

Abstract
The heterodimeric pre-mRNA splicing factor, U2AF (U2 snRNP auxiliary factor), plays a critical role in 3′ splice site selection. Although the U2AF subunits associate in a tight complex, biochemical experiments designed to address the requirement for both subunits in splicing have yielded conflicting results. We have taken a genetic approach to assess the requirement for the Drosophila U2AF heterodimer in vivo. We developed a novel Escherichia coli copurification assay to map the domain on the Drosophila U2AF large subunit (dU2AF50) that interacts with the Drosophila small subunit (dU2AF38). A 28-amino-acid fragment on dU2AF50 that is both necessary and sufficient for interaction with dU2AF38 was identified. Using the copurification assay, we scanned this 28-amino-acid interaction domain for mutations that abrogate heterodimer formation. A collection of these dU2AF50 point mutants was then tested in vivo for genetic complementation of a recessive lethal dU2AF50 allele. A mutation that completely abolished interaction with dU2AF38 was incapable of complementation, whereas dU2AF50 mutations that did not effect heterodimer formation rescued the recessive lethal dU2AF50 allele. Analysis of heterodimer formation in embryo extracts derived from these interaction mutant lines revealed a perfect correlation between the efficiency of subunit association and the ability to complement the dU2AF50 recessive lethal allele. These data indicate that Drosophila U2AF heterodimer formation is essential for viability in vivo, consistent with a requirement for both subunits in splicing in vitro.
Generation of functional mRNA in eukaryotes requires the removal of noncoding sequences (introns) from pre-mRNA by a process termed RNA splicing (17, 24). Pre-mRNA splicing takes place in the spliceosome, a dynamic RNA-protein complex that assembles in a stepwise, ATP-dependent manner on the pre-mRNA (13, 17). The spliceosome is composed of small nuclear ribonucleoprotein particles (snRNPs) and extrinsic (non-snRNP) protein factors. The earliest steps in spliceosome assembly involve the specification of the exon and intron boundaries by U1 and U2 snRNP. U1 snRNP binds the 5′ splice site, and U2 snRNP binds the branch site sequence (13, 17, 19). Since in most cases the first AG dinucleotide downstream of the branch site is used as the 3′ splice site, U2 snRNP defines the 3′ splice site (20, 27). The branch site sequence in metazoan introns is highly degenerate and is not sufficient for U2 snRNP recognition (22). Targeting of U2 snRNP to the branch site requires the extrinsic protein factor U2AF (U2 snRNP auxiliary factor). U2AF binds site specifically to the intron pyrimidine tract located between the branch point sequence and the 3′ splice site and recruits U2 snRNP to the branch site at an early step in spliceosome assembly (22, 36). Regulation of 3′ splice site choice, both positive and negative, can be modulated by U2AF binding to the intron pyrimidine tract (3, 6, 17). Thus, U2AF is a major determinant in 3′ splice site selection.
Human U2AF is a heterodimer composed of large and small subunits (35). Homologs of both U2AF subunits have been identified in Drosophila and Schizosaccharomyces pombe (10, 18, 21, 32, 34). A factor related to the U2AF large subunit, called MUD2p, exists in the budding yeast, Saccharomyces cerevisiae, which appears to have some of the properties of U2AF in terms of its role in branch site selection by U2 snRNP (1, 2, 5). However, no small subunit equivalent has been found in S. cerevisiae (13, 17). Human U2AF consists of a 65-kDa large subunit (hU2AF65) (36) and a 35-kDa small subunit (hU2AF35) (37). The Drosophila U2AF large- and small-subunit homologs are 50 and 38 kDa, respectively (dU2AF50 and dU2AF38) (11, 21). The U2AF large subunit contains an amino-terminal arginine-serine-rich domain (RS), a central domain required for interaction with the small subunit, and three RNA recognition motifs (36).
U2AF can be depleted from nuclear splicing extracts by poly(U)-Sepharose chromatography (34) or anti-hU2AF35 antibodies (39). Both extracts are inactive for splicing but seem to have different requirements for reactivation. Reactivation of the poly(U)-depleted extract can be achieved by the addition of recombinant large subunit alone (hU2AF65 or dU2AF50) (11, 31, 36). Even though the U2AF large and small subunits, purified from mammalian (or Drosophila) cells, associate in a tight complex, the small subunit appears to be dispensable for splicing in this assay. Recombinant hU2AF65 lacking the domain required for interaction with hU2AF35, as defined by the yeast two-hybrid assay, can also reactivate the poly(U)-depleted extract (7). Thus, it is unlikely that reactivation by the large subunit alone is a result of association between trace amounts of the small subunit remaining in the poly(U)-depleted extract and the exogenously added recombinant large subunit. Recently, a biochemical requirement for the small subunit in splicing was observed with an extract immunodepleted of U2AF activity with anti-hU2AF35 antibodies (39). Partial reactivation of the immunodepleted extract could be achieved by the addition of either recombinant hU2AF65 or hU2AF35. Addition of both subunits reconstituted splicing to a level similar to that of the mock-depleted extract (39). The reason for the strikingly different requirements for reactivation of splicing in these two U2AF-depleted extracts remains unresolved.
Consistent with the requirement for the small subunit observed in the immunodepleted extracts, we have previously shown that dU2AF38, like dU2AF50, is an essential gene in Drosophila (21). In the work reported here, we have extended our genetic analysis of Drosophila U2AF to assess the requirement for U2AF heterodimer formation in vivo. A novel Escherichia coli copurification assay was developed to map the domain on dU2AF50 that interacts with dU2AF38. A 28-amino-acid fragment on dU2AF50 that is both necessary and sufficient for interaction with dU2AF38 was identified. This highly conserved domain overlaps and further refines the interaction domain on hU2AF65 determined by the yeast two-hybrid interaction assay (7). Using our copurification assay, we have scanned this 28-amino-acid domain for mutations that disrupt heterodimer formation. One dU2AF50 mutation that completely abrogated interaction with dU2AF38 was identified. To assess the requirement for heterodimer formation in vivo, we have tested a collection of these mutations for complementation of a recessive lethal dU2AF50 allele. Whereas dU2AF50 point mutations that had no effect on heterodimer formation were able to rescue dU2AF50 mutant flies, the mutation that abolished interaction with dU2AF38 was incapable of genetic complementation. Analysis of heterodimer formation in mutant embryo extracts revealed a perfect correlation between the efficiency of subunit association and the ability to complement the dU2AF50 recessive lethal allele. We conclude that Drosophila U2AF heterodimer formation is essential for viability in vivo, consistent with a requirement for both subunits in splicing in vitro.
MATERIALS AND METHODS
Creation of coexpression plasmids.
The dU2AF38 (d38) coding sequence was PCR amplified and inserted into pRSETA (Invitrogen) between the NdeI and PstI sites to generate pRSETA-d38. To increase protein expression levels, an oligonucleotide linker (top strand, 5′ CTAGAGGGTATTAATAATGTATCGATTAAATAAGGAATAACA 3′; bottom strand, 5′ TATGTTATTCCTCCTTATTTAATCGATACATTATTAATACCCT 3′) encoding a small upstream open reading frame and Shine-Dalgarno sequence (23) was inserted between the XbaI and NdeI sites to create pRSETAX-d38. A BamHI fragment containing the kanamycin resistance gene from pUC-4K (Pharmacia) was treated with Klenow fragment DNA polymerase and inserted into the XmnI site in pRSETAX-d38 (disrupting the ampicillin resistance gene) to create pRSETAKX-d38. The dU2AF38 coding sequence from pRSETAKX-d38 was completely sequenced.
The coexpression plasmid that contains two independent promoters is a dimeric plasmid containing an ampicillin-resistant pRSETA-dU2AF50 (11) or dU2AF50-derivative (see below) plasmid and the kanamycin-resistant pRSETAKX-d38 plasmid. The two plasmids were cleaved with AlwNI and ligated. Transformation into DH5α was performed under dilute conditions to reduce cotransformation of self-ligated plasmids. Most kanamycin plus ampicillin-resistant transformants carried heterodimerized plasmids.
The expression vector pRSETA-dU2AF50ΔRS (lacking amino acids 1 to 34) was created by PCR to delete the N-terminal region of dU2AF50. To create dU2AF50ΔRSL, oligonucleotide linkers (top strand, 5′ GATCCGGTACCT 3′; bottom strand, 5′ CCGGAGGTACCG 3′) were annealed and inserted into pRSETA-dU2AF50 between BamHI and BspEII by partial cleavage. To create dU2AF50ΔRS, oligonucleotide linkers (top strand, 5′ GATCCGGTACC 3′; bottom strand, 5′ TCGAGGTACCG 3′) were annealed and inserted into pRSETA-dU2AF50 between the BamHI and XhoI sites. To create dU2AF50ΔL, oligonucleotide linkers (top strand, 5′ TCGAGGGGTACCT 3′; bottom strand, 5′ CCGGAGGTACCCC 3′) were annealed and inserted into pRSETA-dU2AF50 between the XhoI and BspEII sites by partial cleavage. To create the six-histidine [(his)6]-linker, pRSETA-dU2AF50ΔRS was cleaved with BspEII to remove an internal BspEII DNA fragment and religated to create a frameshift in the dU2AF50 coding sequence. The frameshift resulted in a carboxyl-terminal fusion of the following amino acids: GLASQNLHRRSTKLSE.
The hU2AF35 (h35) coding sequence was PCR amplified and inserted into pRSETA (Invitrogen) between the NdeI and HindIII sites to generate pRSETA-h35 (5′ primer, 5′ CCCGGATCCATGGCGGAGTATCTGGCC 3′; 3′ primer, 5′ CCCAAGCTTTCAGAATCGCCCAGATCTAAG 3′). The hU2AF65 (h65) coding sequence from pRSETA-h65 (11) was subcloned into pRSETAKX between the NdeI and EcoRI sites to create pRSETAKX-h65. The two hU2AF subunit expression plasmids were dimerized as described above.
To create the second coexpression plasmid (the bicistron), the dU2AF50 cDNA was inserted into pRSETA (Invitrogen) between the BamHI and HindIII sites to create pdr6. Oligonucleotide linkers containing a consensus Shine-Dalgarno sequence and NdeI and PstI restriction sites (top strand, 5′ AGCTTAGAGGTATTCATATGGAATTCCTGCAG 3′; bottom strand, 5′ AGCTCTGCAGGAATTCCATATGAATACCTCTA 3′) were annealed and inserted into the unique HindIII site in pdr6 to create pdr151. The dU2AF38 cDNA was inserted into pdr151 between the NdeI and PstI sites by partial cleavage of pdr151 to create pdr154.
Creation of point mutations in the dU2AF50 interaction domain.
Point mutations in dU2AF50 were generated by site-directed mutagenesis with uracil-substituted single-stranded DNA (14). The following mutagenic oligonucleotides were used: mutant 1, 5′ GAATCCCGGCGGCGGTGCAGCCGCATAAAGCGACGGCTT 3′; mutant 2, 5′ CGGGGTGATGTGCTCGGCTCCCGCCGCCGGTACATCCCAATAAAG 3′; mutant 3, 5′ GTATTGCATCGGGGTGGCGTGCGCGGCTCCCGGCGGCGGTAC 3′; and mutant 4, 5′ GGACGCCTGCATGGCTGCGGCTTGCGCCGGGGTGATGTGCTCG 3′. Site-directed mutagenesis was performed with single-stranded DNA derived from pdr6. Internal 289-bp XhoI-SphI DNA fragments spanning the interaction domain were sequenced completely to confirm the mutations and subcloned into the bicistron expression vector in a three-way ligation with an SphI-BstEII dU2AF50 DNA fragment from pdr6 and pdr154 cleaved with XhoI and BstEII to create pdr213 (mutant 1), pdr214 (mutant 2), pdr212 (mutant 3), and pdr211 (mutant 4). The 289 XhoI-SphI DNA fragments were also subcloned into the hemagglutinin (HA)-tagged dU2AF50 in vivo expression vector (pdr152, see below) in a similar three-way ligation (pdr152 was cleaved with XhoI and BstEII) to create pdr207–210. NotI DNA fragments from pdr152 and pdr207–210 that contained the dU2AF50 promoter, dU2AF50 coding sequence and dU2AF50 3′ untranslated region were inserted into a unique NotI site in the Drosophila transformation vector pw8 (4).
Creation of epitope-tagged dU2AF50.
The HA-tagged dU2AF50 transgene was created by insertion of an oligonucleotide linker (top strand, 5′ CTAGCTACCCCTATGACGTGCCGGATTACGCCG 3′; bottom strand, 5′ GATCCGGCGTAATCCGGCACGTCATAGGGGTAG 3′) between the NheI and BamHI sites into the dU2AF50 in vivo expression vector (pdr141) (21c) to generate pdr152.
Expression and copurification of U2AF heterodimers.
The U2AF subunits were coexpressed in E. coli BL21 (DE3) pLysS. Cells were grown in Luria broth at 30°C to an optical density at 600 nm at of 0.4, induced by the addition of isopropyl-β-d-thiogalactopyranoside (IPTG) to 0.5 mM, and harvested after 3 to 4 h. All subsequent manipulations were carried out at 4°C. Cells were harvested by centrifugation and resuspended in buffer I (50 mM Tris-HCl [pH 8.0], 1 M NaCl, 5 mM β-mercaptoethanol, 1 mM phenylmethylsulfonyl fluoride [PMSF]). A crude extract was prepared by freeze-thawing the cells, followed by sonication and centrifugation at 30,000 rpm in a Ti70 rotor for 30 min at 4°C. The soluble extract was loaded on an HR5/5 (Pharmacia) Ni2+-nitrilotriacetic acid (NTA)-agarose (Qiagen) column equilibrated with buffer I containing 10% glycerol. Bound protein was washed with buffer I containing 10% glycerol and 20 mM imidazole and eluted in buffer I containing 10% glycerol and 200 mM imidazole. Peak fractions were pooled and frozen in liquid nitrogen and stored at −80°C or diluted to 350 mM NaCl with buffer H (20 mM HEPES [pH 7.6], 1 mM EDTA, 0.5 mM dithiothreitol [DTT], 10% glycerol) and further purified on an HR5/5 Mono S column (Pharmacia) equilibrated with buffer H containing 350 mM NaCl. Bound protein was eluted with a linear NaCl gradient from 350 mM to 1.5 M NaCl. dU2AF50 monomer eluted at ∼550 mM NaCl, and dU2AF heterodimer eluted at ∼900 mM NaCl.
Stoichiometry of the purified dU2AF and dU2AF50 was determined by analytical gel filtration chromatography. Purified proteins (250 μg) were loaded onto a Superose 12 column (Pharmacia) equilibrated with 20 mM HEPES-NaOH (pH 7.6), 250 mM KCl, 10% glycerol, 1 mM DTT, 0.2 mM EDTA, 0.05% Nonidet P-40 (NP-40), 1 mM PMSF. Elution profiles were compared to gel filtration standards (Bio-Rad). dU2AF eluted as a heterodimer, and dU2AF50 eluted as a monomer.
In an attempt to reconstitute dU2AF heterodimers from independent subunit preparations, we mixed individually purified dU2AF50 and dU2AF38 at a concentration of 1 mg/ml and dialyzed the solution overnight in buffer A (20 mM HEPES-NaOH [pH 7.6], 400 mM KCl, 10% glycerol, 1 mM DTT, 0.2 mM EDTA, 0.05% NP-40, 1 mM PMSF) or buffer A with 6 M guanidine-HCl and dialyzed in steps (6, 4, 2, 1, 0.5, and 0 M guanidine-HCl). Heterodimer formation was assayed by coimmunoprecipitation with anti-dU2AF50 antibody resin.
Protein-RNA binding analysis.
Dissociation constants (KDs) for interactions of the dU2AF large subunit and mutant derivatives were determined by use of native gel electrophoresis. Binding reactions were performed in a volume of 10 μl and contained the indicated concentrations of proteins, 0.1 nM 32P-labeled oligonucleotide, 20 mM HEPES-NaOH (pH 7.6), 100 mM KCl, 0.2 mM EDTA, 10% glycerol, 0.5 mM DTT, 50 μg of bovine serum albumin per ml, and 10 mg of heparin per ml. Incubations were continued for 1 h at 4°C. One half the reaction mixture was electrophoresed through a 4% polyacrylamide gel (60:1 ratio of acrylamide-bis–0.5× Tris-Borate-EDTA [pH 8.3]) at 4°C for 100 min at 20 V/cm. RNA binding was quantitated with the use of the Fuji Phosphorimager, and KDs were obtained from protein concentrations at which 50% of MINX RNA was bound (10). The MINX RNA sequence is shown in Fig. 5B.
FIG. 5.

dU2AF50 mutants bind pyrimidine tract RNA with similar affinity to wild-type dU2AF50 (WT). (A) Electrophoretic mobility shift analysis of dU2AF50 and dU2AF50 interaction mutants with the MINX pyrimidine tract. Wild-type dU2AF50 (WT) protein concentrations were 1.25, 0.25, and 0.05 μM (lanes 2 to 4). dU2AF50 Δinteraction domain (ΔI) protein concentrations were 2.5, 0.5, and 0.1 μM (lanes 5 to 7). dU2AF50 mutant 1 (mut.1) protein concentrations were 5, 1, and 0.2 μM (lanes 8 to 10). dU2AF50 mutant 2 (mut.2) protein concentrations were 2.5, 0.5, and 0.1 μM (lanes 12 to 14). dU2AF50 mutant 3 (mut.3) protein concentrations were 5, 1, and 0.2 μM (lanes 15 to 17). dU2AF50 mutant 4 (mut.4) protein concentrations were 10, 2, and 0.4 μM (lanes 18 to 20). Proteins were incubated with 100 pM 32P-labeled RNA oligonucleotide. Protein-RNA complexes (C) and unbound RNA (F) were separated by electrophoresis through a native polyacrylamide gel and visualized by autoradiography. The KDs were determined to be 7.1 × 10−6 M (WT), 9.0 × 10−6 M (ΔI), 8.1 × 10−6 M (mut.1), 8.5 × 10−6 M (mut.2), 8.9 × 10−6 M (mut.3), and 7.0 × 10−6 M (mut.4). (B) Sequence of the MINX pyrimidine tract. (C) An SDS–10% polyacrylamide gel of the recombinant (his)6-tagged dU2AF50 proteins stained with Coomassie blue. Molecular size markers are indicated in kilodaltons.
Genetic analysis of dU2AF50 interaction mutants.
Germ line transformation of HA-tagged dU2AF50 and interaction mutant derivatives into w1118 embryos were as described previously (28). Twenty to thirty independent transformant lines were generated for each mutant, and all autosomal insertion lines were tested for complementation of the recessive lethal dU2AF50 allele, 9-21XR15. y w 9-21XR15 f/Bins (y, w, sn, B) virgin females were mated to w/Y; P(w+; dU2AF50)/+ males. Rescued y w 9-21XR15 f/Y; P(w+; dU2AF50)/+ males were scored, and percent viability was determined by comparison to their unbalanced y w 9-21XR15 f/w; P(w+; dU2AF50)/+ sisters. At least 150 progeny were scored in each complementation cross.
Typically, 50 to 70% of the wild-type dU2AF50 transgene lines are capable of rescuing the 9-21XR15 allele (21c). The dU2AF50 in vivo expression vector is sensitive to genomic insertion site (21b). None of the 13 independent mutant 1 transgene lines tested was able to complement the 9-21XR15 allele. Ten independent mutant 2 transgene lines of the 18 transgene lines tested complemented the 9-21XR15 allele. The rescue ranged from 29 to 100%. The average rescue for the 10 independent rescuing lines was 67%. Eight of the 17 independent mutant 3 transgene lines tested complemented the 9-21XR15 allele. The rescue ranged from 10 to 65%. The average rescue for the eight independent rescuing lines was 41%. None of the 12 independent mutant 4 transgene lines tested complemented the 9-21XR15 allele. Complementation of the 9-21XR15 allele by mutants 2, 3, and 4 nonrescuing transgene lines was observed when the transgene dose was increased. The rescue when mutant 4 transgene dose was doubled ranged from 5 to 21%. The average rescue was 11%. Complementation of the 9-21XR15 allele with an increased transgene dose was performed as follows. y w 9-21XR15 f/Bins (y, w, sn, B); P(w+; dU2AF50)/+ virgin females were crossed to w/Y; P(w+; dU2AF50)/+ males. Rescued y w 9-21XR15 f/Y; P(w+; dU2AF50)/+; P(w+; dU2AF50)/+ males were compared to y w 9-21XR15 f/w; P(w+; dU2AF50)/+; P(w+; dU2AFA50)/+ siblings. All crosses were performed at 25°C on standard Drosophila food.
Immunoblot analysis.
Five adult flies were mashed in a microcentrifuge tube with a blue pestle homogenizer in 100 μl of sodium dodecyl sulfate (SDS) sample buffer and boiled for 3 min to create a whole-fly extract. One-eighth fly equivalent was subjected to electrophoresis through an SDS–10% polyacrylamide gel, transferred to nitrocellulose, and blocked in 5% nonfat milk in phosphate-buffered saline–0.5% Tween-20. The blocked membrane was probed with affinity-purified anti-dU2AF50 or anti-HA antibodies. Primary antibody was detected with horseradish peroxidase-conjugated goat, anti-rabbit (for anti-dU2AF50) or anti-mouse (for anti-HA) immunoglobulin G with enhanced chemiluminescence detection kit as described by the manufacturer (Pierce).
The anti-dU2AF38 and anti-dU2AF50 polyclonal antibodies were generated in rabbits with recombinant (his)6-tagged dU2AF38 and (his)6-tagged dU2AF50 E. coli-expressed proteins as previously described (11). Affinity purification was as described previously (9). Affinity-purified, anti-dU2AF50 antibody was diluted 50,000-fold before incubation. The affinity-purified anti-dU2AF38 was diluted 10,000-fold before incubation. The anti-HA monoclonal antibody (16B12) (Babco) was diluted 1,000-fold before incubation.
Coimmunoprecipitation of U2AF heterodimers.
Coimmunoprecipitations were performed in 0- to 12-h embryo extracts. Embryos were dechorionated in 50% bleach for 2 min, washed thoroughly with H2O followed by 0.05% NP-40, and transferred to microcentrifuge tubes. All subsequent manipulations were performed at 4°C. A total of 100 to 200 μl of embryos was homogenized in 500 μl of lysis buffer (1 M NaCl, 20 mM HEPES-NaOH [pH 7.6], 0.05% NP-40, 0.5 mM PMSF, 0.5 mM EGTA, 0.5 mM EDTA, 0.05 mM DTT, 1× protease inhibitors) in a 2-ml homogenizer (B pestle). Extracts were transferred to microcentrifuge tubes and spun for 10 min. The supernatant was precleared with formalin-fixed Staphylococcus aureus cells, divided into 100-μl aliquots, and incubated overnight with anti-HA (12CA5) or control (anti-KP [Drosophila P element]) antibody bound to protein A trisacryl beads (Pierce). The beads were pelleted, and the immunoprecipitate was washed three times with lysis buffer and resuspended in 25 μl of SDS sample buffer. One-fifth of the pellet was analyzed by immunoblot analysis with affinity-purified anti-dU2AF38 and anti-dU2AF50 antibodies as described above.
RESULTS
Purification of recombinant heterodimers from E. coli requires coexpression.
In an attempt to purify recombinant Drosophila U2AF heterodimers for biochemical analysis, the large and small subunits were separately expressed and purified from E. coli. Although these proteins exist in a tight complex in nuclear extract, we were unable to reconstitute heterodimers from purified recombinant subunits even after denaturation and step renaturation (see Materials and Methods). We reasoned that association of the two subunits might require coexpression. Coexpression of proteins in tissue culture cells has been used successfully for purification of other multimeric complexes (25). Our initial attempts at coexpression of the two U2AF subunits in E. coli by cotransformation of two independent expression plasmids resulted in variable expression and plasmid loss (data not shown). To circumvent the problem of plasmid incompatibility in E. coli (12), we developed two coexpression systems (Fig. 1). In the first, the large- and small-subunit cDNAs were placed under the control of two independent T7 promoters in a single dimeric plasmid (Fig. 1A). Expression of the two subunits in this system was stoichiometric (Fig. 2A, lanes 1, 2, 4, and 12). In the second coexpression system, the two subunits were placed in a bicistron under the control of a single T7 promoter (Fig. 1B). Although expression of the large subunit was much higher than that of the small subunit in this context (Fig. 4A, lanes 2, 3, and 4), subcloning mutant fragments into this expression vector was much more facile than subcloning mutant fragments into the dimeric plasmid used in the first coexpression system. Both vectors were used in this work. In both expression systems, a (his)6 tag was fused to the amino terminus of one of the two U2AF subunits. Recombinant Drosophila or human U2AF heterodimers could be purified by Ni2+-NTA-agarose chromatography (Fig. 2, lanes 3 and 13 and Fig. 4, lane 4). The heterodimer species could be purified away from the excess free (his)6-tagged monomer large subunit by cation-exchange chromatography (data not shown; see Materials and Methods). The stoichiometry of the recombinant heterodimers was confirmed by analytical gel filtration chromatography (see Materials and Methods). The biochemical characterization of these recombinant U2AF heterodimers will be described elsewhere (21d).
FIG. 1.
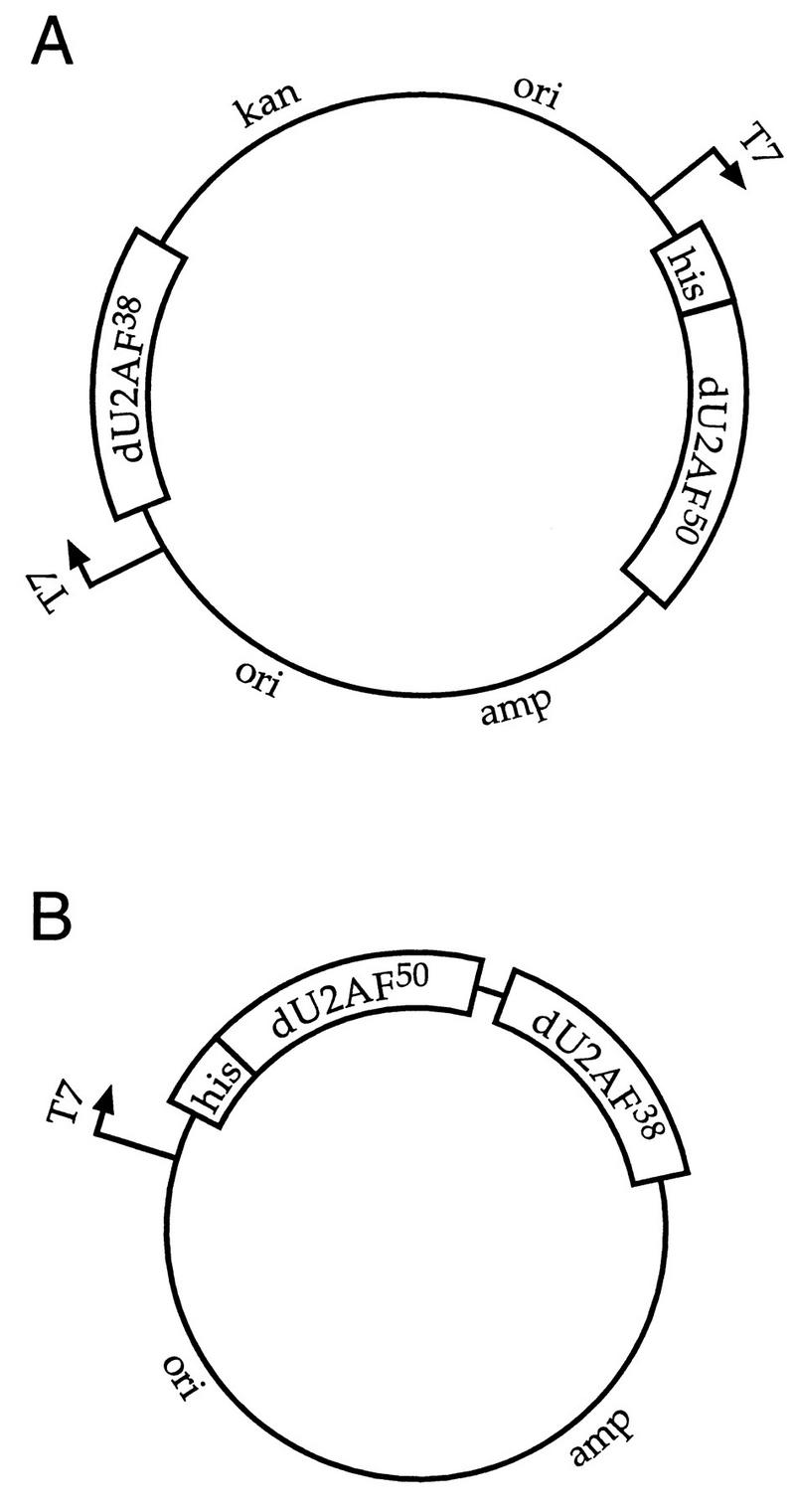
Coexpression plasmids used to purify recombinant U2AF heterodimers. (A) Two independent phage T7 promoters (indicated by arrows) were fused to the large and small U2AF subunits. Only one of the two subunits is (his)6-tagged (his). This expression plasmid is a dimeric plasmid containing two selectable markers, ampicillin resistance (amp) and kanamycin resistance (kan), and two origins of replication (ori). (B) A single phage T7 promoter was fused to a bicistron containing both U2AF subunits. Only one of the two subunits is (his)6 tagged.
FIG. 2.

Mapping the domain on dU2AF50 that is necessary and sufficient for interaction with dU2AF38. (A) An SDS–12% polyacrylamide gel of dU2AF50 wild-type and deletion mutant proteins stained with Coomassie blue. U2AF subunits were coexpressed in E. coli with the dimeric coexpression plasmid. dU2AF50 was (his)6 tagged (His-dU2AF50) in the dU2AF heterodimer, and hU2AF35 was (his)6 tagged in the hU2AF heterodimer (His-hU2AF35). dU2AF heterodimer formation was assessed by copurification of dU2AF38 on Ni2+-NTA-agarose. E. coli lysates from uninduced (−) and induced (+) cells as well as the eluate (EL) from Ni2+-NTA-agarose purification are shown. The position of dU2AF38 is indicated by an arrow. dU2AF38 runs heterogeneously due to carboxyl-terminal proteolysis. The identity of these polypeptides as dU2AF38 was confirmed by immunoblot analysis (data not shown; Fig. 4A and B). A similar heterogeneity was observed when an amino-terminal (his)6-tagged dU2AF38 was purified separately (data not shown). The sizes of the protein molecular size markers are indicated in kilodaltons. WT, wild type. (B) Schematic representation of the results of the copurification interaction assay. The (his)6 tag (His), RS domain (RS), and three RNA recognition motifs (RRM1–3) of dU2AF50 are indicated. The different dU2AF50 domains are not drawn to scale.
FIG. 4.

Identification of point mutations that disrupt heterodimer formation in vitro. (his)6-tagged dU2AF50 or dU2AF50 interaction mutant derivatives were coexpressed with dU2AF38 with the bicistronic coexpression plasmid. Heterodimer formation was assessed by copurification of dU2AF38 on Ni2+-NTA-agarose. E. coli lysates from uninduced (−) and induced (+) cells and the eluate (EL) from Ni2+-NTA-agarose purification were electrophoresed on an SDS–10% polyacrylamide gel and stained with Coomassie blue (A) or transferred to nitrocellulose and probed with affinity-purified anti-dU2AF38 antibodies (B and C). The position of dU2AF38 is indicated with an arrow. Molecular size markers are indicated in kilodaltons.
Identification of the dU2AF50 interaction domain.
Using the dimeric plasmid coexpression system, we mapped the interaction domain on the Drosophila U2AF large subunit. Deletions in (his)6-tagged dU2AF50 were coexpressed with full-length dU2AF38 and assayed for interaction by copurification on Ni2+-NTA-agarose. Deletion of the RS domain (amino acids 1 to 34) on dU2AF50 had no effect on copurification of dU2AF38 (Fig. 2A, lane 5). However, a deletion of the 28-amino-acid linker between the RS domain and the first RNA binding domain of dU2AF50 completely disrupted interaction with dU2AF38 (Fig. 2A, lane 7). This 28-amino-acid fragment, when fused to a (his)6 tag, efficiently copurified the small subunit (Fig. 2A, lane 11). Thus, the linker between the dU2AF50 RS domain and RNA binding domains is both necessary and sufficient for interaction with dU2AF38 (Fig. 2B). Our findings are consistent with and further refine the interaction domain determined for hU2AF65 by the yeast two-hybrid interaction assay (7). This domain is the most highly conserved part of the human and Drosophila large-subunit proteins (10) and is highly conserved in the U2AF large-subunit homologs from S. pombe (18) and Caenorhabditis elegans (38) (Fig. 3).
FIG. 3.

Amino acid sequence comparison of the U2AF large-subunit interaction domains and locations of the alanine-substitution mutations. The 28-amino-acid linker region from four U2AF large-subunit homologs is shown. Amino acid identities and similarities are shown in dark-gray and light-gray boxes, respectively. Dashes denote gaps. Amino acid positions are shown on the right. Triple-alanine substitution mutations (mut.) used to identify residues in dU2AF50 required for heterodimer formation are indicated above the sequence comparison. Amino acid sequence data are from Kanaar et al. (11) (Drosophila), Zamore et al. (36) (human), Zorio et al. (38) (C. elegans), and Potashkin et al. (18) (S. pombe).
Identification of point mutations in dU2AF50 that disrupt heterodimer formation in vitro.
To identify residues in dU2AF50 that are important for heterodimer formation, we scanned the 28-amino-acid linker for mutations that disrupt interaction with dU2AF38. Since association of the U2AF subunits is resistant to high-salt conditions (35), we focused on highly conserved, hydrophobic residues in the interaction domain. Four triple-alanine-substitution mutations were created (Fig. 3) and tested for copurification of dU2AF38 with the bicistronic copurification assay. Mutant 1 (W44A, D45A, and V46A) completely abolished interaction with the small subunit (Fig. 4A, compare lanes 4 and 5). Since dU2AF38 is expressed at substoichiometric levels in the bicistron coexpression plasmid and is difficult to visualize by SDS-polyacrylamide gel electrophoresis (Fig. 4A), we analyzed copurification of the small subunit by immunoblot with anti-dU2AF38 antibodies (Fig. 4B and C). No dU2AF38 was found to copurify with mutant 1 with this more sensitive assay (Fig. 4B, compare lanes 4 and 5). Even though mutants 2 (P48A, P49A, and F51A), 3 (F51A, E52A, and I54A), and 4 (M57A, Y59A, and K60A) all have substitutions of highly conserved residues for alanine, none had a detectable effect on heterodimer formation in E. coli (Fig. 4C, compare lanes 6 to 9). All mutant plasmids expressed approximately equal levels of dU2AF38 and the dU2AF50 mutant proteins (Fig. 4A and B, compare lanes 2 and 3, and 4C, compare lanes 2 to 5, and data not shown).
It was possible that the failure of the interaction domain (linker) deletion mutant and mutant 1 to associate with the small subunit was due to misfolding of the dU2AF50 protein. To assess whether the interaction mutants were affecting the global folding of the dU2AF50 protein, we assayed the recombinant mutant dU2AF50 monomers for RNA binding, since the three RNA binding domains of U2AF make up the majority of the protein and are required for RNA binding (11, 36). The dU2AF50 monomers were separated from the dU2AF heterodimers by ion-exchange chromatography (Fig. 5C, see Materials and Methods), and RNA binding activity was determined by electrophoretic mobility shift analysis (Fig. 5A). The purified, recombinant proteins were incubated for 1 h at various protein concentrations with a 32P-labeled pyrimidine tract RNA oligonucleotide derived from the adeno L1-L2 intron (MINX). The sequence of the MINX pyrimidine tract is shown in Fig. 5B. The four triple-alanine point mutants and the interaction domain deletion mutant all bound the MINX pyrimidine tract with comparable affinity to wild-type dU2AF50. In particular, the dissociation constant of mutant 1 was within 15% of wild-type dU2AF50 (see legend for Fig. 5). We conclude that the dU2AF50 point mutations and the deletion mutation do not effect the global folding of the dU2AF50 protein as assessed by the ability of the soluble, mutant proteins to bind polypyrimidine tract RNA.
U2AF heterodimer formation is essential in vivo.
If U2AF heterodimer formation is required in vivo, then a dU2AF50 mutant that is incapable of interacting with dU2AF38 should fail to complement a recessive lethal dU2AF50 mutation (11, 21). To test the requirement for heterodimer formation in vivo, the triple-alanine mutations were introduced into an HA epitope-tagged dU2AF50 transformation vector. The dU2AF50 transgene was epitope tagged to assess expression levels of the mutant proteins in the presence of endogenous dU2AF50 and to analyze heterodimer formation in embryo extracts (see below). The wild-type, HA-tagged dU2AF50 transgene rescues dU2AF50 mutant flies as efficiently as an untagged transgene (Fig. 6 and unpublished data). A total of 20 to 30 independent transformant lines of each mutant were generated by P element-mediated germ line transformation. All autosomal insertion lines were tested for the ability to complement a recessive lethal dU2AF50 allele. Balanced, heterozygous dU2AF50 mutant females were crossed to males carrying a dU2AF50 transgene. The viability of hemizygous, dU2AF50 mutant male progeny carrying the dU2AF50 transgene was compared to the viability of their unbalanced sisters. Consistent with the ability of mutants 2 and 3 to interact with dU2AF38 in our copurification assay, ∼50% of the dU2AF50 transgene lines that contained these alanine substitution mutations rescued the recessive lethal dU2AF50 allele (see Materials and Methods), and several lines of each mutant could be maintained as stocks in which their sole source of dU2AF50 was the mutant transgene. The average rescues for the mutant 2 and 3 rescuing lines were 67 and 41%, respectively (Fig. 6). The expression levels of the mutant proteins in these transgenic lines were assessed by immunoblot analysis with anti-dU2AF50 and anti-HA antibodies. The mutant protein levels were similar to or lower than the levels in the wild-type, HA-tagged, dU2AF50 transgene lines (Fig. 7, compare lanes 2, 6, and 8 and data not shown).
FIG. 6.

In vivo analysis of dU2AF50 interaction domain mutants. HA-tagged dU2AF50 and mutant-derivative transgenes were tested for complementation of a recessive lethal dU2AF50 allele. The amino acid sequence of the interaction domain (linker) is shown. The alanine substitution mutations are depicted in white. The average rescue of the rescuing transgene lines is shown. Mutant 4 could rescue the dU2AF50 mutant allele only when the transgene was present in two copies. The average rescue when two mutant 4 transgenes were present is shown in parentheses.
FIG. 7.

Protein expression levels of epitope-tagged dU2AF50 mutants. Immunoblot analysis of whole-fly extracts probed with anti-dU2AF50 (α-d50) and anti-HA (α-HA) antibodies. Whole-fly extracts are from w1118, dU2AF50+ (+), flies (lanes 1 and 10), w1118 flies carrying HA-dU2AF50 wild-type (lane 2) or interaction mutant derivatives (lanes 4, 5, 6, and 8) or dU2AF50 mutant flies (d50−) rescued by HA-dU2AF50 wild-type (lane 3) or mutant transgene derivatives (lanes 7 and 9). The presence of the HA epitope on dU2AF50 results in the slower mobility observed. Molecular size markers are indicated in kilodaltons.
Mutant 4 had no detectable effect on heterodimer formation in our copurification assay; yet, none of the 12 independent mutant 4 transgene lines tested were capable of rescuing the recessive lethal dU2AF50 allele. The protein expression levels in these transgene lines were similar to the levels found in rescuing mutant 2 and 3 transgene lines (Fig. 7, compare lane 4 to lanes 6 and 8 and data not shown). To determine whether higher mutant 4 protein levels were required for complementation, independent mutant 4 transgenes were combined and tested for complementation of the dU2AF50 recessive lethal allele (see Materials and Methods). When the mutant 4 transgene dose was doubled, complementation of the dU2AF50 mutant allele was observed. The average rescue when two mutant 4 transgenes were combined was 11% (Fig. 6).
Consistent with the complete disruption of heterodimer formation, mutant 1 was completely incapable of complementing the dU2AF50 recessive lethal allele (Fig. 6). No rescue was observed in the 13 independent mutant 1 transgene lines tested. The mutant 1 protein levels in these transgene lines were similar to mutant 2 and 3 transgene lines that were capable of complementing the dU2AF50 mutant (Fig. 7, compare lane 5 to lanes 6 and 8 and data not shown). To rule out the possibility that the mutant 1 protein levels were not high enough to rescue the dU2AF50 mutant allele, three transgene lines that expressed high levels of mutant 1 protein were combined in pairwise combinations and tested for complementation. Increasing the mutant 1 transgene dose still failed to restore viability to the dU2AF50 mutants (data not shown).
Interaction mutants are impaired in heterodimer formation in embryo extracts.
To assess whether the dU2AF50 point mutations were affecting heterodimer formation in vivo, we analyzed U2AF subunit association in embryo extracts. Embryos (0- to 12-h) were collected from w1118 (dU2AF50+) flies and w1118 flies carrying the wild-type, HA-tagged, dU2AF50 transgene, and dU2AF50 interaction mutant derivatives. Embryo extracts were prepared and incubated with anti-HA antibody or a control antibody of the same isotype. Immunoprecipitation of the HA-tagged dU2AF50 was assayed by immunoblot analysis with anti-dU2AF50 antibodies (Fig. 8A). HA-tagged dU2AF50 was efficiently precipitated with anti-HA antibody from all extracts derived from the HA-tagged dU2AF50 transgene lines (Fig. 8A, lanes 7, 9 to 12) but not from extracts derived from nontransgenic w1118 flies (Fig. 8A, lane 5) and not when the control antibody was used (Fig. 8A, lanes 4, 6, and 8). To facilitate a direct comparison of the dU2AF38 coimmunoprecipitated along with HA-tagged dU2AF50 from the different extracts, the HA antibody was used at substoichiometric concentrations. Under these conditions, equal amounts of HA-tagged dU2AF50 were precipitated from the different extracts (Fig. 8A, compare lanes 7, 9 to 12).
FIG. 8.

Analysis of heterodimer formation by coimmunoprecipitation from embryo extracts. (A) Immunoblot analysis of dU2AF50 and dU2AF38. The embryo extracts used in panel A are from wild-type dU2AF50+ (+) flies that either lack a transgene or contain a wild-type, HA-tagged dU2AF50 transgene (P[HA-d50WT]) or a mutant derivative (P[HA-d50mut]). The transgenes are indicated above the lanes. Lanes 1, 2, 3, and 13 are representative embryo extracts. Lanes 4, 6, and 8 are representative immunoprecipitates from embryo extracts with a control antibody (α-KP). Lanes 5, 7, 9, 10, 11, and 12, are immunoprecipitates from embryo extracts with the anti-HA antibody (α-HA). The immunoprecipitates and extracts were subjected to electrophoresis through an SDS–10% polyacrylamide gel in duplicate and probed with anti-dU2AF50 (α-d50) or anti-dU2AF38 (α-d38) antibodies. (B) Immunoblot analysis of coimmunoprecipitates from embryo extracts derived from rescued dU2AF50 mutant flies. The extracts used in panel B (except lanes 1 and 5) are from dU2AF50 mutant (d50−) fly lines that are rescued by a wild-type HA-dU2AF50 transgene or HA-dU2AF50 mutant derivatives. The transgenes are indicated above the lanes. Lanes 1 to 4 are the embryo extracts, and lanes 5 to 8 are the immunoprecipitates with the anti-HA antibody (α-HA). The extracts and immunoprecipitates were subjected to electrophoresis through an SDS–10% polyacrylamide gel and transferred to nitrocellulose. The membrane was stained with Ponceau S and cut in half at the 45-kDa marker. The top half of the membrane was probed with anti-dU2AF50 antibodies, and the bottom half was probed with anti-dU2AF38 antibodies. The two pieces of nitrocellulose were aligned prior to enhanced chemiluminescence detection. The sizes of the protein molecular size markers are indicated in kilodaltons.
U2AF subunit association was assessed by coimmunoprecipitation of dU2AF38. The presence of the small subunit in the immunoprecipitates was determined by immunoblot analysis with anti-dU2AF38 antibodies. The HA antibody efficiently coimmunoprecipitated dU2AF38 from extracts derived from lines carrying the wild-type, HA-tagged dU2AF50 transgene but not from extracts derived from flies lacking an HA-tagged transgene (Fig. 8A, compare lanes 5 and 7) and not when the control antibody was used (Fig. 8A, lanes 4, 6, and 8). Consistent with the inability of mutant 1 to interact with dU2AF38 in our copurification assay and its inability to complement a recessive lethal dU2AF50 allele, mutant 1 failed to stably associate with dU2AF38 in embryo extracts. dU2AF38 was not detected above background levels in the immunoprecipitate from embryo extracts derived from mutant 1 transgene lines (Fig. 8A, compare lanes 5 and 9). dU2AF38 was coimmunoprecipitated from embryo extracts derived from mutant 2, 3, and 4 transgene lines. The ability of these three mutants to stably associate with dU2AF38 is consistent with the results of our interaction assay (Fig. 4C, lanes 7 to 9), and the efficiency of coimmunoprecipitation perfectly correlates with the efficiency of genetic complementation of the dU2AF50 mutant allele (Fig. 6 and 8A, lanes 7 and 10 to 12). The more stable the association between the HA-tagged dU2AF50 mutant and dU2AF38, the more efficient the complementation of the dU2AF50 mutant allele.
Although mutants 2 and 3 rescued the dU2AF50 recessive lethal allele with relatively high efficiency, both interacted with dU2AF38 less well than wild-type, HA-tagged dU2AF50 (Fig. 8A, compare lanes 10 and 11 to lane 7). It was possible that the endogenous, untagged dU2AF50 protein present in the embryos was competing with the dU2AF50 mutants for interaction with dU2AF38. If this were true, the amount of dU2AF38 coimmunoprecipitated from these extracts might not reflect the amount that could interact with dU2AF38 in the absence of endogenous dU2AF50. To test this hypothesis, we performed coimmunoprecipitations from embryo extracts in which the sole source of dU2AF50 was derived from the HA-tagged transgene. Extracts were prepared from 0- to 12-h embryos derived from dU2AF50− flies carrying an HA-tagged, wild-type transgene or mutant 2 or 3 derivative. Interestingly, even under conditions in which there was no wild-type dU2AF50 to compete with the dU2AF50 mutants for interaction with dU2AF38, subunit association was impaired (Fig. 8B, compare lanes 6, 7, and 8). We conclude that a two- to fourfold reduction in U2AF heterodimer formation has only a modest effect on viability. Taken together, these data indicate that the dU2AF heterodimer is the functional form of U2AF and that mutations that affect heterodimer formation impair U2AF function in vivo.
DISCUSSION
We have described two coexpression systems for purifying U2AF heterodimers from E. coli. These systems may be useful for other heterodimeric complexes that require cotranslation for association, solubility, or stability. Here, these copurification systems were used as an interaction assay to define the domain on the large subunit of Drosophila U2AF that interacts with the small subunit. Although there are many assays available to study domains involved in protein-protein interaction, the advantage of the coexpression copurification assay is that it results in purified protein. This purified protein can then be tested for biochemical activity (Fig. 5).
With this interaction assay, the 28-amino-acid linker between the dU2AF50 RS domain and first RNA binding domain was found to be both necessary and sufficient for interaction with dU2AF38. Our mapping results are in agreement with the interaction domain on hU2AF65 determined by a membrane-immobilized protein interaction assay and further refined by the yeast two-hybrid system (7, 37). Although this 28-amino-acid fragment is highly conserved in the large-subunit homolog from S. pombe (pU2AF59) (Fig. 3) (18), it was found to be necessary but not sufficient for interaction with the S. pombe small-subunit homolog (pU2AF23) (32) in the yeast two-hybrid assay. A truncation of pU2AF59 that retained the linker was not sufficient for interaction with pU2AF23. However, since the expression level of this truncated protein was not examined, it is difficult to interpret this negative result.
We have scanned the 28-amino-acid interaction domain on dU2AF50 for mutations that abolish heterodimer formation. Only one of the four triple-alanine mutants tested failed to interact with dU2AF38 in our copurification assay. When these mutants were then analyzed for subunit association in embryo extracts, all four were found to be impaired in heterodimer formation. It is possible that an impairment in subunit association was masked by the high expression level of (his)6-dU2AF50 relative to dU2AF38 in the bicistronic coexpression vector. If the point mutants had been analyzed with the dimeric coexpression plasmid, in which both subunits are expressed stoichiometrically, subtle differences in subunit association might have been revealed. Nevertheless, the bicistronic coexpression vector proved qualitatively informative, as the triple-alanine mutation that disrupted interaction in our copurification assay was the one that completely abolished heterodimer formation in transgenic embryo extracts.
Interaction between the U2AF subunits is required in vivo.
The dU2AF50 mutant that failed to interact with dU2AF38 in our copurification assay and in embryo extracts was also unable to complement a recessive lethal dU2AF50 allele. These data provide strong evidence that stable association of the U2AF subunits is essential for viability. It is possible that the inability of the dU2AF50 interaction mutant to rescue is due to a reduction in activity or to disruption of an interaction with a protein other than dU2AF38 (7, 29). However, the strong correlation between the ability to form stable heterodimers in embryo extracts and the ability to complement the dU2AF50 recessive lethal allele of the four mutants tested supports our conclusion that U2AF heterodimer formation is required in vivo.
Though we have detected modest splicing defects in dying dU2AF50 mutant larvae, it has not been possible to convincingly show that the cause of lethality in Drosophila U2AF subunit mutants is a splicing defect (21a). This is likely due to the fact that the dying mutant larvae slowly run out of the U2AF protein and/or RNA that was maternally deposited in the mutant embryo. In addition, the splicing of certain introns may be more sensitive to the level of U2AF than others, making detection of a defect in splicing nontrivial. Even with a tight temperature-sensitive allele in certain S. cerevisiae splicing factors, it is not always possible to detect a splicing defect in all introns at the nonpermissive temperature (8a). The accumulated biochemical evidence demonstrating an essential requirement for the U2AF large subunit in constitutive splicing in vitro (11, 36, 39) and the requirement for the S. pombe U2AF large-subunit homolog for splicing in vivo (18) make it likely that the cause of death of dU2AF50 mutants in Drosophila is a defect in splicing. However, we cannot rule out the possibility that U2AF is actually dispensable for splicing in vivo but is required in some unidentified capacity.
Reexamination of the U2AF-depleted extracts.
The requirement for association of the two U2AF subunits in vivo is consistent with a role for both subunits in reconstitution of splicing in the immunodepleted extracts in vitro (39). The convergence of biochemical and genetic data supporting a role for the U2AF heterodimer in splicing prompts a reexamination of the two U2AF-depleted extracts and their different requirements for reactivation.
Biochemical experiments indicate that U1 snRNP bound to the 5′ splice site or splicing factors bound to exonic enhancer elements promote hU2AF65 binding to the intron pyrimidine tract through a network of protein-protein interactions (8, 16). A role for the small subunit in mediating these interactions was suggested by protein-protein interaction studies and UV RNA-protein cross-linking experiments (33, 39). It has been proposed that the essential requirement for the small subunit in splicing is to stabilize hU2AF65 binding to pyrimidine tracts through bridging interactions with constitutive and alternative splicing factors (33, 39). A reduction in RNA binding proteins that could compete with hU2AF65 for pyrimidine tract binding might abrogate the requirement for the small subunit in vitro. This could explain the ability of recombinant hU2AF65 to reactivate constitutive splicing in extracts depleted of U2AF activity by poly(U)-Sepharose chromatography (31, 36). In addition, reactivation of the poly(U)-depleted extracts requires 50 to 100 times more recombinant hU2AF65 (or hU2AF65 lacking the hU2AF35 interaction domain) than U2AF heterodimer purified from nuclear extract (7, 26, 30, 31, 34). The requirement for high concentrations of recombinant hU2AF65 (or dU2AF50) for reactivation has been observed in three independent laboratories with at least two different fusion proteins (11, 15, 31). It is also unlikely that the amino-terminal fusions [glutathione S-transferase or (his)6] are responsible for the reduced activity, since the amino-terminal, HA epitope-tagged dU2AF50 was fully active in vivo. Although this difference in in vitro splicing activity can be explained by a low percentage of active recombinant hU2AF65 or by the presence of inhibitory activities in the protein preparations, it is also consistent with a role for the small subunit in splicing. In fact, even in the immunodepleted extracts, addition of high-enough concentrations of hU2AF65 lacking the ability to interact with the small subunit can bypass the requirement for hU2AF35 (39). (Lower concentrations of wild-type hU2AF65, in the absence of exogenous hU2AF35, will also restore splicing to the immunodepleted extracts, but reactivation in this case might be a consequence of reassociation of recombinant hU2AF65 with endogenous hU2AF35 [39].) Thus, hU2AF65 alone is sufficient to reactivate splicing in either U2AF-depleted extract, but the requirement for the small subunit can be revealed at concentrations of hU2AF65 that do not support reactivation on its own.
The ability of recombinant hU2AF65 to restore splicing to U2AF-depleted extracts, though originally misleading, indicates that U2AF activity—pyrimidine tract binding and U2 snRNP recruitment—can be imparted by the large subunit alone and is consistent with a role of cofactor for the small subunit. This model has both mnemonic and predictive value and is consistent with all the genetic and biochemical experiments performed to date.
ACKNOWLEDGMENTS
We acknowledge members of the Rio and Cline labs for support and encouragement. Thanks go to M. Adams and E. Beall for advice on the immunoprecipitations, to T. Cline for providing a home for the Drosophila experiments, to P. Zuo for discussions, and to M. Adams for critical reading of the manuscript.
This work was initiated by support from the ACS (no. DB112) and was subsequently supported by NIH (R01 HD28063).
REFERENCES
- 1.Abovich N, Liao X C, Rosbash M. The yeast MUD2 protein: an interaction with PRP11 defines a bridge between commitment complexes and U2 snRNP addition. Genes Dev. 1994;8:843–854. doi: 10.1101/gad.8.7.843. [DOI] [PubMed] [Google Scholar]
- 2.Abovich N, Rosbash M. Cross-intron bridging interactions in the yeast commitment complex are conserved in mammals. Cell. 1997;89:403–412. doi: 10.1016/s0092-8674(00)80221-4. [DOI] [PubMed] [Google Scholar]
- 3.Adams M D, Rudner D Z, Rio D C. Biochemistry and regulation of pre-mRNA splicing. Curr Opin Cell Biol. 1996;8:331–339. doi: 10.1016/s0955-0674(96)80006-8. [DOI] [PubMed] [Google Scholar]
- 4.Ashburner M. Drosophila: a laboratory handbook. Cold Spring Harbor, N.Y: Cold Spring Harbor Press; 1989. [Google Scholar]
- 5.Berglund J A, Chua K, Abovich N, Reed R, Rosbash M. The splicing factor BBP interacts specifically with the pre-mRNA branchpoint sequence UACUAAC. Cell. 1997;89:781–787. doi: 10.1016/s0092-8674(00)80261-5. [DOI] [PubMed] [Google Scholar]
- 6.Chabot B. Directing alternative splicing: cast and scenarios. Trends Genet. 1996;12:472–478. doi: 10.1016/0168-9525(96)10037-8. [DOI] [PubMed] [Google Scholar]
- 7.Fleckner J, Zhang M, Valcarcel J, Green M. U2AF65 recruits a novel human DEAD box protein required for assembly of U2 snRNP-branch point interaction. Genes Dev. 1997;11:1864–1872. doi: 10.1101/gad.11.14.1864. [DOI] [PubMed] [Google Scholar]
- 8.Fu X D. The superfamily of arginine/serine-rich splicing factors. RNA. 1995;1:663–680. [PMC free article] [PubMed] [Google Scholar]
- 8a.Guthrie, C. Personal communication.
- 9.Harlow E, Lane D. Antibodies: a laboratory manual. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1988. [Google Scholar]
- 10.Kanaar R, Lee A L, Rudner D Z, Wemmer D E, Rio D C. Interaction of the sex-lethal RNA binding domains with RNA. EMBO J. 1995;14:4530–4539. doi: 10.1002/j.1460-2075.1995.tb00132.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kanaar R, Roche S E, Beall E L, Green M R, Rio D C. The conserved pre-mRNA splicing factor U2AF from Drosophila: requirement for viability. Science. 1993;262:569–573. doi: 10.1126/science.7692602. [DOI] [PubMed] [Google Scholar]
- 12.Kornberg A, Baker T. DNA replication. W. H. New York, N.Y: Freeman and Company; 1992. [Google Scholar]
- 13.Kramer A. The structure and function of proteins involved in mammalian pre-mRNA splicing. Annu Rev Biochem. 1996;65:367–409. doi: 10.1146/annurev.bi.65.070196.002055. [DOI] [PubMed] [Google Scholar]
- 14.Kunkel T A, Roberts J D, Zakour R A. Rapid and efficient site-specific mutagenesis without phenotypic selection. Methods Enzymol. 1987;154:367–382. doi: 10.1016/0076-6879(87)54085-x. [DOI] [PubMed] [Google Scholar]
- 15.MacMillan A M, McCaw P S, Crispino J D, Sharp P A. SC35-mediated reconstitution of splicing in U2AF-depleted nuclear extract. Proc Natl Acad Sci USA. 1997;94:133–136. doi: 10.1073/pnas.94.1.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Manley J L, Tacke R. SR proteins and splicing control. Genes Dev. 1996;10:1569–1579. doi: 10.1101/gad.10.13.1569. [DOI] [PubMed] [Google Scholar]
- 17.Moore M J, Query C C, Sharp P A. Splicing of precursors to mRNAs by the spliceosome. In: Gesteland R F, Atkins J F, editors. The RNA world. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1993. pp. 303–357. [Google Scholar]
- 18.Potashkin J, Naik K, Wentz-Hunter K. U2AF homolog required for splicing in vivo. Science. 1993;262:573–575. doi: 10.1126/science.8211184. [DOI] [PubMed] [Google Scholar]
- 19.Reed R. Initial splice-site recognition and pairing during pre-mRNA splicing. Curr Opin Genet Dev. 1996;6:215–220. doi: 10.1016/s0959-437x(96)80053-0. [DOI] [PubMed] [Google Scholar]
- 20.Reed R. The organization of 3′ splice-site sequences in mammalian introns. Genes Dev. 1989;3:2113–2123. doi: 10.1101/gad.3.12b.2113. [DOI] [PubMed] [Google Scholar]
- 21.Rudner D Z, Kanaar R, Breger K S, Rio D C. Mutations in the small subunit of the Drosophila U2AF splicing factor cause lethality and developmental defects. Proc Natl Acad Sci USA. 1996;93:10333–10337. doi: 10.1073/pnas.93.19.10333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21a.Rudner, D. Z., and D. C. Rio. Unpublished observations.
- 21b.Rudner, D. Z., K. S. Breger, M. Bell, and T. Cline. Unpublished data.
- 21c.Rudner, D. Z., K. S. Breger, and D. C. Rio. Molecular genetic analysis of the heterodimeric splicing factor U2AF: the RS domain on either the large or small Drosophila subunits is dispensable in vivo. Genes Dev., in press. [DOI] [PMC free article] [PubMed]
- 21d.Rudner, D. Z., K. S. Breger, R. Kanaar, M. Adams, and D. C. Rio. Unpublished data.
- 22.Ruskin B, Zamore P D, Green M R. A factor, U2AF, is required for U2 snRNP binding and splicing complex assembly. Cell. 1988;52:207–219. doi: 10.1016/0092-8674(88)90509-0. [DOI] [PubMed] [Google Scholar]
- 23.Schoner B E, Belagaje R M, Schoner R G. Enhanced translational efficiency with two-cistron expression system. Methods Enzymol. 1990;185:94–103. doi: 10.1016/0076-6879(90)85010-l. [DOI] [PubMed] [Google Scholar]
- 24.Sharp P A. Split genes and RNA splicing. Cell. 1994;77:805–815. doi: 10.1016/0092-8674(94)90130-9. [DOI] [PubMed] [Google Scholar]
- 25.Sijbers A M, de Laat W L, Ariza R R, Biggerstaff M, Wei Y F, Moggs J G, Carter K C, Shell B K, Evans E, de Jong M C, Rademakers S, de Rooij J, Jaspers N G, Hoeijmakers J H, Wood R D. Xeroderma pigmentosum group F caused by a defect in a structure-specific DNA repair endonuclease. Cell. 1996;86:811–822. doi: 10.1016/s0092-8674(00)80155-5. [DOI] [PubMed] [Google Scholar]
- 26.Singh R, Valcárcel J, Green M R. Distinct binding specificities and functions of higher eukaryotic polypyrimidine tract-binding proteins. Science. 1995;268:1173–1176. doi: 10.1126/science.7761834. [DOI] [PubMed] [Google Scholar]
- 27.Smith C W J, Patton J G, Nadal-Ginard B. Alternative splicing in the control of gene expression. Annu Rev Genet. 1989;23:527–577. doi: 10.1146/annurev.ge.23.120189.002523. [DOI] [PubMed] [Google Scholar]
- 28.Spradling A C. P element-mediated transformation. In: Roberts D B, editor. Drosophila: a practical approach. Washington, D.C: IRL Press; 1986. pp. 175–197. [Google Scholar]
- 29.Tronchere H, Wang J, Fu X. A protein related to splicing factor U2AF35 that interacts with U2AF65 and SR proteins in splicing of pre-mRNA. Nature. 1997;388:397–400. doi: 10.1038/41137. [DOI] [PubMed] [Google Scholar]
- 30.Valcárcel J, Gaur R K, Singh R, Green M R. Interaction of U2AF65 RS region with pre-mRNA of branch point and promotion base pairing with U2 snRNA. Science. 1996;273:1706–1709. doi: 10.1126/science.273.5282.1706. [DOI] [PubMed] [Google Scholar]
- 31.Valcárcel J, Singh R, Zamore P D, Green M R. The protein Sex-lethal antagonizes the splicing factor U2AF to regulate alternative splicing of transformer pre-mRNA. Nature. 1993;362:171–175. doi: 10.1038/362171a0. [DOI] [PubMed] [Google Scholar]
- 32.Wentz-Hunter K, Potashkin J. The small subunit of the splicing factor U2AF is conserved in fission yeast. Nucleic Acids Res. 1996;24:1849–1854. doi: 10.1093/nar/24.10.1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu J, Maniatis T. Specific interactions between proteins implicated in splice site selection and regulated alternative splicing. Cell. 1993;75:1061–1070. doi: 10.1016/0092-8674(93)90316-i. [DOI] [PubMed] [Google Scholar]
- 34.Zamore P D, Green M R. Biochemical characterization of U2 snRNP auxiliary factor: an essential pre-mRNA splicing factor with a novel intranuclear distribution. EMBO J. 1991;10:207–214. doi: 10.1002/j.1460-2075.1991.tb07937.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zamore P D, Green M R. Identification, purification, and biochemical characterization of U2 small nuclear ribonucleoprotein auxiliary factor. Proc Natl Acad Sci USA. 1989;86:9243–9247. doi: 10.1073/pnas.86.23.9243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zamore P D, Patton J G, Green M R. Cloning and domain structure of the mammalian splicing factor U2AF. Nature. 1992;355:609–614. doi: 10.1038/355609a0. [DOI] [PubMed] [Google Scholar]
- 37.Zhang M, Zamore P D, Carmo-Fonseca M, Lamond A I, Green M R. Cloning and intracellular localization of the U2 small nuclear ribonucleoprotein auxiliary factor small subunit. Proc Natl Acad Sci USA. 1992;89:8769–8773. doi: 10.1073/pnas.89.18.8769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zorio D A R, Lea K, Blumenthal T. Cloning of Caenorhabditis U2AF65: an alternatively spliced RNA containing a novel exon. Mol Cell Biol. 1997;17:946–953. doi: 10.1128/mcb.17.2.946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zuo P, Maniatis T. The splicing factor U2AF35 mediates critical protein-protein interactions in constitutive and enhancer-dependent splicing. Genes Dev. 1996;10:1356–1368. doi: 10.1101/gad.10.11.1356. [DOI] [PubMed] [Google Scholar]