

Abstract
Proliferation of human mammary epithelial cells (HMEC) is limited to a few passages in culture due to an arrest in G1 termed selection or mortality stage 0, M0. A small number of cells spontaneously escape M0, continue to proliferate in culture, and then enter a second mortality stage, M1, at which they senesce. Evidence that M0 involves the Rb pathway comes from the observation that expression of human papillomavirus type 16 E7 alleviates the M0 proliferation block, and we further show that the Rb-binding region of E7 is required to allow cells to bypass M0. In contrast, E6 does not prevent HMEC from entering M0 but, rather, is involved in M1 bypass. Here we show that inactivation of the D-type cyclin-dependent kinase inhibitor p16INK4A is associated with escape from the M0 proliferation block. Early-passage HMEC express readily detectable amounts of p16 protein, whereas normal or E6-expressing HMEC that escaped M0 expressed markedly reduced amounts of p16 mRNA and protein. This initial reduction of p16 expression was associated with limited methylation of the p16 promoter region CpG island. At later passages, a further reduction in p16 expression occurred, accompanied by increased CpG island methylation. In contrast, reduction of p16 expression did not occur in E7-expressing HMEC that bypassed M0, due to inactivation of Rb. These observations in the E6-expressing HMEC correlate well with the finding that CpG island methylation is a mechanism of p16 inactivation in the development of human tumors, including breast cancer.
Since the identification of the retinoblastoma (Rb) gene as a tumor suppressor gene and elucidation of its role in controlling the cell cycle, the Rb pathway has emerged as one of the key targets for inactivation in the development of cancer. Germ line mutations of Rb in families with a predisposition to retinoblastoma and somatic mutations in Rb in a wide variety of sporadic tumors identify Rb as a tumor suppressor (for a review, see reference 54). This, coupled with targeted inactivation of Rb by oncoproteins of the DNA tumor viruses, underscores the importance of Rb in controlling the cell cycle (for a review, see reference 13). The ability of Rb to act as a tumor suppressor is controlled by its phosphorylation by several cyclin-dependent kinases (13). Thus in some cancers, notably breast cancer, disregulation of cyclins D and E may result in inappropriate inactivation of Rb due to increased phosphorylation by cyclin-dependent kinases (CDKs) (8, 30, 61). p16INK4A, originally identified as a protein associating with CDK4 in transformed cells (57), was cloned in a two-hybrid screen for proteins interacting with CDK4 (48). p16 is an inhibitor of the cyclin D-dependent protein kinases CDK4 and CDK6 (20, 48), whose main function appears to be the phosphorylation of Rb (35). Thus, in cells lacking Rb function, overexpression of p16 is not inhibitory (32, 36, 37). Like Rb itself, p16 is a tumor suppressor, as evidenced by its mutation in certain melanoma-prone families and its somatic deletion or mutation in a large percentage of tumors (6, 29, 43–45, 50). Recently, CpG island methylation within the p16 promoter has been identified as a mechanism to eliminate p16 expression in a variety of human tumors (23, 25, 38, 44, 56). Loss of p16 function, like overexpression of cyclins, is predicted to result in higher CDK activity and thus in inappropriate phosphorylation of Rb, effectively inactivating its growth-suppressive effects. All of these observations underscore the important role of the Rb pathway in controlling cell proliferation.
The progression of cells in culture to an immortalized state, although not identical, has many features in common with the development of cancer in vivo. For example, high-risk human papillomaviruses (HPVs) that are associated with anogenital cancer readily immortalize human cells in culture while low-risk HPVs that are associated with benign lesions do not (19, 22, 40). The E6 and E7 genes of the high-risk HPVs inactivate p53 and Rb, respectively, two of the most commonly inactivated tumor suppressors in human cancer (12, 47, 55). HPV-immortalized cells can progress to tumorigenicity by continued passaging or by treatment with carcinogens (17, 26). These tumorigenic derivatives have additional alterations, such as deletions on 18q, in common with cancers (31).
For human mammary epithelial cells (HMEC) to become immortalized, they must overcome several distinct proliferation blocks (15, 49). Initially, the proliferation of HMEC is limited to a few passages in culture due to a proliferation block termed selection or M0 (mortality stage 0), a period when cells become larger and flattened and accumulate in G1 or G0 (15, 51). Expression of E7 in early-passage HMEC allows the cells to bypass M0 (15), suggesting that Rb-related proteins play an important role in the M0 arrest (15). Although E7 is efficient at allowing cells to bypass M0, most of the cells arrest at a later stage and do not become immortalized (15, 53). A subpopulation of normal cells (not expressing HPV oncogenes) can occasionally escape M0 and continue to proliferate until they senesce at M1 (mortality stage 1) (49, 51). Expression of E6 allows these cells to bypass M1 and exhibit an extended life span. The E6-expressing cells eventually enter a crisis period (M2, mortality stage 2), from which immortalized cells emerge (2, 49). Thus, E7 allows HMEC to bypass M0, E6 allows them to bypass M1, and additional changes are thought to allow escape from M2 and to yield immortalized cells (2, 15, 49). In this study, we examined the expression patterns of various cell cycle regulatory proteins to begin to elucidate the mechanisms involved in the M0 arrest. The results suggest a role for the p16 CDK inhibitor protein in the arrest of cells at M0 and indicate that escape from M0 is associated with methylation of the p16 promoter region CpG island and inactivation of p16 expression.
MATERIALS AND METHODS
Cell culture.
HMEC were isolated from tissue specimens from reduction mammoplasties as described by Stampfer (51). HMEC1 and HMEC3 were grown in MEGM (Clonetics) and were described previously (15), while HMEC4, HMEC6, and HMEC9 were grown in DFCI-1 (3). HMEC8 cells were isolated from normal breast tissue from a patient undergoing a mastectomy and grown in DFCI-1. LXSN-based retroviruses expressing HPV oncogenes were used to infect HMEC as soon as possible after establishment of the cultures (11, 19, 39), followed by selection with 100 μg of G418 per ml to eliminate uninfected cells. The cultures were maintained by adding fresh medium every other day and passaging at a ratio of 1:5 before the cultures were confluent. Population doublings were estimated based on 2.25 doublings per 1:5 split. For the purposes of this study, cells were deemed to be immortalized if they achieved 50 passages (>100 population doublings). Cells that were not expressing HPV oncogenes achieved fewer than 25 passages. Cell lines H249 and H1618 (both donated by S. B. Baylin) were used as negative and positive controls, respectively, for p15 and p16 promoter methylation.
Immunoprecipitation of E7.
The cells were labeled with [35S]cysteine and [35S]methionine, and equivalent counts per minute (cpm) were immunoprecipitated with rabbit antisera to HPV-16 E7 or HPV-6 E7 (19).
Western blots.
Whole-cell extracts were prepared in WE buffer, and protein concentrations were determined by the Bio-Rad DC protein assay (15). Protein samples (20 μg) were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and blots were prepared on nitrocellulose or Immobilon-P membranes (Millipore). The following antibodies were used as probes: p53 and p21 (Oncogene Science, clones DO-1 and EA10); Rb, cyclin A, cyclin E, cyclin D1, and p16 (PharMingen, clones G3-245, BF683, HE12, G124-326, and G175-405); and CDK2, CDK4, and p27 (Transduction Laboratories, no. C18520, C18720 and K25020). The original films were scanned on a Sharp JX-325 scanner with Adobe Photoshop software, and, where noted, quantitation was done on the scanned image with ImageQuant software (Molecular Dynamics).
Northern analysis.
Total RNA was isolated by pelleting through CsCl cushions (18). Poly(A)+ RNA was isolated from total RNA by using oligo(dT) cellulose (New England Biolabs). RNA samples were separated on formaldehyde-agarose gels and blotted to Hybond N membranes (Amersham) in 20× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate). After UV cross-linking, the membranes were hybridized in 0.5 M sodium phosphate (pH 7.2)–5% SDS at 68°C and washed in 50 mM sodium phosphate–0.1% SDS at 68°C (18). PE1 probe, containing p16 exon I, was generated by PCR as described previously (38) and labeled with [32P]dCTP by the random-primer method (Boehringer Mannheim). No cross-hybridization between the PE1 probe and p15 RNA was detected when using in vitro-transcribed p16 and p15 test RNAs. The blots were stripped and reprobed with 36B4 as a loading control (33). Quantitation was done on a Molecular Dynamics PhosphorImager with ImageQuant software.
Southern analysis.
Genomic DNA was prepared, digested with EcoRI or BamHI, and separated on 0.8% agarose gels. After being transferred to Hybond N+ (Amersham), the blots were probed for HPV-16 E6 under the conditions described above.
p16 deletion analysis.
To screen the p16 locus (9p21) for possible homozygous deletions or allelic loss, DNA from clonal cell populations was amplified by primer extension preamplification (PEP) (5, 60). Aliquots from each sample were evaluated for STS marker c5.1, located within the p16 gene (29), or polymorphic markers D9S942 and D9S161 in locus-specific PCRs as described previously (6).
p16 promoter methylation analysis.
The PCR-based assay for CpG island methylation of the p16 and p15 promoter regions was carried out as described previously (23, 56). Briefly, this assay consists of sodium bisulfite modification, followed by a PEP reaction and methylation-specific PCR. For some samples, DNA sequencing of the sodium bisulfite-modified genomic DNA was used to screen for methylation not detected by the PCR assay. Briefly, after sodium bisulfite treatment and PEP, the p16 promoter region from −159 to +135 (according to the numbering system of Hara et al. [21]) was amplified with primers 5′-TTTTTAGAGGATTTGAGGGATAGG and 5′-CTACCTAATTCCAATTCCCCTACA under the conditions described by Herman et al. (23). The resulting PCR products were gel purified and cloned (Invitrogen TA cloning kit), and individual clones were sequenced on an ABI sequencer with dye terminators. Based upon non-CpG cytosines, which are expected to be rarely methylated, the bisulfite conversion efficiency was estimated to be at least 99.7%.
RESULTS
Growth characteristics of HMEC in culture—effects of E6 and E7 expression.
LXSN-based retroviruses expressing HPV-16 E6, E7, or E6/E7 were used to infect early-passage HMEC, and the resulting cells were serially passaged. Uninfected, vector-infected, and E6-infected cells all entered M0, as evidenced by reduced proliferation and the appearance of large, flat cells. This usually occurred by passage 7, but the timing was variable depending on the particular HMEC strain. In one uninfected cell strain, HMEC4, a subpopulation of cells spontaneously escaped M0 and then senesced at M1 (approximately passage 20). In all E6-expressing strains, a subpopulation of HMEC escaped from M0 and readily became immortalized. Cells infected with E7 or E6/E7 consistently bypassed M0, failing to show large, flat cells or reduced proliferation. However, HMEC expressing E7 alone did not readily become immortalized; they eventually grew in tight clumps and failed to proliferate beyond passage 25. The E6/E7-expressing HMEC continued to proliferate after bypassing M0 and were immortalized, with little evidence of crisis. Table 1 summarizes these results for six HMEC strains.
TABLE 1.
Effects of E6 and E7 on proliferation of HMEC in culturea
| Cell strain | Vector | M0 escapeb | Immortalizationb |
|---|---|---|---|
| HMEC1c | UN/LXSN | − | − |
| E6 | + (escape) | ND | |
| E7 | + (bypass) | ND | |
| E6/E7 | + (bypass) | + | |
| HMEC3c | UN/LXSN | − | − |
| E6 | + (escape) | ND | |
| E7 | + (bypass) | ND | |
| E6/E7 | + (bypass) | + | |
| HMEC4 | UN | + (escape) | − |
| HMEC6 | UN/LXSN | − | − |
| E6 | + (escape) | + | |
| E7 | + (bypass) | − | |
| E6/E7 | + (bypass) | + | |
| HMEC8 | UN/LXSN | − | − |
| E6 | + (escape) | + | |
| E7 | + (bypass) | − | |
| E6/E7 | + (bypass) | + | |
| HMEC9 | UN/LXSN | − | − |
| E6 | + (escape) | + | |
| E6/E7 | + (bypass) | + |
HMEC strains were infected with the indicated retroviruses or not infected (UN).
The ability of the cells to escape (or bypass) M0 and to yield immortalized cells is indicated. ND, not done. See reference 15 for a description of M0 escape versus M0 bypass.
Results for HMEC1 and HMEC3 are from reference 15.
By using the retrovirus integration site(s) as tags to monitor the fate of individual infected cells, it was possible to estimate the frequency of M0 escape in the E6-expressing HMEC. Genomic DNA was prepared at various stages of culture, and Southern blots were probed for E6 (data not shown). In E6-expressing HMEC6, only two virus integration sites (representing two initially infected cells) were evident after M0 escape (data not shown). Therefore, based on an estimate of between 5,000 and 10,000 cells initially transduced, fewer than 0.1% of the E6-expressing cells proliferated beyond M0. Only one of the two clones was detected in the immortalized culture. In E6-expressing HMEC8, three or four integration sites were detected after M0 escape, again less than 0.1% of the initial population. The pattern of integration sites changed over time, and two sites were detected in the immortalized population, representing either two clones or one clone containing two viral integrations. We also monitored the fate of HMEC6 E6-expressing cells individually cloned after M0 escape. Of 23 clones selected at passage 18, only 4 proliferated to passage 25. These results argue that 99.9% of E6-expressing cells fail to proliferate beyond M0 and that continued selection of clones (and subclones) occurs even among the few cells that do escape M0.
The Rb-binding region of HPV-16 E7 is required to allow HMEC to bypass M0.
The best-characterized activity of HPV-16 E7 is its ability to bind to Rb and related proteins, releasing transcription factor E2F. To determine whether the Rb-binding region of E7 is important for its ability to allow cells to bypass M0, several mutated forms of E7 were tested. HPV-16 E7Δ21–24 has a deletion of 4 amino acids within the LXCXE motif (amino acids 22 to 26 of HPV-16 E7) which have previously been shown to be required for binding of Rb and related proteins (41). HPV-16 E7C24G carries a point mutation within this region and similarly disrupts the binding of Rb and related proteins (4, 10). The HPV-16 E7E26G mutation disrupts Rb binding but leaves intact the ability of E7 to interact with the Rb-related protein p107 (10). HPV-16 E7D21S contains a mutation just outside the LXCXE motif and leaves intact Rb and p107 binding as well as the ability to release E2F from Rb in a gel shift assay (4, 11, 14). HPV-16 E7H2P retains the ability to bind to Rb but is at least partially defective in its ability to dissociate E2F in a gel shift assay (10, 14) and is transformation defective (4). HPV-16 E7Δ6-10 and HPV-16 E7C58G/C91G are able to bind Rb but are transformation defective. In addition, HPV-6 E7 was tested; HPV-6 E7 does bind to Rb but with a greatly reduced efficiency compared to HPV-16 E7 (41).
To compare the abilities of the HPV E7 proteins to allow M0 bypass, early-passage HMEC6 cells were infected with the retroviruses and selected with G418 to eliminate uninfected cells. Initial observation of the cultures indicated that wild-type HPV-16 E7 was active; i.e., few large, flat cells emerged, and the population remained proliferative. HPV-16 E7D21S was only partially active; i.e., the cells did not proliferate as well as those expressing HPV-16 E7, and some large cells emerged. Cells infected with other E7 proteins were similar to vector-infected control cells (Fig. 1A and data not shown).
FIG. 1.
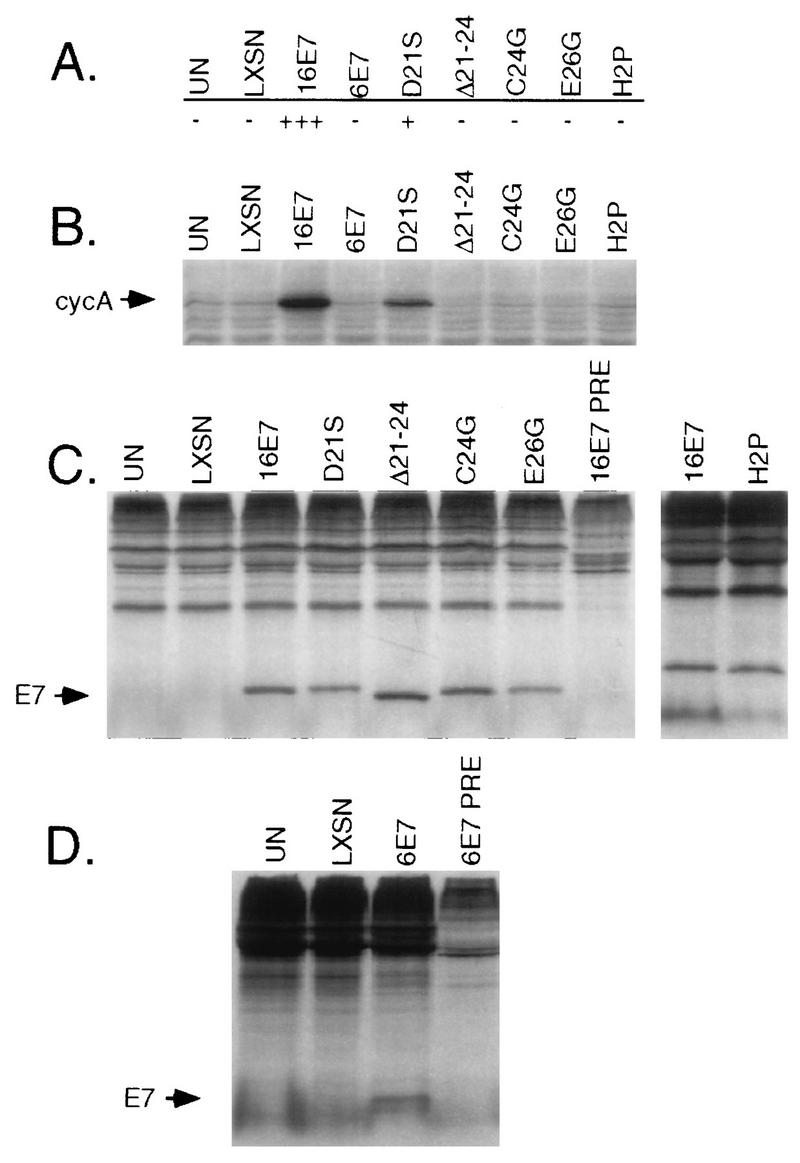
Analysis of different E7 proteins for their abilities to allow M0 bypass in HMEC. (A) Abilities of HMEC expressing different E7 proteins to bypass M0. −, the cells behaved very similarly to uninfected or LXSN-infected controls; +, some activity, but less than that of HPV-16 E7; +++, wild-type HPV-16 E7 activity. Results are shown for uninfected HMEC6 (UN), HMEC6 infected with LXSN retroviral vector (LXSN), or LXSN containing HPV-16 E7, HPV-6 E7, HPV-16 E7D21S, HPV-16 E7Δ21–24, HPV-16 E7C24G, HPV-16 E7E26G, and HPV-16 E7H2P. (B) Western blot for cyclin A in HMEC6 expressing different E7 proteins. (C) Immunoprecipitation with HPV-16 E7 polyclonal antibody of 35S-labeled HMEC4 cells expressing the indicated E7 proteins to demonstrate expression. PRE denotes the use of nonimmunized rabbit serum. (D) Immunoprecipitation as in panel C but with HPV-6 E7 polyclonal antibody.
Cyclin A expression is greatly reduced concomitant with a reduction in the S-phase fraction as early-passage HMEC accumulate in M0 (15) (see below). Therefore, cyclin A expression was used as a marker for M0, independent of cell morphology, to evaluate the mutated E7 proteins. As expected, the uninfected control cells and the LXSN vector-infected cells showed very low levels of cyclin A expression whereas cells expressing HPV-16 E7 continued to express abundant cyclin A (Fig. 1B). Cells expressing HPV-16 E7D21S expressed cyclin A at a higher level than that of the control cells, although only about one-fourth as high as the level observed in the wild-type HPV-16 E7-expressing cells. Cells expressing the other E7 proteins expressed cyclin A at low levels, similar to the vector-infected control cells. These results indicate that the Rb-binding region of HPV-16 E7 is required to allow HMEC to bypass M0 but do not exclude the possibility that other regions of E7 are also required.
To confirm the expression of the various E7 proteins, extracts of HMEC4 cells expressing the E7 proteins were immunoprecipitated with polyclonal E7 antibody. HMEC4 that had escaped from M0 were chosen for this analysis because they had a sufficiently long life span to be infected, selected with G418, and have their proteins labeled. All of the E7 proteins were detected at comparable levels (Fig. 1C and D). Two proteins, HPV-16 E7Δ6-10 and HPV-16 E7C58G/C91G, were detected at lower levels and were not included in the above analysis (data not shown).
Cell cycle-related proteins in early-passage HMEC.
To begin to dissect possible mechanisms involved in the arrest of HMEC at M0, protein extracts were prepared sequentially from passage 3 (early-proliferating cells) to passage 7 (cells mostly arrested at M0). Figure 2 shows the results for HMEC9 cells. Early-passage cells (passage 3) expressed abundant Rb, which was predominantly in the hyperphosphorylated form. As the cells accumulated in M0, the hypophosphorylated form predominated and the overall level of Rb was greatly reduced. Among the cyclins and CDKs involved in Rb phosphorylation, cyclin A expression was dramatically reduced while the levels of cyclins E and D1 remained unchanged. The CDK2 and CDK4 levels were reduced but only at the later passages. Consistent with our previous report (15), neither p53 nor p21 was induced at M0. Similarly, p27 was not induced. In contrast, the level of p16 increased by about 60% during this period. Similar trends were observed for HMEC6 and HMEC8. Although the increase in the p16 level was not dramatic, it was consistent with a possible role in the M0 arrest, given its known ability to inhibit CDK4 and CDK6, two cyclin-dependent kinases that play a central role in Rb phosphorylation.
FIG. 2.
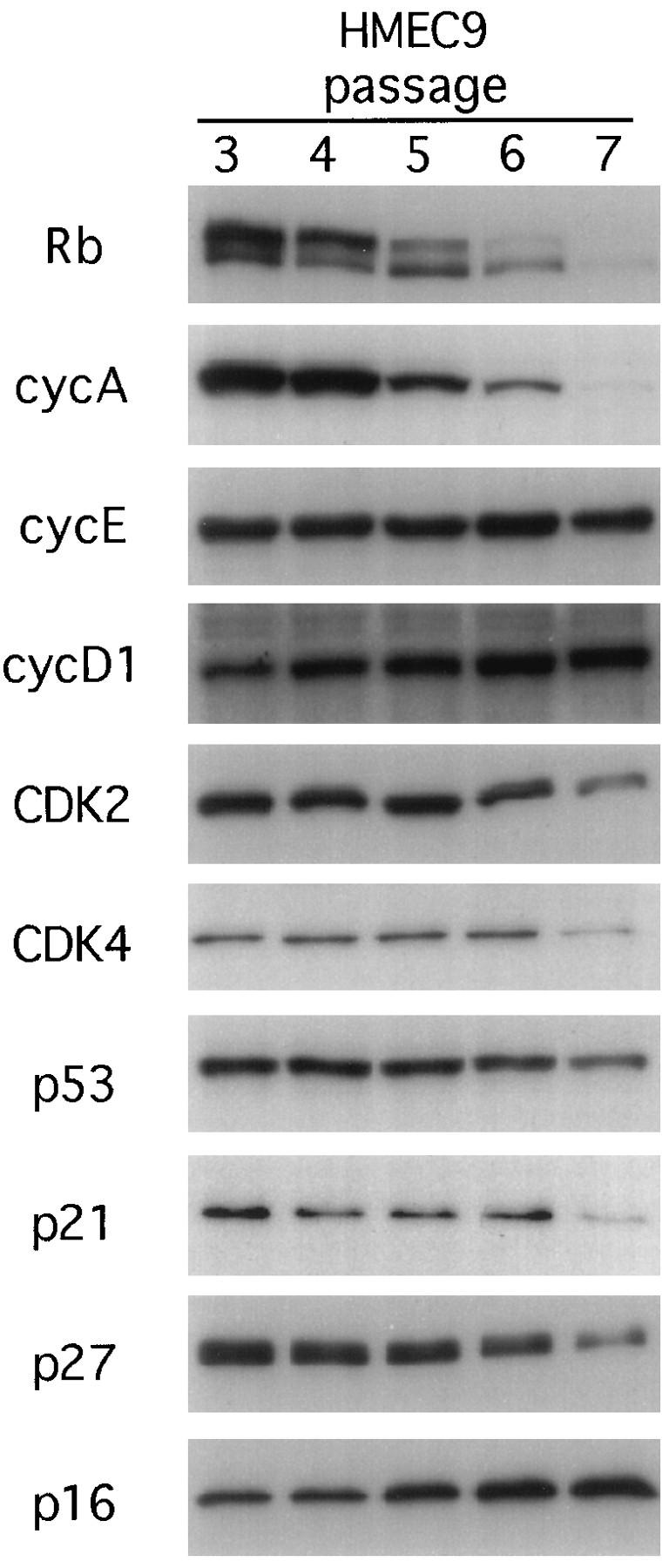
Western blots for cell cycle-related proteins in HMEC9. Cells were harvested early after establishment of the culture (passage 3) and at subsequent passages through passage 7, when the cells were predominantly in M0. The blots were probed with the indicated antibodies.
p16 levels in early-passage and post-M0 HMEC.
Given the above results implicating the Rb pathway in the arrest of cells at M0 and the fact that p16 is commonly deleted in human cancers and cancer cell lines, we compared the levels of p16 in the early-passage HMEC to the levels in post-M0 HMEC. Early-passage, uninfected HMEC1 and HMEC3 expressed p16 that was detectable by Western blotting; however, major differences in p16 expression were detected among the post-M0 cells (Fig. 3). Cells that expressed E7 or E6/E7 and had bypassed M0 showed levels of p16 as high as or higher than the early-passage cells did. However, cells that expressed E6 and had escaped from M0 showed very low levels of p16. A similar pattern of p16 expression was seen in HMEC6 and HMEC9 (see below). The low level of p16 expression in the post-M0 E6-expressing cells was not a direct result of E6 expression, because the E6/E7 expressing cells had abundant p16. Furthermore, expression of E6 in the early-passage HMEC8 did not reduce p16 expression (Fig. 4A). HMEC4 spontaneously escaped M0, and this was associated with a fivefold reduction in p16 levels (Fig. 4A). p16 levels further decreased 2.5-fold with continued passage in culture. As HMEC4 aged and approached M1, the level of p16 remained low. In contrast, the levels of p53, p21, and p27 were increased after M0 and remained high at late passage (Fig. 4A and data not shown). Overall, these results are consistent with a model in which the Rb pathway limits proliferation at M0. In cells that express E7, inactivation of Rb would allow continued proliferation of cells with high levels of p16. In contrast, cells with intact Rb function would arrest at M0, followed by selection for cells having low levels of p16 during M0 escape.
FIG. 3.

Expression of p16 in HMEC1 and HMEC3 before and after M0. Western blots for p16 in uninfected early-passage cells before M0 (E.P.), cells expressing E6 after escaping from M0 (E6), or cells expressing E7 or E6/E7 after bypassing M0 (E7 and E6/E7) are shown.
FIG. 4.

Expression of p16 in HMEC. Passage numbers are indicated above the lanes. M0 usually occurred around passage 7 to 10, as indicated; M1 for uninfected cells usually occurred around passage 20. UN, uninfected HMEC. E6 or E6/E7, HMEC infected with E6- or E6/E7-expressing retrovirus. (A) Western blots for p16 and p27. Numbers in brackets below the lanes indicate the relative amounts of p16. (B) Northern blots for p16 exon 1 with 36B4 loading control. Numbers in brackets below the lanes indicate the relative amounts of p16 mRNA normalized for 36B4. (C) p16 promoter CpG island methylation assay. U and M indicate the primer pair specific for unmethylated and methylated p16 promoter CpG islands, respectively. Molecular size markers (in base pairs) are indicated in the far left lane. H249 and H1618 are lung cancer cell lines used as controls for unmethylated and methylated p16 alleles, respectively.
The patterns of p16 expression in HMEC6, HMEC8, and HMEC9 expressing E6 were examined at various passages after M0 escape. In E6-expressing HMEC6, the p16 level was reduced after M0 escape (passage 12), in contrast to uninfected cells prior to M0 (passage 3) (Fig. 4A). This low level of p16 was maintained through passage 18, was further reduced by passage 25 and reached an undetectable level by passage 59 (a trace of p16 was detected in the passage 29 cells on a longer exposure [data not shown]). E6-expressing HMEC9 showed a pattern of p16 expression very similar to that of the E6-expressing HMEC6 (data not shown). E6-expressing HMEC8 at passage 8 had not escaped M0 and expressed abundant p16 (Fig. 4A). At passage 14, these cells had escaped M0 and showed a reduced level of p16 compared to the passage 8 cells. Surprisingly, the level of p16 was increased again at passages 21 to 32 and then was dramatically reduced to undetectable levels by passage 41. In culture, a mild crisis with noticeable cell death was noted in the E6-expressing HMEC8 at passages 29 through 32; thereafter, the cells formed a more stable population. In contrast, p16 was expressed at high levels in all the late-passage cells expressing E6/E7 (Fig. 4A, HMEC6-E6/E7 passage 66 and data not shown). Note that p27 expression was not decreased during the course of these experiments (Fig. 4A). These results are consistent with the idea that selection against the Rb/p16 pathway beginning around M0 results in the emergence of cells with low levels of p16. In the E6-expressing HMEC8, the emergence of cells with low levels of p16 was delayed. It is interesting that HMEC8 cells were derived from normal breast tissue from a patient undergoing a mastectomy while all the other cells in this study were derived from breast reduction tissue.
p16 gene inactivation occurs at the mRNA level.
To begin to address the mechanism underlying the loss of p16 expression, p16 mRNA levels were analyzed by Northern blotting. In early-passage HMEC4 (passage 5), p16 mRNA was detected as a major band of about 0.8 kb and a minor band at about 1.1 kb (Fig. 4B). The low levels of p16 protein detected in the population that escaped M0 (passages 12 and 18) correlated with reduced levels of p16 mRNA expression.
In HMEC6, p16 mRNA was detected at early passage (passage 3), but it was not detected in the E6-expressing cells at passage 59 (Fig. 4B). E6/E7-expressing HMEC6 expressed an elevated level of p16 mRNA at passage 66 (Fig. 4B). This result was somewhat surprising since the E6/E7-expressing HMEC6 expressed no more p16 protein than the early-passage HMEC6 did. The results with HMEC9 were very similar to those with HMEC6 (data not shown). In E6-expressing HMEC8, p16 mRNA was detected at passage 29, when abundant p16 protein was detected, but was not detected at passage 41, when p16 protein became undetectable (Fig. 4B). These results suggest that the reduction in the level of p16 protein after M0 escape can be explained, at least in part, by alterations at the RNA level.
Deletions in p16 do not explain the loss of p16 expression.
Since p16 gene deletion is known to occur readily in cell lines, various HMEC were cloned and DNA was extracted to check for possible deletions affecting the 9p21 region. Three markers were used for this analysis. The c5.1 STS marker is located within the p16 gene (29). The D9S942 and D9S161 STR markers are located within 9p21 and allow identification of loss of heterozygosity (LOH). Analysis of the early-passage cells showed that all were informative for LOH analysis (data not shown). In HMEC4 that had escaped from M0, 3 of 10 clones contained apparent homozygous deletions affecting the p16 gene at 9p21 while the remaining 7 clones had no deletions (data not shown). In E6-expressing HMEC1 that had escaped M0, no deletions were detected in 10 clones (data not shown). As expected, no deletions were observed among 10 clones of E6/E7-expressing HMEC1 which retained p16 expression (data not shown). To determine whether frequent p16 deletions occurred at later passages, we also tested 10 clones each of HMEC6 and HMEC8 immortalized by E6 (>100 population doublings). Again, no homozygous deletions or LOHs were detected (data not shown). Based on these results, deletion of p16 did not appear to play a major role in the emergence of cells expressing low levels of p16 during M0 escape.
The p16 gene is methylated in E6-expressing HMEC.
Methylation of the CpG island within the promoter region of p16 has been reported as a mechanism of p16 gene inactivation in tumors and cancer cell lines (25, 38, 56). Therefore, a methylation-sensitive PCR assay was used to compare the methylation status of this region between cells which expressed high levels of p16 and those which expressed low levels of p16.
HMEC4 that escaped from M0 exhibited dramatically less p16 than did the early-passage cells. However, no p16 gene methylation was detected by the PCR assay in these cells up to passage 18, which is near the end of their proliferative life span (Fig. 4C). Similarly, no p16 gene methylation was detected in the E6-expressing cells that escaped M0 (HMEC3, HMEC6, and HMEC9), even though these cells expressed dramatically less p16 than did the corresponding early-passage cells (Fig. 4 and data not shown). At later stages of culture, however, cells with p16 CpG island methylation were detected. In E6-expressing HMEC6, methylation was first readily detected by the PCR assay at passage 25, and by passage 59, the p16 gene was predominantly methylated, although a low level (≈10% based on mixing experiments) of unmethylated p16 gene was still detected (Fig. 4C). E6-expressing HMEC9 exhibited a similar pattern; after M0 escape (passage 16), no methylation was detected, but subsequently (passage 44) the cells displayed a mixture of methylated and unmethylated p16 alleles (data not shown). E6-expressing HMEC8 exhibited no evidence of p16 gene methylation up to passage 32, but only methylated alleles were detected by passage 41 (Fig. 4C). Therefore, the change in methylation status coincided with the elimination of p16 protein expression between passages 32 and 41. No p16 gene methylation was detected by the PCR assay in any of the cells expressing E7 alone (Fig. 5A), the three clones of late-passage E6/E7-expressing HMEC8 (Fig. 5B), or the late-passage E6/E7-expressing HMEC9 (data not shown).
FIG. 5.

PCR-based p16 promoter methylation analysis. (A) E7-expressing cells (HMEC1, passage 15; HMEC3, passage 23; HMEC6, passage 16; and HMEC8, passage 24). (B) Clones of E6/E7-expressing HMEC8 (passage 50). (C) E6/E7-expressing HMEC6, pooled population (passage 66), and clones thereof. Primers specific for the unmethylated (U) and methylated (M) p16 promoter regions are indicated. H249 and H1618 are described in the legend to Fig. 4.
Because the PCR-based methylation assay is inherently limited to the detection of methylation within the short regions recognized by the methylation-specific primers, a sequencing approach was used to determine whether methylation was present in regions missed by the PCR assay. After sodium bisulfite modification, individual PCR products of the region from −159 to +135 (according to the numbering system of Hara et al. [21]), which contains 35 potential CpG methylation sites, were cloned, and four clones of each sample were sequenced. The results are summarized in Table 2. In HMEC6, methylation was detected on 0 or 1 CpG site at passage 3, 6 to 14 sites after M0 escape (passage 12), and 19 to 25 sites by passage 25. In E6-expressing HMEC9, 7 to 10 sites were methylated after M0 escape (passage 16) and 9 to 28 of the sites were methylated later (passage 42). In contrast, E6/E7-expressing HMEC9 (passage 32) contained no methylation in the four clones examined. The immortalized cell line H1618, which was used as a positive control for p16 methylation, contained methylation at all 35 CpG sites of all four clones (data not shown).
TABLE 2.
p16 CpG methylation sequencing analysisa
| Stage of cells | HMEC6
|
HMEC9
|
||
|---|---|---|---|---|
| Cloneb | CpG methylationc | Cloneb | CpG methylationc | |
| Early passage without E6 | Passage 3 | NDd | ||
| Clone A | 0 | |||
| Clone B | 1 | |||
| Clone C | 1 | |||
| Clone D | 0 | |||
| After M0 escape with E6 | Passage 12 | Passage 16 | ||
| Clone A | 6 | Clone A | 8 | |
| Clone B | 6 | Clone B | 9 | |
| Clone C | 14 | Clone C | 10 | |
| Clone D | 10 | Clone D | 7 | |
| Later passage with E6 | Passage 25 | Passage 42 | ||
| Clone A | 19 | Clone A | 17 | |
| Clone B | 20 | Clone B | 28 | |
| Clone C | 25 | Clone C | 9 | |
| Clone D | 19 | Clone D | 18 | |
| Later passage with E6/E7 | ND | Passage 32 | ||
| Clone A | 0 | |||
| Clone B | 0 | |||
| Clone C | 0 | |||
| Clone D | 0 | |||
Methylation in the p16 CpG island region from −159 to +135 (according to the numbering system of Hara et al. (21), with +1 being the translational start site), which contains 35 CpG dinucleotides.
Each clone represents an individual PCR product.
Each value is the number of methylated sites out of the 35 CpG sites.
ND, not done.
Surprisingly, a low level of p16 gene methylation was detected by the PCR assay in immortalized E6/E7-expressing HMEC6 (Fig. 5C). To further investigate this methylation, late-passage E6/E7-expressing HMEC6 were cloned and assayed individually for the p16 methylation status. Of 10 clones, 6 (C3, C6, C7, C8, C9, and C10) contained predominantly unmethylated p16, 2 (C1 and C2) contained predominantly methylated p16, and the other 2 (C4 and C5) contained mixtures of methylated and unmethylated alleles (Fig. 5C). Analysis of the viral integration sites of these clones by Southern blotting revealed that clones containing methylated or mixed p16 alleles (C1, C2, C4, and C5) originated from a single parent containing one viral integration. Clones containing unmethylated alleles originated from a single parent containing three viral integrations (C3, C6, C7, C8, and C10) or as many as five or six viral integrations (C9) (data not shown).
To assess whether the methylation was specific for p16, the methylation status of p15INK4B was determined. p15, like p16, is a member of the INK4 family of CDK inhibitors and is very similar to p16 in terms of both amino acid sequence and the ability to specifically inhibit CDK4 and CDK6. It is located next to the p16 gene at chromosomal position 9p21. None of the cells tested showed evidence for p15 gene methylation, including all of those in which p16 gene methylation was detected (data not shown). Therefore, the methylation was specifically selected for in the p16 CpG island.
DISCUSSION
The results presented here show that the Rb-binding region of E7 is required to allow cells to bypass M0, consistent with a role for Rb or related proteins in mediating the M0 proliferation block. Three distinct mutations within the LXCXE Rb-binding motif of HPV-16 E7 (Δ21–24, C24G, and E26G) each resulted in failure to allow M0 bypass. One mutation, HPV-16 E7H2P, which does not disrupt Rb binding, also failed to allow M0 bypass. However, HPV-16 E7H2P is defective in its ability to dissociate E2F from Rb in vitro (14). The reason why HPV-16 E7D21S was less effective than wild-type HPV-16 E7 in allowing M0 bypass despite its having the ability to bind Rb and p107 and dissociate Rb/E2F complexes could be subtle defects in its interaction with Rb-related proteins that are not apparent in the in vitro assays. Alternatively, D21S may be defective in some other activity that is needed for full wild-type HPV-16 E7 function. It is worth noting that the ability of E7 to bind Rb is not sufficient for its ability to transform rodent cells (4) or abrogate growth arrest signals induced by DNA damage, transforming growth factor β, or suprabasal quiescence (11). Conversely, the ability of HPV-16 E7 to bind Rb is not essential for its ability to immortalize human keratinocytes in the context of the full HPV-16 genome (27). Recently, HPV-16 E7 has been shown to inactivate the ability of p27 and p21 to inhibit CDKs (16, 59). Therefore, activities other than binding to Rb are likely to contribute to full wild-type HPV-16 E7 function.
What mechanisms are involved in arrest at M0? Analysis of cell cycle-related proteins revealed that Rb accumulated in the hypophosphorylated form and its level of expression decreased as the early-passage HMEC aged in culture, consistent with its playing a role in blocking proliferation at M0. The level of cyclin A was dramatically reduced as cells arrested at M0, presumably as a consequence of arrest in G1 (15). The levels of CDK2 and CDK4 also decreased but only as a late event. The levels of the CDK inhibitors p21 and p27 declined, while the level of p16 increased. An increase in the level of p16 in aging cells is consistent with the results of other studies with human keratinocytes, uroepithelial cells, and fibroblasts, which show increases in p16 levels as the cells enter senescence (1, 21, 34, 46). Whether the observed increase in the p16 level would be sufficient to arrest the cells or whether additional factors, such as the decrease in the CDK4 level, are important is not clear. Nevertheless, this increase in the p16 level, the emergence of cells having low levels of p16 expression, and the observation that the Rb-binding region of E7 was important to allow cells to bypass M0 point to p16 as being a proliferation-limiting factor at M0.
Other studies have reported loss of p16 expression in cultured cells. Reznikoff et al. (46) reported loss of p16 expression in human uroepithelial cells immortalized by E6 but not those immortalized by E7. In that study, hemizygous p16 deletions appeared to be involved in p16 inactivation. Noble et al. (42) also reported the loss of p16 expression at or before immortalization in fibroblasts derived from a patient with Li-Fraumeni syndrome and human mesothelial cells that had been transfected with HPV-16 E6/E7, although not all cells that lost p16 expression became immortalized. Where examined, the homozygous p16 deletion appeared to account for the lack of p16 expression (42). Loughran et al. (34) reported the loss of p16 expression in immortalized cancer-derived keratinocytes, although two cultures lost p16 expression but did not become immortalized. p16 expression was reactivated by 5-azacytidine treatment, suggesting inactivation by methylation (34). Brenner and Aldaz (7) reported homozygous deletion of p16 in two immortalized human mammary epithelial cell lines and LOH at the p16 locus (9p21) coupled with a nonsense mutation in the remaining allele in another line.
Our study is consistent with the above reports but is unique in several respects. Loss of p16 expression was observed in normal, non-cancer-derived HMEC that had not been immortalized or exposed to viral oncogenes, i.e., HMEC4. Loss of p16 expression was correlated with escape from M0 and was evident at the p16 mRNA level. As in the above studies, loss of p16 expression was not sufficient for immortalization, and these cells underwent senescence. Importantly, senescence of the HMEC4 culture was not associated with reexpression of p16; instead, high levels of p53, p21, and p27 were present. Therefore, two different mechanisms appear to limit the life span of HMEC in culture, the first involving the Rb/p16 pathway and the second involving p53, p21, and possibly p27. This would be consistent with the observation that E7 alleviates M0 by inactivating Rb but does not readily immortalize HMEC whereas E6, by inactivating p53, readily immortalizes HMEC that have previously escaped M0 (2, 49).
In contrast to published reports of experiments with other cell culture systems, we found that methylation, rather than deletion, was associated with p16 inactivation in the HMEC that escaped M0. We had expected to find deletions, especially in the E6-expressing cells, because of the dramatic shutoff of p16 expression and the fact that E6 effectively renders the cells negative for p53, thus creating an environment in which genetic changes would be tolerated. However, no p16 LOH was detected among 30 clones of E6-expressing cells, 20 of which had been in culture for over 100 population doublings. Studies of primary breast tumors indicate that homozygous deletions in p16 are not common (0 of 37 and 0 of 5) (25, 58). In one study, 14 of 24 primary breast tumors revealed LOH or allelic imbalance at the p16 locus (7). However, mutations were not detected in the remaining alleles (7), suggesting that p16 is not the target of these deletions or that another mechanism exists to inactivate the remaining p16 allele. Importantly, Herman et al. (25) found p16 CpG island methylation in 5 of 16 primary breast cancers, suggesting that methylation is a common mechanism of p16 inactivation in vivo. This finding may well explain why p16 LOH is not always coupled to mutation of the remaining allele and why some tumors show no apparent alterations in p16 (56). Our finding that E6-expressing HMEC contained p16 CpG island methylation is consistent with the observations of p16 methylation in primary breast tumors (25). The finding that the p15 CpG island was not methylated in any HMEC suggests that p15 methylation, if it occurred, was not selected for during culture, similar to results from many primary tumors (24, 56).
The cell culture system used allowed a more detailed examination of p16 expression and methylation over time than would be possible in studies of human tumors. Upon escape of cells from M0, p16 expression was reduced at the protein and mRNA levels in E6-expressing HMEC6 and HMEC9 but no p16 deletions were detected. Analysis of the p16 promoter region CpG island for methylation by the PCR-based assay revealed methylation only in the later-passage cells and thus did not explain the initial downregulation of p16. However, a more detailed methylation analysis based on DNA sequencing of all 35 CpG sites in the p16 promoter region from −159 to +135 revealed partial methylation among the cells that escaped M0. At later stages, the E6-expressing cells showed a further decrease in p16 levels and increased CpG island methylation. Sequencing confirmed that at later passages, methylation had extended to sequences recognized by the methylation-specific primers. Evidence that CpG methylation can spread from an initial methylated seed patch comes from studies of spreading within the adenovirus E2A promoter (52). Thus, the increased methylation observed over time could be explained by a spreading mechanism.
It was apparent from the expression data that the majority of the p16 inactivation occurred early after M0 escape, when relatively few CpG sites were methylated, suggesting the possibility that initially, specific methylation sites are more important than the total number of methylated sites in p16 inactivation. Only at later passages was methylation readily detected in the PCR assay. How does the initial methylation event(s) occur? The leading hypothesis is that initial methylation occurs by chance followed by clonal selection of cells with progressively inactivated growth-inhibitory genes (28). The initial methylation event(s) could have occurred in vivo, during culture, or both. Our result that E6-expressing cells are more likely to escape M0 than are uninfected cells raises the question whether E6 expression increases the frequency of unrepaired random methylation events. Currently, there is insufficient data to make an argument either way. However, a recently published report suggests that proliferating-cell nuclear antigen (PCNA) interacts with and possibly regulates the activity of DNA (cytosine-5)-methyltransferase (9). p21WAF1 regulates the PCNA–DNA (cytosine-5)-methyltransferase interaction by competing for binding to PCNA (9). Thus, it is possible that E6, by disrupting the ability of p53 to transactivate p21WAF1 expression, leads to disregulated methylation. E7 has been shown recently to disrupt the interaction between p21WAF1 and PCNA (16) and thus could also potentially lead to disregulated methylation. Clearly, further work is needed to sort out these possibilities.
The finding that p16 continued to be expressed after M0 in cells that express E7 or E6/E7 suggests that E7 can obviate the selection pressure against p16. No p16 methylation was detected in any of the E7-expressing cells or in the majority of E6/E7-expressing cells by the PCR assay. Strikingly, E6/E7-expressing HMEC9 did not contain a single methylated site in our sequencing analysis. However, some clones of E6/E7-expressing HMEC6 did contain methylation. These clones contained a single copy of E6/E7 (whereas others contained multiple copies), suggesting that lower levels of E7 may be insufficient to completely inactivate Rb.
Thus, we suggest the following model to explain inactivation of the Rb/p16 pathway in HMEC. Expression of E7 inactivates Rb directly, allowing cells to bypass M0. In cells not expressing E7, a selection pressure against the Rb/p16 pathway results in the selection of cells expressing low levels of p16 during M0 escape. p16 gene inactivation is associated with limited methylation within the p16 CpG island. This region of methylation may spread over time to result in more complete gene inactivation. A more extensive analysis would be needed to determine whether methylation of specific regions of the CpG island is associated with the initial downregulation of p16 and to conclusively document the spreading of methylation.
The finding of p16 methylation in the E6-expressing cells correlates well with the finding that CpG island methylation is a mechanism of p16 inactivation in the development of human tumors, including breast cancer (25, 38). Thus, the in vitro culture system recapitulates this aspect of breast tumor biology and provides a useful model for further understanding events in neoplastic progression.
ACKNOWLEDGMENTS
We thank Ben Anderson for providing human breast tissue samples, Erik Espling for maintaining stocks of retroviruses, Jens Oliver Funk and Tohru Kiyono for helpful discussions and suggestions, the Biotech Center at FHCRC for sequencing analysis, the Image Analysis Laboratory at FHCRC for help in scanning and quantitation of blots, and Brian J. Reid for comments on the manuscript and interest in this work.
This work was supported by grant RO1 CA 64795 to D.A.G. and pilot project funds from The Seattle Breast Cancer Research Program, R21 CA66186 (principal investigator, D. B. Thomas), to D.A.G. and S.A.F. (both from NCI). The work done by D.J.W. and M.T.B. was carried out in the laboratory of Brian J. Reid and supported by RO1 CA61202, American Cancer Society grant EDT21i, and National Institute of General Medical Sciences Medical Scientist Training Program grant 5T32GM07266.
REFERENCES
- 1.Alcorta D A, Xiong Y, Phelps D, Hannon G, Beach D, Barrett J C. Involvement of the cyclin-dependent kinase inhibitor p16 (INK4a) in replicative senescence of normal human fibroblasts. Proc Natl Acad Sci USA. 1996;93:13742–13747. doi: 10.1073/pnas.93.24.13742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Band V, De Caprio J A, Delmolino L, Kulesa V, Sager R. Loss of p53 protein in human papillomavirus type 16 E6-immortalized human mammary epithelial cells. J Virol. 1991;65:6671–6676. doi: 10.1128/jvi.65.12.6671-6676.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Band V, Sager R. Distinctive traits of normal and tumor-derived human mammary epithelial cells expressed in a medium that supports long-term growth of both cell types. Proc Natl Acad Sci USA. 1989;86:1249–1253. doi: 10.1073/pnas.86.4.1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Banks L, Edmonds C, Vousden K H. Ability of the HPV16 E7 protein to bind RB and induce DNA synthesis is not sufficient for efficient transforming activity in NIH3T3 cells. Oncogene. 1990;5:1383–1389. [PubMed] [Google Scholar]
- 5.Barrett M T, Reid B J, Joslyn G. Genotypic analysis of multiple loci in somatic cells by whole genome amplification. Nucleic Acids Res. 1995;23:3488–3492. doi: 10.1093/nar/23.17.3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barrett M T, Sanchez C A, Galipeau P C, Neshat K, Emond M, Reid B J. Allelic loss of 9p21 and mutation of the CDKN2/p16 gene develop as early lesions during neoplastic progression in Barrett’s esophagus. Oncogene. 1996;13:1867–1873. [PubMed] [Google Scholar]
- 7.Brenner A J, Aldaz C M. Chromosome 9p allelic loss and p16/CDKN2 in breast cancer and evidence of p16 inactivation in immortal breast epithelial cells. Cancer Res. 1995;55:2892–2895. [PubMed] [Google Scholar]
- 8.Buckley M F, Sweeney K J E, Hamilton J A, Sini R L, Manning D L, Nicholson R I, deFazio A, Watts C K W, Musgrove E A, Sutherland R L. Expression and amplification of cyclin genes in human breast cancer. Oncogene. 1993;8:2127–2133. [PubMed] [Google Scholar]
- 9.Chuang L S-H, Ian H-I, Koh T-W, Ng H-H, Xu G, Li B F L. Human DNA-(cytosine-5) methyltransferase-PCNA complex as a target for p21WAF1. Science. 1997;277:1996–2000. doi: 10.1126/science.277.5334.1996. [DOI] [PubMed] [Google Scholar]
- 10.Davies R, Hicks R, Crook T, Morris J, Vousden K. Human papillomavirus type 16 E7 associates with a histone H1 kinase and with p107 through sequences necessary for transformation. J Virol. 1993;67:2521–2528. doi: 10.1128/jvi.67.5.2521-2528.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Demers G W, Espling E, Harry J B, Etscheid B G, Galloway D A. Abrogation of growth arrest signals by human papillomavirus type 16 E7 is mediated by sequences required for transformation. J Virol. 1996;70:6862–6869. doi: 10.1128/jvi.70.10.6862-6869.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dyson N, Howley P M, Munger K, Harlow E. The human papilloma virus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science. 1989;243:934–937. doi: 10.1126/science.2537532. [DOI] [PubMed] [Google Scholar]
- 13.Ewen M E. The cell cycle and the retinoblastoma protein family. Cancer Metastasis Rev. 1994;13:45–66. doi: 10.1007/BF00690418. [DOI] [PubMed] [Google Scholar]
- 14.Foster, S. A., and D. A. Galloway. 1996. Unpublished data.
- 15.Foster S A, Galloway D A. Human papillomavirus type 16 E7 alleviates a proliferation block in early passage human mammary epithelial cells. Oncogene. 1996;12:1773–1779. [PubMed] [Google Scholar]
- 16.Funk J O, Waga S, Harry J B, Espling E, Stillman B, Galloway D A. Inhibition of CDK activity and PCNA-dependent DNA replication by p21 is blocked by interaction with the HPV 16 E7 oncoprotein. Genes Dev. 1997;11:2090–2100. doi: 10.1101/gad.11.16.2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Garrett L R, Perez-Reyes N, Smith P P, McDougall J K. Interaction of HPV-18 and nitrosomethylurea in the induction of squamous cell carcinoma. Carcinogenesis. 1993;14:329–332. doi: 10.1093/carcin/14.2.329. [DOI] [PubMed] [Google Scholar]
- 18.Glisin V, Crkvenjakov R, Byus C. Ribonucleic acid isolated by cesium chloride centrifugation. Biochemistry. 1974;13:2633. doi: 10.1021/bi00709a025. [DOI] [PubMed] [Google Scholar]
- 19.Halbert C L, Demers G W, Galloway D A. The E7 gene of human papillomavirus type 16 is sufficient for immortalization of human epithelial cells. J Virol. 1991;65:473–478. doi: 10.1128/jvi.65.1.473-478.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hannon G J, Beach D. p15INK4B is a potential effector of TGF-β-induced cell cycle arrest. Nature. 1994;371:257–261. doi: 10.1038/371257a0. [DOI] [PubMed] [Google Scholar]
- 21.Hara E, Smith R, Parry D, Tahara H, Stone S, Peters G. Regulation of p16CDKN2 expression and its implications for cell immortalization and senescence. Mol Cell Biol. 1996;16:859–867. doi: 10.1128/mcb.16.3.859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hawley-Nelson P, Vousden K H, Hubbert N L, Lowy D R, Schiller J T. HPV 16 E6 and E7 proteins cooperate to immortalize primary human foreskin keratinocytes. EMBO J. 1989;8:3905–3910. doi: 10.1002/j.1460-2075.1989.tb08570.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Herman J G, Graff J R, Myohanen S, Nelkin B D, Baylin S B. Methylation-specific PCR: a novel assay for methylation status of CpG islands. Proc Natl Acad Sci USA. 1996;93:9821–9826. doi: 10.1073/pnas.93.18.9821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Herman J G, Jen J, Merlo A, Baylin S B. Hypermethylation-associated inactivation indicates a tumor suppressor role for p15INK4B. Cancer Res. 1996;56:722–727. [PubMed] [Google Scholar]
- 25.Herman J G, Merlo A, Mao L, Lapidus R G, Issa J P J, Davidson N E, Sidransky D, Baylin S B. Inactivation of the CDKN2/p16/MTS1 gene is frequently associated with aberrant DNA methylation in all common human cancers. Cancer Res. 1995;55:4525–4530. [PubMed] [Google Scholar]
- 26.Hurlin P J, Kaur P, Smith P P, Perez-Reyes N, Blanton R A, McDougall J K. Progression of human papillomavirus type 18-immortalized human keratinocytes to a malignant phenotype. Proc Natl Acad Sci USA. 1991;88:570–574. doi: 10.1073/pnas.88.2.570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jewers R J, Hildebrandt P, Ludlow J W, Kell B, McCance D J. Regions of human papillomavirus type 16 E7 oncoprotein required for immortalization of human keratinocytes. J Virol. 1992;66:1329–1335. doi: 10.1128/jvi.66.3.1329-1335.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jones P A. DNA methylation errors and cancer. Cancer Res. 1996;56:2463–2467. [PubMed] [Google Scholar]
- 29.Kamb A, Gruis N A, Weaver-Feldhaus J, Liu Q, Harshman K, Tavtigian S V, Stockert E, Day R S, Johnson B E, Skolnick M H. A cell cycle regulator potentially involved in genesis of many tumor types. Science. 1994;264:436–440. doi: 10.1126/science.8153634. [DOI] [PubMed] [Google Scholar]
- 30.Keyomarsi K, Pardee A B. Redundant cyclin overexpression and gene amplification in breast cancer cells. Proc Natl Acad Sci USA. 1993;90:1112–1116. doi: 10.1073/pnas.90.3.1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Klingelhutz A J, Smith P P, Garrett L R, McDougall J K. Alteration of the DCC tumor-suppressor gene in tumorigenic HPV-18 immortalized human keratinocytes transformed by nitrosomethylurea. Oncogene. 1993;8:95–99. [PubMed] [Google Scholar]
- 32.Koh J, Enders G H, Dynlacht B D, Harlow E. Tumour-derived p16 alleles encoding proteins defective in cell-cycle inhibition. Nature. 1995;375:506–510. doi: 10.1038/375506a0. [DOI] [PubMed] [Google Scholar]
- 33.Laborda J. 36B4 cDNA used as an estradiol-independent mRNA control is the cDNA for human acidic phosphoprotein PO. Nucleic Acids Res. 1991;19:3998. doi: 10.1093/nar/19.14.3998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Loughran O, Malliri A, Owens D, Gallimore P H, Stanley M A, Ozanne B, Frame M C, Parkinson E K. Association of CDKN2A/p16INK4A with human head and neck keratinocyte replicative senescence: relationship of dysfunction to immortality and neoplasia. Oncogene. 1996;13:561–568. [PubMed] [Google Scholar]
- 35.Lukas J, Bartkova J, Rohde M, Strauss M, Bartek J. Cyclin D1 is dispensable for G1 control in retinoblastoma gene-deficient cells independently of cdk4 activity. Mol Cell Biol. 1995;15:2600–2611. doi: 10.1128/mcb.15.5.2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lukas J, Parry D, Aagaard L, Mann D J, Bartkova J, Strauss M, Peters G, Bartek J. Retinoblastoma-protein-dependent cell-cycle inhibition by the tumor suppressor p16. Nature. 1995;375:503–506. doi: 10.1038/375503a0. [DOI] [PubMed] [Google Scholar]
- 37.Medema R H, Herrera R E, Lam F, Weinberg R A. Growth suppression by p16ink4 requires functional retinoblastoma protein. Proc Natl Acad Sci USA. 1995;92:6289–6293. doi: 10.1073/pnas.92.14.6289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Merlo A, Herman J G, Mao L, Lee D J, Gabrielson E, Burger P C, Baylin S B, Sidransky D. 5′ CpG island methylation is associated with transcriptional silencing of the tumor suppressor p16/CDKN2/MTS1 in human cancers. Nat Med. 1995;1:686–692. doi: 10.1038/nm0795-686. [DOI] [PubMed] [Google Scholar]
- 39.Miller A D, Rosman G J. Improved retroviral vectors for gene transfer and expression. BioTechniques. 1989;7:980–990. [PMC free article] [PubMed] [Google Scholar]
- 40.Munger K, Phelps W C, Bubb V, Howley P M, Schlegel R. The E6 and E7 genes of the human papillomavirus type 16 together are necessary and sufficient for transformation of primary human keratinocytes. J Virol. 1989;63:4417–4421. doi: 10.1128/jvi.63.10.4417-4421.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Munger K, Werness B A, Dyson N, Phelps W C, Harlow E, Howley P M. Complex formation of the human papillomavirus E7 proteins with the retinoblastoma tumor suppressor gene product. EMBO J. 1989;8:4099–4105. doi: 10.1002/j.1460-2075.1989.tb08594.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Noble J R, Rogan E M, Neumann A A, Maclean K, Bryan T M, Reddel R R. Association of extended in vitro proliferative potential with loss of p16INK4 expression. Oncogene. 1996;13:1259–1268. [PubMed] [Google Scholar]
- 43.Nobori T, Miura K, Wu D J, Lois A, Takabayashi K, Carson D A. Deletions of the cyclin-dependent kinase-4 inhibitor gene in multiple human cancers. Nature. 1994;368:753–756. doi: 10.1038/368753a0. [DOI] [PubMed] [Google Scholar]
- 44.Otsuki T, Clark H M, Wellmann A, Jaffe E S, Raffeld M. Involvement of CDKN2 (p16INK4A/MTS1) and p15INK4B/MTS2 in human leukemias and lymphomas. Cancer Res. 1995;55:1436–1440. [PubMed] [Google Scholar]
- 45.Reed A, Califano J, Cairns P, Westra W H, Jones R M, Koch W, Ahrendt S, Eby Y, Sewell D, Nawroz H, Bartek J, Sidransky D. High frequency of p16 (CDKN2/MTS-1/INK4A) inactivation in head and neck squamous cell carcinoma. Cancer Res. 1996;56:3630–3633. [PubMed] [Google Scholar]
- 46.Reznikoff C A, Yeager T R, Belair C D, Savelieva E, Puthenveettil J A, Stadler W M. Elevated p16 at senescence and loss of p16 at immortalization in human papillomavirus 16 E6, but not E7, transformed human uroepithelial cells. Cancer Res. 1996;56:2886–2890. [PubMed] [Google Scholar]
- 47.Scheffner M, Werness B A, Huibregtse J M, Levine A J, Howley P M. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell. 1990;63:1129–1136. doi: 10.1016/0092-8674(90)90409-8. [DOI] [PubMed] [Google Scholar]
- 48.Serrano M, Hannon G J, Beach D. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature. 1993;366:704–707. doi: 10.1038/366704a0. [DOI] [PubMed] [Google Scholar]
- 49.Shay J W, Wright W E, Brasiskyte D, Van Der Haegen B A. E6 of human papillomavirus type 16 can overcome the M1 stage of immortalization in human mammary epithelial cells but not in human fibroblasts. Oncogene. 1993;8:1407–1413. [PubMed] [Google Scholar]
- 50.Spruck C H, III, Gonzalez-Zulueta M, Shibata A, Simoneau A R, Lin M-F, Gonzales F, Tsai Y, Jones P A. p16 gene in uncultured tumors. Science. 1994;370:183–184. doi: 10.1038/370183a0. [DOI] [PubMed] [Google Scholar]
- 51.Stampfer M R. Isolation and growth of human mammary epithelial cells. J Tissue Culture Methods. 1985;9:107–115. [Google Scholar]
- 52.Toth M, Lichtenberg U, Doerfler W. Genomic sequencing reveals a 5-methylcytosine-free domain in active promoters and the spreading of preimposed methylation patterns. Proc Natl Acad Sci USA. 1989;86:3728–3732. doi: 10.1073/pnas.86.10.3728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wazer D E, Liu X-L, Chu Q, Gao Q, Band V. Immortalization of distinct human mammary epithelial cell types by human papillomavirus 16 E6 or E7. Proc Natl Acad Sci USA. 1995;92:3687–3691. doi: 10.1073/pnas.92.9.3687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Weinberg R A. Tumor suppressor genes. Science. 1991;254:1138–1146. doi: 10.1126/science.1659741. [DOI] [PubMed] [Google Scholar]
- 55.Werness B A, Levine A J, Howley P M. Association of human papillomavirus types 16 and 18 E6 proteins with p53. Science. 1990;248:76–79. doi: 10.1126/science.2157286. [DOI] [PubMed] [Google Scholar]
- 56.Wong D J, Barrett M T, Stoger R, Emond M J, Reid B J. p16INK4A promoter is hypermethylated at a high frequency in esophageal adenocarcinomas. Cancer Res. 1997;57:2619–2622. [PubMed] [Google Scholar]
- 57.Xiong Y, Zhang H, Beach D. Subunit rearrangement of the cyclin-dependent kinases is associated with cellular transformation. Genes Dev. 1993;7:1572–1583. doi: 10.1101/gad.7.8.1572. [DOI] [PubMed] [Google Scholar]
- 58.Xu L, Sgroi D, Sterner C J, Beauchamp R L, Pinney D M, Keel S, Ueki K, Rutter J L, Buckler A J, Louis D N, Gusella J F, Ramesh V. Mutational analysis of CDKN2 (MTS1/p16ink4) in human breast carcinomas. Cancer Res. 1994;54:5262–5264. [PubMed] [Google Scholar]
- 59.Zerfass-Thome K, Zwerschke W, Mannhardt B, Tindle R, Botz J W, Jansen-Durr P. Inactivation of the cdk inhibitor p27KIP1 by the human papillomavirus type 16 E7 oncoprotein. Oncogene. 1996;13:2323–2330. [PubMed] [Google Scholar]
- 60.Zhang L, Cui X, Schmitt K, Hubert R, Navidi W, Arnheim N. Whole genome amplification from a single cell: implications for genetic analysis. Proc Natl Acad Sci USA. 1992;89:5847–5851. doi: 10.1073/pnas.89.13.5847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zwijsen R M L, Klompmaker R, Weintjens E B H G M, Kristel P M P, van der Burg B, Michalides R J A M. Cyclin D1 triggers autonomous growth of breast cancer cells by governing cell cycle exit. Mol Cell Biol. 1996;16:2554–2560. doi: 10.1128/mcb.16.6.2554. [DOI] [PMC free article] [PubMed] [Google Scholar]