

Abstract
This review is focused on hybrid molecules defined as chemical entities with two or more structural domains, as antimalarial drug‐candidates, over the past 25 years. Due to their different pharmacophores, such hybrids can interact with a single biological target by different and complementary mechanisms; they can also act simultaneously on several targets having complementary biological functions (dual mode of action), and can theoretically reduce the selection of parasite drug‐resistance. This review is not an exhaustive report of all hybrid drugs tested on malaria parasites but a selection of hybrids with pharmacologically relevant antiplasmodial properties and original chemical structures. The choice of pharmacophore synthons and junction arms is obviously decisive. Among the large varieties of hybrid drugs published, emoquine‐1 appears at the moment as a promising antimalarial drug candidate, considering 1) its high activities on several multidrug‐resistant Plasmodium lab strains and field isolates, 2) its capacity to eliminate the quiescent forms of the artemisinin‐resistant parasites, and 3) its curative properties in a malaria mouse model. Such molecules confirm the synergistic effect of hybrid compounds compared to the combination of the pharmacophores leading to novel chemical structures that meet the critical parameters for new antimalarial drugs.
Keywords: artemisinin resistance, drug resistance, hybrid drugs, malaria, therapy
Among hybrid molecules currently developed as antimalarial drug candidates, emoquine‐1 exhibits high activity against all the multidrug‐resistant Plasmodium strains tested up to now, including artemisinin‐resistant quiescent parasites, critical parameters for promising antimalarial drugs. It is also curative in mouse malaria. None of its fragments exhibits such activity, thus emphasizing the potency of hybrid strategy.

1. Introduction
Despite huge and constant efforts to prevent and to treat malaria, this tropical disease remains one major health concern in the World.[ 1 ] Besides the development of vaccines, small molecules constitute the main arsenal for the treatment of malaria. The isolation and characterization of artemisinin from Artemisia annua by Tu in the 1960s[ 2 ] has been at the origin of highly efficient treatments against Plasmodium falciparum parasites based on artemisinin derivatives or in combination with other antimalarial drugs, and participated to the malaria death decrease observed in the 2000s.[ 3 ] In a pioneer work, it has been suggested that the mechanism of action of artemisinin was related to the activation of the trioxane entity, leading to an alkylation process.[ 4 ] The alkylation of heme by artemisinin was then demonstrated, leading to identify the heme‐artemisinin adducts, first with a synthetic metalloporphyrin, then with heme itself, and later, in mice treated with artemisinin.[ 5 , 6 ] The understanding of the artemisinin mechanism of action led to a large reflection to create new antimalarial agents with the antiplasmodial properties of artemisinin but with less risk to select parasite resistance. As for other diseases such as AIDS, tuberculosis, or cancers, a new rational strategy was thus to combine active molecules with independent modes of action to prevent the emergence of resistance.[ 7 ] Therefore, hybrid molecules with a potential dual activity were proposed to improve the pharmacological properties of both molecules, facilitate treatment compliance, and propose original chemical synthesis to limit cross‐resistance with drugs already on the market.[ 8 ]
It appears that the first hybrid drugs as antimalarials started with the work of Boman et al. on hybrid peptides which unfortunately exhibited weak antiplasmodial activity.[ 9 ] Then, inspired by previous work on bleomycin, an efficient anticancer drug that possesses three different structural entities (a DNA binding domain, an iron‐chelating moiety and a cell‐penetration entity),[ 10 ] trioxaquines, containing a 1,2,4‐trioxane structure as in artemisinin linked to a 4‐aminoquinoline reminiscent of chloroquine, were developed in 2000 (for the initial work, see ref. [11], and for review articles see [12, 13]). Since the early 2010s, research has been focused on developing antimalarial drugs able to bypass artemisinin resistance, which presupposes the best possible understanding of the underlying mechanisms.[ 14 ]
2. Bleomycin, a Paradigm of Hybrid Drug with Dual Activity
The studies about the mechanism of action of bleomycin (BLM, Figure 1 ),[ 15 , 16 , 17 , 18 ] an efficient anticancer agent, are at the root of interest of Meunier and his collaborators for hybrid drugs. In fact, bleomycin, a glycopeptide isolated from Streptomyces verticillus, discovered in 1962, has been used to treat several types of cancer from the early 1970s. The cytotoxic activity of this natural drug against mammalian cells is based on efficient oxidative breakage of DNA in cancer cells, a reaction that requires iron, dioxygen, and a reducing agent as necessary cofactors. This drug has three distinct structural domains, each having their own biological role. The L‐gulose‐D‐mannose carbohydrate residue allows cell penetration, the bithiazole residue with its cationic side chain (a trialkylsulfonium in BLM A2, or a guanidinium at physiological pH, in BLM B2) binds to the GC‐rich sequences of DNA, and five nitrogen atoms are involved in an iron‐binding site.[ 15 , 18 , 19 ] The key step of BLM‐induced oxidative DNA cleavage is the hydroxylation of the C4’=H bond of the deoxyribose units by a high‐valent iron‐oxo BLM‐FeV=O complex, an activated species reminiscent of that of cytochrome P450. In vitro, Fe‐BLM can be activated by P450 mimics based on hydrogen peroxide, or by water‐soluble single oxygen atom donors such as potassium hydrogen persulfate (KHSO5) to provide the same reactivity.[ 19 ]
Figure 1.

Representation of bleomycin with its three different structural domains (asterisks indicate the nitrogen atoms involved in metal binding). R stands for NH=(CH2)3=S+(CH3)2 in bleomycin A2, and for NH=(CH2)4=NH=C(=CN)=NH2 in bleomycin B2.
Based on the results obtained with bleomycin, the concept of hybrid molecule in drug design has been extended to many different therapeutic areas. These chemical entities with several structural domains, due to their two (or more) pharmacophores can interact with a single biological target by different and complementary mechanisms, or can also act simultaneously on several targets having different biological functions (dual mode of action) (Figure 2 ).
Figure 2.

Schematic representation of three different possible modes of interaction of hybrid molecules: A) one single target (for double‐edged molecules), B) two independent targets (each entity of the hybrid molecule acts with its target), and C) two related targets (both entities of the hybrid molecule act at the same time on two connected targets). Redrawn from ref. [12]. In each case, the linker can be cleavable or not in physiological conditions.
3. Hybrid Molecules Based on Marketed Antimalarial Drug Fragments
For the last three decades, the main malaria treatments were based on artemisinins, then on a combination of an artemisinin derivative with an antimalarial drug from another chemical class, the so‐called artemisinin‐based combination therapies (ACT). Until the 2000s, the goal was to target chloroquine‐resistant Plasmodium with synthetic analogs of artemisinin with improved pharmaco‐kinetic/‐dynamic properties, and/or with an easier availability at low cost. For that purpose, semisynthetic artemisinin derivatives, or synthetic peroxides, were designed and evaluated. In this field, even if they are not strictly speaking hybrids, the 1,2,4‐trioxolane series, OZ277 and, more recently, OZ439 are worth being cited and were considered promising drugs.[ 20 , 21 , 22 ] 1,2,4,5‐Tetraoxane derivatives were also developed.[ 23 , 24 , 25 ] However, to prevent artemisinin resistance before it arose, efforts were also focused on the design of hybrid molecules, which link two antiplasmodial entities having independent modes of action and/or different pharmacological targets.[ 26 ]
The most active molecules of each series of hybrids, here selected for their relevant pharmacological and antiplasmodial properties and their original chemical structures, are depicted in Table 1 , along with the antimalarial drugs used as sources of inspiration.
Table 1.
Selection of hybrid molecules designed as potential antimalarial drugs.

|
|
(Abbreviations: ATQ: atovaquone (clinically approved), ART: artemisinin derivative (clinically approved), MF: mefloquine (clinically approved), TCS: triclosan (approved antiseptic), PQ: primaquine (clinically approved), NHC: N‐heterocyclic carbene, proca: procainamide, FAS‐II: fatty acid synthase‐II, HDAC: histone deacetylase, TrxR: thioredoxine reductase, DNMT: DNA methyl transferase, GR: glutathione reductase)
3.1. Hybrids Based on Quinolines and Endoperoxides
3.1.1. Heme Metabolism as Target
During the infection of erythrocytes, P. falciparum digests a large part of the host hemoglobin to collect amino acids for its own protein production and to make space in the host red blood cell. The released Fe(II)‐heme, able to catalytically reduce dioxygen, is a damaging source of reactive oxygen species (aka ROS), especially hydroxyl radicals HO•. To avoid poisoning by this redox‐active iron complex, the parasite efficiently polymerizes heme to hemozoin, the “malaria pigment” that is a crystalline, redox‐inactive polymer of Fe(III)‐heme.[ 27 , 28 , 29 ] Upon treatment with artemisinin, the reductive cleavage of the peroxide bond of the drug initiated by iron(II)‐heme results in alkylation of heme by the drug, leading to the formation of covalent heme‐drug adducts.[ 5 , 26 , 30 , 31 ] These adducts completely inhibit the polymerization of heme in vitro, suggesting that soluble redox‐active heme derivatives accumulate in parasites treated by artemisinin, and are responsible for a lethal oxidative stress.[ 32 ] These heme‐artemisinin covalent adducts have been detected in malaria‐infected mice after treatment with artemisinin, indicating that heme can act both as trigger and target of artemisinin in vivo.[ 6 ]
Furthermore, the antimalarial chloroquine, containing a 4‐aminoquinoline moiety, has a high affinity for ferric porphyrins (K d = 3.5 nM) as a result of a strong π‐stacking between the quinoline ring and the porphyrin macrocycle in vitro.[ 33 , 34 ] This interaction between the aromatic quinoline residue and the iron‐protoporphyrin IX liberated by the digestion of hemoglobin, was considered responsible for disruption of hemozoin formation and, consequently, for the antimalarial efficacy of quinoline‐based drugs.[ 35 , 36 ] Artemisinin and chloroquine are sharing heme metabolism as a common target. So, many hybrid molecules containing an artemisinin derivative covalently connected by various linkers to an (iso)quinoline moiety have been reported to have good in vitro antimalarial activity.[ 37 ] An artemisinin–quinine hybrid was also reported to exhibit an activity superior to that of artemisinin alone, quinine alone, or a 1:1 mixture of artemisinin and quinine, thus validating the hybrid strategy. [37e]
3.1.2. Trioxaquines
With the aim of targeting Plasmodium heme metabolism by two different and complementary mechanisms, namely, alkylation and π−π interaction, hybrid trioxaquines were built with a 1,2,4‐trioxane moiety with heme‐alkylating property, like in artemisinin, covalently linked to a 4‐aminoquinoline, having heme stacking ability, like in chloroquine (Table 1a).[ 11 , 26 , 38 , 39 , 40 ] Several trioxaquines were found highly active in vitro against various P. falciparum strains, including chloroquine‐resistant human isolates.[ 41 ] In terms of mechanism of action, trioxaquine DU1302 totally inhibits in vitro the polymerization of heme into β‐hematin, a synthetic analog of hemozoin,[ 28 ] even more than chloroquine.[ 32 ] It also effectively alkylates heme in vitro and in malaria‐infected mice,[ 42 , 43 ] thus confirming the complementarity of its two pharmacophores targeting heme.
Based on these promising results, the improved derivative PA1103 (Table 1a) showed, in addition, in vivo activity by oral route in P. vinckei‐infected mice, a high activity in humanized mice infected with P. falciparum, and had also a good drug profile (preliminary absorption, metabolism, and safety parameters).[ 44 ]
At that time, the emergence and spread of artemisinin‐resistant P. falciparum was just beginning in Southeast Asia,[ 45 , 46 ] and the experimental P. falciparum artemisinin‐resistant line F32‐ART was selected in vitro to understand the mechanisms of this resistance.[ 47 ] At the molecular level, resistance of Plasmodium is induced by a mutation in the gene encoding for the Kelch protein 13 (K13), and is phenotypically associated to a quiescence state.[ 48 , 49 , 50 ]
Unfortunately, a strong cross‐resistance was evidenced between trioxaquines and artemisinins, despite the hybrid structure of trioxaquines based on trioxane and quinoline moieties. Moreover, a drug‐sensitive P. falciparum strain cultured under a dose‐escalating regimen of trioxaquine DU1302, became resistant to both artemisinin and trioxaquines, and contained the same K13 mutation that was previously found in the F32‐ART line.[ 51 ] So, the hybrid structure of trioxaquines was not sufficient to avoid the in vitro selection of resistant parasites. It was also reported that K13 mutations compromise the activity of the 1,2,4‐trioxolane derivatives (=ozonides),[ 52 ] thus preventing the further development of antimalarials based on a peroxide structure as pharmacophore.
Noteworthy, because Plasmodium and Schistosoma, both blood human parasites having a similar hemoglobin metabolism leading to the formation of hemozoin, trioxaquines were also tested against Schistosoma and several trioxaquines, especially PA1259, were found highly active on S. mansoni.[ 53 ]
3.1.3. Trioxaferroquines
Among the antimalarial drug candidates, ferroquine (Fq, Table 1b,b’) can also be considered as a hybrid molecule with a ferrocenyl moiety inserted within the side chain of chloroquine.[ 54 ] This molecule (SSR97193), developed by Sanofi‐Aventis, has not progressed beyond phase II clinical trials since 2013 in monotherapy[ 55 ] and the clinical trial in association with the 1,2,4‐trioxane OZ439 (artefenomel) was also stopped (portfolio MMV: Medecines for Malaria Venture).
Moreover, the concept of hybrid molecules has been extended to design a series of trioxaferroquines and isotrioxaferroquines, some of them being depicted in Table 1b,b’. These compounds were made with a ferroquine‐like skeleton covalently linked to a 1,2,4‐trioxane entity.[ 56 ] Trioxaferroquine and isotrioxaferroquine were found highly active against chloroquine‐resistant P. falciparum strains (IC50 values in the range 16–43 nM), like artemisinin and Fq. In addition, all the corresponding trioxaferrocenes, deprived of the quinoline fragment (=1,2,4‐trioxane covalently linked to ferrocene), were inactive, with IC50 values up to one order of magnitude greater than that of the corresponding trioxaferroquine. So, the covalent bonding between quinoline and trioxaferrocene significantly enhanced the activity of both fragments, despite the fact that chloroquine itself was totally inactive on these strains. The results clearly indicate the importance of the 4‐aminoquinoline moiety of trioxaferroquines for their antimalarial efficiency, even in the case of chloroquine‐resistant strains.
When mice infected with P. vinckei were orally treated with trioxaferroquine (10 mg kg−1 day−1), rapid parasite clearance occurred, with parasitemia below detectable level on day 4. However, the parasite recrudescence observed indicated that this hybrid was not curative per os, which is the preferred administration route for antimalarial drugs.[ 56 ]
3.2. Hybrids Based on Naphthoquinone and 4‐Aminoquinoline
3.2.1. Plasmodium Mitochondrion as a Target
Despite ART resistance spreading, ACTs are still the cornerstone of antimalarial therapy. However, upon artemisinin exposure, artemisinin‐resistant parasites stop their proliferation by entering into a quiescent state characterized by minimal metabolism, and they resume their proliferation after the drug is eliminated.[ 47 ] Importantly, the quiescence state is characterized by a drastically lowered metabolism that allows the parasites to resist to almost all of antimalarial drugs.[ 57 ] However, quiescent parasites maintain mitochondrial activity.[ 58 , 59 ] In this way, atovaquone (Table 1i, ATQ), which competes with ubiquinone to inhibit mitochondrial cytochrome bc1 complex and blocks the parasite respiratory chain, remained effective in an artemisinin‐resistant parasite line.[ 57 , 60 , 61 ] Unfortunately, because of its pharmacokinetics, its price, and the easy and quick selection of resistance in the field, atovaquone is unusable as such.[ 62 ]
3.2.2. Hybrids Containing an Atovaquone Fragment
In an attempt to take advantage of the mitochondrial target of atovaquone, several hybrids containing atovaquone or a part of it, were synthesized and evaluated.
The hybrid drugs ATQ‐MQ and ATQ‐TCS (Table 1h,i, respectively) containing a ATQ pharmacophore covalently linked to mefloquine (MQ) or triclosan (TCS) were synthesized.[ 63 ] Mefloquine was chosen because it is one of the few partner drugs used in ACTs reported to be active on quiescent artemisinin‐resistant parasites.[ 64 ] Triclosan inhibits the lipid metabolism in Plasmodium apicoplast, and was reported to delay the recrudescence of quiescent parasites after dihydroartemisinin treatment.[ 59 ] So, both ATQ‐MQ and ATQ‐TCS may behave as dual drugs, potentially targeting mitochondria with ATQ and the heme‐ or fatty acid‐metabolism with the second moiety, respectively.
In vitro, these hybrids exhibited sub‐nanomolar antiproliferative activities, (IC50 = 0.6 nM for ATQ‐TCS and 0.6–1.1 nM for ATQ‐MQ), equal or slightly below the activity of the corresponding pharmacophores alone or in combination. Moreover, the cytotoxicity of these hybrids was very low against human cells, resulting in very high selectivity indexes. These hybrids exhibited no cross‐resistance with artemisinin. Interestingly, the ATQ‐TCS hybrid was also found active against parasites in the quiescent stage, but unfortunately significantly less than the corresponding ATQ + TCS combination. These features might be due to the low solubility of this hybrid.[ 63 ]
To disrupt the mitochondrial activity of artemisinin‐resistant parasites, the 1,4‐naphthoquinone moiety of atovaquone was associated to a tetradentate copper chelator based on a 8‐aminoquinoline ring and a diamine side chain from the TDMQ series, yielding the Atokel series[ 65 ] (Table 1c). In fact, the TDMQ ligands, initially designed to regulate copper homeostasis in the brain of patients with Alzheimer's disease,[ 66 , 67 , 68 ] have a high affinity for Cu(II). So, it was hypothesized that Atokels might target Plasmodium mitochondria with their naphthoquinone ring, and that the TDMQ fragment might be able to recruit copper ions in situ and, consequently, create an oxidative stress within mitochondria which are rich in oxygen and electrons. Atokel‐13 (Table 1c) exhibited a disappointing antiplasmodial activity, with an IC50 = 620 nM on the artemisinin‐resistant strain of P. falciparum, F32‐ART, while the IC50 value of atovaquone itself is 0.8 nM. Noteworthy, no atokel derivative bearing a free hydroxyl functionality at C3 of the naphthoquinone was obtained, while the deprotonated hydroxyl group at C3’ of the drug (pKa = 6.9) is considered important for interaction of atovaquone with the Rieske protein of cyt bc1.[ 69 ] So, the lack of this C3’=OH group in the Atokel series might be, at least in part, responsible for the low antimalarial activity of these compounds. It should be noted that other compounds based on a naphthoquinone without hydroxyl substitution at C3 exhibited no significant antimalarial activity.[ 70 ]
3.2.3. Hybrids Containing a 4‐Aminoquinoline Moiety
Similarly, a series of hybrid drugs containing a 4‐amino‐7‐chloroquinoline moiety linked to a N‐alkyl‐3‐hydroxypyridin‐4‐one residue was reported (Table 1d). [71a] The aminoquinoline residue was expected to inhibit hemozoin biocrystallization (similarly to chloroquine), while the chelating hydroxypyridinone should inhibit parasite growth by iron chelation. Interestingly, several molecules of this series exhibited IC50 values in the range 10–80 nM on both chloroquine‐sensitive and chloroquine‐resistant P. falciparum strains. [71a] Unfortunately, to the best of our knowledge, these compounds have not been evaluated on artemisinin‐resistant parasites. Conjugation of 4‐amino‐7‐chloroquinoline with a 3‐substituted‐2‐methyl‐1,4‐naphthoquinone residue, which is an inhibitor of Plasmodium glutathione reductase, had been reported previously (Table 1e). [71b] These hybrids exhibited IC50 values in the range 23–36 nM.
In the last two decades, many research groups have reported hybridization of the 4‐amino‐7‐chloroquinoline core with diverse heterocyclic fragments such as semicarbazone, Mannich bases, chalcones, 9‐aminoacridine, imipramine, etc.[ 72 ] More recently, 4‐amino‐7‐chloroquinoline pyrano[2,3‐c]pyrazoles [73a] or pyrimidines [73b] also yielded compounds exhibiting IC50 values in the range 10–80 nM against the P. falciparum chloroquine‐sensitive and chloroquine‐resistant strains. These results confirm that hybrids containing the 4‐amino‐7‐chloroquinoline motif may retain high antiproliferative activity against chloroquine‐resistant strains (see below, paragraph on emoquine and quinoxyzines). but their activity has not been tested in parasites carrying resistance to other 4‐aminoquinolines (piperaquine or amodiaquine).
Conversely, hybrids based on a 4,6‐diphenyl pyrimidine linked either to a 4‐aminoquinoline[ 74 ] or to a cinnamoyl residue[ 75 ] exhibited lower activities (320 and 180 nM, respectively), as well as the 4‐aminoquinoline‐urea‐benzothiazole hybrid (IC50 ranging from 330 to 970 nM).[ 76 ] These results confirm that a pertinent choice of the pharmacophore associated to the 4‐aminoquinoline moiety, and of the linker, are crucial to design efficient antimalarials.
4. Hybrid Molecules Containing Metals
4.1. Hybrids Containing Iron
Due to its ferrocenyl moiety, ferroquine (Fq, Table 1b,b’) can be considered as a hybrid molecule, as detailed above. Its antimalarial activity has been proposed to be related to the redox activity of the ferrocenyl moiety. [54c]
4.2. Hybrids Containing Gold(I)
4.2.1. 4‐Aminoquinoline–Gold(I)
Among metallodrugs against malaria, a complex of chloroquine (CQ) coordinating Au(I) at the N1 position was found active on chloroquine‐resistant P. falciparum cultured in vitro (IC50 = 5 nM) and in mice infected by P. berghei, the parasite inhibition being due to the presence of both CQ and gold.[ 77 ] Along the same lines, a “ternary hybrid” containing a chloroquine and a primaquine linked through Au(I) connected at their respective terminal aliphatic amine was recently reported (CQPQ‐gold(I), Table 1j).[ 78 ] This hybrid displayed in vitro antimalarial activity against P. falciparum asexual blood stages, with IC50 values of 28 and 166 nM against chloroquine‐susceptible and chloroquine‐resistant P. falciparum strains, respectively. Interestingly, CQPQ‐gold(I) is also active against liver stages, with IC50 of 0.6 μM on P. berghei‐infected cells. These results could not be obtained when CQ and primaquine (PQ) were combined, proving the superiority of the CQPQ‐gold(I) hybrid. CQPQ‐gold(I) inhibits β‐hematin formation, glutathione depletion, and thioredoxin reductase binding, thereby explaining its multistage antiplasmodial activity.[ 78 ]
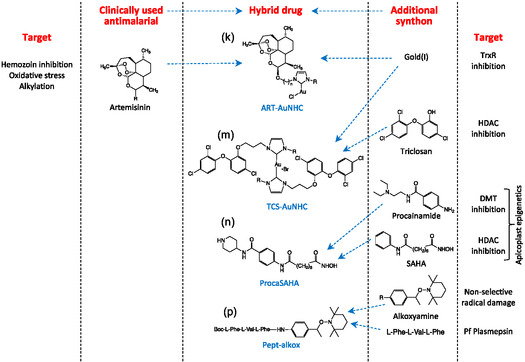
4.2.2. Artemether‐Au(I)‐N‐Heterocyclic Carbene and Triclosan‐Au(I)‐N‐Heterocyclic Carbine
More recently, with the aim to overcome P. falciparum artemisinin resistance, hybrid compounds containing gold(I) N‐heterocyclic carbene (NHC) complexes, expected to inhibit the mitochondrial thioredoxin reductase, were synthesized. The second pharmacophore was either artemether (ART‐AuNHC, Table 1k) [79a] or triclosan (TCS‐Au‐NHC, Table 1m). [79b]
In the ART‐Au‐NHC series, the best compound has an IC50 value of 9 nM comparable to that of the antimalarial reference drug artemether (IC50 = 6.1 nM), and several other hybrids displayed activities below 50 nM, associated with a low cytotoxicity against human cells. Yet, cross‐resistance between artemisinin and these hybrid molecules was evidenced by the recrudescence assay. [79a] These data are in agreement with previously obtained results that highlighted the risk of parasites cross‐resistance between artemisinins and all endoperoxide‐based compounds.[ 51 , 80 ]
Hybrids triclosan‐Au(I)‐carbene were obtained by complexation of Au(I) by two equivalents of a TCS–NHC ligand where the TCS and NHC entities are covalently linked through the phenol function of TCS. These complexes exhibit the usual linear C=Au(I)=C geometry, with TCS moieties in trans position with respect to the Au(NHC)2 plane. These derivatives displayed a moderate antiplasmodial activity (at best IC50 = 120 nM) but a weak selectivity (SI = 7.5). Interestingly, one of these TCS‐Au‐NHC complexes did not exhibit cross‐resistance with artemisinin.
5. Hybrid Molecules Targeting Epigenetic Enzymes
Drugs targeting epigenetic mechanisms that are essential for Plasmodium life cycle and survival (namely, epidrugs) are emerging as potential antimalarial drugs. Various epidrugs were recently reported to have significant antiproliferative activity in vitro against different stages of the P. falciparum and P. vivax life cycle (IC50 < 100 nM).[ 81 ] Importantly, these drugs targeting histone methylation or acetylation as well as DNA methylation, were also active against parasites in a quiescence state induced and maintained by dihydroartemisinin, and decreased the ability of artemisinin‐resistant parasites to recover after artemisinin exposure. These results suggest the potential of epidrugs as starting points for new possible drug candidates.[ 82 ] Hybrids can also be designed to target epigenetic enzymes called transferases, catalyzing the transfer of a chemical mark (like a methyl or an acetyl group) on histones or DNA (called “writers”), because their active site contains two pockets: one binding the methyl‐ or acetyl‐donor and the other bind the amino acid or the cytosine to be modified. Thus, designing inhibitors targeting both pockets, attached by a linker, is a starting point for epidrugs.
5.1. DNA‐Methyltransferase Inhibitors Hybrids
Hybrid molecules designed to target DNA methyltransferases are based on a quinoline moiety (to mimic the adenosine from the methyl donor S‐adenosylmethionine) and a moiety to mimic the cytosine (either a quinazoline in the “bisubstrate” series or 2‐aminopyrimidine moiety in SGI‐1027). Both compounds have IC50 values < 100 nM against P. falciparum asexual blood stages and SGI‐1027 has a submicromolar activity against early and late gametocytes, [81b,c] as well as against quiescent artemisinin‐resistant parasites.[ 82 ] The “bisubstrate” compound 20 can cure P. berghei infected mice at a dose of 10 mg kg−1 day−1. [83a]
5.2. Procainamide–Suberoylanilide Hydroxamic Acid (SAHA)
Hybrid derivatives that chemically combine the pan‐histone deacetylase inhibitor SAHA with the DNA methyltransferase inhibitor procainamide were synthesized (Table 1n). [83b] Compared to the single drugs, these molecules showed a higher activity in Plasmodium and a potent inhibition of human HDAC6, but no cytotoxicity in human cell lines. The Proca‐SAHA hybrids are fully active against multidrug‐resistant P. falciparum isolates from Cambodia, the epicenter of malaria drug‐resistance, which makes them promising drug candidates. They target transmission of the parasite by inhibiting the formation of male gametes. These compounds are slow‐acting and have an additive effect in combination with fast‐acting artemisinins, suggesting the potential of a combination between epidrugs and the clinically used dihydroartemisinin. The lead compound decreases parasitemia in mice in a model of severe malaria. In view of these results, this hybrid molecule might offer an original alternative to therapeutic failures of current antimalarial therapy. [83b]
6. Hybrid Molecules as Prodrugs
Prodrugs were designed, containing an alkoxyamine and a peptide fragment (Table 1p). Recognition and hydrolysis of the peptide moiety by specific parasite proteases involved in hemoglobin digestion should free the unstable alkoxyamine fragment that spontaneously release potentially damaging the alkyl radical within the parasite. Despite the moderate antiplasmodial activity of these hybrids in the classical antiproliferative assay (sub‐micromolar IC50 values), in‐depth assays evidenced that the antiparasitic activity was due to a cytostatic effect rather than a truly antiparasitic profile leading to new pharmacomodulations.[ 84 ]
7. Selection of a Highly Efficient Recent Hybrid which Avoids Cross‐Resistance
7.1. Quinoxyzines and Emoquines (Table 1g,f)
Despite widespread resistance of P. falciparum to chloroquine, it has been known for a long time that 4‐aminoquinoline analogs with a side chain at C4 shorter or longer than that of chloroquine are often active against multiresistant P. falciparum,[ 85 ] including clinical isolates that failed to respond to the treatment with artesunate.[ 86 ] Consequently, the 4‐amino‐7‐chloroquinoline scaffold may still be of interest and, in recent years, hybrid compounds containing a 4‐amino‐7‐chloroquinoline fragment substituted at C4 were designed as potential drugs active against multiresistant parasites including artemisinin‐resistant Plasmodium.
As the second pharmacophore, chlorcyclizine was considered, leading to quinoxyzines as well as emodin, leading to emoquines (Table 1g,f, respectively). Emodin (6‐methyl‐1,3,8‐trihydroxyanthraquinone, Table 1) is a naturally occurring anthraquinone derivative that exhibits a wide spectrum of pharmacological properties including antimicrobial activity[ 87 ] and inhibition of several viruses.[ 87 , 88 ] Importantly, the p‐naphthoquinone ring of emodin is reminiscent of the structure of the antimalarial atovaquone. Despite the fact that emodin itself has no antiplasmodial efficacy, some emodin derivatives exhibit a weak antiplasmodial activity with IC50 values in the low micromolar range on P. falciparum.[ 89 , 90 ] On another hand, chlorcyclizine was selected as possible partner for hybrid molecules. This moeity is the active fragment of hydroxyzine and related diphenylmethyl‐piperazine derivatives, which are known as antagonists of H1 histamine receptor, resulting in their antihistaminic and anxiolytic properties. These chlorcyclizine derivatives also exhibit virucidal activity against several DNA and RNA viruses,[ 91 ] and antibacterial and antileshmanial activities.[ 92 ]
Several emoquine and quinoxyzine derivatives were synthesized, bearing different linkers between the two pharmacophores.[ 93 ] As an example, the convergent synthesis of emoquine‐1 is summarized in Figure 3 .
Figure 3.

Convergent synthesis of emoquine‐1 by coupling of emodin functionalized at C3 by a ω‐bromoethoxy substituent, with 7’‐chloro‐4’‐diethylaminoquinoline. The emodin and quinoline fragments whose antimalarial activity has been compared with that of emoquine are shown at the bottom of the figure.
Two of these hybrid molecules, emoquine‐1 and (R)‐quinoxyzine (Table 1f,g, respectively), showed a high and specific activity against P. falciparum (IC50 ≤ 100 nM) with high selectivity indexes with respect to human cells (SI = 220 and 140, respectively). This strong antiplasmodial activity was retained on Cambodian clinical isolates of P. falciparum resistant to both artemisinins and chloroquine. These hybrids are also active on artemisinin‐resistant parasites at the quiescent stage. Moreover, emoquine‐1 even retained its high antiplasmodial activity on the atovaquone‐resistant P. falciparum strain. These results confirm the broad‐range antiplasmodial activity of emoquine‐1, with high selectivity for Plasmodium, and no cross‐resistance with artemisinins, chloroquine, and atovaquone.[ 93 ]
With regard to the structure–activity relationship, emodin itself, or emodin fragment (Figure 3) and norchlorcyclizine did not exhibit any significant antimalarial activity (IC50 > 10 μM). Conversely, the quinoline fragment (Figure 3) exhibited a potent and selective antimalarial activity (IC50 = 54 nM on chloroquine‐sensitive but artemisinin‐resistant strain). Consequently, the quinoline fragment is essential for the antiplasmodial activity of both hybrid molecules, emoquine‐1 and (R)‐quinoxyzine. However, the quinoline fragment is inactive on the P. falciparum isolate that is resistant to both atovaquone and chloroquine (IC50 = 2.3 μM).[ 93 ] So, the emodin fragment of this hybrid, despite being inactive as independent element, may be essential to avoid cross‐resistance to major quinoline‐type antiplasmodial drugs. In addition, substitution of the linear α,β‐diethylamine side chain at C4’ of the quinoline fragment (Figure 3), by a piperazine moiety, resulted in a drastically lower antimalarial activity.[ 93 ] This may be due to a lower electron delocalization and, consequently, a change of the pKa value of the 4’‐aminoquinoline moiety, resulting in a lower accumulation within the parasites.[ 94 ] In a mouse model of malaria, emoquine‐1 was efficient after oral or intraperitoneal treatment and presented a curative dose at 10 mg kg−1 day−1 intraperitoneally (no recrudescence at 30 days) and ED50 value per os (25 mg kg−1 day−1). These results are very promising to develop this preferred administration route[ 93 ] and underline emoquine‐1 as a promising drug candidate, even on artemisinin‐resistant strains.
These results confirm the real advantage of such hybrid molecules with two pharmacophores and a correctly tuned linker for the design of new antiplasmodial agents.
8. Choice of the Linker Connecting Pharmacophores
The design and the structural choice of the linker in hybrid molecules is an important parameter to modulate their pharmacological properties, as underlined by Tsogoeva and coll.[ 95 ] In fact, the nature and length of the linker can play a variety of roles, from contributing to the interaction of the hybrid molecule with its sites of action when they are identified, to modulating lipophilicity (and, therefore, membrane crossing), pharmacokinetic properties or metabolism. Generally speaking, the chemical structure of the linker may induce a specific behavior. For example, polyoxymethylene or alkyl amines are expected to improve aqueous solubility. When in vivo stability of the hybrid drug is required, ester or amide linkers will be avoided because of the high and ubiquitous activity of esterases; conversely, these chemical functionalities can be used in linkers designed to be cleavable.
More precisely in the case of antimalarials, there are only a handful of hybrid drugs reasonably active. So, linkers cannot be compared independently of pharmacophores. As experimental results evidenced in Table 1, the preferred linker was different in different series. For example, in the trioxaquine series, the best linker was 1,4‐diaminocyclohexane as in PA1103 selected from about 120 different trioxaquines;[ 44 ] but trioxaquine DU1302, bearing a α,β‐diethylamine linker, also exhibited IC50 values below 20 nM.[ 38 ] In the case of emoquine‐1,[ 93 ] most of the activity was lost when the linear α,β‐diethylamine side chain at C4’ of the quinoline was replaced by a quite similar but more rigid 1,4‐piperazine residue. So, there is no “magic” general linker, but its structure and its length are crucial and should be optimized for each particular series of hybrid drugs, along with the choice of pharmacophores.
9. Conclusion
Artemisinin‐based combination therapies are still the cornerstone of antimalarial therapy, despite artemisinin‐resistant parasite strains emergence and/or spread in Asia, Guyana, and Africa. To avoid dramatic malaria mortality due to increasing failure of the current treatments, research for new drugs is an urgent need.
Development of hybrid molecules, based on chemical entities with two (or more) different structural domains (pharmacophores) having complementary biological functions, not necessarily on the same target, is an interesting avenue. In fact, the different pharmacophores can act synergistically. Moreover, the combination of two pharmacophores in a single molecule theoretically reduces the risk of parasite drug‐resistance selection.
In fact, the sole hybrid nature of molecules is not a panacea, nor a guarantee of their efficacy. Obviously, the choice of synthons and linker arms is crucial, without underestimating the possible contribution of a little bit of luck in the choice of pharmacophores and linkers.
10. Summary and Outlook
Hybrid drugs can exhibit efficient antiplasmodial activity, despite each fragment being inactive as an independent element. This short review on hybrid drugs targeting malaria parasites emphasizes the crucial role of the different chemical entities, but also of the linker that can modulate the biological activity of the pharmacophores themselves. Among possible active fragments, the 4‐amino‐7‐chloroquinoline motif usually retains high antiproliferative activity against chloroquine‐resistant strains. The more active of these hybrid molecules, emoquine‐1, showed a high and specific activity against P. falciparum without toxicity against mammalian cells. This strong antiplasmodial activity was retained on clinical isolates of P. falciparum resistant to both artemisinins and chloroquine, or to atovaquone. This hybrid is thus active on artemisinin‐resistant parasites, confirming the lack of cross‐resistance with artemisinins, and also against the quiescent stage. Since emoquine‐1 is curative in a mouse malaria model, this compound, whose activity can clearly be assigned to its hybrid structure, appears as a very promising antimalarial drug‐candidate.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgements
The authors gratefully acknowledge all colleagues, postdoctoral fellows, Ph.D. and master students, and collaborators who are or have been involved in the research of hybrid molecules as potential drugs. The authors also acknowledge Dr Etienne Joly for fruitful discussions.
Biographies
Anne Robert (Ph.D. Toulouse, 1985) is currently director of research at the “Laboratoire de Chimie de Coordination du CNRS” in Toulouse. Her work has been focused on the role of endogenous redox metals in biology (involved in biomimetic oxidations or mechanism of action of antimalarial drugs) and redox metal homeostasis. It includes the design of new antimalarial drugs and metal‐specific drug candidates for treatment of Alzheimer's disease or Wilson's disease.

Bernard Meunier is emeritus director of research at the “Laboratoire de Chimie de Coordination du CNRS” in Toulouse with a focus on bio‐inorganic chemistry (metalloenzymes) and medicinal chemistry (cancer, Alzheimer's and Wilson's diseases). He is member of the French Academy of Sciences, National Academy of Pharmacy, and Polish Academy of Sciences. He is currently a guest professor at the Guangdong University of Technology. He has been associate professor at the “École Polytechnique” from 1993 to 2006.

Françoise Benoit‐Vical (Ph.D., 1997) is director of research at Inserm (French National Institute of Health and Medical Research) and manages the CNRS (French National Institute of Scientific Research) group “Antimalarial molecules and pharmacological approaches” at the “Laboratoire de Chimie de Coordination du CNRS” in Toulouse, France. Her areas of interest concern new therapeutic solutions in malaria, mode of action of antiplasmodial molecules, study of resistance mechanisms developed by the Plasmodium parasite, and pharmacomodulation work in medicinal chemistry.

Contributor Information
Anne Robert, Email: anne.robert@lcc-toulouse.fr.
Bernard Meunier, Email: bmeunier@lcc-toulouse.fr.
Françoise Benoit‐Vical, Email: francoise.vical@inserm.fr.
Data Availability Statement
Research data are not shared.
References
- 1. World Health Organization , Global malaria program news update, https://www.who.int/teams/global‐malaria‐programme/reports/world‐malaria‐report‐2024, (accessed: December 2024).
- 2. Tu Y., Angew. Chem. Int. Ed. 2016, 55, 10210. [DOI] [PubMed] [Google Scholar]
- 3.a) Price R. N., Nosten F., Luxemburger C., van Vugt M., Phaipun L., Chongsuphajaisiddhi T., White N. J., Trans. R. Soc. Trop. Med. Hyg. 1997, 5, 574; [DOI] [PubMed] [Google Scholar]; b) Price R. N., Nosten F., Luxemburger C., Kuile F. O., Paiphun L., Chongsuphajaisiddhi T., White N. J., Lancet 1996, 9016, 1654; [DOI] [PubMed] [Google Scholar]; c) White N. J., Nosten F., Looareesuwan S., Watkins W. M., Marsh K., Snow R. W., Kokwaro G., Ouma J., Hien T. T., Molyneux M. E., Taylor T. E., Newbold C. I., Ruebush T. K., Danis M., Greenwood B. M., Anderson R. M., Olliaro P., Lancet 1999, 353, 1965; [DOI] [PubMed] [Google Scholar]; d) van der Pluijm R. W., Tripura R., Hoglund R. M., Pyae Phyo A., Lek D., Islam A., Anvikar A. R., Satpathi P., Satpathi S., Kumari Behera P., Tripura A., Baidya S., Onyamboko M., Hoang Chau N., Sovann Y., Suon S., Sreng S., Mao S., Oun S., Yen S., Amaratunga C., Chutasmit K., Saelow C., Runcharern R., Kaewmok W., Thi Hoa N., Viet Thanh N., Hanboonkunupakarn B., Callery J. J., Kumar Mohanty A., et al., Lancet 2020, 395, 1345.32171078 [Google Scholar]
- 4. Meshnick S. R., Taylor T. E., Kamchonwongpaisan S., Microbiol. Rev. 1996, 60, 301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.a) Robert A., Meunier B., J. Am. Chem. Soc. 1997, 119, 5968; [Google Scholar]; b) Robert A., Cazelles J., Meunier B., Angew. Chem. Int. Ed. 2001, 40, 1954. [DOI] [PubMed] [Google Scholar]
- 6. Robert A., Benoit‐Vical F., Claparols C., Meunier B., Proc. Natl. Acad. Sci. 2005, 102, 13676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. White N., Philos. Trans. R. Soc. Lond. B 1999, 354, 739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Agarwal D., Gupta R. D., Awasthi S. K., Antimicrob. Agents Chemother. 2017, 61, e00249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Boman H. G., Wade D., Boman I. A., Wåhlint B., Merrifield R. B., FEBS Lett. 1989, 259, 103. [DOI] [PubMed] [Google Scholar]
- 10. Schroeder B. R., Ghare M. I., Bhattacharya C., Paul R., Yu Z., Zaleski P. A., Bozeman T. C., Rishel M. J., Hecht S. M., J. Am. Chem. Soc. 2014, 136, 13641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dechy‐Cabaret O., Benoit‐Vical F., Robert A., Meunier B., ChemBioChem 2000, 1, 281. [DOI] [PubMed] [Google Scholar]
- 12. Meunier B., Acc. Chem. Res. 2008, 41, 69. [DOI] [PubMed] [Google Scholar]
- 13. Walsh J. J., Bell A., Curr. Pharm. Des. 2009, 15, 2970. [DOI] [PubMed] [Google Scholar]
- 14. Okombo J., Fidock D. A., Nat. Rev. Microbiol. 2024, 23, 178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sausville E. A., Stein R. W., Horwitz S. B., Biochemistry 1978, 17, 2746. [DOI] [PubMed] [Google Scholar]
- 16. Pratviel G., Bernadou J., Meunier B., Biochem. Pharmacol. 1989, 38, 133. [DOI] [PubMed] [Google Scholar]
- 17. Hecht S. M., J. Nat. Prod. 2000, 63, 158. [DOI] [PubMed] [Google Scholar]
- 18. Burger R. M., Chem. Rev. 1998, 98, 1153.11848928 [Google Scholar]
- 19. Pratviel G., Bernadou J., Meunier B., Angew. Chem. Int. Ed. Engl. 1995, 34, 746. [Google Scholar]
- 20. Vennerstrom J. L., Arbe‐Barnes S., Brun R., Charman S. A., Chiu F. C. K., Chollet J., Dong Y., Dorn A., Hunziker D., Matile H., McIntosh K., Padmanilayam M., Santo Tomas J., Scheurer C., Scorneaux B., Tang Y., Urwyler H., Wittlin S., Charman W. N., Nature 2004, 430, 900. [DOI] [PubMed] [Google Scholar]
- 21. Creek D. J., Charman W. N., Chiu F. C., Prankerd R. J., Dong Y., Vennerstrom J. L., Charman S. A., Antimicrob. Agents Chemother. 2008, 52, 1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dong Y., Wang X., Kamaraj S., Bulbule V. J., Chiu F. C., Chollet J., Dhanasekaran M., Hein C. D., Papastogiannidis P., Morizzi J., Shackleford D. M., Barker H., Ryan E., Scheurer C., Tang Y., Zhao Q., Zhou L., White K. L., Urwyler H., Charman W. N., Matile H., Wittlin S., Charman S. A., Vennerstrom J. L., J. Med. Chem. 2017, 60, 2654. [DOI] [PubMed] [Google Scholar]
- 23. Ellis G. L., Amewu R., Sabbani S., Stocks P. A., Shone A., Stanford D., Gibbons P., Davies J., Vivas L., Charnaud S., Bongard E., Hall C., Rimmer K., Lozanom S., Jesus M., Gargallo D., Ward S. A., O'Neill P. M., J. Med. Chem. 2008, 5, 2170. [DOI] [PubMed] [Google Scholar]
- 24. O’Neill P., Amewu R., Charman S. A., Sabbani S., Gnädig N. F., Straimer J., Fidock D. A., Shore E. R., Roberts N. L., Wong M. H.‐L., Hong W. D., Pidathala C., Riley C., Murphy B., Aljayyoussi G., Javier Gamo F., Sanz L., Rodrigues J., Gonzalez Cortes C., Herreros E., Angulo‐Barturén I., Jiménez‐Díaz M. B., Ferrer Bazaga S., Santos Martínez‐Martínez M., Campo B., Sharma R., Ryan E., Shackleford D. M., Campbell S., Smith D. A., et al., Nat. Commun. 2017, 8, 15159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kumar A., Agarwal D., Sharma B., Devi Gupta R., Kumar Awasthi S., ACS Omega 2024, 9, 31611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Meunier B., Robert A., Acc. Chem. Res. 2010, 43, 1444. [DOI] [PubMed] [Google Scholar]
- 27. Francis S. E., Sullivan D. J., Goldberg D. E., Annu. Rev. Microbiol. 1997, 51, 97. [DOI] [PubMed] [Google Scholar]
- 28. Pagola S., Stephens P. W., Bohle D. S., Kosar A. D., Madsen S. K., Nature 2000, 404, 307. [DOI] [PubMed] [Google Scholar]
- 29. Klar P. B., Waterman D. G., Gruene T., Mullick D., Song Y., Gilchrist J. B., Owen C. D., Wen W., Biran I., Houben L., Regev‐Rudzki N., Dzikowski R., Marom N., Palatinus L., Zhang P., Leiserowitz L., Elbaum M., ACS Cent. Sci. 2024, 10, 1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Robert A., Meunier B., Chem. Eur. J. 1998, 4, 1287. [Google Scholar]
- 31. Laurent S. A.‐L., Robert A., Meunier B., Angew. Chem. Int. Ed. 2005, 44, 2060. [DOI] [PubMed] [Google Scholar]
- 32. Loup C., Lelièvre J., Benoit‐Vical F., Meunier B., Antimicrob. Agents Chemother. 2007, 51, 3768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Moreau S., Perly B., Chachaty C., Deleuze C., Biochim. Biophys. Acta 1985, 840, 107. [DOI] [PubMed] [Google Scholar]
- 34. Egan T. J., Marques H. M., Coord. Chem. Rev. 1999, 190–192, 493. [Google Scholar]
- 35. Chou A. C., Chevli R., Fitch C. D., Biochemistry 1980, 19, 1543. [DOI] [PubMed] [Google Scholar]
- 36.a) Sullivan D. J., Gluzman I. Y., Russell D. G., Goldberg D. E., Proc. Natl. Acad. Sci. 1996, 93, 11865; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Sullivan D. J., Matile H., Riedley R. G., Goldberg D. E., J. Biol. Chem. 1998, 273, 31103. [DOI] [PubMed] [Google Scholar]
- 37.a) Çapcı A., Lorion M. M., Wang H., Simon N., Leidenberger M., Borges Silva M. C., Moreira D. R. M., Zhu Y., Meng Y., Chen J. Y., Lee Y. M., Friedrich O., Kappes B., Wang J., Ackermann L., Tsogoeva S. B., Angew. Chem. Int. Ed. 2019, 58, 13066; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Feng L.‐S., Xu Z., Chang L., Li C., Yan X.‐F., Gao C., Ding C., Zhao F., Shi F., Wu X., Med Res Rev. 2020, 40, 931; [DOI] [PubMed] [Google Scholar]; c) Kouznetsov V., Gomez‐Barrio A., Eur. J. Med. Chem. 2009, 44, 3091; [DOI] [PubMed] [Google Scholar]; d) Kumar A., Srivastava K., Kumar S. R., Siddiqi M. I., Puri S. K., Sexana J. K., Chauhan P. M. S., Eur. J. Med. Chem. 2011, 46, 676; [DOI] [PubMed] [Google Scholar]; e) Walsh J. J., Coughlan D., Heneghan N., Gaynor C., Bell A., Bioorg. Med. Chem. Lett. 2007, 17, 3599. [DOI] [PubMed] [Google Scholar]
- 38. Dechy‐Cabaret O., Benoit‐Vical F., Loup C., Robert A., Gornitzka H., Bonhoure A., Vial H., Magnaval J.‐F., Séguéla J.‐P., Meunier B., Chem. Eur. J. 2004, 10, 1625. [DOI] [PubMed] [Google Scholar]
- 39. Robert A., Dechy‐Cabaret O., Cazelles J., Meunier B., Acc. Chem. Res. 2002, 35, 167. [DOI] [PubMed] [Google Scholar]
- 40. Benoit‐Vical F., Lelièvre J., Berry A., Deymier C., Dechy‐Cabaret O., Cazelles J., Loup C., Robert A., Magnaval J.‐F., Meunier B., Antimicrob. Agents Chemother. 2007, 51, 1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Basco L. K., Dechy‐Cabaret O., Ndounga M., Meche F. S., Robert A., Meunier B., Antimicrob. Agents Chemother. 2001, 45, 1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Laurent S. A.‐L., Loup C., Mourgues S., Robert A., Meunier B., ChemBioChem 2005, 6, 653. [DOI] [PubMed] [Google Scholar]
- 43. Bousejra‐El Garah F., Claparols C., Benoit‐Vical F., Meunier B., Robert A., Antimicrob. Agents Chemother. 2008, 52, 2966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Coslédan F., Fraisse L., Pellet A., Guillou F., Mordmüller B., Kremsner P. G., Moreno A., Mazier D., Maffrand J.‐P., Meunier B., Proc. Natl. Acad. Sci. 2008, 105, 17579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Noedl H., Se Y., Schaecher K., Smith B. L., Socheat D., Fukuda M. M., New Engl. J. Med. 2008, 359, 2619. [DOI] [PubMed] [Google Scholar]
- 46. Dondorp A. M., Nosten F., Yi P., Das D., Phyo A. P., Tarning J., Lwin K. M., Ariey F., Hanpithakpong W., Lee S. J., Ringwald P., Silamut K., Imwong M., Chotivanich K., Lim P., Herdman T., An S. S., Yeung S., Singhasivanon P., Day N. P., Lindegardh N., Socheat D., White N. J., New Engl. J. Med. 2009, 361, 455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Witkowski B., Lelièvre J., Barragán M. J., Laurent V., Su X. Z., Berry A., Benoit‐Vical F., Antimicrob. Agents Chemother. 2010, 54, 1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Straimer J., Gnädig N. F., Witkowski B., Amaratunga C., Duru V., Ramadani A. P., Dacheux M., Khim N., Zhang L., Lam S., Gregory P. D., Urnov F. D., Mercereau‐Puijalon O., Benoit‐Vical F., Fairhurst R. M., Ménard D., Fidock D. A., Science 2015, 347, 428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ariey F., Witkowski B., Amaratunga C., Beghain J., Langlois A. C., Khim N., Kim S., Duru V., Bouchier C., Ma L., Lim P., Leang R., Duong S., Sreng S., Suon S., Chuor C. M., Bout D. M., Ménard S., Rogers W. O., Genton B., Fandeur T., Miotto O., Ringwald P., Le Bras J., Berry A., Barale J. C., Fairhurst R. M., Benoit‐Vical F., Mercereau‐Puijalon O., Ménard D., Nature 2014, 505, 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Witkowski B., Khim N., Chim P., Kim S., Ke S., Kloeung N., Chy S., Duong S., Leang R., Ringwald P., Dondorp A. M., Tripura R., Benoit‐Vical F., Berry A., Gorgette O., Ariey F., Barale J. C., Mercereau‐Puijalon O., Ménard D., Antimicrob. Agents Chemother. 2013, 57, 914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Paloque L., Witkowski B., Lelièvre J., Ouji M., Ben Haddou T., Ariey F., Robert A., Augereau J.‐M., Ménard D., Meunier B., Benoit‐Vical F., J. Antimicrob. Chemother. 2018, 73, 395. [DOI] [PubMed] [Google Scholar]
- 52. Macintyre F., Adoke Y., Tiono A. B., Duong T. T., Mombo‐Ngoma G., Bouyou‐Akotet M., Tinto H., Bassat Q., Issifou S., Adamy M., Demarest H., Duparc S., Leroy D., Laurijssens B. E., Biguenet S., Kibuuka A., Tshefu A. K., Smith M., Foster C., Leipoldt I., Kremsner P. G., Phuc B. Q., Ouedraogo A., Ramharter M., BMC Med. 2017, 15, 181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Thétiot‐Laurent S. A.‐L., Boissier J., Robert A., Meunier B., Angew. Chem. Int. Ed. 2013, 52, 7936. [DOI] [PubMed] [Google Scholar]
- 54.a) Biot C., Glorian G., Maciejewski L. A., Brocard J. S., J. Med. Chem. 1997, 40, 3715; [DOI] [PubMed] [Google Scholar]; b) Dubar F., Khalife J., Brocard J., Dive D., Biot C., Molecules 2008, 13, 2900; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Dubar F., Slomianny C., Khalife J., Dive D., Kalamou H., Guérardel Y., Grellier P., Biot C., Angew. Chem. Int. Ed. 2013, 52, 7690. [DOI] [PubMed] [Google Scholar]
- 55. McCarthy J. S., Rückle T., Djeriou E., Cantalloube C., Ter‐Minassian D., Baker M., O'Rourke P., Griffin P., Marquart L., Hooft van Huijsduijnen R., Möhrle J. J., Malar. J. 2016, 15, 469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Bellot F., Coslédan F., Vendier L., Brocard J., Meunier B., Robert A., J. Med. Chem. 2010, 53, 4103. [DOI] [PubMed] [Google Scholar]
- 57. Ménard S., Ben Haddou T., Ramadani A. P., Ariey F., Iriart X., Beghain J., Bouchier C., Witkowsk B., Berry A., Mercereau‐Puijalon O., Benoit‐Vical F., Emerg. Infect. Dis. 2015, 21, 1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Peatey C. L., Chavchich M., Chen N., Gresty K. J., Gray K. A., Gatton M. L., Waters N. C., Cheng Q., J. Infect. Dis. 2015, 212, 426. [DOI] [PubMed] [Google Scholar]
- 59. Chen N., LaCrue A. N., Teuscher F., Waters N. C., Gatton M. L., Kyle D. E., Cheng Q., Antimicrob. Agents Chemother. 2014, 58, 4773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Glazunov V. P., Ya Yakubovskaya A., Pokhilo N. D., Bochinskaya N. V., Anufriev V. P., Russ. Chem. Bull. Int. Ed. 2003, 52, 198. [Google Scholar]
- 61. Mok S., Stokes B. H., Gnädig N. F., Ross L. S., Yeo T., Amaratunga C., Allman E., Solyakov L., Bottrill A. R., Tripathi J., Fairhurst R. M., Llinás M., Bozdech Z., Tobin A. B., Fidock D. A., Nat. Commun. 2021, 12, 530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kessl J. J., Meshnick S. R., Trumpower B. L., Trends Parasitol. 2007, 23, 494. [DOI] [PubMed] [Google Scholar]
- 63. Ouji M., Nguyen M., Mustière R., Jimenez T., Augereau J.‐M., Benoit‐Vical F., Deraeve C., Bioorg. Med. Chem. Lett. 2021, 39, 127884. [DOI] [PubMed] [Google Scholar]
- 64. Reyser T., Paloque L., Ouji M., Nguyen M., Ménard S., Witkowski B., Augereau J.‐M., Benoit‐Vical F., J. Antimicrob. Chemother. 2020, 75, 2826. [DOI] [PubMed] [Google Scholar]
- 65. Li Y., Loureiro A., Nguyen M., Laurent M., Bijani C., Benoit‐Vical F., Robert A., Liu Y., Meunier B., ChemistryOpen 2022, 11, e202200064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Robert A., Liu Y., Nguyen M., Meunier B., Acc. Chem. Res. 2015, 48, 1332. [DOI] [PubMed] [Google Scholar]
- 67. Zhang W., Huang D., Huang M., Huang J., Wang D., Liu X., Nguyen M., Vendier L., Mazères S., Robert A., Liu Y., Meunier B., ChemMedChem 2018, 13, 684. [DOI] [PubMed] [Google Scholar]
- 68. Zhao J., Shi Q., Tian H., Li Y., Liu Y., Xu Z., Robert A., Liu Q., Meunier B., ACS Chem. Neurosci. 2021, 12, 140. [DOI] [PubMed] [Google Scholar]
- 69. Birth D., Kao W. C., Hunte C., Nat. Commun. 2014, 5, 4029. [DOI] [PubMed] [Google Scholar]
- 70. Koumpoura C. L., Nguyen M., Bijani C., Vendier L., Salina E. G., Buroni S., Degiacomi G., Cojean S., Loiseau P. M., Benoit‐Vical F., García‐Sosa A. T., Robert A., Baltas M., ACS Omega 2022, 7, 35635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.a) Andayi W. A., Egan T. J., Gut J., Rosenthal P. J., Chibale K., ACS Med. Chem. Lett. 2013, 4, 642; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Davioud‐Charvet E., Delarue S., Biot C., Schwöbel B., Boehme C. C., Müssigbrodt A., Maes L., Sergheraert C., Grellier P., Heiner Schirmer R., Becker K., J. Med. Chem. 2001, 44, 4268. [DOI] [PubMed] [Google Scholar]
- 72. Oliveira R., Miranda D., Magalhães J., Capela R., Perry M. J., O’Neill P. M., Moreira R., Lopes F., Bioorg. Med. Chem. 2015, 23, 5120. [DOI] [PubMed] [Google Scholar]
- 73.a) Ravindar L., Ng Y. H., Feroz S. R., Norazmi N. A. Z. B., Ali A. H., Hasbullah S. A., Ismail N., Agustar H. K., Lau Y. L., Hassan N. I., Eur. J. Med. Chem. 2024, 279, 116828; [DOI] [PubMed] [Google Scholar]; b) Ravindar L., Hasbullah S. A., Rakesh K. P., Raheem S., Ismail N., Linge L. Y., Hassan N. I., Bioorg. Med. Chem. Lett. 2024, 114, 129992. [DOI] [PubMed] [Google Scholar]
- 74. Kayamba F., Malimabe T., Ademola I. K., Pooe O. J., Kushwaha N. D., Mahlalela M., van Zyl R. L., Gordon M., Mudau P. T., Zininga T., Shonhai A., Nyamori V. O., Karpoormath R., Eur. J. Med. Chem. 2021, 217, 113330. [DOI] [PubMed] [Google Scholar]
- 75. Kayamba F., Karpoormath R., Obakachi V. A., Mahlalela M., Banda D., van Zyl R. L., Lala S., Zininga T., Shonhai A., Baba Shaika B., Pooe O. J., Eur. J. Med. Chem. 2025, 281, 116944. [DOI] [PubMed] [Google Scholar]
- 76. Oyeneyin O. E., Moodley R., Mashaba C., Garnie L. F., Omoboyowa D. A., Rakodi G. H., Maphoru M. V., Balogun M. O., Hoppe H. C., Egan T. J., Tukulula M., Helyion 2024, 10, e38434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Navarro M., Pérez H., Sánchez‐Delgado R. A., J. Med. Chem. 1997, 40, 1937. [DOI] [PubMed] [Google Scholar]
- 78.a) de Souza Pereira C., Costa Quadros H., Magalhaes Moreira D. R., Castro W., Ivisson Santos De Deus Da Silva R., Botelho Pereira Soares M., Fontinha D., Prudêncio M., Schmitz V., Dos Santos H. F., Gendrot M., Fonta I., Mosnier J., Pradines B., Navarro M., ChemMedChem 2021, 16, 662; [DOI] [PubMed] [Google Scholar]; b) de Souza Pereira C., Costa Quadros H., Aboagye S. Y., Fontinha D., D’Alessandro S., Byrne M. E., Gendrot M., Fonta I., Mosnier J., Moreira D. R. M., Basilico N., Williams D. L., Prudêncio M., Pradines B., Navarro M., Pharmaceutics 2022, 14, 1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.a) Ouji M., Barnoin G., Fernández Álvarez Á., Augereau J.‐M., Hemmer C., Benoit‐Vical F., Gornitzka H., Molecules 2020, 25, 2817; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ouji M., Bourgeade Delmas S., Fernández Álvarez Á., Augereau J.‐M., Valentin A., Hemmert C., Gornitzka H., Benoit‐Vical F., ChemistrySelect 2020, 5, 619. [Google Scholar]
- 80. Straimer J., Gnädig N. F., Stokes B. H., Ehrenberger M., Crane A. A., Fidock D. A., MBio 2017, 8, e00172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.a) Maher S. P., Bakowski M. A., Vantaux A., Flannery E. L., Andolina C., Gupta M., Antonova‐Koch Y., Argomaniz M., Cabrera‐Mora M., Campo B., Chao A. T., Chatterjee A. K., Cheng W. T., Chuenchob E., Cooper C. A., Cottier K., Galinski M. R., Harupa‐Chung A., Ji H., Joseph S. B., Lenz T., Lonardi S., Matheson J., Mikolajczak S. A., Moeller T., Orban A., Padín‐Irizarry V., Pan K., Péneau J., Prudhomme J., et al., eLife 2024, 13, RP98221; [Google Scholar]; b) Coetzee N., von Grüning H., Opperman D., van der Watt M., Reader J., Birkholtz L. M., Sci Rep. 2020, 10, 2355; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Vanheer L. N., Zhang H., Lin G., Kafsack B. F. C., Antimicrob. Agents Chemother. 2020, 64, e02523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Reyser T., Paloque L., Nguyen M., Augereau J.‐M., Fuchter M. J., Lopez M., Arimondo P. B., Hassell‐Hart S., Spencer J., Di Stefano L., Benoit‐Vical F., Pharmaceutics 2023, 15, 2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.a) Nardella F., Halby L., Hammam E., Erdmann D., Cadet‐Daniel V., Peronet R., Ménard D., Witkowski B., Mecheri S., Scherf A., Arimondo P. B., ACS Cent Sci. 2020, 6, 16; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Nardella F., Halby L., Dobrescu I., Viluma J., Bon C., Claes A., Cadet‐Daniel V., Tafit A., Roesch C., Hammam E., Erdmann D., Mairet‐Khedim M., Peronet R., Mecheri S., Witkowski B., Scherf A., Arimondo P. B., J. Med. Chem. 2021, 64, 10403. [DOI] [PubMed] [Google Scholar]
- 84. Embo‐Ibouanga A. W., Nguyen M., Paloque L., Coustets M., Joly J.‐P., Augereau J.‐M., Vanthuyne N., Bikanga R., Coquin N., Robert A., Audran G., Boissier J., Mellet P., Benoit‐Vical F., Marque S. R. A., Molecules 2024, 29, 1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Ridley R. G., Hofheinz W., Matile H., Jaquet C., Dorn A., Masciadri R., Jolidon S., Richter W. F., Guenzi A., Girometta M. A., Urwyler H., Huber W., Thaithong S., Peters W., Antimicrob. Agents Chemother. 1996, 40, 1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. De D., Krogstad F. M., Cogswell F. B., Krogstad D. J., Am. J. Trop. Med. Hyg. 1996, 55, 579. [DOI] [PubMed] [Google Scholar]
- 87. Dong X., Fu J., Yin X., Cao S., Li X., Lin L., Ni J., Phytother. Res. 2016, 30, 1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Ho T. Y., Wu S. L., Chen J. C., Li C. C., Hsiang C. Y., Antiviral Res. 2007, 74, 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Li Y., Touret F., de Lamballerie X., Nguyen M., Laurent M., Benoit‐Vical F., Robert A., Liu Y., Meunier B., Org. Biomol. Chem. 2023, 21, 7382. [DOI] [PubMed] [Google Scholar]
- 90. Pandeti S., Gunjan S., Paidipelli S., Tripathi R., Tadigoppula N., Nat. Prod. Chem. Res. 2014, 2, 1000150. [Google Scholar]
- 91.a) Chakrabarty A. N., Mookerjee M., Dastidar S. G., Int. J. Antimicrob. Agents 2000, 14, 215; [DOI] [PubMed] [Google Scholar]; b) He S., Lin B., Chu V., Hu Z., Hu X., Xiao J., Wang A. Q., Schweitzer C. J., Li Q., Imamura M., Hiraga N., Southall N., Ferrer M., Zheng W., Chayama K., Marugan J. J., Liang T. J., Sci. Transl. Med. 2015, 7, 282ra49; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Mingorance L., Friesland M., Coto‐Llerena M., Pérez‐del‐Pulgar S., Boix L., López‐Oliva J. M., Bruix J., Forns X., Gastaminza P., Antimicrob. Agents Chemother. 2014, 58, 3451; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Reznikov L. R., Norris M. H., Vashisht R., Bluhm A. P., Li D., Liao Y. J., Brown A., Butte A. J., Ostrov D. A., Biochem. Biophys. Res. Commun. 2021, 538, 173; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Kornhuber J., Hoertel N., Gulbins E., Mol. Psychiatry 2022, 27, 307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Roy D., Panda G., ACS Omega 2020, 5, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Li Y., Nguyen M., Laurent M., Wein S., Paloque L., Kessavdjee‐Djouma S., Witkowski B., Musset L., Augereau J.‐M., Cerdan R., Robert A., Liu Y., Meunier B., Benoit‐Vical F., J. Med. Chem. 2025, 68, 3559. [DOI] [PubMed] [Google Scholar]
- 94. Warhurst D. C., Craig J. C., Adagu I. S., Guy R. K., Madrid P. B., Fivelman Q. L., Biochem. Pharmacol. 2007, 73, 1910. [DOI] [PubMed] [Google Scholar]
- 95. Sampath Kumar H. M., Herrmann L., Tsogoeva S. B., Bioorg. Med. Chem. Lett. 2020, 30, 127514. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Research data are not shared.