells expressing nN-GFP, the ER network was drastically altered. Instead of a distinct ER network, large aggregates of red fluorescence were present, often in the proximity of the nucleus (panels 3 and 4). The green fluorescence colocalized with these aggregates. The fluorescence associated with GFP-nNΔHR, in which the upstream hydrophobic region was deleted, clearly coincided with the ER-dsRed2 fluorescence associated with the perinuclear and cortical ER (Fig. 7, panels 5 and 6). In contrast, deletion of the amphipathic helix from GFP-nN (GFP-nN-HR) prevented its association with membranes (Fig. 7, panel 7). These results indicated that the putative amphipathic helix is involved in membrane association.
FIG. 7.

Subcellular localization of GFP fusion proteins containing the N-terminal portion of NTB in N. benthamiana cells. N. benthamiana leaf epidermal cells were transfected by biolistic delivery with plasmids allowing the expression of the GFP fusion proteins indicated on the left and the ER-dsRed2 marker. Each panel represent a single slice of individual transfected cells examined by confocal microscopy 24 h after transfection. The green and red channels show the fluorescence associated with the GFP fusion proteins and ER-dsRed2 fluorescence, respectively. The merge panels represent the digitally superimposed images. Panel 2, 4, and 6 are close-up views of the area shown in the white square in panels 1, 3, and 5, respectively. The bars in the merge panels represent 10 μm.
In vitro analysis of the topology of the N-terminal region of NTB-VPg in the membranes.
To further analyze the ability of various elements in the N-terminal region of NTB to promote membrane association, we used in vitro membrane association assays (60). To examine the topology of the N terminus of NTB in the membrane, we introduced a glycosylation site immediately upstream of the NTB sequence. This glycosylation site was shown by others to be recognized efficiently by canine microsomal membranes in vitro when placed upstream of a transmembrane domain and when it is translocated in the luminal side of the membrane (39). In construct G-nN2, the first 250 aa of NTB were fused in frame with this glycosylation signal (Fig. 8A). A protein of the expected size was produced in a coupled in vitro transcription-translation system (Fig. 8B, lane 1). Addition of canine microsomal membranes to the reaction mixture resulted in the production of an additional protein with slower mobility (lane 2, shown by the diamond). Treatment of the reaction mixture with PNGase F resulted in the disappearance of this additional protein, indicating that it was a glycosylated form of G-nN2 (lane 3). Similar results were obtained with construct G-nN, which included only the first 80 aa of NTB, corresponding to the region of NTB included in the GFP-nN protein used for the in planta experiments (lanes 10 to 12). A larger form of G-nN was also observed in the upper region of the gel which probably corresponds to a dimer (its apparent mobility was 28 kDa, compared to 13 kDa for the monomeric form of the protein). The 28-kDa protein was glycosylated in the presence of membranes (lanes 10 to 12, shown by the diamond in the upper region of the gel), raising the possibility that dimerization of G-nN occurred in the membrane environment. Deletion of the upstream hydrophobic region (G-nN2ΔHR) did not prevent recognition of the glycosylation site (lanes 4 to 6), while deletion of the putative amphipathic helix (G-nN2ΔAH3) drastically decreased the extent of glycosylation (lanes 7 to 9). These results suggested that aa 654 to 686, containing the putative amphipathic helix, directed the translocation of the N terminus of NTB to the luminal face of the membranes in vitro.
FIG. 8.

Topological analysis of the N terminus of NTB using an introduced N-glycosylation site. (A) Schematic representation of truncated proteins containing the N-terminal domain of NTB fused to an inserted artificial N-glycosylation site. Amino acids inserted at the N terminus of the NTB domain are shown with the shaded area (introduced N-glycosylation site represented by the letter Y). The black triangle represents the predicted amphipathic helix, and the asterisk represents the upstream hydrophobic region. The sequence of amino acids inserted in frame with the N terminus of NTB is shown at the bottom with the consensus N-glycosylation site underlined. The name of each protein is shown on the left. Numbers in parentheses indicate the amino acids at the N and C termini of the region of P1 contained in each fusion protein along with the predicted molecular mass of the proteins. G-nN2ΔAH3 contained the same region of NTB as G-nN2, with the exception that aa 654 to 686 were deleted. (B) In vitro membrane association assays of truncated proteins containing the N-terminal domain of NTB. Proteins were translated in the presence (+) or the absence (−) of canine microsomal membranes (MM). After translation, the in vitro translation products were treated with PNGase F as indicated above each lane. The translation products were separated by SDS-PAGE (12% polyacrylamide for lanes 1 to 9 and 18% polyacrylamide for lanes 10 to 12) and detected by autoradiography. Diamonds indicate the glycosylated forms of the protein monomers or dimers. Migration of molecular mass standards is shown on the left for lanes 1 to 9 and on the right for lanes 10 to 12. (C) Schematic representation of proteins containing either the NTB-VPg domains or the NTB domain fused to an inserted artificial N-glycosylation site. The NTB domain is shown by the open box, and the VPg domain is shown by the black box. N-glycosylation sites in inserted amino acids at the N terminus of NTB or in the VPg domain are shown by the letter Y above the shaded box and the black box, respectively. In the T/A mutant (shown in G-NVT/A), the glycosylation site in the VPg domain is inactivated. (D) In vitro membrane association assays of WT or mutated versions of NTB-VPg (NV) and NTB (N). The G-NV, G-NVT/A, and G-N proteins contained the inserted artificial N-glycosylation site at the N terminus, as shown in panel C. NVT/A and G-NVT/A were mutated at the conserved N-glycosylation site present in the VPg domain. Proteins were translated in the presence (+) or in the absence (−) of canine microsomal membranes (MM). The translation products were separated by SDS-PAGE (10% polyacrylamide) and detected by autoradiography. Diamonds indicate the single- and double-glycosylated forms of the proteins. Migration of molecular mass standards is shown on the left.
We next wished to confirm this result in the context of the entire NTB-VPg protein. We have previously shown that the C terminus of the protein is translocated in the lumen, resulting in the recognition of a glycosylation site in the VPg domain. The T1229/A mutation was introduced into NTB-VPg to produce NVT/A, which did not contain a glycosylation site. The glycosylation site described above was then fused in frame with the N terminus of NV (consisting of WT NTB-VPg), NVT/A, or N (consisting of the entire NTB domain without the VPg domain), to produce G-NV, G-NVT/A, and G-N, respectively (Fig. 8C). As shown previously, WT NV was glycosylated in the presence of the canine microsomal membranes (Fig. 8D, lanes 1 and 2) (60). Glycosylation was prevented after mutation of the VPg glycosylation site (NVT/A, lanes 3 and 4) or deletion of the VPg domain (N, lanes 9 and 10). These results confirmed that the VPg domain is translocated in the lumen in the context of the entire NTB-VPg protein. Glycosylated forms of the proteins were also observed after translation of G-NV, G-NVT/A, and G-N in the presence of membranes (lanes 5 to 8 and lanes 11 and 12), suggesting that the artificial glycosylation site at the N terminus of NTB was translocated in the lumen. Interestingly, two forms of glycosylated proteins were observed for G-NV (lane 6). One form comigrated with the glycosylated form of proteins containing a single glycosylation site, while the other form had a slower mobility and was apparently glycosylated at two sites. This protein contains two glycosylation sites: the N-terminal introduced glycosylation site and the C-terminal VPg glycosylation site. This result suggests that several topologies of the protein coexisted, one of which implied the translocation of both the N and C termini of the protein in the lumen.
Subcellular fractionation of a truncated protein containing the C-terminal or N-terminal region of NTB-VPg fused to the HA epitope tag.
The results presented above and previously (60) suggest that the C-terminal and N-terminal regions of NTB contain elements that can target GFP fusion proteins to intracellular membranes in planta and can promote membrane association of the native NTB-VPg or smaller truncated proteins in vitro. We next wished to confirm that these membrane association elements were also active in planta when fused to smaller epitope tags. In initial experiments, we tested a fusion protein containing the entire NTB and VPg domains and the HA epitope tag (YPYDVPDYA). Unfortunately, we were not able to detect the fusion protein in extracts from agroinfiltrated plants using anti-HA or anti-NTB antibodies (data not shown). We then tested smaller fusion proteins containing the cNV3 or nN truncated protein (cNV3-HA and nN-HA, Fig. 9A). G-nN-HA was a derivative of G-nN described above and included the HA epitope and the artificial glycosylation site at the N terminus of the NTB domain. The concentration of the fusion proteins in plant extracts was low, and detection of specific proteins by the anti-HA antibody was hindered by the presence of large amounts of a 14-kDa protein from plants in postnuclear (S3) and cytoplasmic (S30) fractions (data not shown). To circumvent this problem, HA fusion proteins were purified from S3, S30, and P30 fractions using an anti-HA affinity matrix in the presence of mild detergents. Using this method, we detected specific proteins in S3 fractions derived from plants expressing G-nN-HA and cNV3-HA but not in those derived from plants expressing nN-HA (Fig. 9B and data not shown). Both G-nN-HA and cNV3-HA were found predominantly in the P30 fractions, although a portion of the cNV3-HA protein was also present in the S30 fraction. Membrane flotation assays were conducted using a reconcentrated membrane-enriched fraction (P100 fraction, resuspended in 1/10 of the original volume of the P30 fraction). Both proteins separated at the interface between the 65% and 10% sucrose layers, confirming that they were membrane associated (fraction 9, Fig. 9C). The subcellular fractionation behavior of the cNV3-HA and G-nN-HA proteins was very similar to that of the corresponding GFP fusion proteins (GFP-cNV3 and GFP-nN, Fig. 3 and 6).
FIG. 9.
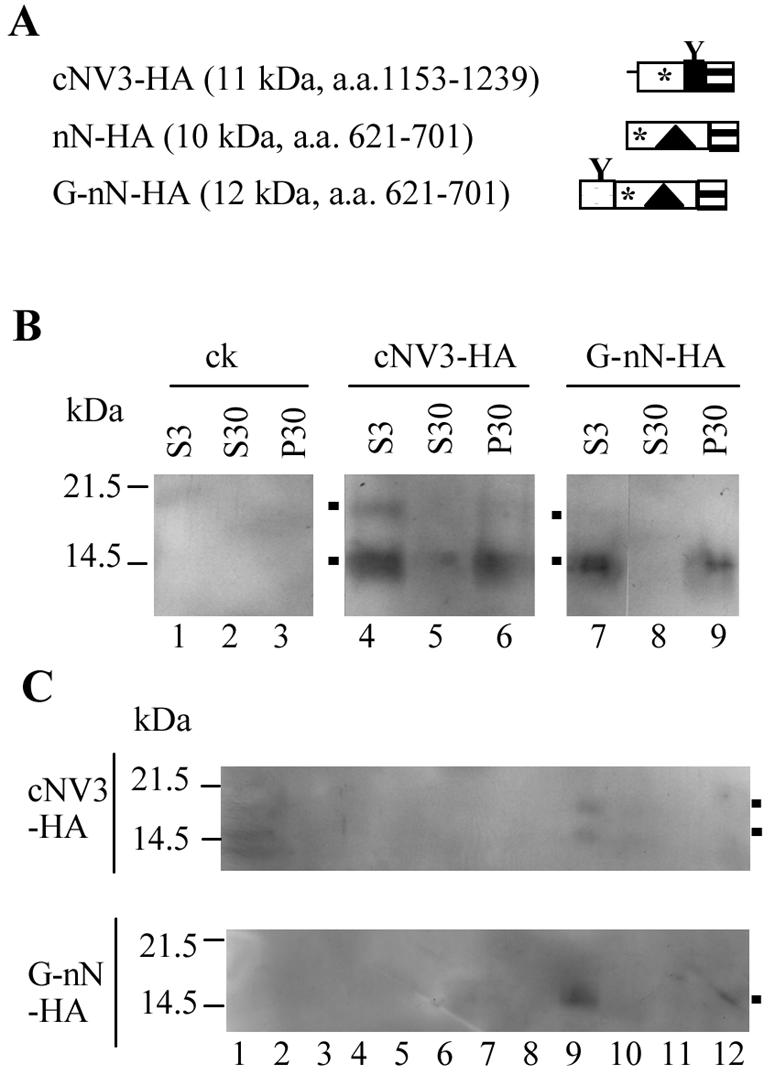
Immunodetection of the N-terminal and C-terminal regions of NTB-VPg fused to epitope tags. (A) Schematic representation of fusion proteins. The boxes with horizontal hatches represent HA epitope tags. The open and black boxes represent the NTB and VPg domains, respectively (with the putative amphipathic helix shown by the triangle and hydrophobic regions shown by the asterisk). The shaded box represents the artificial glycosylation site as described in Fig. 8. The predicted molecular mass of each fusion protein is indicated in parentheses along with the amino acids of the P1 polyprotein included in the proteins. (B) Immunodetection of GFP fusion proteins. HA fusion proteins present in postnuclear (S3), soluble (S30), and membrane-enriched (P30) fractions derived from agroinfiltrated plants were purified by HA affinity chromatography as described in Materials and Methods. Proteins were separated by SDS-PAGE (16% polyacrylamide) and immunodetected with anti-HA antibodies. Migration of molecular mass standards is shown on the left. The positions of various forms of the G-nN-HA and cNV3-HA proteins are indicated by the black squares on the left of the gels. ck, negative control agroinfiltrated with pBIN-p19 only. (C) Membrane flotation assays. P100 fractions were deposited at the bottom of a sucrose step gradient. HA fusion proteins present in 25 μl of fractions 1 to 12 collected from the bottom of the gradient were separated by SDS-PAGE as described above and immunodetected with anti-HA antibodies.
Several forms of the cNV3-HA and G-nN-HA proteins were detected. In the case of cNV3-HA, these proteins had apparent molecular masses of 14 and 18 kDa. Both proteins were found in association with intracellular membranes, suggesting that the 18-kDa protein is a glycosylated form of the 14-kDa protein (fraction 9, Fig. 9C). In the case of G-nN-HA, a protein of approximately 13 kDa was predominant. Small amounts of a 17-kDa protein were also occasionally detected (Fig. 9B, lane 7), raising the possibility that glycosylation of G-nN-HA also occurred. Unfortunately, the low concentration of this protein in the extracts did not allow us to conduct deglycosylation assays that could have confirmed this suggestion.
DISCUSSION
The results presented in this report demonstrate that the ToRSV NTB-VPg protein, the putative membrane anchor for the replication complex, has the ability to associate with intracellular membranes in planta when fused to GFP. Deletion of hydrophobic domains in the N-terminal and C-terminal regions of NTB-VPg prevented the membrane association (GFP-mN, Fig. 1 and 2). Although we cannot exclude the possibility that fusion to the GFP alters the natural subcellular localization of NTB-VPg, it is noteworthy that similar ER-associated localization was observed for fusion proteins with C-terminal or N-terminal fusions to the GFP. The results presented in this study are also consistent with previous observations that NTB-VPg is an integral membrane protein associated with the ER in ToRSV-infected plants and that it has the ability to independently associate with canine microsomal membranes (which consist predominantly of ER-derived membranes) in vitro (30, 60). Taken together, these results suggest that membrane association elements within NTB-VPg are functional in the context of GFP fusion proteins. In fact, fusion of the truncated cNV3 protein to GFP (GFP-cNV3 and cNV3-GFP fusion proteins) did not prevent the recognition of the naturally occurring VPg glycosylation site, suggesting that these fusion proteins were inserted in the proper orientation in the membrane (see below). The nN and cNV3 truncated proteins also had similar subcellular fractionation behavior when fused to GFP or to the HA epitope tag.
The C-terminal region of NTB contains a previously characterized hydrophobic domain that includes a transmembrane helix. This domain directs the translocation of VPg in the ER lumen in vitro and in ToRSV-infected plants (30, 60). In this report, we provide several lines of evidence confirming that the hydrophobic domain functions as an ER-targeting domain in vivo. First, WT GFP-cNV3 and cNV3-GFP colocalized at least partially with ER markers and were detected in membrane-enriched fractions. Deletion of the hydrophobic domain (ΔTM1 mutant) prevented the membrane association. Second, glycosylated forms of the WT proteins were present in the membrane-enriched fractions, confirming that they were associated with intracellular membranes and that the VPg domain was translocated in the lumen. Sensitivity of the glycosylation to Endo H indicated that the glycosylated proteins were retained in the ER, suggesting that ER retention signals are present in the C-terminal region of NTB-VPg. The characterization of these ER retention signals will be the subject of further studies.
Although it is clear that the hydrophobic region within the C-terminal region of NTB can act as an ER-targeting domain, the membrane association of GFP-cNV3 and cNV3-GFP was only partial, as evidenced by the diffusion of a portion of the green fluorescence within the nucleus and the presence of a portion of the proteins immunodetected by the GFP antibodies in the soluble fraction. A similar phenomenon was observed with cNV3-HA, although the proportion of protein in the S30 fraction was smaller than with the corresponding GFP fusion proteins. One possible explanation is that targeting of these truncated proteins to ER membranes is inefficient, at least in the context of the GFP fusions. Another possible explanation is that degradation of the fusion proteins releases soluble fragments with GFP activity. A truncated 31-kDa protein, detected in the S30 and P30 fractions of plants expressing cNV3-GFP, may correspond to such a cleavage product (Fig. 3B, lanes 5 and 6). In the case of GFP-cNV3, the predominant 36-kDa protein detected in the S30 and P30 fractions may also correspond to a cleavage fragment for the following reasons. First, it was smaller than the detected cNV3-GFP protein even though the predicted molecular masses of the two fusion proteins are identical (Fig. 3B, lanes 1 and 2 and lanes 5 and 6). Second, the 36-kDa protein did not comigrate with a 38-kDa protein produced after deglycosylation treatment of the GFP-cNV3 membrane-enriched fraction (Fig. 3C, lanes 2 to 4). Interestingly, we have previously shown that cNV3 is cleaved by a membrane-associated proteinase in vitro (60). Although it is possible that this cleavage is responsible, at least in part, for the release of GFP-cNV3 and cNV3-GFP in the cytosolic fraction, further work is necessary to conclusively determine whether membrane-associated cleavage of cNV3 (and full-length NTB-VPg) occurs in plants.
Our results demonstrate that the N-terminal putative amphipathic helix can promote efficient targeting to intracellular membranes in planta and in vitro. The putative role of an upstream hydrophobic region is not clear at this point, as it was not able to promote membrane association in the absence of the amphipathic helix. In addition, deletion of this region did not prevent the membrane association of proteins that contained the amphipathic helix. However, our results do not exclude the possibility that the upstream hydrophobic region influences the exact positioning of the amphipathic helix in the membrane.
The results of the glycosylation site mapping revealed that the N terminus of NTB can be translocated in the membrane lumen in vitro in the context of truncated proteins containing only the N-terminal amphipathic helix or in the context of the entire NTB-VPg protein. Amphipathic helices usually orient parallel to the membrane with the hydrophobic face toward the membrane and the hydrophilic face toward the cytoplasm, unless they form membrane-embedded oligomers through the barrel stave mechanism and assemble into aqueous pores or ion channels (5, 49). The barrel stave mechanism is derived from studies of pore-forming cytolytic amphipathic peptides and implies the formation of trans-bilayer oligomers of the amphipathic helices with their hydrophobic sides facing the lipid bilayer and their hydrophilic sides oriented toward the water-filled pore. Using SDS-PAGE, we have observed larger forms of truncated proteins containing the amphipathic helix, which may correspond to oligomers of the protein (Fig. 8 and 9). Membrane-dependent glycosylation of one of these putative oligomeric forms was observed in vitro (Fig. 9). These observations provide support for the suggestion that oligomerization of the amphipathic helix promotes its translocation in the membrane. It is not known whether translocation of the N terminus of NTB-VPg in the membrane lumen occurs in vivo. Further experimentation is necessary to confirm that the amphipathic helix has the ability to oligomerize in the membrane environment and to determine whether the N terminus of NTB-VPg is translocated in the membrane lumen in vivo.
The formation of aqueous pores is often associated with permeabilization of the membrane, and several viral proteins (viroporins) have been found to destabilize and permeabilize membranes (29). In animal viruses, viroporins have been suggested to play a role in the release of virus particles from infected cells. Many viroporins are located predominantly at the intracellular membranes and have the ability to promote intracellular membrane remodeling, which is a prerequisite for viral replication (29). Some viroporins, such as the human immunodeficiency virus type 1 VPu and influenza virus M2 proteins, induce the formation of intracellular ion channels, although the role of these channels in the virus replication cycle is not clear (29). Viroporins have not been identified so far in plant viruses. The Cowpea mosaic comovirus 60K protein (which contains the NTB and VPg domains and is related to the ToRSV NTB-VPg protein) is cytotoxic in plants and induces drastic morphological changes of the ER membranes. It can also induce cell lysis when expressed in insect cells (11). The domains of the 60K protein involved in these cytotoxic effects have not been characterized. It is noteworthy, however, that a putative amphipathic helix has been identified at the N terminus of the 60K protein (11). In this study, we did not observe severe changes in the structure of the ER in cells expressing GFP fusion proteins containing the entire ToRSV NTB-VPg protein, although aggregates of the ER-dsRed2 fluorescence were occasionally observed (Fig. 2, panels 5 and 6). One possibility is that the levels of expression of these proteins were not sufficient to significantly alter the ER structure. Interestingly, high levels of expression of a fusion protein containing the N-terminal amphipathic helix (nN-GFP) in plant cells resulted in drastic modification of the morphology of the ER. However, we cannot exclude the possibility that the interaction of the amphipathic helix with the membranes is influenced by the presence of the GFP domain. In fact, modification of the ER structure was not observed to the same extent in cells expressing a fusion protein containing the amphipathic helix fused to the C terminus of GFP (GFP-nN). The ability of the ToRSV NTB-VPg protein to modify and possibly permeabilize membranes will be the subject of further studies.
Multiple interactions between transmembrane helices within the membrane environment have been documented for a large number of polytopic membrane proteins (18, 26), suggesting that interactions between the ToRSV NTB-VPg amphipathic and transmembrane helices are at least theoretically possible. The current two-stage model for membrane insertion and folding of polytopic membrane proteins suggests that individual transmembrane helices are initially inserted into the membrane independently, followed by a second stage in which transmembrane helices interact with each other in the membrane environment to give higher-order folding and oligomerization (18, 26). An example of this is provided by the poliovirus 2B protein, a viroporin which forms aqueous pores driven by the tetramerization of an amphipathic helix and further stabilized by intramolecular hydrophobic interactions between the amphipathic helix and an adjacent transmembrane helix and by intermolecular hydrophobic interactions between the transmembrane helices of the four 2B monomers (1). According to the two-stage model for membrane protein folding, we propose an updated model for the insertion of NTB-VPg in the membrane (Fig. 10). In this model, NTB-VPg initially interacts with the membranes as a monomer, with the transmembrane helix traversing the membrane and the amphipathic helix parallel to the membranes (step 1). The ability of each of these elements to oligomerize could result in the formation of an aqueous pore with the hydrophilic side of the amphipathic helix lining the pore (step 2). Possible interactions between the hydrophobic face of the amphipathic helix and the transmembrane helix within a monomer unit and between the transmembrane helices of the various monomers could result in the formation of an α-loop-α motif that spans the bilayer and helps stabilize the structure. In this model, both the N and C termini of NTB-VPg are oriented to the luminal face of the membranes while the central region of NTB is oriented toward the cytoplasm and accessible for protein-protein interactions with other viral or host proteins present in the replication complex.
FIG. 10.

Updated model for the insertion of the NTB-VPg protein into ER membranes. The protein initially associates as a monomer within the membrane (part 1) with the N-terminal amphipathic helix (represented by the light gray cylinder) parallel to the membrane and the C-terminal transmembrane helix (represented by the dark gray cylinder) traversing the membrane. In a second step (part 2), the protein oligomerizes through the N-terminal amphipathic helix and possibly also the C-terminal transmembrane helix to form an aqueous pore with an α-loop-α conformation. Hydrophobic interactions between the N-terminal amphipathic helix and the C-terminal transmembrane helix may help stabilize the formation of the pore. To simplify the drawing, only two monomers are shown interacting in the structure. However, formation of the pore would imply the interaction of at least four NTB-VPg molecules. The VPg domain is represented by the black box. The region containing the conserved motif for the NTB-binding domain is also shown (shaded box).
Acknowledgments
We thank D. Rochon (Pacific Agri-Food Research Centre [PARC] Summerland) for TBSV cDNA clones, M. Chrispeels (University of California, San Diego) for anti-Bip antibodies, and M. Weis (PARC Summerland) for help with the confocal microscope. We are grateful to I. Jupin and J. F. Laliberté for critical reading of the manuscript.
This work was supported in part by an NSERC discovery grant awarded to H.S.
REFERENCES
- 1.Agirre, A., A. Barco, L. Carrasco, and J. L. Nieva. 2002. Viroporin-mediated membrane permeabilization. Pore formation by nonstructural poliovirus 2B protein. J. Biol. Chem. 277:40434-40441. [DOI] [PubMed] [Google Scholar]
- 2.Agol, V. I., A. V. Paul, and E. Wimmer. 1999. Paradoxes of the replication of picornaviral genomes. Virus Res. 62:129-147. [DOI] [PubMed] [Google Scholar]
- 3.Aldabe, R., A. Barco, and L. Carrasco. 1996. Membrane permeabilization by poliovirus proteins 2B and 2BC. J. Biol. Chem. 271:23134-23137. [DOI] [PubMed] [Google Scholar]
- 4.Aldabe, R., and L. Carrasco. 1995. Induction of membrane proliferation by poliovirus proteins 2C and 2BC. Biochem. Biophys. Res. Commun. 206:64-76. [DOI] [PubMed] [Google Scholar]
- 5.Bechinger, B. 1999. The structure, dynamics and orientation of antimicrobial peptides in membranes by multidimensional solid-state NMR spectroscopy. Biochim. Biophys. Acta 1462:157-183. [DOI] [PubMed] [Google Scholar]
- 6.Brass, V., E. Bieck, R. Montserret, B. Wolk, J. A. Hellings, H. E. Blum, F. Penin, and D. Moradpour. 2002. An amino-terminal amphipathic alpha-helix mediates membrane association of the hepatitis C virus nonstructural protein 5A. J. Biol. Chem. 277:8130-8139. [DOI] [PubMed] [Google Scholar]
- 7.Brignati, M. J., J. S. Loomis, J. W. Wills, and R. J. Courtney. 2003. Membrane association of VP22, a herpes simplex virus type 1 tegument protein. J. Virol. 77:4888-4898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Buck, K. W. 1996. Comparison of the replication of positive-stranded RNA viruses of plants and animals. Adv. Virus Res. 47:159-251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carette, J. E., K. Guhl, J. Wellink, and A. Van Kammen. 2002. Coalescence of the sites of cowpea mosaic virus RNA replication into a cytopathic structure. J. Virol. 76:6235-6243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carette, J. E., M. Stuiver, J. Van Lent, J. Wellink, and A. Van Kammen. 2000. Cowpea mosaic virus infection induces a massive proliferation of endoplasmic reticulum but not Golgi membranes and is dependent on de novo membrane synthesis. J. Virol. 74:6556-6563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carette, J. E., J. van Lent, S. A. MacFarlane, J. Wellink, and A. van Kammen. 2002. Cowpea mosaic virus 32- and 60-kilodalton replication proteins target and change the morphology of endoplasmic reticulum membranes. J. Virol. 76:6293-6301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carrere-Kremer, S., C. Montpellier-Pala, L. Cocquerel, C. Wychowski, F. Penin, and J. Dubuisson. 2002. Subcellular localization and topology of the p7 polypeptide of hepatitis C virus. J. Virol. 76:3720-3730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carrier, K., F. Hans, and H. Sanfacon. 1999. Mutagenesis of amino acids at two tomato ringspot nepovirus cleavage sites: effect on proteolytic processing in cis and in trans by the 3C-like protease. Virology 258:161-175. [DOI] [PubMed] [Google Scholar]
- 14.Chen, J., and P. Ahlquist. 2000. Brome mosaic virus polymerase-like protein 2a is directed to the endoplasmic reticulum by helicase-like viral protein 1a. J. Virol. 74:4310-4318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cho, M. W., N. Teterina, D. Egger, K. Bienz, and E. Ehrenfeld. 1994. Membrane rearrangement and vesicle induction by recombinant poliovirus 2C and 2BC in human cells. Virology 202:129-145. [DOI] [PubMed] [Google Scholar]
- 16.Ciervo, A., F. Beneduce, and G. Morace. 1998. Polypeptide 3AB of hepatitis A virus is a transmembrane protein. Biochem. Biophys. Res. Commun. 249:266-274. [DOI] [PubMed] [Google Scholar]
- 17.Cuconati, A., A. Molla, and E. Wimmer. 1998. Brefeldin A inhibits cell-free, de novo synthesis of poliovirus. J. Virol. 72:6456-6464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Curran, A. R., and D. M. Engelman. 2003. Sequence motifs, polar interactions and conformational changes in helical membrane proteins. Curr. Opin. Struct. Biol. 13:412-417. [DOI] [PubMed] [Google Scholar]
- 19.Datta, U., and A. Dasgupta. 1994. Expression and subcellular localization of poliovirus VPg-precursor protein 3AB in eukaryotic cells: evidence for glycosylation in vitro. J. Virol. 68:4468-4477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Davis, S. J., and R. D. Vierstra. 1998. Soluble, highly fluorescent variants of green fluorescent protein (GFP) for use in higher plants. Plant Mol. Biol. 36:521-528. [DOI] [PubMed] [Google Scholar]
- 21.DeGrado, W. F., H. Gratkowski, and J. D. Lear. 2003. How do helix-helix interactions help determine the folds of membrane proteins? Perspectives from the study of homo-oligomeric helical bundles. Protein Sci. 12:647-665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Deleage, G., C. Combet, C. Blanchet, and C. Geourjon. 2001. ANTHEPROT: an integrated protein sequence analysis software with client/server capabilities. Comput. Biol. Med. 31:259-267. [DOI] [PubMed] [Google Scholar]
- 23.den Boon, J. A., J. Chen, and P. Ahlquist. 2001. Identification of sequences in Brome mosaic virus replicase protein 1a that mediate association with endoplasmic reticulum membranes. J. Virol. 75:12370-12381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.dos Reis Figueira, A., S. Golem, S. P. Goregaoker, and J. N. Culver. 2002. A nuclear localization signal and a membrane association domain contribute to the cellular localization of the Tobacco mosaic virus 126-kDa replicase protein. Virology 301:81-89. [DOI] [PubMed] [Google Scholar]
- 25.Echeverri, A. C., and A. Dasgupta. 1995. Amino terminal regions of poliovirus 2C protein mediate membrane binding. Virology 208:540-553. [DOI] [PubMed] [Google Scholar]
- 26.Engelman, D. M., Y. T. Chen, C. N. Chin, A. R. Curran, A. M. Dixon, A. D. Dupuy, A. S. Lee, U. Lehnert, E. E. Matthews, Y. K. Reshetnyak, A. Senes, and J.-L. Popot. 2003. Membrane protein folding: beyond the two stage model. FEBS Lett. 555:122-125. [DOI] [PubMed] [Google Scholar]
- 27.Gaire, F., C. Schmitt, C. Stussi-Garaud, L. Pinck, and C. Ritzenthaler. 1999. Protein 2A of grapevine fanleaf nepovirus is implicated in RNA2 replication and colocalizes to the replication site. Virology 264:25-36. [DOI] [PubMed] [Google Scholar]
- 28.Gazina, E. V., J. M. Mackenzie, R. J. Gorrell, and D. A. Anderson. 2002. Differential requirements for COPI coats in formation of replication complexes among three genera of Picornaviridae. J. Virol. 76:11113-11122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gonzalez, M. E., and L. Carrasco. 2003. Viroporins. FEBS Lett. 552:28-34. [DOI] [PubMed] [Google Scholar]
- 30.Han, S., and H. Sanfacon. 2003. Tomato ringspot virus proteins containing the nucleoside triphosphate binding domain are transmembrane proteins that associate with the endoplasmic reticulum and cofractionate with replication complexes. J. Virol. 77:523-534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hirokawa, T., S. Boon-Chieng, and S. Mitaku. 1998. SOSUI: classification and secondary structure prediction system for membrane proteins. Bioinformatics 14:378-379. [DOI] [PubMed] [Google Scholar]
- 32.Hofmann, K., and W. Stoffel. 1993. TMbase—a database of membrane spanning protein segments. Biol. Chem. Hoppe-Seyler 347:166. [Google Scholar]
- 33.Jones, D. T., W. R. Taylor, and J. M. Thornton. 1994. A model recognition approach to the prediction of all-helical membrane protein structure and topology. Biochemistry 33:3038-3049. [DOI] [PubMed] [Google Scholar]
- 34.Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680-685. [DOI] [PubMed] [Google Scholar]
- 35.Lama, J., and L. Carrasco. 1996. Screening for membrane-permeabilizing mutants of the poliovirus protein 3AB. J. Gen. Virol. 77(Pt. 9):2109-2119. [DOI] [PubMed] [Google Scholar]
- 36.Lundin, M., M. Monne, A. Widell, G. Von Heijne, and M. A. Persson. 2003. Topology of the membrane-associated hepatitis C virus protein NS4B. J. Virol. 77:5428-5438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Maley, F., R. B. Trimble, A. L. Tarentino, and T. H. Plummer, Jr. 1989. Characterization of glycoproteins and their associated oligosaccharides through the use of endoglycosidases. Anal. Biochem. 180:195-204. [DOI] [PubMed] [Google Scholar]
- 38.Moradpour, D., R. Gosert, D. Egger, F. Penin, H. E. Blum, and K. Bienz. 2003. Membrane association of hepatitis C virus nonstructural proteins and identification of the membrane alteration that harbors the viral replication complex. Antiviral Res. 60:103-109. [DOI] [PubMed] [Google Scholar]
- 39.Nilsson, I. M., and G. von Heijne. 1993. Determination of the distance between the oligosaccharyltransferase active site and the endoplasmic reticulum membrane. J. Biol. Chem. 268:5798-5801. [PubMed] [Google Scholar]
- 40.Ritzenthaler, C., C. Laporte, F. Gaire, P. Dunoyer, C. Schmitt, S. Duval, A. Piequet, A. M. Loudes, O. Rohfritsch, C. Stussi-Garaud, and P. Pfeiffer. 2002. Grapevine fanleaf virus replication occurs on endoplasmic reticulum-derived membranes. J. Virol. 76:8808-8819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rost, B., R. Casadio, and P. Fariselli. 1996. Refining neural network predictions for helical transmembrane proteins by dynamic programming. Proc. Int. Conf. Intell. Syst. Mol. Biol. 4:192-200. [PubMed] [Google Scholar]
- 42.Rott, M. E., A. Gilchrist, L. Lee, and D. Rochon. 1995. Nucleotide sequence of tomato ringspot virus RNA1. J. Gen. Virol. 76(Pt. 2):465-473. [DOI] [PubMed] [Google Scholar]
- 43.Rust, R. C., L. Landmann, R. Gosert, B. L. Tang, W. Hong, H. P. Hauri, D. Egger, and K. Bienz. 2001. Cellular COPII proteins are involved in production of the vesicles that form the poliovirus replication complex. J. Virol. 75:9808-9818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Salonen, A., T. Ahola, and L. Kaariainen. 2005. Viral RNA replication in association with cellular membranes. Curr. Top. Microbiol. Immunol. 285:139-173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sanfacon, H. 1995. Nepoviruses, p. 129-141. In R. P. Singh, U. S. Singh, and K. Kohmoto (ed.), Pathogenesis and host specificity in plant diseases. Vol. III. Viruses and viroids. Pergamon Press, Oxford, United Kingdom. [Google Scholar]
- 46.Schaad, M. C., P. E. Jensen, and J. C. Carrington. 1997. Formation of plant RNA virus replication complexes on membranes: role of an endoplasmic reticulum-targeted viral protein. EMBO J. 16:4049-4059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schlegel, A., T. H. Giddings, Jr., M. S. Ladinsky, and K. Kirkegaard. 1996. Cellular origin and ultrastructure of membranes induced during poliovirus infection. J. Virol. 70:6576-6588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schwartz, M., J. Chen, W. M. Lee, M. Janda, and P. Ahlquist. 2004. Alternate, virus-induced membrane rearrangements support positive-strand RNA virus genome replication. Proc. Natl. Acad. Sci. USA 101:11263-11268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shai, Y. 1999. Mechanism of the binding, insertion and destabilization of phospholipid bilayer membranes by alpha-helical antimicrobial and cell non-selective membrane-lytic peptides. Biochim. Biophys. Acta 1462:55-70. [DOI] [PubMed] [Google Scholar]
- 50.Sonnhammer, E. L., G. von Heijne, and A. Krogh. 1998. A hidden Markov model for predicting transmembrane helices in protein sequences. Proc. Int. Conf. Intell. Syst. Mol. Biol. 6:175-182. [PubMed] [Google Scholar]
- 51.Suhy, D. A., T. H. Giddings, Jr., and K. Kirkegaard. 2000. Remodeling the endoplasmic reticulum by poliovirus infection and by individual viral proteins: an autophagy-like origin for virus-induced vesicles. J. Virol. 74:8953-8965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sun, F., Y. Xiang, and H. Sanfacon. 2001. Homology-dependent resistance to tomato ringspot nepovirus in plants transformed with the VPg-protease coding region. Can. J. Plant Pathol. 23:292-299. [Google Scholar]
- 53.Teterina, N. L., K. Bienz, D. Egger, A. E. Gorbalenya, and E. Ehrenfeld. 1997. Induction of intracellular membrane rearrangements by HAV proteins 2C and 2BC. Virology 237:66-77. [DOI] [PubMed] [Google Scholar]
- 54.Towner, J. S., T. V. Ho, and B. L. Semler. 1996. Determinants of membrane association for poliovirus protein 3AB. J. Biol. Chem. 271:26810-26818. [DOI] [PubMed] [Google Scholar]
- 55.Towner, J. S., M. M. Mazanet, and B. L. Semler. 1998. Rescue of defective poliovirus RNA replication by 3AB-containing precursor polyproteins. J. Virol. 72:7191-7200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tusnady, G. E., and I. Simon. 1998. Principles governing amino acid composition of integral membrane proteins: applications to topology prediction. J. Mol. Biol. 283:489-506. [DOI] [PubMed] [Google Scholar]
- 57.Voinnet, O., S. Rivas, P. Mestre, and D. Baulcombe. 2003. An enhanced transient expression system in plants based on suppression of gene silencing by the p19 protein of tomato bushy stunt virus. Plant J. 33:949-956. [DOI] [PubMed] [Google Scholar]
- 58.von Heijne, G. 1992. Membrane protein structure prediction. Hydrophobicity analysis and the positive-inside rule. J. Mol. Biol. 225:487-494. [DOI] [PubMed] [Google Scholar]
- 59.Wang, A., K. Carrier, J. Chisholm, A. Wieczorek, C. Huguenot, and H. Sanfacon. 1999. Proteolytic processing of tomato ringspot nepovirus 3C-like protease precursors: definition of the domains for the VPg, protease and putative RNA-dependent RNA polymerase. J. Gen. Virol. 80(Pt. 3):799-809. [DOI] [PubMed] [Google Scholar]
- 60.Wang, A., S. Han, and H. Sanfacon. 2004. Topogenesis in membranes of the NTB-VPg protein of Tomato ringspot nepovirus: definition of the C-terminal transmembrane domain. J. Gen. Virol. 85:535-545. [DOI] [PubMed] [Google Scholar]
- 61.Wang, A., and H. Sanfacon. 2000. Proteolytic processing at a novel cleavage site in the N-terminal region of the tomato ringspot nepovirus RNA-1-encoded polyprotein in vitro. J. Gen. Virol. 81:2771-2781. [DOI] [PubMed] [Google Scholar]
- 62.Yamaga, A. K., and J. H. Ou. 2002. Membrane topology of the hepatitis C virus NS2 protein. J. Biol. Chem. 277:33228-33234. [DOI] [PubMed] [Google Scholar]
- 63.Zhang, S. C., R. Ghosh, and H. Jeske. 2002. Subcellular targeting domains of Abutilon mosaic geminivirus movement protein BC1. Arch. Virol. 147:2349-2363. [DOI] [PubMed] [Google Scholar]
- 64.Zhang, S. C., C. Wege, and H. Jeske. 2001. Movement proteins (BC1 and BV1) of Abutilon mosaic geminivirus are cotransported in and between cells of sink but not of source leaves as detected by green fluorescent protein tagging. Virology 290:249-260. [DOI] [PubMed] [Google Scholar]