

Abstract
We examined alternative and classical complement activation induced by whole bacilli of Mycobacterium bovis BCG and Mycobacterium tuberculosis products. After exposure to BCG, there were higher levels of the terminal complement complex in sera from Indian tuberculosis patients than in sera from healthy controls. The addition of BCG with or without EGTA to these sera indicated that approximately 70 to 85% of the total levels of the terminal complement complex was formed by classical activation. Sera from Indian tuberculosis patients contained more antibody to lipoarabinomannan (LAM) than sera from healthy Indians. Levels of anti-LAM immunoglobulin G2 (IgG2), but not anti-LAM IgM, correlated positively with classical activation induced by BCG in the sera. By flow cytometry, deposition of C3 and terminal complement complex on bacilli incubated with normal human serum was demonstrated. The anticomplement staining was significantly reduced in the presence of EGTA and EDTA. Flow cytometry also revealed the binding of complement to BCG incubated with rabbit anti-LAM and then with factor B-depleted serum. This indicates that classical activation plays a major role in complement activation induced by mycobacteria and that anti-LAM IgG on the bacilli can mediate this response. Classical complement activation may be important for the extent of phagocytosis of M. tuberculosis by mononuclear phagocytes, which may influence the course after infection.
Mycobacterium tuberculosis is a facultative intracellular parasite, and several studies have focused upon the mechanisms by which mycobacteria enter mononuclear phagocytes. The complement system plays a major role in opsonizing mycobacteria for cellular uptake. It has been shown that monocyte complement receptors (CR) mediate the phagocytosis of M. tuberculosis and Mycobacterium bovis BCG coated with C3 by alternative complement activation (17, 26). Phenolic glycolipid 1, which is found in abundance on Mycobacterium leprae, fixes C3 via serum antibody (Ab) binding and classical-pathway complement activation and mediates phagocytosis by monocytes (27). The authors have also shown that serum from healthy adults contains Ab to lipomannan, lipoarabinomannan (LAM), and arabinogalactan (28), and it is well established that anti-M. tuberculosis Ab occur in both tuberculous and nontuberculous individuals (1). Production of such Ab in the latter group may be influenced by BCG vaccination widely used against tuberculosis to induce cell-mediated protection against the disease or by exposure to epitopes shared by avirulent environmental mycobacteria and M. tuberculosis. Moreover, the presence of complement-activating Ab to avirulent mycobacteria that cross-react with M. tuberculosis may be decisive for the development of localized instead of disseminated tuberculosis (6).
Complement activation culminates in the formation of the terminal complement complex (TCC). The presence of TCC containing C5b-9 with or without vitronectin (24) on the bacterial surface may explain the reported uptake of bacilli via monocyte vitronectin receptors (25). The soluble terminal complement activation product C5a is a potent chemotaxin and stimulator and may recruit activated host monocytes that can be invaded. Recently, the binding of M. tuberculosis to CR3 expressed in Chinese hamster ovary cells was reported to be predominantly nonopsonic (7). Previously, we have shown that antigen (Ag) 85C of M. bovis BCG and M. tuberculosis promotes monocyte CR3-mediated uptake of beads coated with mycobacterial products (13). Interestingly, 85C could be a ligand for the non-iC3b-binding epitope in CR3 found to bind M. tuberculosis to macrophages (29). In addition, several other ligands and receptors, unrelated to complement, are known to participate in the uptake of mycobacteria in mononuclear phagocytes (2, 12, 25, 30).
We wanted to study complement activation induced by BCG and M. tuberculosis Ag in sera from nontuberculous and tuberculous subjects. Especially, we wished to investigate classical complement activation and its relationship to the specificity of anti-M. tuberculosis Ab. Therefore, sera from healthy subjects and tuberculosis patients were exposed to mycobacteria and differences in soluble complement activation products in the sera were examined by an enzyme-linked immunosorbent assay (ELISA) specific for neoepitopes in the activation products (11, 21). Ab to mycobacteria in the populations were identified, and the levels were determined by ELISA. In addition, deposition of complement on BCG exposed to different sera and Ab was studied by flow cytometry.
(This work was presented at the Sixth European Meeting on Complement in Human Disease, Innsbruck, Austria 12 to 15 March, 1997.)
MATERIALS AND METHODS
Plasma.
Blood from healthy Norwegians (n = 5) was collected in heparinized or EDTA-containing (10 mM final concentration) Vacutainer tubes. Plasma was obtained after centrifugation, split into aliquots, and immediately frozen at −70°C.
Sera.
Normal human serum (NHS) from healthy Norwegians (n = 20) was obtained from whole blood coagulated at room temperature and immediately separated by centrifugation at 2,300 × g for 10 min and frozen at −70°C in aliquots, either individually or pooled. Factor B-depleted serum (A506F17901) was purchased from Quidel (San Diego, Calif.), and C2-deficient serum was obtained from a male patient with discoid lupus erythematosus and recurrent infections (3). Ninety-seven sera from Indian tuberculosis patients (Bombay, India) were screened for Ab activity against LAM of M. tuberculosis culture fluid by Western blotting, and 17 of these sera with either high or low levels of anti-LAM activity (Fig. 1) were selected for further study of complement activation properties. These sera were obtained from 7 female and 10 male outpatients (ages 13 to 50) from a suburban slum of Bombay. The patients all had pulmonary tuberculosis as judged by sputum examination and chest X ray. The median duration of disease was 6 weeks (range, 2 to 14 weeks), and none had a previous history of tuberculosis. In addition, another three such patient sera were used in one TCC dose-response experiment. Serum was also obtained from 11 healthy Indians. Blood samples were obtained by vein puncture and maintained at 37°C for 60 min or at 20°C overnight. Sera were then separated and dispersed in 200-μl aliquots and stored at −20°C. They were transported to the site of the study on dry ice, and there they were received in a frozen state and further stored at −20°C until usage. All the plasma and sera, except for those from one Norwegian (donor 16) and one Indian patient, were from BCG-vaccinated individuals. To minimize spontaneous complement activation, the plasma and sera were kept on ice before performing the experiments.
FIG. 1.

Western blot after SDS-PAGE of M. tuberculosis culture fluid (lanes 1 to 6) or sonicate (lane 7). Blots resulting from incubation with Indian normal serum (lane 1), two tuberculosis patient sera with low levels of anti-LAM (lanes 2 and 3), two tuberculosis patient sera with high levels of anti-LAM (lanes 4 and 5), and rabbit anti-LAM diluted 1:1,000 (lanes 6 and 7) are shown. The positions of molecular mass markers (kD, kilodaltons) are indicated at the right.
Ags.
Ags used were M. bovis BCG Copenhagen (substrain 1331), a void fraction of BCG Tokyo (substrain 172) culture fluid gel filtered on a Sephacryl S-1000 column (Pharmacia LKB Biotechnology AB, Uppsala, Sweden), M. tuberculosis sonicate and culture fluid, and highly purified LAM obtained by a novel method (12a). Soluble Ags were obtained after cultivation of bacilli on wholly synthetic Sauton medium.
Abs.
Rabbit polyclonal Ab (PAb) to the B Ag of the 85 complex of BCG and M. tuberculosis, rabbit immunoglobulin G (IgG) PAb purified by affinity chromatography using a LAM-containing solid matrix (12a), mouse monoclonal antibody (MAb) bH6 to a neoepitope on C3 activation products (11), mouse MAb to C6 (clone 9C4) (21), and mouse MAb aE11 to a neoepitope on polymerized C9 and specific for TCC (20) were produced in our laboratories. Rabbit anti-human C3c PAb was from Behring, Marburg, Germany, and mouse MAb IgG2a to Aspergillus niger glucose oxidase was from Dako Immunoglobulins, Copenhagen, Denmark. The labeled Abs were horseradish peroxidase (HRP)–sheep anti-human IgG and IgM (Dako), HRP–mouse anti-human IgG1 to -4 (Zymed, San Francisco, Calif.), HRP–donkey anti-rabbit Ig (Amersham International, Amersham, United Kingdom), HRP–sheep anti-human Ig (Amersham), mouse MAb to human C6 biotinylated in our laboratory, fluorescein isothiocyanate (FITC)–rabbit anti-human C3c (Dako), FITC–goat anti-rabbit IgG, and phycoerythrin (PE)–goat anti-mouse Ig (Southern Biotechnology Associates, Inc., Birmingham, Ala.).
Miscellaneous reagents.
Reagents used were zymosan A, bovine serum albumin (BSA), Tween 20, EDTA, and EGTA (Sigma) and gelatin (Difco, Detroit, Mich.).
Experiments.
One-tenth- to 1,000-μg/ml final concentrations of zymosan A, whole BCG bacilli (lyophilized vaccine), and M. tuberculosis sonicate or culture fluid were incubated with heparin- or EDTA-plasma or serum (75% final concentration), with or without a 10 mM final concentration of EGTA, for 0 to 120 min in Eppendorf tubes at 37°C under head-on-end rotation. The individual normal and patient sera were diluted 1/10 in the Norwegian NHS (n = 20) in order to maintain a constant complement source. Three patient sera used in initial experiments (Fig. 5) were diluted 1/10 to 1/1,000 in another NHS pool (n = 5), which also was used as control serum in experiments with C2-deficient and B-depleted sera. Controls were sera incubated similarly with phosphate-buffered saline (PBS) instead of the agents listed above, and the final volume was usually 300 μl. Lyophilized BCG bacilli (5 × 106 viable bacilli/tube; Statens Seruminstitut, Copenhagen, Denmark), zymosan, and M. tuberculosis sonicate or culture fluid were diluted in PBS to the appropriate concentrations. After incubation the tubes were immediately centrifuged in an Eppendorf centrifuge at maximum speed (16,000 × g) for 20 min, and 200 μl of the supernatant was removed and frozen at −70°C until analysis for C3bc (collective designation for C3b, iC3b, and C3c) and TCC concentrations.
FIG. 5.

Dose-response curve for TCC formation in three Indian tuberculosis patient sera (IP1, -2, or -3) with either strong (◊ and □) or weak (▵) anticarbohydrate activity, diluted 1/1,000 to 1/10 in the NHS pool (n = 5) and incubated with BCG bacilli as described in the legend for Fig. 4. The mean values for PBS or zymosan controls incubated with a 1/100 dilution of IP1, IP3, and IP2 in NHS were 16, 15, and 16 or 324, 286, and 290 AU of TCC/ml, respectively. The data points represent means of triplicate experiments.
In one experiment dilutions of affinity-purified rabbit anti-LAM IgG or normal rabbit IgG were incubated with BCG bacilli (final concentration, 250 μg/ml) or PBS in NHS as described above with or without 10 mM EGTA or EDTA and the TCC concentration in the supernatant was measured.
Analyses. (i) SDS-PAGE with immunoblotting.
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was performed with the Pharmacia system for horizontal electrophoresis in a Multifor II electrophoresis unit, model 2117 (Pharmacia), by using precast polyacrylamide gels and ExcelGel SDS gradient 8-18 as described previously (33). Briefly, 10 μl of M. tuberculosis culture fluid or sonicate (13 μg) was run in SDS-PAGE gel, transferred to 0.2-μm-thick nitrocellulose paper (Schleicher & Schuell, Dassel, Germany), incubated with sera (1:200) from Indian tuberculosis patients or healthy Indians overnight at room temperature, and developed with HRP–anti-human Ig. Controls were rabbit anti-LAM Abs.
(ii) Complement activation assays.
Complement activation was assessed in two ELISAs specific for neoepitopes exposed on activation products but not expressed in native complement factors. The first was specific for C3 activation products, thus evaluating the initial part of the complement cascade (11). In brief, polystyrene plates were coated with a mouse MAb (bH6) as the catching Ab, being specific for a C3 neoepitope (C3bc) expressed on C3b, iC3b, and C3c, but not on native C3. The Ag in the second layer was plasma or serum diluted 1/300 in PBS containing 10 mM EDTA and 0.2% Tween 20. Rabbit PAb anti-human C3c was used in the third layer. Finally, a peroxidase-conjugated donkey anti-rabbit Ig was added. The substrate was 2,2′-azino-di-(3-ethylbenzthiazoline sulfonic acid) (ABTS). The plates were read on a Dynatech MR 7000 (Alexandria, Va.) and referred to a standard of zymosan-activated serum defined to contain 1,000 arbitrary units (AU)/ml. The second similar assay detected the SC5b-9 TCC (21). In brief, polystyrene plates were coated with a mouse MAb (aE11) specific for a neoepitope expressed on activated C9 (20). A biotinylated mouse MAb to human C6 was used in the third layer, and finally peroxidase-labeled streptavidin was added. The rest of the assay was performed as described for the C3 activation assay.
(iii) ELISA for Ab identification.
Abs to LAM were determined by ELISA. Polystyrene plates were coated overnight with purified LAM (1 μg/well), blocked with 5 mg of BSA per ml and 1% gelatin, and washed four times. For the second layer the test sera, diluted 1/50 in 0.2% BSA–0.05% gelatin–0.2% Tween 20, were incubated for 1 h at 37°C. The plates were washed four times and incubated with HRP-labeled rabbit anti-human Ig, anti-IgG or anti-IgM alone, or the HRP-labeled mouse MAb to the IgG subclasses, which had been diluted 1/1,000 and kept for 45 min at 37°C. The plates were developed with H2O2 in ABTS and analyzed as described above. Negative controls were wells without test sera, but otherwise treated as described above.
Abs to Ag 85B or to the BCG Tokyo culture fluid void fraction were measured in an ELISA similar to that for anti-LAM by using plates coated at 0.5 μg/well with highly purified Ag 85B (22) or an appropriate dilution of the BCG Tokyo culture fluid void fraction, respectively.
The binding of rabbit anti-LAM Abs to the M. tuberculosis sonicate (coat: 0.1 μg/well) was tested in a manner similar to that described above (ELISA) but with HRP–anti-rabbit Ig in the second Ab layer.
(iv) Flow cytometry.
Flow cytometry analysis of M. bovis BCG was performed with a FACScan model 440 (Becton Dickinson Immunocytometry Systems, Mountain View, Calif.) fluorescence-activated cell sorter. Lyophilized BCG Copenhagen substrain 1331 was suspended in PBS to 1 mg/ml, and 75 μl of the suspension was mixed with 225 μl of NHS diluted 1:25 in PBS and incubated for 1 h at 37°C under head-on-end rotation. The NHS dilution of 1:25 was chosen because it gave the most C3c binding in titration experiments. All incubations were performed under these conditions. Bacilli were pelleted by centrifugation and washed twice in PBS to remove unbound material. The bacilli were then incubated with FITC-labeled rabbit anti-C3c Ig diluted 1:500 or with mouse MAb aE11 (anti-TCC) diluted 1:5,000. Bacilli stained with aE11 were washed and incubated with PE-labeled, human-absorbed, goat anti-mouse Ig diluted 1:400. The subclass negative control to aE11 was mouse MAb IgG2a to A. niger glucose oxidase. EGTA or EDTA was used at a 10 mM final concentration in diluted sera. Heat-inactivated serum was made by incubation at 56°C for 30 min. In experiments with factor B-depleted NHS, bacilli were preincubated with affinity-purified rabbit anti-LAM diluted 1:100 or protein A-purified normal rabbit Ig diluted 1:100. The final volume of all incubations was 300 μl in Eppendorf tubes. All sera and primary Ab solutions were sterile filtered before usage to avoid particulate matter other than bacilli.
Statistics.
Nonparametric statistics were used throughout: the Mann-Whitney U test (M-W-U) and the Wilcoxon rank sign test were used for examination of the difference in values of one parameter between two independent and two dependent groups, respectively, and the Spearman rank correlation coefficient (ρ) was used for correlations between values of two parameters in the same population. The statistical analysis program was GB-STAT (Dynamics Microsystems, Silver Spring, Md.), and P values <0.05 were considered significant.
RESULTS
Complement activation in plasma incubated with BCG bacilli or M. tuberculosis sonicate or culture fluid.
Whole intact BCG bacilli or M. tuberculosis sonicate or culture fluid was incubated with normal human plasma in EDTA or heparin for 1 h at 37°C and subsequently assayed for C3bc and TCC concentrations (Fig. 2a and b) in the supernatant. In heparin-plasma there was a dose-dependent increase in concentration of the activation products for all three agents (Fig. 2a and b) and for zymosan controls, which reached maximums of 566 AU of C3bc/ml and 749 AU of TCC/ml at a zymosan concentration of 1 mg/ml. In EDTA-plasma there was no detectable TCC, but the formation of 51 and 79 AU of C3bc/ml in the presence of 1 mg of whole BCG or BCG sonicate per ml, respectively, was observed.
FIG. 2.

Dose-response curves for C3bc (a) and TCC (b) formation induced by whole BCG bacilli or M. tuberculosis sonicate (Mtson) or culture fluid (Mtcf). Dilutions of each agent in PBS were incubated with 75% pooled normal human plasma in heparin (HP) or EDTA (EP) (n = 5) for 1 h at 37°C under agitation and centrifuged, and C3bc and TCC levels were measured in the supernatants by ELISA. For experiments with BCG the concentrations 1, 10, and 100 μg/ml were instead 1.5, 15.0, and 150.0 μg of BCG/ml, respectively. The mean values for PBS controls in heparin- or EDTA-plasma were 37.6 or 10.1 AU of C3bc/ml and 6.8 or 1.4 AU of TCC/ml, respectively. Each data point represents the mean of triplicate experiments.
When 1 mg of BCG bacilli or M. tuberculosis sonicate or culture fluid per ml was incubated in heparin-plasma at 37°C, there was a linear time-dependent increase in the formation of C3bc (Fig. 3a) and TCC (Fig. 3b). Heparin-plasma exposed to 1 mg of zymosan per ml gave a 10-fold-greater increase in TCC values, which reached a plateau after 1 h. The TCC values in the EDTA-plasmas (Fig. 3b) and in heparin-plasma incubated with PBS (data not shown) were around 1 AU/ml.
FIG. 3.

BCG bacilli, M. tuberculosis sonicate (Mtson) or culture fluid (Mtcf), or zymosan (Zym) at 1 mg/ml was added to heparin- or EDTA-plasma pools (n = 5) and incubated for 0 to 120 min at 37°C. The C3bc (a) and TCC (b) levels were measured in the supernatants. Note that TCC values for zymosan in heparin-plasma must be multiplied by 10. The mean TCC values for zymosan, BCG, and M. tuberculosis sonicate or culture fluid incubated in EDTA-plasma (EP) were around 1 AU/ml. The mean values for PBS incubated in heparin (HP)- or EDTA-plasma for 2 h were 67 or 20 AU of C3bc/ml and 5.6 or <0.5 AU of TCC/ml, respectively. The results are means of two experiments in triplicate.
Complement activation in normal, C2-deficient, or factor B-depleted serum incubated with BCG bacilli.
To examine classical and alternative complement activation, BCG bacilli or zymosan was incubated for 1 h in normal, C2-deficient, or factor B-depleted serum followed by an assay for C3bc and TCC formation. When more than 100 μg of BCG bacilli per ml was added, there was a sharp increase in C3bc and TCC concentrations in all the sera in the following order: NHS > C2-deficient serum > factor B-depleted serum (Fig. 4). The data suggest that as much as 67% of the C3bc formation induced by BCG bacilli in serum from healthy individuals may be mediated by classical complement activation. However, the sera may vary with respect to the concentration of antibodies, and complement components other than C2 and B and are therefore not readily comparable. By contrast, virtually all complement activation induced by zymosan occurred via the alternative pathway (data not shown).
FIG. 4.

Dose-response curve for C3bc and TCC formation in NHS (n = 5), C2-deficient serum (C2 defS), or factor B-depleted serum (B depS) incubated for 1 h at 37°C with BCG bacilli. Note that C3bc values must be multiplied by 10. The values for PBS controls in NHS, in C2-deficient serum, and in B-depleted serum were 154, 132, and 245 AU of C3bc/ml and 16.0, 8.9, and 9.0 AU of TCC/ml, respectively. The data represent means of two experiments in triplicate.
Western blotting.
Since the surfaces of mycobacteria are largely covered by carbohydrates and since anticarbohydrate specificities typically appear as smears in Western blotting after SDS-PAGE of mycobacterial Ag, this technique was used to screen sera from tuberculosis patients for the presence of Abs to mycobacterial surface Ags. Anti-LAM Ab activity is depicted in Western blotting as a smear between 30 and 40 kDa. Ninety-seven sera from Indian tuberculosis patients and 11 sera from healthy Indians were studied for Ab activity against LAM in M. tuberculosis culture fluid. Seventeen patient sera with high or low levels of anti-LAM activity in Western blotting were selected for further study of complement activation properties. Figure 1 shows representative examples of serum from one healthy Indian and patient sera with high or low levels of anti-LAM activity, as well as a rabbit PAb against LAM. The patient sera also had considerable Ab activity against protein Ags in the M. tuberculosis culture fluid such as the Ag 85 complex at 30.0 to 32.5 kDa (32).
TCC formation in sera from healthy individuals and tuberculosis patients exposed to BCG bacilli.
In one initial experiment sera from two Indian patients with strong smears and one with weaker smears against M. tuberculosis culture fluid as determined by Western blotting (data not shown) were diluted in the Norwegian NHS pool and incubated with BCG bacilli for 1 h at 37°C. In contrast with what was found for the latter serum, 1/10 dilutions of the former two sera induced more than a twofold increase in TCC values compared with higher dilutions (Fig. 5). All dilutions in NHS of the serum with the weaker anticarbohydrate smear produced TCC levels similar to those obtained with BCG and NHS alone.
When complement activation in different serum populations was examined after 1/10 dilutions in the NHS pool and exposure to BCG, the TCC values in Indian patient sera were found to be significantly higher than those in sera from healthy Indians or Norwegians (M-W-U; P = 0.0407 and 0.0024, respectively) (Fig. 6). Normal Norwegian sera did not differ from normal Indian sera with respect to TCC formation (M-W-U; P = 0.33). This suggests the presence of complement activation properties in patient sera and lower activity in normal sera.
FIG. 6.

Total (toTCC) or classical (clTCC)-pathway-mediated TCC formation induced by BCG bacilli (250 μg/ml) incubated for 1 h at 37°C with sera from healthy Norwegians (NN) (n = 20), healthy Indians (IN) (n = 11), or Indian tuberculosis patients (IP) (n = 17) that were diluted 1/10 in the Norwegian NHS pool (n = 20) (total serum concentration, 75%). Experiments were performed in the presence or absence of 10 mM EGTA. After subtracting values for PBS controls from each serum, levels of TCC formed by classical complement activation were calculated by subtracting TCC values in EGTA sera from TCC values in the respective non-EGTA sera. The results are given as mean values of triplicate experiments. The range of PBS values in experiments with EGTA for healthy Norwegians, healthy Indians, and patients were 3 to 29, 4 to 16, and 10 to 31 AU of TCC/ml, respectively; corresponding ranges without EGTA were 15 to 23, 19 to 35, and 19 to 58 AU of TCC/ml, respectively.
To inhibit classical-pathway complement activation, 10 mM EGTA was added to the sera in a parallel setup before incubation with BCG bacilli. The differences between total TCC values and TCC values measured in the presence of EGTA should therefore represent TCC formed by classical complement activation. Although these values were significantly lower than the respective total TCC values in each group (Wilcoxon sign rank test; P < 0.0005), TCC formed by classical complement activation represents a major proportion (68 to 87%) of the total TCC formation in these populations (Fig. 6). There was a higher degree of classical-pathway complement activation in sera from healthy Indians than in sera from healthy Norwegians (M-W-U; P = 0.0157), which is best explained by a higher level of exposure to mycobacterial Ag in India. However, no statistically significant difference in classical complement activation between sera from healthy Indian or Norwegian individuals and Indian patient sera (M-W-U; P = 0.57 and 0.16, respectively) was found. Larger and more representative populations are needed for demonstration of these putative differences.
Identification of Ab specificities in the Indian sera.
Different ELISAs were constructed to identify Abs in the patient sera probably responsible for some of the complement activation observed in Fig. 5 and 6. One ELISA employed wells coated with the void fraction of gel-filtered BCG culture fluid, containing cell wall-related Ags. It showed higher levels of Abs to these molecules in Indian patients than in healthy Indian subjects (M-W-U; P = 0.0323) (Fig. 7). Then, an anti-LAM ELISA with either anti-Ig, anti-IgG or anti-IgM, or, subsequently, anti-IgG subclasses as the detection Abs was developed. It revealed higher levels of total IgG, but not higher levels of IgM or anti-LAM, in Indian patients than in healthy individuals (M-W-U; P ≤ 0.0323 or 0.45, respectively) (Fig. 7). Anti-LAM IgG in the sera was mostly IgG1 and IgG2, with little or no activity in the IgG3 and IgG4 subclasses. In addition, the sera were examined in an anti-M. tuberculosis Ag 85B ELISA, which also showed higher Ab levels in Indian patients than in normal sera (M-W-U; P = 0.0118) (Fig. 7).
FIG. 7.

ELISAs were performed for the quantification of Ab to LAM, Ag 85B, and the void fraction of gel-filtered BCG Tokyo culture fluid in healthy Norwegian (NN) (n = 20), healthy Indian (IN) (n = 11), and tuberculosis patient (IP) (n = 17) sera. The plates were coated with 1.0 or 0.5 μg of LAM or 85B, respectively, or a 1/10 dilution of BCG Tokyo void fraction; they were then incubated with a 1/50 dilution of the sera in PBS and developed with HRP-labeled rabbit anti-human IgG plus IgM and H2O2 in ABTS. The negative-control mean values for LAM, Ag 85B and BCG Tokyo void fraction ELISAs run without sera were 13, 10, and 48 optical density (OD) units, respectively.
Anti-LAM IgG levels correlated positively with TCC levels produced by classical complement activation (ρ = 0.4919, P = 0.0079) (Fig. 8), whereas anti-LAM IgM levels did not (ρ = −0.02, P = 0.92). Anti-LAM IgG1 and IgG2 levels correlated positively with classical complement activation in the Indian sera, but only significantly for anti-LAM IgG2 (ρ = 0.3049, P = 0.11 and ρ = 0.4806, P = 0.0096, respectively). By contrast, a negative, but not statistically significant, correlation was found between anti-85B values and classical TCC formation (ρ = −0.3717, P = 0.0515). This indicates that Ab to a major soluble secreted mycobacterial Ag is not involved in complement activation. There was no correlation between the levels of anti-LAM and anti-85B Ab in Indians (ρ = 0.1585, P = 0.41). For anti-85B (M-W-U; P = 0.0003), but not anti-LAM, Abs (M-W-U; P = 0.1857), there was a difference between sera from healthy Norwegians and Indians.
FIG. 8.

Nonparametric correlation plot (ρ = 0.4942, P = 0.0075) for IgG Ab to LAM versus TCC formation via classical complement activation in the various populations tested.
Complement activation by rabbit anti-LAM Ab and BCG.
Since (i) there were higher levels of Ab to LAM in patient than in healthy sera, (ii) the Ab levels correlated with classical complement activation induced by BCG, and (iii) rabbit Ab to LAM bound M. tuberculosis sonicate in Western blotting (Fig. 1) and dose dependently in an ELISA (ρ = 1; data not shown), we tested whether rabbit anti-LAM incubated with BCG and serum activate complement. First, we found that TCC was formed in the supernatant of NHS, but not in the presence of EGTA or when PBS was substituted for BCG (126, 41, or 54 AU/ml, respectively). Second, flow cytometry with FITC–anti-C3c was performed on bacilli incubated with either rabbit anti-LAM or normal rabbit IgG and then with B-depleted serum. The first experiment showed positive staining, and the next showed reduced staining (Fig. 9). The addition of EGTA to B-depleted serum in the first experiment showed negative staining (Fig. 9). There was positive staining with anti-TCC of BCG treated with anti-LAM and B-depleted serum (data not shown). When BCG cells were incubated directly with normal serum there was positive staining with FITC–anti-C3c and PE–anti-mouse Ig after incubation with anti-TCC (MAb aE11), which was reduced significantly by the addition of EGTA and EDTA (Fig. 10 a and b). Titration experiments revealed the most positive staining with serum diluted 1:25 (Fig. 9 and 10) and little staining with undiluted serum (data not shown). Other controls were heat-inactivated serum and an irrelevant MAb control to anti-TCC (aE11), which gave little or no staining. Since EGTA abolished most of the anti-complement staining, the flow cytometry experiments illustrated that most of the complement deposition on the bacilli was caused by classical complement activation.
FIG. 9.
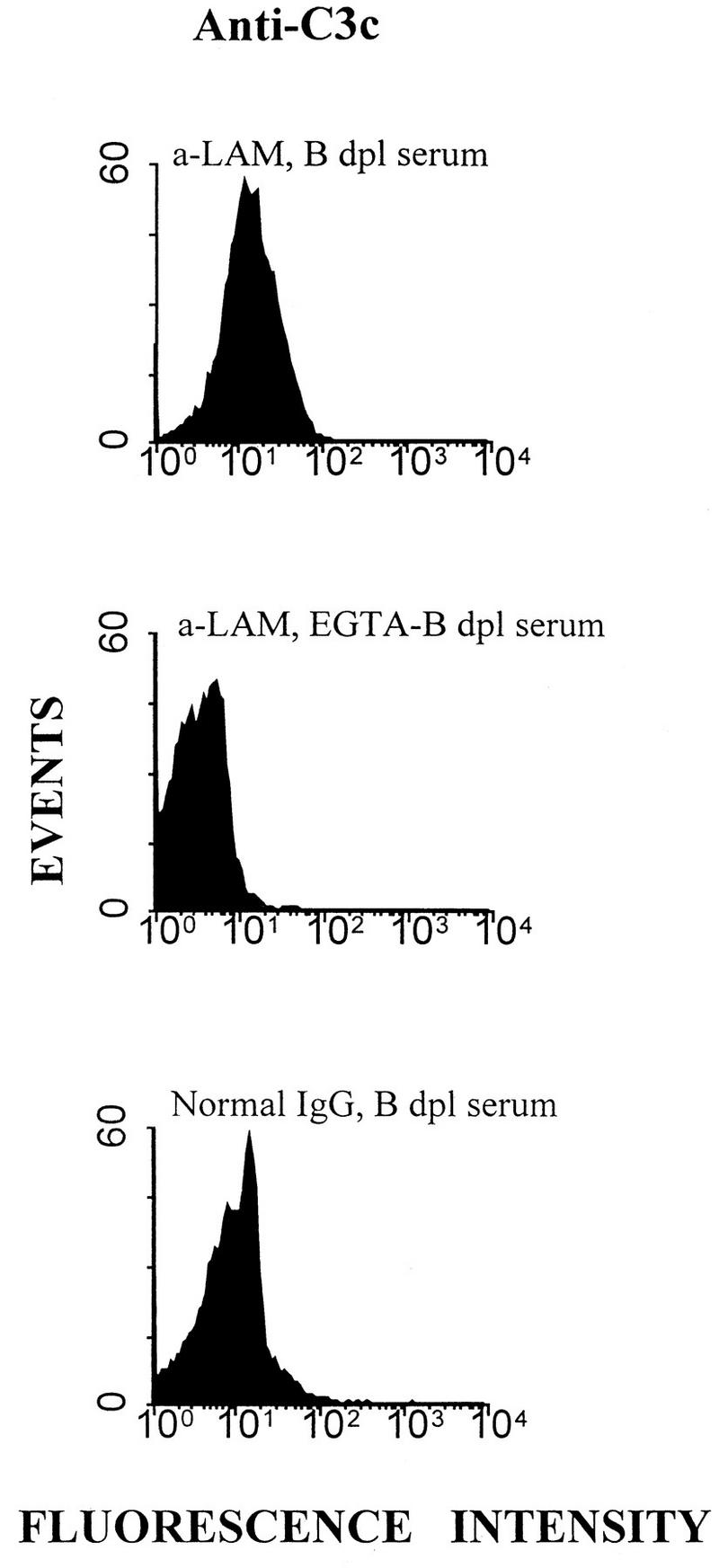
Flow cytometry for the assay of C3 deposition on BCG. Bacilli were incubated with affinity-purified rabbit anti-LAM IgG or normal rabbit Ig, washed twice, and exposed to a dilution of B-depleted (dpl) serum with or without EGTA for 1 h at 37°C and then washed again. The conjugate was FITC-labeled anti-C3c. The values for median channel fluorescence intensity in the histograms from top to bottom were 14.33, 3.28, and 9.31.
FIG. 10.
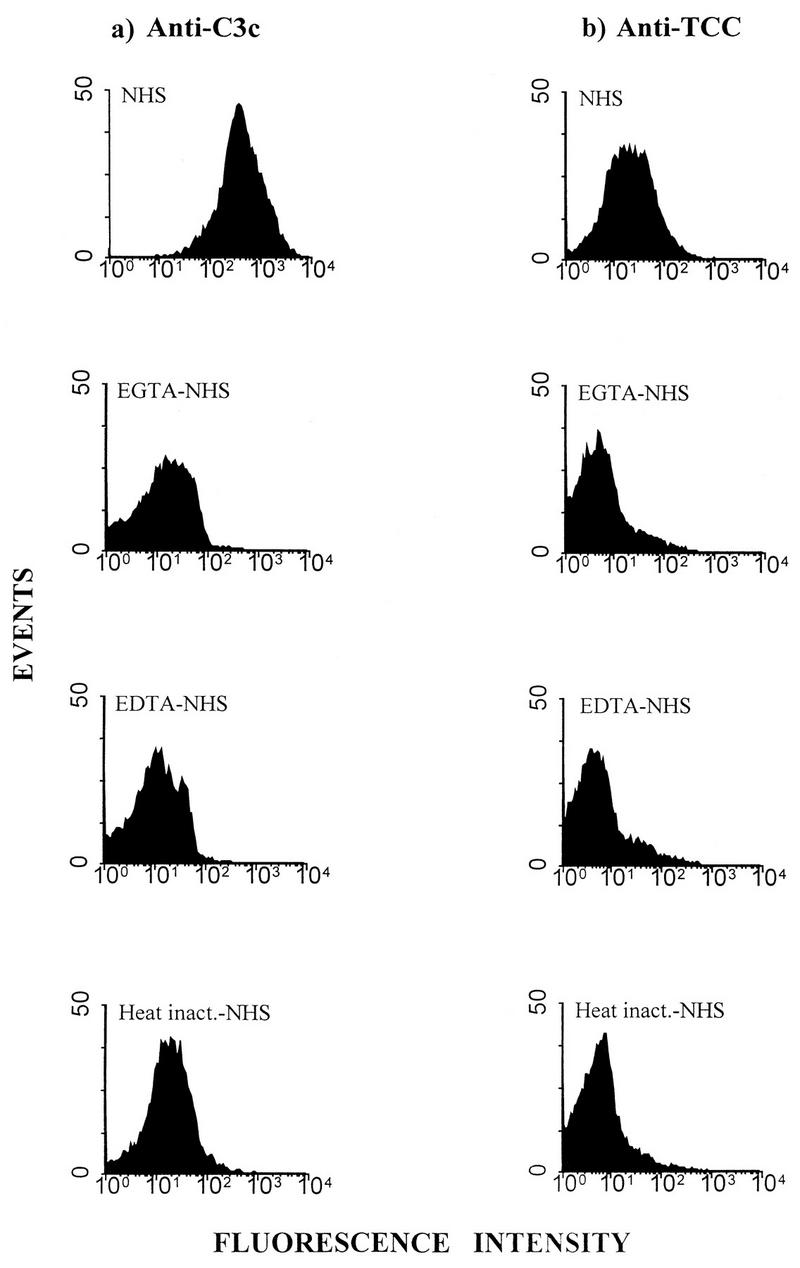
Flow cytometry for assay of C3 (a) and TCC (b) deposition on BCG bacilli incubated directly with a dilution of NHS with or without EGTA or EDTA or with heat-inactivated serum for 1 h at 37°C. The bacilli were washed and stained with either FITC–anti-C3c, or PE–anti-mouse Ig after incubation with anti-TCC (aE11). The values for median channel fluorescence intensity in the histograms from top to bottom in panel a were 433.23, 12.41, 10.70, and 18.60; from top to bottom in panel b they were 20.91, 4.22, 4.26, and 5.23. The isotype control for MAb aE11, normal mouse IgG2a, stained negatively, with a median channel fluorescence of 3.43 (data not shown).
DISCUSSION
Our results show that M. tuberculosis and BCG are able to activate both the initial and terminal complement pathways. Previously, only the initial C3-fixing properties of mycobacteria had been demonstrated (27, 28). The experiments (Fig. 4) with BCG bacilli and serum depleted of factor B to abrogate alternative-pathway complement activation suggest that a major part of the complement activation induced by BCG in normal serum occurs via the classical pathway. This was supported by the dose-response experiments with patient sera containing high levels of anticarbohydrate activity and fluid phase TCC measurement and by data obtained by EGTA chelation of Ca2+ but not Mg2+, a condition in which the classical but not the alternative pathway is inhibited. Although the classical complement activation induced by BCG in the patient population was not significantly greater than such activation in the healthy population, levels of Ab reacting with BCG culture fluid as well as with purified LAM were higher in the patient population. Sera from larger populations would probably have shown a significant difference in classical complement activation after exposure to BCG bacilli.
Since M. tuberculosis, in contrast with BCG, is too hazardous to handle openly in the laboratory, live nondenatured BCG bacilli were used in this study. We propose that data obtained with BCG are applicable to M. tuberculosis because the structures of the surfaces of the bacilli are very similar and because LAM on both BCG and M. tuberculosis has been characterized in detail, thereby showing that the two main types, ara-LAM and man-LAM, are present (31).
Since IgG2 usually activates complement less efficiently than IgG1 and IgG3, it was somewhat surprising to find that anti-LAM IgG2 levels, but not IgG1 levels, correlated significantly with classical complement activation in Indian sera exposed to BCG. However, the finding is compatible with the high concentration of LAM on BCG and M. tuberculosis (30), the high density of Ag epitopes required for complement activation by IgG2 Ab (19), and the preferential elicitation of the IgG2 subclass by polysaccharide Ag (23). Recently, IgG2 was also shown to be the predominant subclass of anti-LAM Ab in human immunodeficiency virus-negative Zambian tuberculosis patients (8). The correlation coefficient (ρ = 0.4919; Fig. 8) may suggest that the contribution of anti-LAM IgG to the observed BCG-induced classical complement activation in the Indian sera was 50%. Other antimycobacteria specificities, especially to carbohydrates with lower molecular weights than LAM, may also be of importance. Since intracellular pathogens make use of other receptors in addition to CR3 to enter host cells (4), Fcγ receptors may also participate in monocyte uptake of M. tuberculosis. However, Fcγ receptors are shown to be of little importance in the phagocytosis of M. leprae (26) and therefore probably play a minor role in the phagocytosis of M. tuberculosis as well.
In the present study we used assays based on complement activation-dependent epitopes in C3 and TCC to determine the extent of complement activation induced by BCG in different sera. These are, however, soluble complement activation products and an indirect measurement of what goes on on the surface of the bacilli. Except for Schlesinger and Horwitz’s finding of phenolic glycolipid 1 as the main C3-fixing molecule on M. leprae (27), the question about complement deposition on mycobacteria has not been addressed thoroughly. Therefore, we also examined the binding of complement to the bacilli by flow cytometry. In the present paper C3 and TCC deposition on mycobacteria is, to our knowledge, visualized for the first time. The experiments demonstrated that most of the complement on the bacilli was deposited due to classical complement activation, which confirms our findings in the fluid phase after incubation of BCG with serum. The high level of dilution of serum used for fluorescence-activated cell sorter analysis of BCG probably reduces the concentration of alternative-pathway factors more than classical-pathway components, giving little potential for alternative-pathway complement activation. However, with near-undiluted normal serum (75%) there was almost no anti-C3c staining of BCG, which points to the little importance of alternative-pathway complement activation for the deposition of C3 on the bacilli. If the concentration of complement proteins in interstitial fluid is similar to its 10 to 20% concentration of serum proteins and if one takes into account the preference of mycobacteria for tissue host macrophages, the study data would explain the deposition of C3 on BCG incubated only in diluted serum. However, the reason behind this phenomenon remains obscure.
A third complement activation pathway that is initiated by the binding of mannan-binding lectin (MBL) (15) to carbohydrates has been described (14). Mannan is a major constituent of the mycobacterial surface, and the lectin pathway may be important for complement activation induced by mycobacteria. In addition, MBL can bind to the C1q receptor on leukocytes and function as an opsonin for enhanced phagocytosis of mycobacteria (16, 18). We examined the levels of MBL in the Indians by a sandwich ELISA and compared MBL levels in subjects presumably heterozygous for deletions in the MBL gene that have been described previously (10). We found significantly higher levels of MBL in Indian tuberculosis patients than in healthy Indians (data not shown), which may suggest induction of MBL expression by tuberculosis in subjects heterozygous for the deletion in the MBL gene. However, there was no correlation between MBL levels and BCG-induced TCC formation, suggesting that the lectin pathway was not much involved in complement activation by the mycobacteria. The importance of MBL for complement activation or phagocytosis of mycobacteria needs to be investigated further.
Gram-positive bacteria and acid-fast rods like M. tuberculosis are not lysed by complement. Obviously, the bacilli use the complement system as a means to enter host leukocytes. Once inside the cellular phagosomes, mycobacteria may escape detection by the immune system for many years. It is unknown whether C3 fixation by M. tuberculosis and monocyte complement receptor-mediated phagocytosis is detrimental to the host or not. The binding of complement-activating Ab and possibly MBL to the bacilli would enhance C3 fixation on the bacilli and their uptake in monocytes. The suggested limited dissemination in childhood tuberculosis by Abs to LAM and other mycobacterial Ags could be interpreted as being due to increased monocyte phagocytosis of tubercle bacilli. One may speculate whether higher levels of complement-activating Ab to mycobacteria after exposure to avirulent mycobacteria could be one explanation of why BCG vaccination is less effective against tuberculosis with increasing age (5). In contrast to BCG, avirulent mycobacteria could possibly induce Ab production alone and increased phagocytosis of M. tuberculosis bacilli on later encounter without an adequate cell-mediated immune response. In fact, one theory of why BCG vaccination in children is less effective against M. tuberculosis (but not M. leprae) (9) in Africa (Malawi) than in Europe (England) is that the population in southern countries is more prone to exposure to avirulent mycobacteria. This exposure in itself, by inducing a humoral response with high frequencies of complement-activating Ab to mycobacteria and possible augmented phagocytosis by monocytes, could also be one of several reasons why there is more tuberculosis in Africa than in Europe.
ACKNOWLEDGMENTS
We thank Kamal P. Harver and Nergis Mistry at The Foundation for Medical Research, Bombay, India, for providing the Indian sera as part of the NORAD IND040 Institutional Cooperation Agreement. The excellent technical assistance by Ingunn Gihle, Gunni Ulvund, and Irene Hatlehol Andreassen is highly appreciated.
This work was supported by grants from the Anders Jahre Fund for the Promotion of Science, the Research Council of Norway (project no. 105026/710), and the European Community (project no. TS*-CT94-0001).
REFERENCES
- 1.Bardana E J, Jr, McClatchy J K, Farr R S, Minden P. Universal occurrence of antibodies to tubercle bacilli in sera from non-tuberculous and tuberculous individuals. Clin Exp Immunol. 1973;13:65–77. [PMC free article] [PubMed] [Google Scholar]
- 2.Bermundez L E, Young L S, Enkel H. Interaction of Mycobacterium avium complex with human macrophages: roles of membrane receptors and serum proteins. Infect Immun. 1991;59:1697–1702. doi: 10.1128/iai.59.5.1697-1702.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Braathen L R, Bratlie A, Teisberg P. HLA genotypes in a family with a case of homozygous C2 deficiency and discoid lupus erythematosus. Acta Dermato-Venereol. 1986;66:419–422. [PubMed] [Google Scholar]
- 4.Bullock W E, Wright S D. Role of the adherence promoting receptors, CR3, LFA-1, and p150,95, in binding of Histoplasma capsulatum by human macrophages. J Exp Med. 1987;165:195–210. doi: 10.1084/jem.165.1.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Christensen O, Bjartveit K, Dahlstrom G. Tuberculosis situation in the Scandinavian countries. Scand J Respir Dis Suppl. 1978;102:19–40. [PubMed] [Google Scholar]
- 6.Costello A M, Kumar A, Narayan V, Akbar M S, Ahmed S, Abou-Zeid C, Rook G A, Stanford J, Moreno C. Does antibody to mycobacterial antigens, including lipoarabinomannan, limit dissemination in childhood tuberculosis? Trans R Soc Trop Med Hyg. 1992;86:686–692. doi: 10.1016/0035-9203(92)90192-f. [DOI] [PubMed] [Google Scholar]
- 7.Cywes C, Godenir N L, Hoppe H C, Scholle R R, Steyn L M, Kirsch R E, Ehlers M R W. Nonopsonic binding of Mycobacterium tuberculosis to human complement receptor type 3 expressed in Chinese hamster ovary cells. Infect Immun. 1996;64:5373–5383. doi: 10.1128/iai.64.12.5373-5383.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Da Costa C T, Khaolkar-Young S, Elliott A M, Wasunna K M, McAdam K P. Immunoglobulin G subclass responses to mycobacterial lipoarabinomannan in HIV-infected and non-infected patients with tuberculosis. Clin Exp Immunol. 1993;91:25–29. doi: 10.1111/j.1365-2249.1993.tb03348.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fine P E the Karonga Prevention Trial Group. Randomized controlled trial of single BCG, repeated BCG, or combined BCG plus killed Mycobacterium leprae vaccine, for prevention of leprosy and tuberculosis in Malawi. Lancet. 1996;348:17–24. [PubMed] [Google Scholar]
- 10.Garred P, Madsen H O, Hofmann B, Svejgaard A. Increased frequency of homozygosity of abnormal mannan-binding-protein-gene alleles in patients with suspected immunodeficiency. Lancet. 1995;346:941–943. doi: 10.1016/s0140-6736(95)91559-1. [DOI] [PubMed] [Google Scholar]
- 11.Garred P, Mollnes T E, Lea T. Quantification in enzyme-linked immunosorbent assay of a C3 neoepitope expressed on activated human complement factor C3. Scand J Immunol. 1988;27:329–335. doi: 10.1111/j.1365-3083.1988.tb02354.x. [DOI] [PubMed] [Google Scholar]
- 12.Gaynor C D, McCormack F X, Voelker D R, McGowan S E, Schlesinger L S. Pulmonary surfactant protein A mediates enhanced phagocytosis of Mycobacterium tuberculosis by a direct interaction with human macrophages. J Immunol. 1995;155:5343–5351. [PubMed] [Google Scholar]
- 12a.Hamasur, B., and S. B. Svenson. Unpublished data.
- 13.Hetland G, Wiker H G. Antigen 85C on Mycobacterium bovis, BCG and M. tuberculosis promotes monocyte-CR3-mediated uptake of microbeads coated with mycobacterial products. Immunology. 1994;82:445–449. [PMC free article] [PubMed] [Google Scholar]
- 14.Ikeda K, Sannoh T, Kawasaki N, Kawasaki T, Yamashina I. Serum lectin with known structure activates complement through the classical pathway. J Biol Chem. 1987;262:7451–7454. [PubMed] [Google Scholar]
- 15.Kawasaki T, Etoh R, Yamashina I. Isolation and characterization of a mannan-binding protein in association with a novel C1s-like serine protease. Biochem Biophys Res Commun. 1978;81:1018–1024. doi: 10.1016/0006-291x(78)91452-3. [DOI] [PubMed] [Google Scholar]
- 16.Kuhlman M, Joiner K, Ezekowitz R A B. The human mannose-binding protein functions as an opsonin. J Exp Med. 1989;169:1733–1745. doi: 10.1084/jem.169.5.1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Launois P, Niang M, Dieye A, Sarthou J L, Rivier F, Millan J. Human phagocyte respiratory burst by Mycobacterium bovis BCG and M. leprae: functional activation by BCG is mediated by complement and its receptors on monocytes. Int J Lepr Mycobacterial Dis. 1992;60:225–233. [PubMed] [Google Scholar]
- 18.Malhotra R, Thiel S, Reid K B M, Sim R B. Human leukocyte C1q receptor binds other soluble proteins with collagen domains. J Exp Med. 1990;172:955–959. doi: 10.1084/jem.172.3.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Michaelsen T E, Garred P, Aase A. Human IgG subclass pattern of inducing complement-mediated cytolysis depends on antigen concentration and to a lesser extent on epitope patchiness, antibody affinity and complement concentration. Eur J Immunol. 1991;21:11–16. doi: 10.1002/eji.1830210103. [DOI] [PubMed] [Google Scholar]
- 20.Mollnes T E, Lea T, Frøland S S, Harboe M. Quantification of the terminal complement complex in human plasma by an enzyme-linked immunosorbent assay based on monoclonal antibodies against a neoantigen of the complex. Scand J Immunol. 1985;22:197–202. doi: 10.1111/j.1365-3083.1985.tb01871.x. [DOI] [PubMed] [Google Scholar]
- 21.Mollnes T E, Redl H, Høgåsen K, Bengtsson A, Garred P, Speilberg L, Lea T, Oppermann M, Götze O, Schlag G. Complement activation in septic baboons detected by neoepitope-specific assays for C3b/iC3b/C3c, C5a and the terminal C5b-9 complement complex (TCC) Clin Exp Immunol. 1993;91:295–300. doi: 10.1111/j.1365-2249.1993.tb05898.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nagai S, Wiker H G, Harboe M, Kinimoto M. Isolation and partial characterization of major protein antigens in the culture fluid of Mycobacterium tuberculosis. Infect Immunol. 1991;59:372–382. doi: 10.1128/iai.59.1.372-382.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Perlmutter R M, Hansburg D, Briles D E, Nicoletti R A, Davie J M. Subclass restriction of murine anti-carbohydrate antibodies. J Immunol. 1978;121:566–572. [PubMed] [Google Scholar]
- 24.Podack E R, Kolb W P, Müller-Eberhard H J. The C5b-9 complex: formation, isolation and inhibition of its activity by lipoprotein and the S-protein of human serum. J Immunol. 1978;120:1841–1848. [PubMed] [Google Scholar]
- 25.Rao S P, Ogata K, Catanzaro A. Mycobacterium avium-M. intracellulare binds to the integrin receptor αVβ3 on human monocytes and monocyte-derived macrophages. Infect Immun. 1993;61:663–670. doi: 10.1128/iai.61.2.663-670.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schlesinger L S, Bellinger-Kawahara C G, Payne N R, Horwitz M A. Phagocytosis of Mycobacterium tuberculosis is mediated by human monocyte complement receptors and complement component C3. J Immunol. 1990;144:2771–2780. [PubMed] [Google Scholar]
- 27.Schlesinger L S, Horwitz M A. Phenolic glycolipid-1 of Mycobacterium leprae binds complement component C3 in serum and mediates phagocytosis by human monocytes. J Exp Med. 1991;174:1031–1038. doi: 10.1084/jem.174.5.1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schlesinger L S, Horwitz M A. A role for natural antibody in the pathogenesis of leprosy: antibody in nonimmune serum mediates C3 fixation to the Mycobacterium leprae surface and hence phagocytosis by human mononuclear phagocytes. Infect Immun. 1994;62:280–289. doi: 10.1128/iai.62.1.280-289.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stokes R W, Haidl I D, Jefferies W A, Speert D P. Mycobacteria-macrophage interactions: macrophage phenotype determines the nonopsonic binding of Mycobacterium tuberculosis to murine macrophages. J Immunol. 1993;151:7067–7076. [PubMed] [Google Scholar]
- 30.Stokes R W, Speert D P. Lipoarabinomannan inhibits nonopsonic binding of Mycobacterium tuberculosis to murine macrophages. J Immunol. 1995;155:1361–1369. [PubMed] [Google Scholar]
- 31.Venisse A, Berjeaud J-M, Chaurand P, Gilleron M, Puzo G. Structural features of lipoarabinomannan from Mycobacterium bovis BCG. J Biol Chem. 1993;268:12401–12411. [PubMed] [Google Scholar]
- 32.Wiker H G, Nagai S, Harboe M, Lundqvist L. A family of cross-reactive proteins secreted by Mycobacterium tuberculosis. Scand J Immunol. 1992;36:307–319. doi: 10.1111/j.1365-3083.1992.tb03104.x. [DOI] [PubMed] [Google Scholar]
- 33.Wiker H G, Nagai S, Hewinson R G, Russell W P, Harboe M. Heterogenous expression of the related MBP70 and MBP83 proteins distinguish various substrains of Mycobacterium bovis, BCG and Mycobacterium tuberculosis H37Rv. Scand J Immunol. 1996;43:374–380. doi: 10.1046/j.1365-3083.1996.d01-61.x. [DOI] [PubMed] [Google Scholar]