

Abstract
The major surface glycoprotein (MSG) of Pneumocystis carinii f. sp. carinii consists of a heterogeneous family of proteins that are encoded by approximately 100 unique genes. A genomic expression library was screened with a panel of MSG-specific monoclonal antibodies (MAbs) to identify conserved and rare epitopes. All of the antibodies reacted with epitopes that are encoded within the 5′ end of MSG. The results from the expression screening identified antibodies that recognize highly conserved, moderately conserved, and rare epitopes. Four MAbs (MAbs RA-F1, RA-E7, RA-G10, and RB-E3) reacted with a maltose binding protein–MSG-B fusion protein (MBPMSG-B41–1065) by immunoblotting and enzyme-linked immunosorbent assay. Three of the MAbs (MAbs RA-F1, RA-G10, and RA-E7) reacted with the same continuous epitope that was localized to amino acids 278 to 290 of MSG-B. Comparison of the sequence of the RA-F1-, RA-G10-, and RA-E7-reactive epitope to the deduced amino acid sequences of multiple MSGs demonstrated that it is highly conserved. The reactivity of RB-E3 with MSG-B was shown to be dependent on amino acids 184 to 192, which may comprise a portion of a discontinuous epitope.
Pneumocystis carinii f. sp. hominis is an important cause of pneumonia in patients with human immunodeficiency virus infection, cancer, and organ transplantation and in other immunocompromised hosts. The host factors that predispose individuals to the development of P. carinii f. sp. hominis pneumonia involve impaired cellular and humoral immunity; however, the specific immune defects are poorly understood. All forms of P. carinii that have been examined contain a 95,000- to 140,000-kDa glycoprotein, termed either the major surface glycoprotein (MSG) or glycoprotein A (gpA), which plays an important role in the immunobiology of the organism (12, 17, 20, 34, 42, 50). MSG is highly immunogenic and stimulates the production of various cytokines, contains protective B- and T-cell epitopes, and facilitates the interaction of P. carinii with host cells (9, 30, 33, 43, 44, 54).
MSG actually consists of a heterogeneous family of proteins encoded by about 100 genes. DNA sequence analysis of MSG cDNAs and/or genes has demonstrated that the gene family encodes many distinct MSG isoforms, which are very similar in size but whose sequences vary (10, 18, 21, 26, 48).
Transcription of MSG genes appears to occur only within a single telomeric expression site (7, 39, 49). The isolation of multiple unique MSG cDNAs from a single P. carinii f. sp. carinii-infected rat lung suggests that multiple MSG mRNAs and proteins may be present at the same time within a population of P. carinii f. sp. carinii (26). The ability to alter the expression of different MSG molecules raises the possibility of antigenic variation occurring in P. carinii f. sp. carinii.
MSG appears to be involved in both the cellular and the humoral immune responses of the host to P. carinii f. sp. carinii. Spleen cells isolated from rats environmentally exposed to P. carinii f. sp. carinii proliferate in response to MSG, and sera from these animals contain MSG-specific antibodies (44). MSG is also recognized by humans exposed to P. carinii f. sp. hominis. MSG-specific antibodies have been detected in approximately 30 to 70% of human serum specimens that contain P. carinii f. sp. hominis antibodies (28, 31). Animals generate a vigorous immune response against MSG upon immunization with purified antigens or P. carinii f. sp. carinii preparations (7). Passive immunotherapy with MSG-specific monoclonal antibodies (MAbs) and/or hyperimmune polyclonal antiserum has also been shown to modulate P. carinii infection in ferrets, rats, and mice with severe combined immunodeficiency, supporting the significance of MSG in the host response to P. carinii (13, 14, 35). The involvement of MSG in the host immune response suggests that immunization with it may provide protection against P. carinii infection. Because multiple isoforms of MSG can be expressed within a population of P. carinii, it will be important to identify immunoreactive regions common to all MSGs.
Several groups have described the production and characterization of P. carinii-specific MAbs (11, 16, 19, 23, 24, 27, 29). MSG-specific MAbs have also used to distinguish rat P. carinii f. sp. carinii and P. carinii f. sp. ratti strains at a phenotypic level (47). A MAb specific to MSG purified from P. carinii f. sp. hominis has been used in the development of a new method of diagnosis of P. carinii f. sp. hominis pneumonia by radioimmunodetection (15).
In the study described in this paper we have characterized a panel of MSG-specific MAbs on the basis of the frequency with which their epitopes are encoded and expressed within a population of P. carinii f. sp. carinii. This analysis demonstrated the presence of conserved epitopes that appear to be present on the majority of MSGs and epitopes that appear to be present on a restricted number of MSGs. A single MSG isoform was expressed in a bacterial vector and was used to identify the epitopes recognized by two of the MAbs that recognize conserved epitopes. Detailed characterization of MSG-specific MAb reactivities and identification of epitopes recognized by the MAbs will greatly improve their usefulness in the analysis of the expression of different MSG isoforms.
MATERIALS AND METHODS
Pneumocystis nomenclature.
The Pneumocystis nomenclature proposed at the 3rd International Workshop on Pneumocystis in Cleveland, Ohio, in June 1994 will be used in this report (32). Provisional tripartite names denoting the mammal of origin were given to the organism populations isolated from various mammalian hosts. Pneumocystis isolated from rats will be referred to as either P. carinii f. sp. carinii, designated the prototype, or P. carinii f. sp. ratti, designated a variant; the organism from human beings will be referred to as P. carinii f. sp. hominis.
Source of organisms.
P. carinii f. sp. carinii pneumonia was induced by corticosteroid treatment of rats, and the organisms were recovered from infected lungs and were quantitated as described previously (4). Briefly, infected lungs were removed en bloc, minced, and homogenized. The homogenate was centrifuged at 1,000 × g for 10 min at 4°C, and the resulting pellet was treated with 0.85% ammonium chloride to lyse the erythrocytes. The pellet was washed twice, and the organisms were resuspended in phosphate-buffered saline.
Source of antibodies.
MSG-specific MAbs were produced and characterized as described previously (27). Three different P. carinii antigen preparations were used to produce the MAbs used in this study: (i) P. carinii f. sp. carinii, (ii) MSG purified from P. carinii f. sp. carinii, and (iii) detergent-solubilized P. carinii f. sp. hominis (Table 1). Mice were immunized three times subcutaneously with the antigen preparations, and antigen preparations were then injected intraperitoneally 3 days prior to fusion. Spleen cells from the mice were fused to a myeloma cell line, and hybrids were selected by previously described methods. The resulting hybridomas were screened for P. carinii-specific reactivity by immunoblotting or enzyme-linked immunosorbent assay (ELISA). Polyclonal antibodies to P. carinii f. sp. carinii MSG were prepared by immunizing rabbits with purified MSG. Polyclonal rabbit anti-maltose binding protein (anti-MBP) serum was obtained from New England Biolabs, Beverly, Mass. Enzyme-conjugated antibodies were obtained from Kirkegaard & Perry, Gaithersburg, Md.
TABLE 1.
Characterization of MSG-specific MAbs and frequency of epitope expression
| MAb | Immunogena | Reactivityb
|
No. of positive plaques | ||
|---|---|---|---|---|---|
| P. carinii f. sp. carinii MSG | P. carinii f. sp. hominis MSG | MBPMSG41–1065 | |||
| RA-F1 | Rat MSG | + | + | + | 738 |
| RB-E3 | Rat Pc | + | − | + | 286 |
| RA-C6 | Rat MSG | + | − | − | 46 |
| RB-C8 | Rat Pc | + | + | − | 112 |
| RB-2F9 | Rat Pc | + | + | − | 307 |
| HB-G6 | Human Pc | + | + | − | 3 |
| RA-C7 | Rat MSG | + | − | − | 23 |
| RA-C1 | Rat MSG | + | − | − | 55 |
| RA-C11 | Rat MSG | + | − | − | 6 |
| RB-F8 | Rat Pc | + | − | − | 7 |
| RA-G10 | Rat MSG | + | + | + | NDc |
| RA-E7 | Rat MSG | + | + | + | ND |
Rat MSG, P. carinii f. sp. carinii MSG purified by electroelution from an SDS-polyacrylamide gel; Rat Pc, SDS-solubilized P. carinii f. sp. carinii; Human Pc, SDS-solubilized P. carinii f. sp. hominis.
Determined by immunoblotting reactivity.
ND, not done.
Hybridization and screening of the λgt11 genomic expression library.
The bacteriophage λgt11 genomic library was generated by insertion of randomly sheared P. carinii f. sp. carinii DNA into λgt11, and hybridizations were performed by standard methods (36). The λgt11 library was screened with MAbs as described previously (10). To determine the number of plaques to be screened, the library was first evaluated by hybridization of the λgt11 library to four different single-copy gene probes and to an MSG gene probe. The single-copy gene probes used were the TATA box binding protein (40), thymidylate synthase (8), alpha-tubulin (53), and 55-kDa antigen (37). Approximately 20,000 plaques were screened with each gene probe. Four positive plaques were detected with the 55-kDa antigen gene probe, and five positive plaques were found with the other gene probes. These data suggested that 4,000 to 5,000 plaques contain one genome equivalent in this library. This is reasonably close to the theoretical value calculated from the size of the genome, which is approximately 107 bp (5). A genome of this size would be contained in 4 × 103 phages, each carrying a 2.5-kb insert. The λgt11 library was generated with randomly sheared P. carinii f. sp. carinii DNA that ranged in size from 1 to 4 kb.
Analysis of MAb RA-F1-reactive λgt11 clones.
Four clones reactive with MAb RA-F1 were plaque purified by standard techniques (36). The reactivities of each of these clones were determined with the other MAbs as described previously. Phage DNA from all four clones was isolated by the Wizard lambda prep kit (Promega, Madison, Wis.). Partial sequences from both insert ends were obtained by using a PCR sequencing kit with the λgt11 forward and reverse primers by following the manufacturer’s standard protocol (Life Technologies, Inc., Gaithersburg, Md.). The sequences from each clone were aligned with the DNANALYZE program (51) to MSG-B (41) to determine the location of the beginning and end of each of the inserts. Primer C5 (5′-CATGAAAGACTTGAGAAATGT-3′) was used in the PCR cycle sequencing of clones 1 and 4 in order to compare sequences from the same area from all four RA-F1-reactive clones.
The epitope regions of clones 1 and 3 were amplified with primers C3 (5′-GTAACATCCTTCCCTCAAC-3′) and C7 (5′-GTCTTGTCCCTTTTTATAGCA-3′), and the epitope regions of clones 2 and 4 were amplified with primers C5.2 (5′-TAGTCCTGTCAAGCCGA-3′) and C7. The products were ligated into pGemT (Promega) according to the manufacturer’s instructions, and the inserts were sequenced by the DNA core facility of the Department of Molecular Genetics, Biochemistry, and Microbiology, University of Cincinnati College of Medicine.
Cloning of the msg-b gene.
A gene encoding a single species of MSG, designated msg-b, was amplified from Rp3-1 template DNA; Rp3-1 is a 16-kb fragment containing repetitive elements cloned from the P. carinii f. sp. carinii genome (38, 39, 41). Primers msg1+ and msg3801− (Table 2) were designed to amplify the entire msg-b gene. MunI sites were engineered into these primers in order to ligate the products directly into an EcoRI site. PCR was performed by standard methods. Reactions were amplified for two cycles of 94°C for 1 min, 45°C for 1 min, and 72°C for 3 min and were then amplified for 30 cycles of 94°C for 1 min, 50°C for 1 min, and 72°C for 3 min. Amplified DNA was purified by using the Wizard PCR Preps system (Promega) according to the manufacturer’s instructions. Ligation of the MunI-cut msg-b PCR product into the EcoRI site of pMALc2 was unsuccessful. Therefore, restriction sites that were compatible with sites in the pMALc2 polylinker were identified within the msg-b sequence. Digestion of an AvaI site present at base 124 and a HindIII site located downstream of the stop codon at base 3366 allowed ligation of the msg-b PCR product between the SalI and HindIII sites of the pMALc2 polylinker. This construct, designated pMAL/msg-b123–3366, contains the entire msg-b gene lacking the initial 122 bp ligated in frame with the MBP that is encoded by pMALc2.
TABLE 2.
Primers used to amplify target regions of MSG-B for cloning and expression in pMAL-c2
| Primer | Location (nucleotides)a | Sequenceb | Restriction site |
|---|---|---|---|
| msg1+ | 1–20 | AATTGATGGCACGGCCGGTTAAGAGG | MunI |
| msg375+ | 370–391 | GGATCCTGTGTCAAGTTGAGG | BamHI |
| msg515+ | 497–520 | GCCTAGTGTTAAGCTCGAGAAGGCG | XbaI |
| msg552− | 538–555 | TCCAAGCTTACTCGGATCAAGGCA | HindIII |
| msg579+ | 579–594 | GAGCTCGGTTGAAGTTTGCCGG | SacI |
| msg586− | 575–597 | GGCCGGCAAGCTTCAACCAATT | HindIII |
| msg834− | 819–835 | TTCAAGCTTCTTCTTTTTCAGCCTTT | HindIII |
| msg872− | 861–884 | CCCGACTTCAAGCTTGGTAGATCC | HindIII |
| msg965− | 950–965 | CCTAAGCTTAACGCTTTTTCGCAT | HindIII |
| msg1049− | 1034–1052 | TTTAAGCTTTCGCAGTTTTCGGTTT | HindIII |
| msg3801− | 3762–3801 | ACTGTACAATTGTCATCCATTTCAAATCGTCTTTCAATG | MunI |
Relative to MSB-B sequence.
Nucleotides in boldface denote restriction enzyme site.
Exonuclease digestion-generated MSG-B truncations.
In order to produce a linear construct with 5′ and 3′ 4-base overhanging ends, a 24-bp oligonucleotide containing a KpnI site was cloned into the 3′ end of the pMAL/msg-b123–3366 construct. After digestion with KpnI and SalI (Promega), the linearized DNA was subjected to unidirectional digestion with exonuclease III (ExoIII; New England Biolabs) as described previously (2, 6).
Amplification and cloning of internal msg-b fragments.
Eight overlapping MBPMSG-B fusion proteins were produced by amplifying the target regions of msg-b by PCR and cloning the products into pMAL-c2. Primers were either constructed to correspond to flanking regions of the target MSG-B region or to sequences within pMAL-c2 (Table 2). PCR products were digested with AvaI and HindIII and were then ligated into the polylinker of pMAL-c2 between the SalI and HindIII sites. Direct cloning of some of the PCR products into pMAL-c2 was unsuccessful; therefore, these products were initially cloned into a PCR cloning vector, pGEM-T (Promega). Ligation and transformation of pGEM-T was carried out as described by the manufacturer. The PCR products were excised from pGEM-T by restriction digestion. The inserts were gel purified and ligated into the polylinker of pMAL-c2 digested with the corresponding restriction enzymes.
The recombinant pMAL/msg-b clones were sequenced by a modified double-stranded DNA sequencing technique (Sequenase, version 2.0, U.S. Biochemicals). A mutation was detected in pMAL/msg-b579–966. A point mutation at bp 966 (T→A) created a stop codon that resulted in the expression of a fusion protein that extends from amino acids 193 to 322 with a predicted molecular weight of 60,000 (MBPMSG193–322). Analysis of the DNA sequence of pMAL/msg-b375–576 revealed following base 576 a frameshift mutation that altered the MSG-B amino acid sequence starting at amino acid 192 and that created a stop codon at base 709. Thus, the resulting fusion protein had a predicted molecular weight of 62,957, but the MSG-B amino acid sequence extended only from residues 125 to 192 (MBPMSG125–192). The remaining six constructs were cloned in the correct reading frame with MBP and contained no mutations.
Fusion protein expression and purification.
The pMAL/msg-b plasmids were used to transform electrocompetent Escherichia coli in an Electroporator II apparatus (Invitrogen, San Diego, Calif.) according to the manufacturer’s instructions. Positive transformants were picked and grown overnight at 37°C in 25-ml cultures of Luria-Bertani liquid broth supplemented with 50 μg of ampicillin per ml (LB-amp). On the following day, the overnight cultures were used to inoculate new 1-liter cultures of LB-amp which were then grown to an optical density at 600 nm (OD600) of 0.5 to 0.7. Fusion protein production was induced by the addition of isopropyl-β-d-thiogalactopyranoside to a final concentration of 0.5 mM. After 3 h of induction at 37°C, the cultures were centrifuged at 4,000 × g for 20 min and bacterial pellets were reconstituted in 25 ml of amylose column buffer (20 mM Tris-HCl [pH 7.0], 200 mM NaCl, 1 mM EDTA).
After freezing overnight at −20°C, cell suspensions were thawed and sonicated for five cycles of 15-s bursts followed by 15 s on ice. Sonicates were centrifuged at 10,000 × g for 30 min, and MBPMSG-B fusion proteins were purified from the supernatants by using an amylose resin column (New England Biolabs) according to the manufacturer’s instructions.
SDS-PAGE and immunoblotting.
Proteins were solubilized in 2× lysis buffer (125 mM Tris-HCl [pH 6.8], 4.0% sodium dodecyl sulfate (SDS), 20% glycerol, 10% β-mercaptoethanol) for analysis by SDS-polyacrylamide gel electrophoresis (PAGE) (22, 25). The proteins were transferred electrophoretically from the SDS-polyacrylamide gel to a nitrocellulose membrane, and immunoblotting was performed as described previously (25, 45).
ELISA.
Purified fusion proteins from the deletion constructs were diluted to 20 μg/ml in coating buffer from the ELISAmate reagent kit (Kirkegaard & Perry), and 50 μl was used to sensitize 96-well polyvinyl ELISA plates (Dynatech, Chantilly, Va.) overnight at 4°C. All subsequent steps were performed at room temperature. The wells were washed twice with ELISAmate wash buffer and were blocked with ELISAmate bovine serum albumin blocking reagent for 2 h. The wells were washed four times with wash buffer, incubated with the MAb for 2 h, washed four times with wash buffer, and then incubated with goat anti-mouse immunoglobulin G–horseradish peroxidase conjugated antibody (Kirkegaard & Perry) at a 1:1,000 dilution in ELISAmate diluent buffer for 2 h. The wells were washed four times in wash buffer and developed with ELISAmate ABTS peroxidase substrate. The optical densities of the wells were monitored at 405 nm on a 96-well plate reader (Bio-Tek Instruments, Winooski, Vt.).
RESULTS
Assessment of MAb epitope prevalence in the MSG family.
A panel of MAbs known to react with MSG had been generated previously (27). To estimate the number of MSG isoforms recognized by each of these MAbs, we used a genomic expression library that had been made by insertion of random fragments from the P. carinii f. sp. carinii genome into λgt11. A genomic expression library could be used because previous studies had shown that MSG genes lack introns (41). It was determined that 4,000 to 5,000 plaques contain one genome equivalent in this library. However, because the inserts in the library are in random orientations and reading frames with respect to the vector DNA sequence, only one of six MSG gene fragments would be expected to be capable of expressing an MSG epitope. Therefore, 24,000 plaques would be expected to express every unique epitope at least once.
Ten previously described MAbs (MAbs RA-F1, RB-E3, RA-C6, RB-C8, RB-F9, HB-G6, RA-C7, RA-C1, RA-C11, and RB-F8) were each used to screen 300,000 plaques in the expression library (27). Table 1 shows that the number of reactive plaques ranged from three positive plaques with MAb HB-G6 to 738 positive plaques with MAb RA-F1.
The plaque data suggested that MAb RA-F1 recognized an epitope found on many MSGs. To examine this possibility, four RA-F1-reactive clones were chosen for further analysis. After plaque purification, the insert size was determined by EcoRI digestion of the phage DNA. The four phages contained different inserts, which ranged in size from 1.3 kb (clone 2) to 4 kb (clone 1) (Fig. 1). Analysis of the DNA sequence showed that each clone contained a different MSG gene (data not shown).
FIG. 1.

Physical maps and MAb reactivities of four λgt11 clones each carrying a fragment from a different MSG gene. The lines show the sizes of the inserts carried in the four phage clones compared to the size of a previously characterized cloned msg gene (msg-b). The numbers at the ends of each line indicate the amino acid residues encoded by each msg gene fragment. The reactivity (+) or nonreactivity (−) of the plaques produced by each clone is indicated for each of eight MAbs.
To further characterize these RA-F1-positive phages, their reactivities were assessed with the 11 other MAbs. Four of these 11 MAbs (MAbs RA-C7, HB-G6, RA-C11, and RB-F8) did not react with any of the clones, which is consistent with the fact that each of these four MAbs recognized few plaques in the λgt11 library screen (fewer than 23 plaques) (Table 1). The remaining seven MAbs each reacted with at least one RA-F1-positive phage (Fig. 1). Three MAbs (MAbs RA-F1, RA-E7, and RA-G10) reacted with all four clones, suggesting that they recognize the same epitope, and subsequent studies (described below) showed that this was the case. The reactivities of MAbs RA-F1, RA-E7, RA-G10, RA-C1, RA-C6, and RB-C8 with clone 2 indicates that the epitopes for these MAbs are contained within the region from amino acids 227 to 660. The location of the epitopes for MAbs RB-E3 and RB-F9 could be localized to the first 600 amino acids of MSG on the basis of their reactivities with clone 3.
Localization of two MSG-specific MAb epitopes.
To facilitate identification of the MAb epitopes, the msg-b isoform was produced in a bacterial expression system as described in the Materials and Methods. The MBPMSG-B fusion protein (MBPMSG-B41–1065) was assayed by immunoblot analysis for its reactivity to the 12 MAbs (data not shown), and it was found to be reactive with MAbs RA-F1, RA-E7, RA-G10, and RB-E3 (Fig. 1).
An epitope mapping strategy was designed to monitor the loss of reactivity of the MAbs with truncated forms of the MBPMSG-B fusion protein. MAb RA-E7 was chosen to represent the RA-F1, RA-E7, and RA-G10 group because of the availability of sufficient quantities of this MAb for epitope mapping studies. RB-E3 was selected because it appeared to recognize a unique epitope. The regions of MSG-B recognized by MAbs RA-E7 and RB-E3 were initially localized by expression of truncated MBPMSG-B fusion proteins produced by digestion of pMAL/msg-b124–3366 with ExoIII. MAbs RA-E7 and RB-E3 remained reactive with the two shortest ExoIII-generated truncations that stopped at MSG-B amino acids 563 (MBPMSG41–563) and 373 (MBPMSG-B41–373) (Fig. 2 and 3, lanes 3 and 4). These results indicated that the epitopes were contained in the region from amino acids 41 to 373. Further attempts to produce shorter proteins by ExoIII digestion were unsuccessful.
FIG. 2.
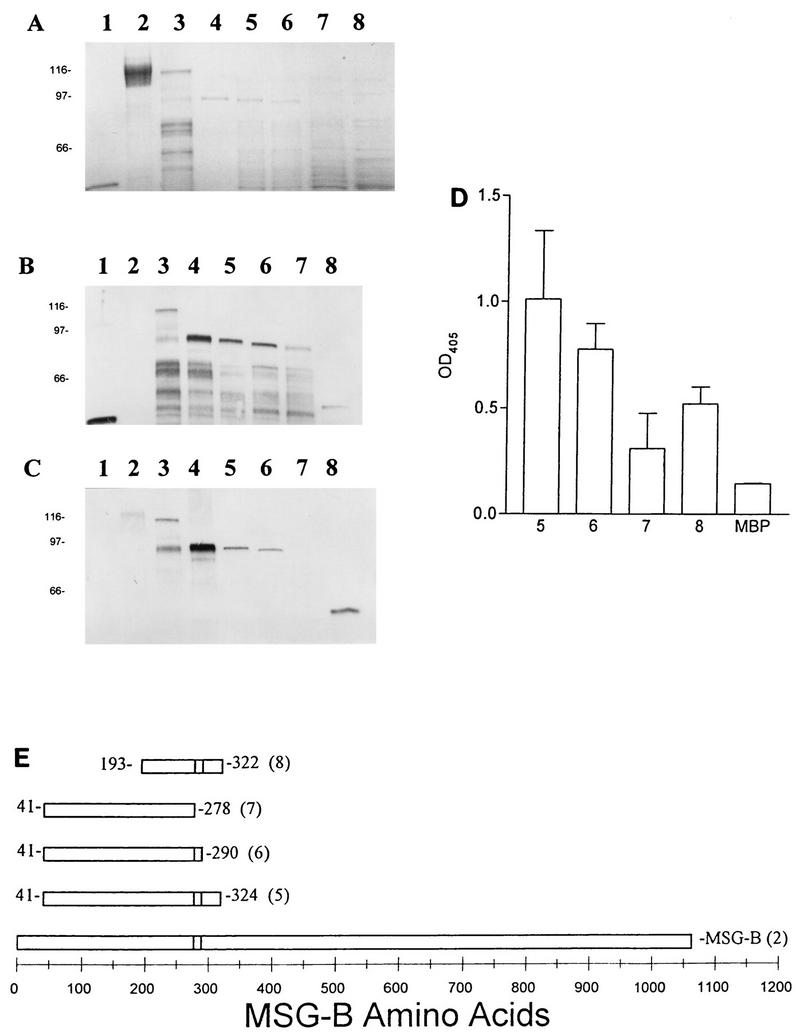
Mapping of MAb RA-E7 reactivity with MBPMSG truncated fusion proteins by SDS-PAGE and immunoblotting. (A) Coomassie blue-stained gel. (B) Immunoblot with anti-MBP polyclonal antisera. (C) Immunoblot with RB-E7. Lanes 1, MBP; lanes 2, native MSG; lanes 3, MBPMSG41–561; lanes 4, MBPMSG41–373; lanes 5, MBPMSG41–324; lanes 6, MBPMSG41–290; lanes 7, MBPMSG41–278; lanes 8, MBPMSG193–322. (D) ELISA analysis of MAb RA-E7 reactivity with MBPMSG-B truncated fusion proteins. Wells of 96-well microtiter plates were coated with the fusion proteins or MBP alone. The numbers correspond to the lanes described above. (E) Schematic of mapping of the epitope reactive with MAb RA-E7. The numbers in parentheses correspond to the lanes described above.
FIG. 3.
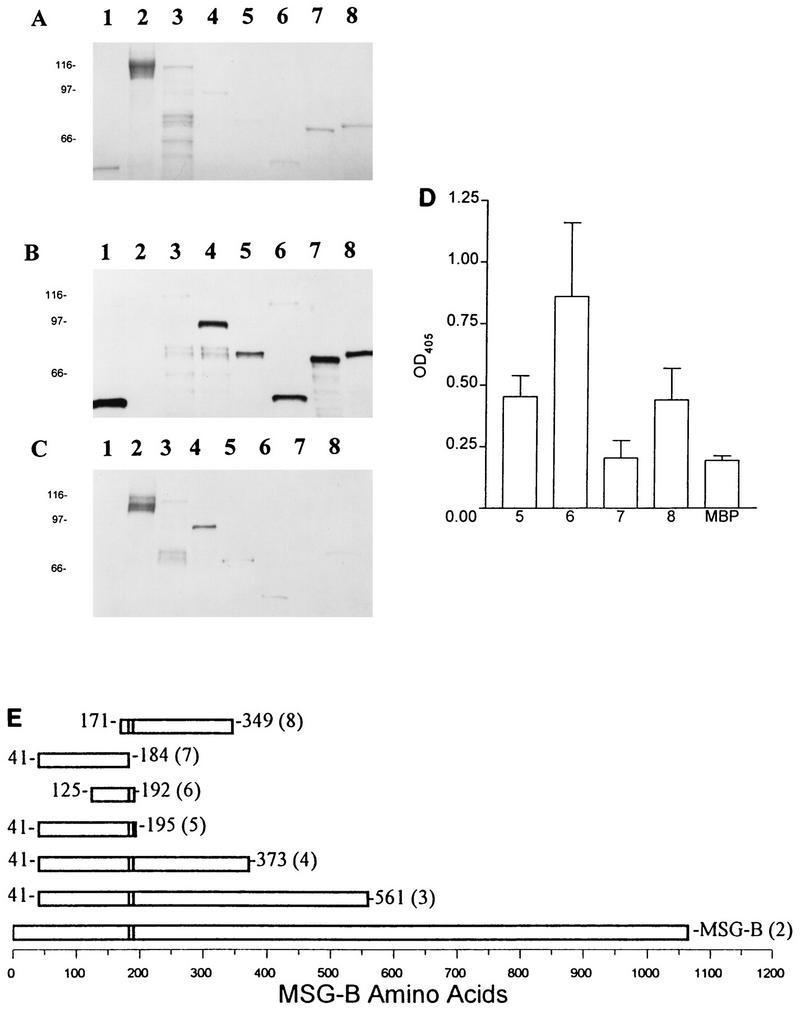
Mapping of MAb RB-E3 reactivity with MBPMSG truncated fusion proteins by SDS-PAGE, immunoblotting, and ELISA. (A) Coomassie blue-stained gel. (B) Immunoblot with anti-MBP polyclonal antisera. (C) Immunoblot with RB-E3. Lanes 1, MBP; lanes 2, native MSG; lanes 3, MBPMSG41–561; lanes 4, MBPMSG41–373; lanes 5, MBPMSG41–195; lanes 6, MBPMSG125–192; lanes 7, MBPMSG41–184; lanes 8, MBPMSG171–349. (D) ELISA analysis of MAb RB-E3 reactivity with MBPMSG-B truncated fusion proteins. Wells of 96-well microtiter plates were coated with the fusion proteins or MBP alone. The numbers correspond to the lanes described above. (E) Schematic of mapping of the epitope reactive with MAb RB-E3. The numbers in parentheses correspond to the lanes described above.
To identify the epitopes recognized by MAbs RB-E3 and RA-E7, MBPMSG-B fusion proteins that covered amino acids 41 to 373 were prepared by amplifying the target regions of msg-b by PCR and cloning the products into pMAL-c2. Evaluation of the fusion proteins expressed from the PCR-generated pMAL/msg-b constructs by SDS-PAGE (Fig. 2A and 3A) and immunoblotting with polyclonal antisera against MBP (Fig. 2B and 3B) demonstrated that they were of the predicted size.
The epitope for RA-E7 was mapped to MSG-B amino acids 279 to 290 by monitoring the loss of MAb reactivity of the fusion proteins by immunoblotting (Fig. 2C) and ELISA (Fig. 2D). The epitope was deduced from variations in reactivity between MBPMSG41–278 and MBPMSG41–290. The MBPMSG41–278 construct contains MSG-B amino acids 41 to 278, and MBPMSG41–290 covers amino acids 41 to 290. The loss of reactivity of MAb RA-E7 with MBPMSG41–278 identifies the reactive amino acids as amino acids 279 to 290 (Fig. 2E). A construct with a large 5′ deletion (MBPMSG193–587) also reacted with MAb RA-E7, indicating that the upstream portion of MSG-B from amino acids 41 to 193 is not required for recognition by MAb RA-E7. This finding was also supported by the reactivity of phage clone 2 with MAb RA-E7; clone 2 contained MSG amino acids 227 to 660 (Fig. 1). MAbs RA-G10 and RA-F1 demonstrated reactivity patterns with the truncated fusion proteins identical to that of MAb RA-E7, confirming that all three MAbs recognize the same epitope (data not shown).
MAb RB-E3 recognized all of the PCR-generated MBPMSG-B fusion proteins except for MBPMSG41–184 by immunoblotting (Fig. 3C) and ELISA (Fig. 3D). Comparison of the nonreactive MBPMSG41–184 fusion protein and the reactive MBPMSG125–192 fusion protein demonstrated that amino acids 185 to 192 are involved in the recognition of MSG-B by MAb RB-E3 (Fig. 3E).
The reactivity of MAb RB-E3 with MBPMSG125–192 was not dependent on upstream amino acids 125 to 171, as demonstrated by the recognition of the MBPMSG171–349 fusion protein by MAb RB-E3. The localization of the reactivity of MAb RB-E3 to amino acids 184 to 192 is also supported by the phage clone data, which suggest that this epitope is contained within the first 223 amino acids of MSG-B (Fig. 1).
Analysis of the epitope regions in additional MSG isoforms.
The availability of reactive and nonreactive phages provided a means of comparing the epitopes in these clones to the epitopes identified in MSG-B. The regions corresponding to those encoding the epitope in MSG-B reactive with MAbs RA-F1, RA-G10, and RA-E7 were amplified from phage clones 2 and 4, both of which produced plaques that were reactive with MAb RA-E7. The PCR products were cloned into pGEM-T, and the DNA was sequenced. Analysis of the deduced amino acid sequence revealed that the sequence of clone 2 matched the MSG-B sequence exactly and the sequence of clone 4 matched at 10 of 11 of the MSG-B amino acids (Fig. 4A). This finding supports the idea that the region reactive with MAb RA-E7 is highly conserved among MSG isoforms. The 12-amino-acid binding site for MAb RA-E7 was also well conserved among five additional MSG sequences predicted from previously published DNA sequences (Fig. 4A). A similar analysis of the putative epitope reactive with MAb RB-E3 yielded a more complex picture (Fig. 4B). While only clone 3 produced reactive phage plaques, the predicted amino acid sequences of both clones 1 and 3 were similar to the MSG-B sequence, and within the epitope there were no obvious amino acid substitutions that may account for the variation of reactivity between clones 1 and 3. Therefore, amino acids 185 to 192 appear to be essential for the reactivity of MAb RB-E3 with MSG-B, but additional amino-terminal residues may be involved in the presentation of the epitope to the antibody.
FIG. 4.

Alignment of MSG-B epitope region reactive with MAbs RA-E7 and RB-E3 with seven additional deduced MSG amino acid sequences. The deduced amino acid sequence of the epitope regions from five previously described P. carinii f. sp. carinii MSGs and from the four phage clones were visually aligned with the MAb-reactive epitopes identified on MSG-B. (A) RA-E7 epitope; (B) RB-E3 epitope. a, from reference 48; b, from reference 21; c, deduced amino acids from λgt11 clones.
DISCUSSION
The identification of conserved and variable epitopes on MSG molecules provides a method of studying the expression of different isoforms through the use of either polyclonal antiserum produced against peptides or MSG-specific MAbs. Previously, three different MSG variants were identified in a single lobe of an infected lung with epitope-specific polyclonal antisera by immunohistochemistry (1). In another study two MSG-specific MAbs (MAbs RA-C6 and RA-C11) were able to identify antigenic differences between genetically distinct P. carinii f. sp. carinii and P. carinii f. sp. ratti populations and within a genetically defined population of P. carinii f. sp. carinii. Two additional MAbs (MAbs RA-F1 and RA-C7) reacted with all P. carinii f. sp. carinii and P. carinii f. sp. ratti populations examined (47).
In this study MSG-specific MAbs were initially characterized on the basis of the frequency with which their epitopes are encoded within the P. carinii f. sp. carinii genome by screening an expression library made by insertion of randomly sheared genomic P. carinii f. sp. carinii DNA. The MAbs could be separated into four groups on the basis of the number of plaques that each one recognized. MAb RA-F1 appeared to react with a conserved epitope on the basis of its reactivity with 738 plaques. MAbs RB-E3, RB-2F9, and RB-C8 each recognized between 100 and 300 plaques, suggesting that these epitopes are encoded in multiple MSG genes. The three MAbs in the third group (MAbs RA-C6, RA-C7, and RA-C1) reacted with 46, 23, and 55 plaques, respectively. This group of MAbs appeared to recognize a less conserved epitope. The final group of MAbs (MAbs HB-G6, RA-C11, and RB-F8) recognized a rare epitope on the basis of the low number (three to seven) of plaques that they detected.
In addition to characterizing the frequency with which MAb epitopes are expressed in the Pneumocystis genome, analysis of reactive λgt11 phage established that the MAbs react with MSG. Characterization of the MSG-specific MAbs was previously based on their reactivity with a 116,000-molecular-weight glycoprotein that was specific to Pneumocystis (27). The reactivities of the MAbs with MSG were confirmed through DNA sequence analysis of the λgt11 clones. The four reactive clones that were analyzed by DNA sequencing all contained pieces of DNA that were homologous with previously identified msg genes.
The reactivities of four MAbs with recombinant MSG provided a method for mapping their epitopes. The epitope reactive with MAbs RA-F1, RA-G10, and RA-E7 was localized to a highly conserved region in the amino-terminal end of MSG-B and was identified as amino acids 278 to 290. Comparison of the identified epitope with deduced amino acids from previously cloned msg genes, cDNAs, or cloned regions of MSG from reactive λgt11 phage demonstrated that this sequence is highly conserved. The reactivity of MAb RB-E3 was also localized in the amino-terminal portion of MSG-B approximately 100 amino acids upstream from the epitope reactive with MAbs RA-F1, RA-G10, and RA-E7. Amino acids 185 to 192 of MSG-B were determined to be required for reactivity with MAb RB-E3. Comparison of the amino acid sequence of the epitope reactive with MAb RB-E3 with that of previously cloned msg genes demonstrated that this region is not as well conserved as the epitope reactive with MAbs RA-F1, RA-G10, and RA-E7. Alignment of the amino acids in the RB-E3-reactive epitope regions from a reactive λgt11 phage and a nonreactive λgt11 phage did not reveal the presence of critical residues that would be essential for reactivity.
There are two basic types of epitopes, continuous and discontinuous (3, 46). Continuous epitopes consist of short linear sequences of amino acids. Discontinuous epitopes involve distant residues brought together by protein folding. The immunoblotting, ELISA, and sequence comparison data indicate that MSG-B amino acids 279 to 290 represent the epitope reactive with MAbs RA-F1, RA-G10, and RA-E7, and deletion of these amino acids clearly results in the loss of MAb reactivity. In addition, the high degree of conservation of the epitope among other MSGs indicates that it is a continuous epitope. The functional identification of the RB-E3-reactive epitope by immunoblotting and ELISA suggests that it is continuous; however, the lack of a correlation of MAb reactivity with the conservation of specific amino acids indicates that additional residues may be required for high-affinity binding of MAb RB-E3. The decreased reactivity of the MBPMSG-B171–349 fusion protein also indicates that additional amino acids removed from the epitope region are required for antibody recognition. The presence of a cysteine at residue 187 that could be involved in the folding of MSG through the formation of disulfhydryl bonds is noteworthy. The location of cysteine residues is maintained among all MSGs analyzed to date (26, 52). The conservation of the cysteine positions suggests the importance of disulfhydryl bonds in maintaining a conserved higher-order structure that could be critical to the function of MSG.
MSG has been implicated in the binding of P. carinii to host cells and molecules. The extracellular MSG domains involved in these interactions have not been identified, and little is known about the position or orientation of MSG within the cell membrane or cell wall of P. carinii. As described previously, MAbs RA-E7, RA-G10, RA-F1, RB-E3, RA-C1, RA-C6, RB-C8, and RB-F9 react with the surface of P. carinii f. sp. carinii by immunofluorescence (27). Localization of the MAb reactivity within the first 600 amino acids demonstrates that at least portions of the amino terminus of MSG are exposed on the surface of P. carinii f. sp. carinii. These results suggest that the surface-exposed amino terminus of MSG may also be involved in binding to host proteins.
The presence of hundreds of MSG genes in the genome suggests that P. carinii is capable of undergoing a form of antigenic variation and that the ability to alter this abundant surface protein is critical to its survival. The MAbs characterized in these studies will provide useful tools for analyzing the expression of particular MSG isoforms on P. carinii.
ACKNOWLEDGMENTS
This work was supported by the Medical Research Service of the U.S. Department of Veterans Affairs and Public Health Service contract AI 25139 and grant AI 36701 from the National Institutes of Health.
REFERENCES
- 1.Angus C W, Tu A, Vogel P, Qin M, Kovacs J A. Expression of variants of the major surface glycoprotein of Pneumocystis carinii. J Exp Med. 1996;183:1229–1234. doi: 10.1084/jem.183.3.1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ausubel F M, Brent B, Kingston R E, Moore D D, Seidman J G, Smith J A, Struhl K, editors. Current protocols in molecular biology. New York, N.Y: Greene Publishing Associates and Wiley-Interscience; 1991. [Google Scholar]
- 3.Barlow D J, Edwards M S, Thornton J M. Continuous and discontinuous protein antigenic determinants. Nature. 1986;322:747–748. doi: 10.1038/322747a0. [DOI] [PubMed] [Google Scholar]
- 4.Cushion M T, Ruffolo J J, Walzer P D. Analysis of the developmental stages of Pneumocystis carinii, in vitro. Lab Invest. 1988;58:324–331. [PubMed] [Google Scholar]
- 5.Cushion M T, Zhang J, Kaselis M, Giuntoli D, Stringer S L, Stringer J R. Evidence for two genetic variants of Pneumocystis carinii coinfecting laboratory rats. J Clin Microbiol. 1993;31:1217–1223. doi: 10.1128/jcm.31.5.1217-1223.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Doherty T M, Booth R J, Love S G, Gibson J J, Harding D R, Watson J D. Characterization of an antibody-binding epitope from the 18-kDa protein on Mycobacterium leprae. J Immunol. 1989;142:1691–1695. [PubMed] [Google Scholar]
- 7.Edman J C, Hatton T W, Nam M, Turner R, Mei Q, Angus C W, Kovacs J A. A single expression site with a conserved leader sequence regulates variation of expression of the Pneumocystis carinii family of major surface glycoprotein genes. DNA Cell Biol. 1996;15:989–999. doi: 10.1089/dna.1996.15.989. [DOI] [PubMed] [Google Scholar]
- 8.Edman U, Edman J C, Lundgren B, Santi D V. Isolation and expression of the Pneumocystis carinii thymidylate synthase gene. Proc Natl Acad Sci USA. 1989;86:6503–6507. doi: 10.1073/pnas.86.17.6503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fisher D J, Gigliotti F, Zauderer M, Harmsen A G. Specific T-cell response to a Pneumocystis carinii surface glycoprotein (gp120) after immunization and natural infection. Infect Immun. 1991;59:3372–3376. doi: 10.1128/iai.59.10.3372-3376.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garbe T R, Stringer J R. Molecular characterization of clustered variants of genes encoding major surface antigens of human Pneumocystis carinii. Infect Immun. 1994;62:3092–3101. doi: 10.1128/iai.62.8.3092-3101.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gigliotti F. Host species-specific antigenic variation of a mannosylated surface glycoprotein of Pneumocystis carinii. J Infect Dis. 1992;165:329–336. doi: 10.1093/infdis/165.2.329. [DOI] [PubMed] [Google Scholar]
- 12.Gigliotti F, Ballou L R, Hughes W T, Mosley B D. Purification and initial characterization of a ferret Pneumocystis carinii surface antigen. J Infect Dis. 1988;158:848–854. doi: 10.1093/infdis/158.4.848. [DOI] [PubMed] [Google Scholar]
- 13.Gigliotti F, Garvy B A, Harmsen A G. Antibody-mediated shift in the profile of glycoprotein A phenotypes observed in a mouse model of Pneumocystis carinii pneumonia. Infect Immun. 1996;64:1892–1899. doi: 10.1128/iai.64.6.1892-1899.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gigliotti F, Hughes W T. Passive immunoprophylaxis with specific monoclonal antibody confers partial protection against Pneumocystis carinii pneumonitis in animal models. J Clin Invest. 1988;81:1666–1668. doi: 10.1172/JCI113503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goldenberg D M, Sharkey R M, Udem S, Vagg R, Levine G M, Conte P, Swayne L C, Hansen H J, Cunniff D, Anton J, et al. Immunoscintigraphy of Pneumocystis carinii pneumonia in AIDS patients. J Nucl Med. 1994;35:1028–1034. [PubMed] [Google Scholar]
- 16.Graves D C, McNabb S J, Ivey M H, Worley M A. Development and characterization of monoclonal antibodies to Pneumocystis carinii. Infect Immun. 1986;51:125–133. doi: 10.1128/iai.51.1.125-133.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Graves D C, McNabb S J, Worley M A, Downs T D, Ivey M H. Analyses of rat Pneumocystis carinii antigens recognized by human and rat antibodies by using Western immunoblotting. Infect Immun. 1986;54:96–103. doi: 10.1128/iai.54.1.96-103.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Haidaris P J, Wright T W, Gigliotti F, Haidaris C G. Expression and characterization of a cDNA clone encoding an immunodominant surface glycoprotein of Pneumocystis carinii. J Infect Dis. 1992;166:1113–1123. doi: 10.1093/infdis/166.5.1113. [DOI] [PubMed] [Google Scholar]
- 19.Kovacs J A, Halpern J L, Lundgren B, Swan J C, Parrillo J E, Masur H. Monoclonal antibodies to Pneumocystis carinii: identification of specific antigens and characterization of antigenic differences between rat and human isolates. J Infect Dis. 1989;159:60–70. doi: 10.1093/infdis/159.1.60. [DOI] [PubMed] [Google Scholar]
- 20.Kovacs J A, Halpern J L, Swan J C, Moss J, Parrillo J E, Masur H. Identification of antigens and antibodies specific for Pneumocystis carinii. J Immunol. 1988;140:2023–2031. [PubMed] [Google Scholar]
- 21.Kovacs J A, Powell F, Edman J C, Lundgren B, Martinez A, Drew B, Angus C W. Multiple genes encode the major surface glycoprotein of Pneumocystis carinii. J Biol Chem. 1993;268:6034–6040. [PubMed] [Google Scholar]
- 22.Laemmli U K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 23.Lee C H, Bolinger C D, Bartlett M S, Kohler R B, Wilde III C E, Smith J W. Production of monoclonal antibody against Pneumocystis carinii by using a hybrid of rat spleen and mouse myeloma cells. J Clin Microbiol. 1986;23:505–508. doi: 10.1128/jcm.23.3.505-508.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Linder E, Lundin L, Vorma H. Detection of Pneumocystis carinii in lung-derived samples using monoclonal antibodies to an 82 kDa parasite component. J Immunol Methods. 1987;98:57–62. doi: 10.1016/0022-1759(87)90435-2. [DOI] [PubMed] [Google Scholar]
- 25.Linke M J, Cushion M T, Walzer P D. Properties of the major antigens of rat and human Pneumocystis carinii. Infect Immun. 1989;57:1547–1555. doi: 10.1128/iai.57.5.1547-1555.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Linke M J, Smulian A G, Stringer J R, Walzer P D. Characterization of multiple unique cDNAs encoding the major surface glycoprotein of rat-derived Pneumocystis carinii. Parasitol Res. 1994;80:478–486. doi: 10.1007/BF00932694. [DOI] [PubMed] [Google Scholar]
- 27.Linke M J, Smulian A G, Yoshihara P, Walzer P D. Production and characterization of monoclonal antibodies specific for the major surface glycoprotein of Pneumocystis carinii. J Eukaryot Microbiol. 1994;41:99S–100S. [PubMed] [Google Scholar]
- 28.Lundgren B, Lebech M, Lind K, Nielsen J O, Lundgren J D. Antibody response to a major human Pneumocystis carinii surface antigen in patients without evidence of immunosuppression and in patients with suspected atypical pneumonia. Eur J Clin Microbiol Infect Dis. 1993;12:105–109. doi: 10.1007/BF01967583. [DOI] [PubMed] [Google Scholar]
- 29.Matsumoto Y, Amagai T, Yamada M, Imanishi J, Yoshida Y. Production of a monoclonal antibody with specificity for the pellicle of Pneumocystis carinii by hybridoma. Parasitol Res. 1987;73:228–233. doi: 10.1007/BF00578509. [DOI] [PubMed] [Google Scholar]
- 30.O’Riordan D M, Standing J E, Limper A H. Pneumocystis carinii glycoprotein A binds macrophage mannose receptors. Infect Immun. 1995;63:779–784. doi: 10.1128/iai.63.3.779-784.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Peglow S L, Smulian A G, Linke M J, Pogue C L, Nurre S, Crisler J, Phair J, Gold J W, Armstrong D, Walzer P D. Serologic responses to Pneumocystis carinii antigens in health and disease. J Infect Dis. 1990;161:296–306. doi: 10.1093/infdis/161.2.296. [DOI] [PubMed] [Google Scholar]
- 32.Pneumocystis Working Group. Nomenclature of Pneumocystis. J Eukaryot Microbiol. 1994;41:121S–122S. [PubMed] [Google Scholar]
- 33.Pottratz S T, Paulsrud J, Smith J S, Martin W J., II Pneumocystis carinii attachment to cultured lung cells by pneumocystis gp 120, a fibronectin binding protein. J Clin Invest. 1991;88:403–407. doi: 10.1172/JCI115318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Radding J A, Armstrong M Y, Ullu E, Richards F F. Identification and isolation of a major cell surface glycoprotein of Pneumocystis carinii. Infect Immun. 1989;57:2149–2157. doi: 10.1128/iai.57.7.2149-2157.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Roths J B, Sidman C L. Single and combined humoral and cell-mediated immunotherapy of Pneumocystis carinii pneumonia in immunodeficient scid mice. Infect Immun. 1993;61:1641–1649. doi: 10.1128/iai.61.5.1641-1649.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sambrook J, Fritsch E F, Maniatis T. Molecular cloning: a laboratory manual. 2nd ed. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 37.Smulian A G, Theus S A, Denko N, Walzer P D, Stringer J R. A 55 kDa antigen of Pneumocystis carinii: analysis of the cellular immune response and characterization of the gene. Mol Microbiol. 1993;7:745–753. doi: 10.1111/j.1365-2958.1993.tb01165.x. [DOI] [PubMed] [Google Scholar]
- 38.Stringer S L, Hong S T, Giuntoli D, Stringer J R. Repeated DNA in Pneumocystis carinii. J Clin Microbiol. 1991;29:1194–1201. doi: 10.1128/jcm.29.6.1194-1201.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sunkin S M, Stringer J R. Translocation of surface antigens to a unique telomeric expression site in Pneumocystis carinii. Mol Microbiol. 1996;19:283–295. doi: 10.1046/j.1365-2958.1996.375905.x. [DOI] [PubMed] [Google Scholar]
- 40.Sunkin S M, Stringer J R. Transcription factor genes from rat Pneumocystis carinii. J Eukaryot Microbiol. 1995;42:12–19. doi: 10.1111/j.1550-7408.1995.tb01534.x. [DOI] [PubMed] [Google Scholar]
- 41.Sunkin S M, Stringer S L, Stringer J R. A tandem repeat of rat-derived Pneumocystis carinii genes encoding the major surface glycoprotein. J Eukaryot Microbiol. 1994;41:292–300. doi: 10.1111/j.1550-7408.1994.tb01509.x. [DOI] [PubMed] [Google Scholar]
- 42.Tanabe K, Takasaki S, Watanabe J, Kobata A, Egawa K, Nakamura Y. Glycoproteins composed of major surface immunodeterminants of Pneumocystis carinii. Infect Immun. 1989;57:1363–1368. doi: 10.1128/iai.57.5.1363-1368.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Theus S A, Andrews R P, Steele P, Walzer P D. Adoptive transfer of lymphocytes sensitized to the major surface glycoprotein of Pneumocystis carinii confers protection in the rat. J Clin Invest. 1995;95:2587–2593. doi: 10.1172/JCI117960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Theus S A, Linke M J, Andrews R P, Walzer P D. Proliferative and cytokine responses to a major surface glycoprotein of Pneumocystis carinii. Infect Immun. 1993;61:4703–4709. doi: 10.1128/iai.61.11.4703-4709.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci USA. 1979;76:4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.VanRegenmortel M. Structural and functional approaches to the study of protein antigenicity. Immunol Today. 1989;10:266–272. doi: 10.1016/0167-5699(89)90140-0. [DOI] [PubMed] [Google Scholar]
- 47.Vasquez J, Smulian A G, Linke M J, Cushion M T. Antigenic differences associated with genetically distinct Pneumocystis carinii from rats. Infect Immun. 1996;64:290–297. doi: 10.1128/iai.64.1.290-297.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wada M, Kitada K, Saito M, Egawa K, Nakamura Y. cDNA sequence diversity and genomic clusters of major surface glycoprotein genes of Pneumocystis carinii. J Infect Dis. 1993;168:979–985. doi: 10.1093/infdis/168.4.979. [DOI] [PubMed] [Google Scholar]
- 49.Wada M, Sunkin S M, Stringer J R, Nakamura Y. Antigenic variation by positional control of major surface glycoprotein gene expression in Pneumocystis carinii. J Infect Dis. 1995;171:1563–1568. doi: 10.1093/infdis/171.6.1563. [DOI] [PubMed] [Google Scholar]
- 50.Walzer P D, Linke M J. A comparison of the antigenic characteristics of rat and human Pneumocystis carinii by immunoblotting. J Immunol. 1987;138:2257–2265. [PubMed] [Google Scholar]
- 51.Wernke G (Cincinnati, Ohio) DNANALYZE, version 5.2. 1988. [Google Scholar]
- 52.Wright T W, Simpson-Haidaris P J, Gigliotti F, Harmsen A G, Haidaris C G. Conserved sequence homology of cysteine-rich regions in genes encoding glycoprotein A in Pneumocystis carinii derived from different host species. Infect Immun. 1994;62:1513–1519. doi: 10.1128/iai.62.5.1513-1519.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang J, Stringer J R. Cloning and characterization of an alpha-tubulin-encoding gene from rat-derived Pneumocystis carinii. Gene. 1993;123:137–141. doi: 10.1016/0378-1119(93)90553-f. [DOI] [PubMed] [Google Scholar]
- 54.Zimmerman P E, Voelker D R, McCormack F X, Paulsrud J R, Martin W J., II 120-kD surface glycoprotein of Pneumocystis carinii is a ligand for surfactant protein A. J Clin Invest. 1992;89:143–149. doi: 10.1172/JCI115554. [DOI] [PMC free article] [PubMed] [Google Scholar]