

Abstract
Regulation of phosphoinositide 3-kinase (PI 3-kinase) can occur by binding of the regulatory p85 subunit to tyrosine-phosphorylated proteins and by binding of the p110 catalytic subunit to activated Ras. However, the way in which these regulatory mechanisms act to regulate PI 3-kinase in vivo is unclear. Here we show that several growth factors (basic fibroblast growth factor [bFGF], platelet-derived growth factor [PDGF], and epidermal growth factor [EGF; to activate an EGF receptor-Ret chimeric receptor]) all activate PI 3-kinase in vivo in the neuroectoderm-derived cell line SKF5. However, these growth factors differ in their ability to activate PI 3-kinase-dependent signaling. PDGF and EGF(Ret) treatment induced PI 3-kinase-dependent lamellipodium formation and protein kinase B (PKB) activation. In contrast, bFGF did not induce lamellipodium formation but activated PKB, albeit to a small extent. PDGF and EGF(Ret) stimulation resulted in binding of p85 to tyrosine-phosphorylated proteins and strong Ras activation. bFGF, however, induced only strong activation of Ras. In addition, while RasAsn17 abolished bFGF activation of PKB, PDGF- and EGF(Ret)-induced PKB activation was only partially inhibited and lamellipodium formation was unaffected. Interestingly, in contrast to activation of only endogenous Ras (bFGF), ectopic expression of activated Ras did result in lamellipodium formation. From this we conclude that, in vivo, p85 and Ras synergize to activate PI 3-kinase and that strong activation of only endogenous Ras exerts a small effect on PI 3-kinase activity, sufficient for PKB activation but not lamellipodium formation. This differential sensitivity to PI 3-kinase activation could be explained by our finding that PKB activation and lamellipodium formation are independent PI 3-kinase-induced events.
After growth factor binding to a receptor tyrosine kinase, a variety of signaling events are induced. Among these are the activation of the small GTPase Ras and the activation of phosphoinositide 3-kinase (PI 3-kinase). The active GTP-bound form of Ras binds to several effector molecules, including (i) family members of the serine-threonine kinase Raf1, which results in the activation of the kinases extracellular signal-regulated kinase (ERK) and mitogen-activated protein/ERK kinase (MEK) (29); (ii) Ral guanine nucleotide exchange factor (RalGEF) family members, which leads to the activation of the small GTPase Ral (15); and, interestingly, (iii) members of the PI 3-kinase family (35). PI 3-kinase phosphorylates phosphatidylinositol (PI) lipids on the 3′ position of the inositol ring, and growth factor-induced PI 3-kinase activation leads to an increase in the levels of PI-3,4,5 triphosphate (PI-3,4,5P3) and PI-3,4P2 (21, 41). PI 3-kinase has been implicated in a large variety of biological responses to growth factors; these include actin reorganization (lamellipodium formation), apoptosis protection, glucose uptake, activation of kinases (p70s6k, protein kinase B [PKB], and atypical PKCs), and inactivation of glycogen synthase kinase-3 (GSK-3) (for a review, see reference 44). Thus far, the only clear direct downstream target identified for these lipids is PKB (also known as c-Akt or Rac kinase) (3, 17). This serine-threonine kinase has a pleckstrin homology domain, which is the target for PI-3,4P2 lipids in vitro and in vivo (16, 22, 26). In addition, phosphorylation is essential for activation (1–3). This phosphorylation is mediated at least in part by an upstream kinase that recently has been cloned (PDK1) (2). PKB mediates the effect of PI 3-kinase on the survival of cells from apoptosis (13, 23); this is likely to be a direct effect, since PKB has been shown to phosphorylate the BCL-2 family member BAD (7). Activation of p70s6k (3) and inactivation of GSK-3 (6) are also likely to be mediated by PKB. Inactivation of GSK-3 appears to be direct, since GSK-3 is a substrate for PKB in vitro. Activation of p70s6k is probably not direct and will involve one or more intermediate steps. However, for both GSK-3 and p70s6k, evidence thus far comes from experiments with constitutively active PKB, and more definite proof awaits the results of experiments with dominant negative PKB. As mentioned, PI 3-kinase also mediates other biological responses such as the induction of Rac-dependent lamellipodium formation (18, 31). However, whether PKB mediates this effect or whether another target for the 3′-phosphorylated PI lipids is involved is still elusive.
Two different mechanisms of activation of PI 3-kinase have been described. First, activation can occur by binding of the 85-kDa regulatory subunit of PI 3-kinase to tyrosine-phosphorylated residues, for instance those present on activated receptor tyrosine kinases (43), and, second, activation can occur by binding of constitutively active Ras to the 110-kDa catalytic subunit (35, 37). In vivo, both interactions result in increased PI-3,4,5P3 and PI-3,4P2 formation, and evidence exists that inhibition of either of the two mechanisms affects PI 3-kinase activation (14, 18, 24–26, 35). However, the interpretation of these results has been complicated by the observation that (i) PI 3-kinase may also act upstream from Ras (19); (ii) other closely related small GTPases such as R-Ras, Cdc42, and Rac may bind and activate PI 3-kinase as well (27, 42, 48); and (iii) almost all the studies analyzed the role of ectopically (over)expressed oncogenic Ras.
Given the potential role of Ras in regulating PI 3-kinase activity, Ras has also been implicated in the regulation of PI 3-kinase-directed processes, such as Rac-mediated lamellipodium formation and the activation of PKB, but the picture is still rather unclear. Introduction of constitutively active Ras in Swiss 3T3 cells induces the formation of lamellipodia in a Rac-dependent manner (34); however, surprisingly, the formation of these lamellipodia is insensitive to inhibitors of PI 3-kinase but is fully inhibited by dominant negative PI 3-kinase (Δp85) (31). This suggests that Ras may activate isotypes of PI 3-kinase that are not sensitive to LY294002 or wortmannin. Furthermore, it was shown that endogenous Ras is not essential for growth factor-induced lamellipodium formation (30, 34), suggesting that constitutively active Ras-induced and growth factor-induced lamellipodium formation are independent pathways. For PKB, one report showed that platelet-derived growth factor (PDGF)-induced activation is dependent on endogenous Ras (17) whereas others reported that PKB can be activated independently of endogenous Ras (3, 26). Furthermore, one report showed that constitutively active Ras could not induce PKB activation (17) while others showed the opposite (26, 27).
We therefore analyzed the regulation of PI 3-kinase, through Ras and p85, by measuring 3′-phosphorylated PI lipid formation as well as by using PKB activation and lamellipodium formation as readouts for PI 3-kinase activation. The latter was done to study how potential differences in PI 3-kinase regulation might translate into a different biological response. As a model system, we used neuroectoderm-derived SKF5 cells. These cells stably express an epidermal growth factor (EGF) receptor-Ret chimeric receptor (HERRet) and are responsive to EGF, PDGF, and basic fibroblast growth factor (bFGF) (45–47). We found that (i) activation of endogenous Ras is neither necessary nor sufficient for the induction of lamellipodia but is sufficient for the activation of PKB and (ii) Rac-dependent lamellipodium formation and PKB activation are independent downstream effects of PI 3-kinase. From these results, we conclude that PKB activation and lamellipodium formation are independent PI 3-kinase-mediated events that are differentially regulated by endogenous Ras.
MATERIALS AND METHODS
Cell lines and inhibitors.
The HERRet-expressing stable transfectant of the human SK-N-MC neuroepithelioma cell line (SKF5 cell line) (47) was cultured in DF12 medium supplemented with 10% fetal calf serum. Wortmannin and LY294002 were purchased from Sigma and used at 50 nM and 50 μM, respectively. Pretreatment with these agents was carried out for 10 min.
In vitro PI 3-kinase assay, Western blotting, and determination of Ras activation.
The materials and methods for the PI 3-kinase assay (46), Western blotting, and experiments to determine endogenous Ras activity have been described previously (9, 47).
Transient transfections.
For transient expression of the constitutively active and dominant negative mutants of Ras, cells were transfected after 2 days of culture on coverslips, by the standard calcium precipitation technique. Per coverslip, 0.5 μg of GTPase-encoding expression plasmid together with 0.5 μg of carrier DNA was applied. After overnight incubation, the precipitate was removed and the cells were allowed to recover for 24 h. The cells were serum starved for 16 h and stimulated as indicated (see the figures). For PKB experiments, cells were cultured in 5-cm petri dishes and transfected overnight with a total of 5 μg/dish, consisting of 0.5 μg of HA-PKB expression vector per dish and optionally 2 μg of mutant GTPase per dish, supplemented with carrier DNA in the form of an irrelevant expression plasmid. The precipitate was removed, and the cells were allowed to recover for 24 h, including overnight serum starvation.
Immunofluorescence.
After stimulation of (transfected) cells grown on glass coverslips, the cells were fixed for 20 min in 4% paraformaldehyde and permeabilized with 0.1% Triton X-100 in phosphate-buffered saline (PBS), and nonspecific binding was blocked by incubation for 45 min with 0.5% bovine serum albumin in PBS (all at 4°C). For detection of RasAsn17 and RasLeu61, the anti-Ras monoclonal antibody Y13-238 (Oncogene Science) was used. For detection of GAG-PKB-expressing cells, a polyclonal anti-PKB antibody was used, and HA-PKB-CAAX was detected with a monoclonal anti-HA antibody (12CA5). Incubations with the primary antibody were carried out for 2 h at room temperature and were followed by three rinses in PBS. Subsequently, the coverslips were incubated for 2 h with goat anti-mouse-conjugated CY3 (Jackson Laboratories) or goat anti-rabbit-conjugated tetramethylrhodamine-5′-isocyanate (TRITC; Sigma) antiserum to reveal the primary antibody, in combination with fluorescein isothiocyanate (FITC)-coupled phalloidin to stain polymerized actin. After three washes with PBS, the coverslips were embedded in Immumount (Shandon) and analyzed with a Labophot fluorescence microscope (Nikon).
Determination of PKB activity.
The in vitro kinase assay to determine PKB kinase activity has been described previously (3). Briefly, HA-PKB-transfected cells were lysed after growth factor stimulation and either endogenous PKB was immunoprecipitated with a rabbit polyclonal antiserum or, for transfected cells, HA-PKB proteins were immunoprecipitated with an antihemagglutinin (HA) antibody (12CA5). HA-PKB kinase activity was determined by incubation of immunoprecipitated PKB with [γ-32P]ATP and the kinase substrate histone 2B. Histone 2B phosphorylation was determined by autoradiography following sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) separation of the kinase reaction mixture. The fold induction of PKB activity was determined with a phosphorimager (Storm).
Analysis of PI synthesis.
Subconfluent cultures of SKF5 cells were serum starved overnight and labeled in vivo for 2 h at 37°C with [32P]orthophosphate. The cells were stimulated for 2 min with growth factors and lysed in 1 M HCl, and the lipids were extracted. The lipids were deacylated with methylamine and analyzed by high-pressure liquid chromatography as described by Rodriguez-Viciana et al. (35).
RESULTS
PI 3-kinase activation in SKF5 cells.
PI 3-kinase plays a key role in a variety of biological responses such as the rearrangement of the actin cytoskeleton, glucose uptake, apoptosis protection, and the regulation of kinases such as PKB, p70s6k, and GSK-3. Treatment with any growth factor that activates PI 3-kinase does not always result in the activation of the whole plethora of PI 3-kinase-dependent signaling. The most notable example is the observation that while the stimulation of glucose uptake by insulin is PI 3-kinase mediated, other factors such as PDGF and interleukin-4, which also activate PI 3-kinase, do not stimulate glucose uptake (20). To study growth factor-dependent variations in PI 3-kinase signaling and to explore whether the observed variation might shed light on the regulation of PI 3-kinase, we first determined PI 3-kinase activity in the neuroectoderm-derived SKF5 cell line following treatment with various growth factors. To do so, we measured the in vivo levels of PI-3,4P2 and PI-3,4,5P3 in [32P]orthophosphate-labeled cells before and after stimulation (Fig. 1). Treatment with PDGF and bFGF, which activate endogenous receptors, and treatment with EGF, which activates a stably transfected EGF receptor-Ret chimeric receptor (HERRet) (39, 47) [referred to here as EGF(Ret)], all induced an increase in the levels of both lipids, indicating activation of PI 3-kinase by these factors. However, there was a clear difference in the magnitude of the response; PDGF induced an average fivefold increase in the levels of both lipids whereas EGF(Ret) induced only a twofold (PI-3,4,5P3) to threefold (PI-3,4P2) increase. The increase induced by bFGF was slightly smaller than that observed for EGF and averaged a 1.5-fold increase.
FIG. 1.

Growth factor-induced PI 3-kinase activity. EGF(Ret), bFGF, and PDGF induce different amounts of phosphorylated PIs as measured by in vivo labeling and analysis of total lipids. Serum-starved subconfluent cultures of SKF5 cells were labeled in vivo with [32P]orthophosphate and were left untreated (control) or were stimulated for 2 min with 40 ng of EGF per ml or 20 ng of bFGF or PDGF per ml, followed by isolation of total lipids. Radioactive lipids were analyzed by high-pressure liquid chromatography. Shown is the fold induction of the PI-3,4P2 and PI-3,4,5P3 synthesis above control values. Error bars indicate the standard deviations.
PI 3-kinase-mediated signaling: lamellipodium formation and PKB activation.
To study whether the above-described quantitative differences in growth factor-induced 3′-phosphorylated PI lipid formation cause a difference in the activation of PI 3-kinase-dependent signaling pathways, we analyzed lamellipodium formation in these cells. As expected from our previous work (46), EGF(Ret) induced the formation of lamellipodia (Fig. 2A) in a PI 3-kinase-dependent manner (i.e., sensitive to wortmannin and LY294002). The same was observed after treatment of the cells with PDGF (Fig. 2A). Lamellipodium formation was observed within 5 min and was sensitive to both wortmannin and LY294002 (data not shown). For the formation of both EGF(Ret)- and PDGF-induced lamellipodia, the activation of the small GTPase Rac is both necessary and sufficient whereas neither Cdc42 nor Rho is involved (data not shown). Surprisingly, treatment of SKF5 cells with bFGF did not induce the formation of lamellipodia as determined both by fluorescent labelling of polymerized actin (Fig. 2A) and by time-lapse video microscopy (data not shown). Apparently, the increase in the levels of 3′-phosphorylated PI lipids following bFGF treatment is not sufficient to induce lamellipodium formation. Also, lysophosphatidic acid (LPA) and the phorbol ester 12-O-tetradecanoylphorbol-13-acetate (TPA) did not induce lamellipodium formation in these cells (data not shown). For TPA, this result was unexpected, since in Swiss 3T3 cells TPA is a very strong inducer of lamellipodium formation (34).
FIG. 2.

Growth factor-induced lamellipodium formation and PKB activation in SKF5 cells. (A) EGF(Ret) and PDGF, but not bFGF, induce lamellipodium formation. Serum-starved SKF5 cells grown on coverslips were left untreated (control) or were stimulated for 10 min with 40 ng of EGF per ml or 20 ng of bFGF or PDGF per ml. Polymerized actin was stained with FITC-conjugated phalloidin. Lamellipodia are indicated by white arrows. Bar, 10 μm. (B) EGF(Ret), bFGF, and PDGF differentially induce PKB activity. SKF5 cells were serum starved overnight and were either left untreated (−) or treated for 5 min with 40 ng of EGF per ml (E) or 20 ng of bFGF (F) or PDGF (P) per ml. PKB activity was determined in an in vitro kinase assay on endogenous PKB immunoprecipitated by a rabbit polyclonal anti-PKB serum with histone 2B (H2B) as a substrate. Shown is an autoradiogram of a representative experiment after SDS-PAGE analysis of the kinase reaction.
Next, growth factor-induced activation of endogenous PKB was measured by immunoprecipitation with a PKB-specific antiserum followed by an in vitro kinase assay with histone 2B as a substrate (Fig. 2B). Similar to the formation of lamellipodia, EGF(Ret) and PDGF both induced a clear activation of endogenous PKB activity. Surprisingly, and in contrast to the lack of lamellipodium formation, bFGF did induce PKB activity, albeit to a small extent. LPA did not induce PKB activation in SKF5 cells (data not shown). As expected from previous work (3, 17), pretreatment of SKF5 cells with wortmannin or LY294002 resulted in complete inhibition of growth factor-induced PKB activity (data not shown). Identical results were obtained when PKB activity was measured following transfection of HA-tagged PKB and immunoprecipitation with the 12CA5 monoclonal antibody (see below). Clear activation by PDGF and EGF(Ret) and low but reproducible activation by bFGF were observed.
Thus, bFGF reveals a difference in PI 3-kinase-dependent signaling toward PKB and lamellipodium formation.
Regulation of PI 3-kinase signaling: tyrosine phosphorylation.
To further understand the difference in PI 3-kinase-dependent signaling induced by bFGF on the one hand and PDGF and EGF(Ret) on the other, we first tried to understand the mechanism of PI 3-kinase activation in SKF5 cells in greater detail. To this end, we analyzed in vitro PI 3-kinase activity present in antiphosphotyrosine immunoprecipitates (Fig. 3A). PI 3-kinase activity, as measured in this way, is stimulated by EGF(Ret) and PDGF, and this induction roughly correlates with the increase observed in the levels of PI-3P lipids. However, after treatment with bFGF, we could not detect a reproducible increase in PI 3-kinase activity associated with antiphosphotyrosine immunoprecipitates. This is also reflected by the appearance of the 85-kDa regulatory subunit in antiphosphotyrosine immunoprecipitates after EGF(Ret) and PDGF stimulation. No p85 was detectable in antiphosphotyrosine precipitates after bFGF stimulation (Fig. 3B). These results suggest that PDGF and EGF(Ret) use a mechanism of PI 3-kinase activation that involves p85 tyrosine phosphorylation or binding to tyrosine-phosphorylated proteins (e.g., receptor) and that this is either absent or not detectable after treatment with bFGF.
FIG. 3.

Growth factor-induced, phosphotyrosine-associated PI 3-kinase activity. (A) EGF(Ret) and PDGF, but not bFGF, induce PI 3-kinase activity as measured in an in vitro kinase assay. Serum-starved subconfluent cultures of SKF5 cells were left untreated (−) or were stimulated for 5 min with 40 ng of EGF per ml or 20 ng of bFGF or PDGF per ml, followed by immunoprecipitation of tyrosine-phosphorylated proteins. Immunoprecipitates were subjected to a kinase assay in the presence of [γ-32P]ATP and phosphatidylinositol. Shown is an autoradiogram of the kinase reaction after thin-layer chromatography. The position of the 3′-phosphorylated phosphatidylinositol (PI-3P) is indicated. All experiments were quantified and averaged (±standard error of the mean); the result is represented by bars (fold induction). The number of three independent experiments were used for each point. (B) EGF(Ret) and PDGF, but not bFGF, induce the association of the p85 subunit of PI 3-kinase with tyrosine-phosphorylated proteins. Serum-starved subconfluent cultures of SKF5 cells were left untreated or were stimulated for 5 min with 40 ng of EGF per ml (E) or 20 ng of bFGF (F) or PDGF (P) per ml. In the upper panel, tyrosine-phosphorylated proteins were immunoprecipitated and analyzed by Western blotting with a monoclonal antibody to the p85 subunit of PI 3-kinase. In the lower panel, in the same cell lysates the activity of ERK2 was analyzed by Western blotting with a polyclonal antibody to ERK2. The positions of the active (ppERK2) and inactive (ERK2) forms of the kinase are indicated.
Regulation of PI 3-kinase signaling: activation of PI 3-kinase by oncogenic Ras.
Next we investigated a possible role for Ras in the activation mechanism of PI 3-kinase. We first analyzed whether activation of Ras in the SKF5 cells would result in the activation of PI 3-kinase-dependent signaling. To do so, we again took lamellipodium formation and PKB activation as readouts for PI 3-kinase activation and investigated the effect of ectopic expression of constitutively active Ras (RasLeu61) on these readouts. As shown in Fig. 4A, RasLeu61 induced the formation of lamellipodia in the absence of growth factor stimulation in transient-transfection experiments. This induction was completely sensitive to the PI 3-kinase inhibitors LY294002 (Fig. 4A) and wortmannin (data not shown), both of which reverse lamellipodium formation within 10 min after addition.
FIG. 4.
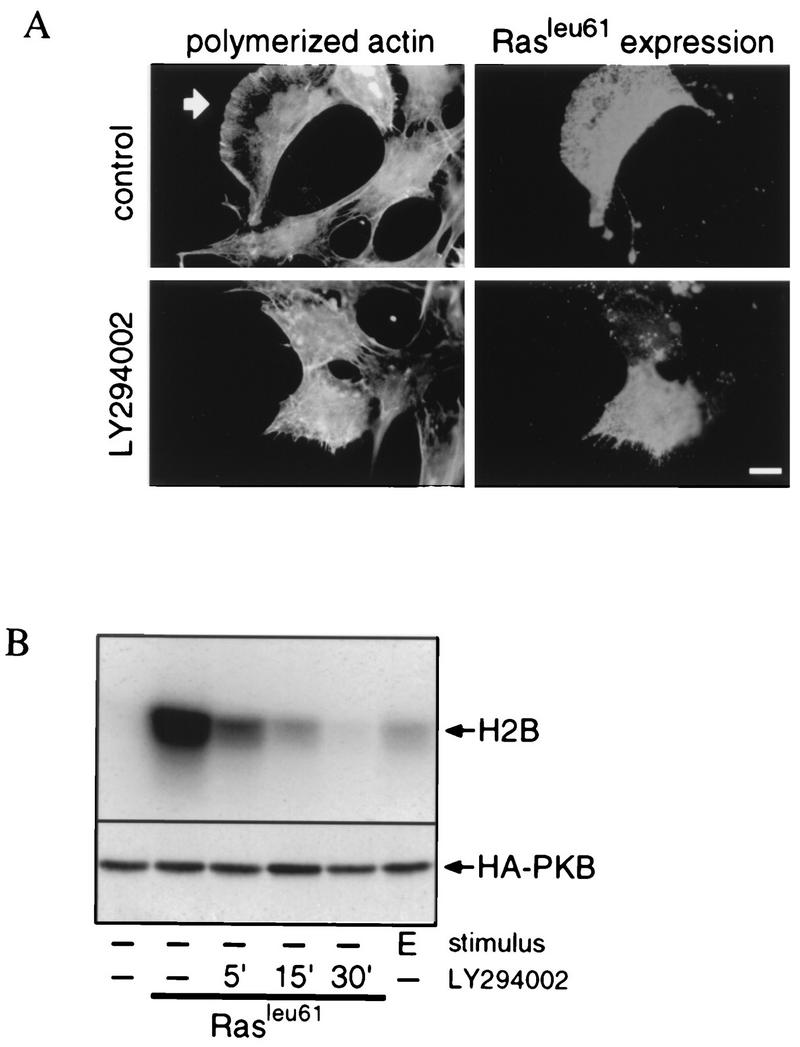
Constitutively active Ras induces PI 3-kinase-mediated lamellipodium formation and PKB activation. (A) Constitutively active Ras-induced lamellipodium formation is dependent on PI 3-kinase activity. SKF5 cells were transiently transfected with a RasLeu61 expression plasmid. After overnight serum starvation, the cells were either left untreated (control) or treated for 10 min with LY294002. Polymerized actin was stained with FITC-conjugated phalloidin, and RasLeu61 expression was revealed with a Ras-specific monoclonal antibody. The arrow indicates lamellipodia. Bar, 10 μm. (B) RasLeu61-induced PKB activation is dependent on PI 3-kinase activity. SKF5 cells were transiently transfected with HA-PKB and RasLeu61 expression plasmids. After overnight serum starvation, the cells were either left untreated or treated for the indicated times with LY294002. PKB activity was determined in an in vitro kinase assay on immunoprecipitated HA-PKB with histone 2B (H2B) as a kinase substrate. Shown is an autoradiogram of a representative experiment after SDS-PAGE separation of the proteins (upper panel). Expression of HA-PKB was controlled by 12CA5 immunoblotting of samples taken before immunoprecipitation (lower panel).
Coexpression of RasLeu61 and PKB also resulted in increased PKB activation, which is PI 3-kinase dependent (Fig. 4B). However, in contrast to lamellipodium formation, the kinetics of downregulation of PKB activity is much slower. PKB activity returned to the basal level only after a 30-min treatment with LY294002 (Fig. 4B) or wortmannin (data not shown). From these results, we conclude that in the SKF5 cells active Ras can induce PI 3-kinase-mediated events, which is compatible with the suggestion that PI 3-kinase is a direct effector of Ras (35).
Regulation of PI 3-kinase signaling: involvement of endogenous Ras.
To see whether activation of endogenous Ras mediates PI 3-kinase activation by growth factors, in particular bFGF, we measured growth factor-induced activation of endogenous Ras. First, Ras activation was measured quantitatively by determining the ratio of GTP to GDP bound to endogenous Ras. After treatment of the cells for 5 min, a clear and comparable increase in the level of RasGTP was observed with EGF(Ret), bFGF, and PDGF (Fig. 5A). Second, we determined the kinetics of endogenous Ras activation with glutathione S-transferase (GST)-Raf-RBD as an activation-specific probe (9). In this assay, active GTP-bound Ras is isolated specifically by binding to GST-Raf-RBD followed by Western blot detection. Both EGF(Ret) and bFGF displayed an initial peak in Ras activity followed by a sustained second phase. In contrast, PDGF induced only the first peak of Ras activation and after approximately 30 min the Ras activity returned to the basal level (Fig. 5B). However, during the time course of the lamellipodium formation and the activation of PKB, there is no clear difference in the activation of endogenous Ras induced by the three different growth factors. As a further control, we also measured Ras activation after LPA stimulation (Fig. 5B). This ligand also stimulated Ras activation, albeit to a significantly lower level than the other three ligands did. Given the inability of bFGF to induce lamellipodium formation, the strong induction of endogenous Ras by bFGF is not sufficient to induce PI 3-kinase-dependent formation of lamellipodia.
FIG. 5.
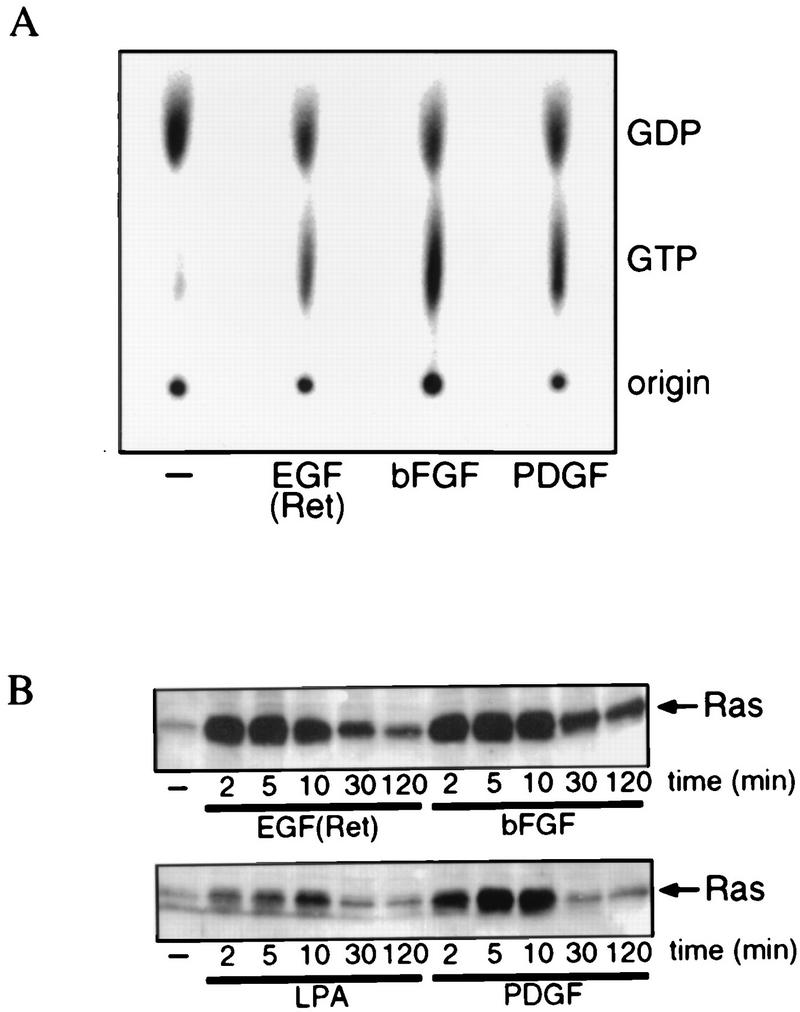
Growth factor-induced Ras activation. (A) EGF(Ret), bFGF, and PDGF induce Ras activation. Serum-starved subconfluent cultures of SKF5 cells were labeled in vivo with [32P]orthophosphate and then treated for 5 min with 40 ng of EGF per ml or 20 ng of bFGF or PDGF per ml, and Ras-bound guanine nucleotides were isolated. Shown is an autoradiogram of the extracted guanine nucleotides after thin-layer chromatography. The positions of labeled GDP and GTP are indicated. The percentages of Ras in the GTP-bound state are as follows: control, 16%; EGF(Ret), 35%; bFGF, 48%; PDGF, 39%. (B) Kinetics of EGF(Ret)-, bFGF-, PDGF- and LPA-induced Ras activation. Serum-starved subconfluent cultures of SKF5 cells were stimulated for the indicated times with 40 ng of EGF per ml, 20 ng of bFGF or PDGF per ml, or 1 μM LPA. GTP-bound Ras was specifically precipitated with GST-Raf-RBD and analyzed by Western blotting.
Next, to see whether endogenous Ras is necessary for PI 3-kinase-dependent signaling, we introduced dominant negative RasAsn17 into SKF5 cells by transient transfection, to investigate whether activation of endogenous Ras is involved in growth factor-induced activation of PI 3-kinase-mediated events. As shown in Fig. 6A, RasAsn17 expression did not affect EGF(Ret)- or PDGF-induced formation of lamellipodia. In addition, RasAsn17 partially affected EGF(Ret)- and PDGF-induced PKB activation (Fig. 6B). This is unlikely to result from incomplete inhibition of Ras activation, since under identical conditions the activation of mitogen-activated protein kinase by these growth factors was completely blocked (Fig. 6C). However, the small increase in PKB activity observed after bFGF stimulation was completely inhibited by RasAsn17 (Fig. 6B).
FIG. 6.
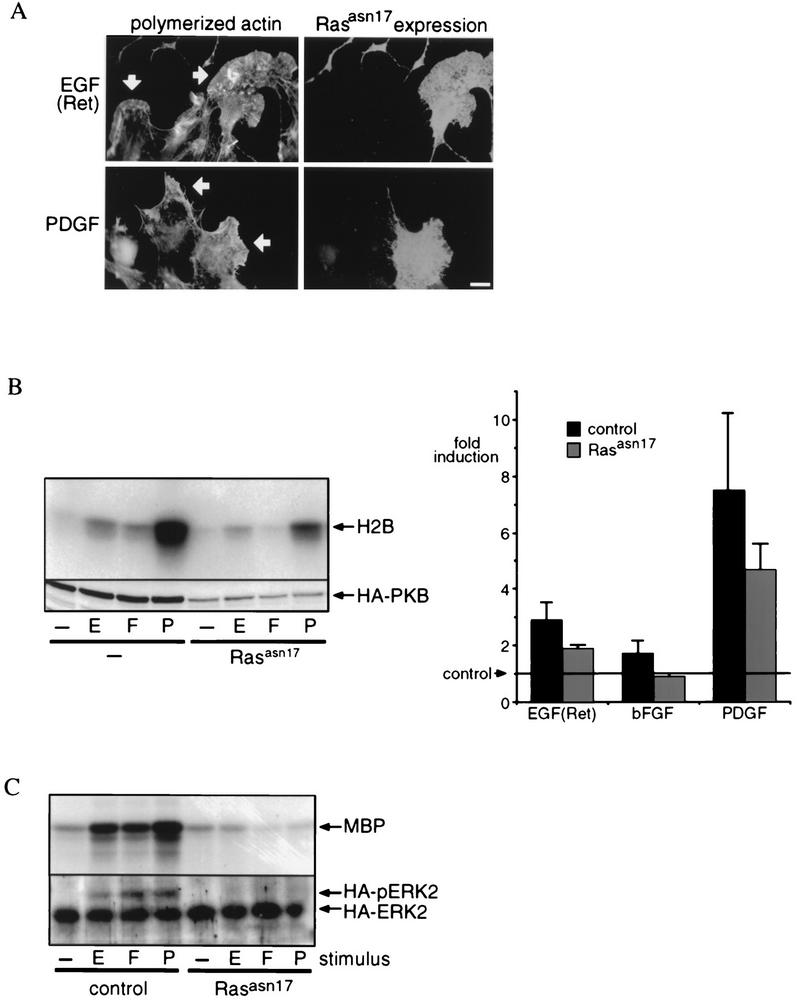
Role of endogenous Ras in lamellipodium formation and PKB activation. (A) Dominant negative Ras does not inhibit lamellipodium formation. SKF5 cells were transiently transfected with dominant negative RasAsn17. The cells were serum starved and were treated with 40 ng of EGF per ml or 20 ng of PDGF per ml. RasAsn17 expression was revealed with an anti-Ras monoclonal antibody, and polymerized actin was stained with phalloidin-FITC. Arrows indicate lamellipodia. Bar, 10 μm. (B) Dominant negative Ras inhibits bFGF-induced activation but partially affects EGF(Ret)- or PDGF-induced PKB activation. SKF5 cells were transiently transfected with HA-PKB and RasAsn17 expression plasmids. After overnight serum starvation, the cells were either left untreated (−) or treated for 5 min with 40 ng of EGF (E) per ml or 20 ng of bFGF (F) or PDGF (P) per ml. PKB activity was determined in an in vitro kinase assay on immunoprecipitated HA-PKB with histone 2B (H2B) as a substrate. The upper panel shows an autoradiogram of a representative kinase assay after SDS-PAGE analysis of the kinase reaction. Expression of HA-PKB was controlled by 12CA5 immunoblotting of samples taken before immunoprecipitation (lower panel). All experiments were quantified and averaged (±standard error of the mean); the results are represented by the bar diagram (fold induction). Five independent experiments were used for each point. (C) RasAsn17 completely blocks Ras-dependent signaling. SKF5 cells were transiently transfected with HA-MAP kinase and RasAsn17 expression plasmids. After overnight serum starvation, the cells were either left untreated or treated for 5 min with 40 ng of EGF per ml or 20 ng of bFGF or PDGF per ml. HA-MAP kinase activity was determined in an in vitro kinase assay on immunoprecipitated HA-MAP kinase with myelin basic protein (MBP) as a substrate (upper panel). Before immunoprecipitation, a sample was taken to control for the expression of HA-MAP kinase. Proteins were separated with a gel system that reveals an electrophoretic mobility shift of phosphorylated (activated) HA-MAP kinase followed by 12CA5 immunoblotting. The positions of active (HA-pERK2) and inactive (HA-ERK2) MAP kinase are indicated (lower panel).
From these results, we may conclude that the activation of Ras is apparently necessary and sufficient to induce PKB activation following bFGF treatment but that following EGF(Ret) and PDGF treatment, p21ras-dependent and -independent pathways to activate PI 3-kinase are operational. Since we observed p85 tyrosine phosphorylation and/or association with activated receptors for EGF(Ret) and PDGF but not bFGF, it is likely that this may represent such a (redundant) Ras-independent pathway.
PKB activation and Rac-dependent lamellipodium formation represent separate PI 3-kinase-dependent pathways.
Evidence is accumulating that PKB is a direct target for the PI-3,4,5P3 and/or PI-3,4P2 lipid generated by PI 3-kinase. Therefore, the most likely possibility would be that PKB functions downstream of PI 3-kinase to mediate Rac-induced lamellipodium formation. However, the observation that compared to EGF(Ret) and PDGF, bFGF induces a weak activation of PKB and no lamellipodium formation appears to argue against this possibility and could suggest an alternative mechanism whereby Rac regulates PKB activity. We addressed this question by coexpressing constitutively active and dominant negative mutants of both Rac and PKB in SKF5 cells (Fig. 7). For PKB, we used a GAG-PKB construct similar to v-Akt (3) as a constitutively active mutant. This construct has ligand-independent activity that in SKF5 cells roughly equals PDGF-induced PKB activity (Fig. 7A). For a dominant negative mutant, we used CAAX-PKB. Unlike PKB that is N-terminally tagged by a membrane localization signal (e.g., GAG-PKB [3] and myr-PKB [1]), the addition of a C-terminal membrane localization signal (e.g., the CAAX box of Ki-Ras) results in an inactive kinase. This is most likely due to the inability to phosphorylate Ser473 located at the extreme C terminus of PKB (1, 2a). In SKF5 cells, coexpression of CAAX-PKB (no tag) with HA-tagged PKB resulted in almost complete inhibition of growth factor-induced HA-PKB activity (Fig. 7A). The residual PKB activity is less than that observed with bFGF treatment.
FIG. 7.
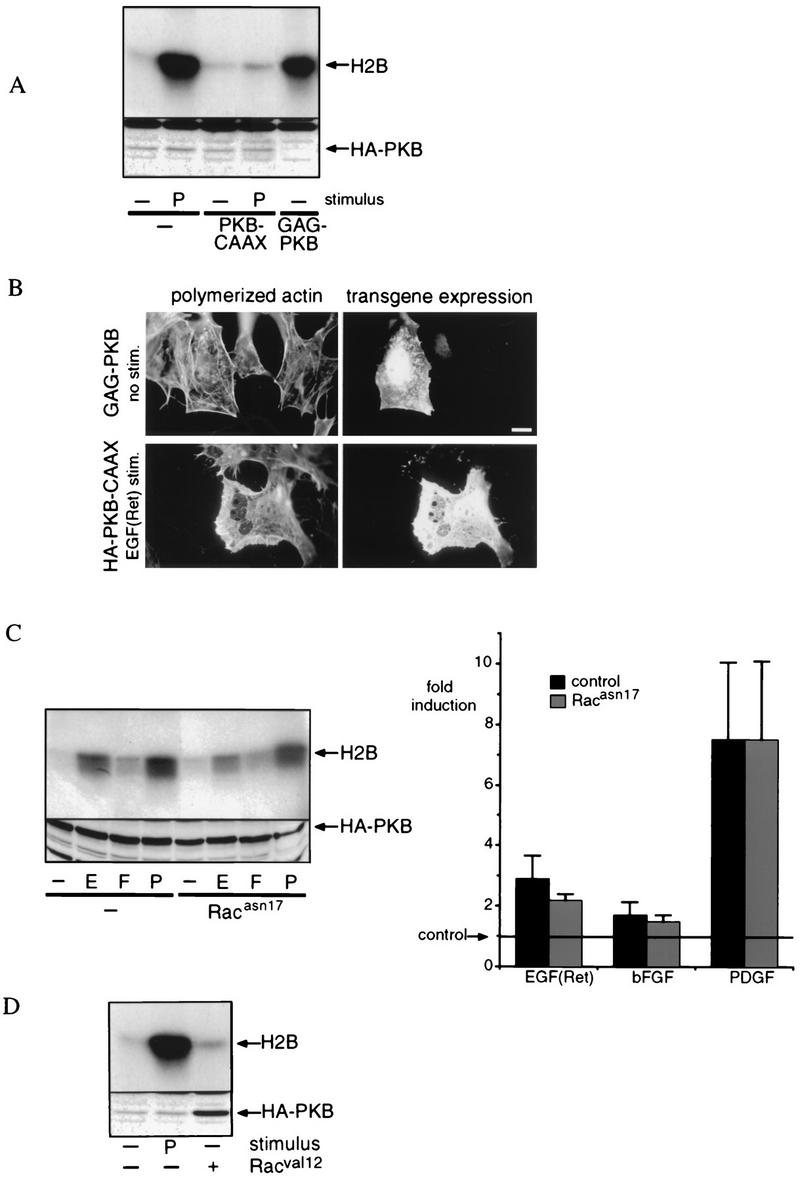
Rac and PKB represent separate PI 3-kinase-dependent signaling pathways. (A) Characterization of constitutively active and dominant negative PKB. SKF5 cells were transiently transfected with GAG-PKB (2 μg) or with HA-PKB (0.5 μg) and either left unstimulated (−) or stimulated with 20 ng of PDGF (P) per ml. To test for dominant negative function, 0.5 μg of HA-PKB was cotransfected with 5 μg of CAAX-PKB (no HA tag). Shown is an autoradiogram representative of three independent experiments. The expression of HA-PKB was controlled by 12CA5 immunoblotting of samples taken before immunoprecipitation (lower panel). (B) Constitutively active PKB does not induce lamellipodium formation, and dominant negative PKB does not interfere with EGF(Ret)-induced lamellipodium formation. SKF5 cells were transiently transfected with constitutively active GAG-PKB or with dominant negative HA-PKB-CAAX, as indicated. The cells were serum starved overnight and were left untreated (GAG-PKB) or stimulated for 10 min with 40 ng of EGF per ml (HA-PKB-CAAX). GAG-PKB expression was revealed with an anti-PKB polyclonal antibody, HA-PKB-CAAX expression was revealed with a monoclonal anti-HA antibody, and polymerized actin was stained with phalloidin-FITC. Bar, 10 μm. (C) Dominant negative Rac does not inhibit growth factor-induced PKB activation. SKF5 cells were transiently transfected with HA-PKB and RacAsn17 expression plasmids. After overnight serum starvation, the cells were either left untreated or treated for 5 min with 40 ng of EGF (E) per ml or 20 ng of bFGF (F) or PDGF per ml. PKB activity was determined in an in vitro kinase assay on immunoprecipitated HA-PKB with histone 2B (H2B) as a substrate. The upper panel shows an autoradiogram of a representative kinase assay after SDS-PAGE analysis of the kinase reaction. Expression of HA-PKB was controlled by 12CA5 immunoblotting of samples taken before immunoprecipitation (lower panel). All experiments were quantified and averaged (±standard error of the mean); the result of this is represented by the bar diagram (fold induction). Four independent experiments were used for each point. (D) Constitutively active Rac does not induce PKB activity. SKF5 cells were transiently transfected with HA-PKB and, as indicated, with a RacVal12 expression plasmid. After overnight serum starvation, the cells were either left untreated (−) or were treated for 5 min with 20 ng of PDGF (P) per ml. PKB activity was determined in an in vitro kinase assay on immunoprecipitated HA-PKB with histone 2B (H2B) as a kinase substrate. The upper panel shows an autoradiogram of a representative kinase assay after SDS-PAGE analysis of the kinase reaction. Expression of HA-PKB was controlled by 12CA5 immunoblotting of samples taken before immunoprecipitation (lower panel).
The expression of constitutively active GAG-PKB did not result in lamellipodium formation (Fig. 7B), suggesting that PKB activation is not sufficient to induce Rac activation and subsequent lamellipodium formation. In addition, expression of the dominant negative mutant CAAX-PKB did not interfere with EGF(Ret) (Fig. 7B)- or PDGF (data not shown)-induced lamellipodium formation. Thus, from these experiments, it is apparent that PKB activation is neither sufficient nor necessary for lamellipodium formation in these cells.
Alternatively, Rac could be involved in the activation of PKB. However, expression of dominant negative RacAsn17 did not interfere with growth factor-induced PKB activation (Fig. 7C) and expression of constitutively active RacVal12 did not result in a significant increase of PKB activity (Fig. 7D). We therefore conclude that Rac and PKB function in separate signaling pathways with PI 3-kinase as a common activator.
DISCUSSION
Using the SKF5 cell line, we have investigated if and how activation of PI 3-kinase by different growth factors results in differential usage of PI 3-kinase-dependent signaling. Here we show that bFGF treatment, in contrast to EGF(Ret) and PDGF treatment, does not result in the activation of all PI 3-kinase-dependent signaling (e.g., no lamellipodium formation), although all three can activate PI 3-kinase. We show that activation of PKB and lamellipodium formation represent separate PI 3-kinase-dependent pathways, and we provide evidence that the mechanism whereby PI 3-kinase is activated is critical in determining whether a PI 3-kinase-dependent signaling pathway is activated.
Regulation of PI 3-kinase activation.
Activation of PI 3-kinase can occur through distinct mechanisms, binding of p85 to tyrosine-phosphorylated proteins or peptides that results in a conformational change and subsequent activation of p110 (33, 38) or the binding of p110 to activated Ras (35, 37). Since the substrate for active PI 3-kinase is present at the plasma membrane, translocation to the membrane also has to occur, and artificial recruitment of p110 to the membrane (CAAXp110) results in partial activation of p110 (19, 26). However, since Ras and the activated receptor are already membrane localized, this prerequisite is already fulfilled. Finally, PI 3-kinase activity can be negatively modulated by serine phosphorylation of the p85 subunit (4, 11).
First, we compared p85 binding to tyrosine-phosphorylated proteins following growth factor treatment. We could not detect this binding following bFGF treatment, in contrast to EGF(Ret) and PDGF treatment, suggesting that this represents a variation between these growth factors in the activation of PI 3-kinase. However, to see whether this difference also underlies the difference in the ability to stimulate PI 3-kinase-dependent signaling, we next studied whether there might be differences in PI 3-kinase regulation by Ras as well.
To be able to assess the involvement of Ras in PI 3-kinase-mediated signaling, we studied two PI 3-kinase-dependent responses: lamellipodium formation and PKB activation. First, we studied the effect of ectopic expression of oncogenic RasLeu61. Expression of RasLeu61 induced both PKB activation and lamellipodium formation in a completely PI 3-kinase dependent manner (i.e., sensitive to the inhibitors wortmannin and LY294002), showing that in these cells no PI 3-kinase-independent pathway(s) are used to activate PKB and lamellipodium formation. This is in contrast to, for instance, constitutively active Ras-induced lamellipodium formation in Swiss 3T3 cells and PAE cells. In these two cell types, constitutively active Ras-induced lamellipodium formation is only partially sensitive to PI 3-kinase inhibitors (31, 36). This may be due to the involvement of additional PI 3-kinases, which are insensitive to wortmannin and LY294002, since in PAE cells a dominant negative mutant of the 85-kDa regulatory subunit of PI 3-kinase does inhibit constitutively active Ras-induced lamellipodium formation (36). Alternatively, active Ras may induce PI 3-kinase-independent pathways that result in lamellipodium formation. Apparently, neither alternative operates or is present in SKF5 cells. This suggestion is corroborated by the observation that the phorbol ester TPA does not induce lamellipodium formation in SKF5 cells although TPA is a strong but PI 3-kinase inhibitor-insensitive inducer of lamellipodia in Swiss 3T3 cells (31). Since certain PKC isotypes (e.g., PKCζ) have been implicated as Ras effector molecules, this suggests that Ras may induce lamellipodium formation via a PKC-dependent, LY294002/wortmannin-insensitive pathway as well (12).
Differential regulation of PI 3-kinase by endogenous Ras.
The results with RasLeu61 show that in SKF5 cells PI 3-kinase may function as a downstream target of active Ras and suggest a simple model which predicts that any growth factor that induces activation of endogenous Ras will also induce lamellipodium formation and PKB activation. In SKF5 cells, PDGF, bFGF, and EGF(Ret) all activate endogenous Ras to similar GTP levels and, indeed, they all activate PKB, although bFGF does so rather weakly. Surprisingly, bFGF, unlike PDGF and EGF(Ret), does not induce lamellipodium formation.
This anomaly could be explained by assuming that bFGF, in contrast to PDGF or EGF(Ret), either provides an additional signal that induces a pathway dominant over Ras signaling or functions in inhibiting lamellipodium formation. A short pretreatment with bFGF does not inhibit EGF(Ret)- or PDGF-induced lamellipodium formation, indicating that bFGF does not induce an inhibitory signal for this pathway. From this, it may be concluded that apparently, unlike ectopic expression of active Ras, strong activation of endogenous Ras is not sufficient to induce the activation of all PI 3-kinase-mediated effects in SKF5 cells.
The question whether activation of endogenous Ras can result in the activation of PI 3-kinase-dependent signaling differs from the question whether activation of endogenous Ras is necessary. Here, we show that inhibition of endogenous Ras activation does not result in inhibition of EGF(Ret)- and PDGF-induced lamellipodium formation, in agreement with the absence of any effect of RasAsn17 on PDGF-induced membrane ruffling in Swiss 3T3 fibroblasts and insulin- and HGF-induced membrane ruffling in KB cells (30, 34). Also, EGF(Ret)- and PDGF-induced activation of PKB is only partially affected by RasAsn17. It should, however, be noted that a lack of effect of RasAsn17 expression does not necessarily imply that endogenous Ras is not involved or that Ras-independent pathways are present. Indeed, RasAsn17 does not block TPA-induced ERK2 activation, whereas both overexpression of p120GAP (32) and expression of the Raf-RBD do (28, 40). However, both Raf-RBD (3) and membrane-targeted p120GAP (46a), even coexpressed with RasAsn17, do not inhibit growth factor-induced PKB activation, suggesting that the residual PKB activation in the presence of RasAsn17 is indeed due to Ras-independent PI 3-kinase regulation.
In the case of bFGF, the role of Ras in the activation of PI 3-kinase appears essential, since bFGF-induced HA-PKB activation is completely blocked by coexpression of RasAsn17. Thus, the mechanism whereby bFGF activates PI 3-kinase clearly differs from that used by EGF(Ret) and PDGF.
From these results, we draw the following conclusions regarding the activation mechanism of PI 3-kinase in these cells. (i) Activation of receptor tyrosine kinases, such as EGF(Ret) and the PDGF receptor, activates PI 3-kinase and PI 3-kinase-mediated events by activation of Ras and binding of the 85-kDa regulatory subunit to phosphotyrosine residues of the receptor tyrosine kinase. In this case, (in)activation of endogenous Ras has only a partial effect on PI 3-kinase-mediated events. (ii) Activation of receptor tyrosine kinases that do not activate PI 3-kinase by direct binding, such as the bFGF receptor, can alternatively use only endogenous Ras-mediated activation. Activation of PI 3-kinase only through Ras appears weak compared to PI 3-kinase activation by both Ras and p85. This is indicated by the lack of lamellipodium formation and the low induction of PI 3-kinase activity following bFGF activation. This is corroborated by the finding that inhibition of Ras activation does not result in an inhibition of lamellipodium formation and only partially inhibits PKB activation, following EGF(Ret) and PDGF treatment. In these cases, the “p85 pathway” can induce an increase in PI 3-kinase activity sufficient for signaling toward PKB and lamellipodia. The above-described in vivo regulation is supported by in vitro data that show a synergism in PI 3-kinase activation between RasGTP and p85 bound to tyrosine-phosphorylated proteins or peptides (37). Furthermore, it has been shown that RasAsn17 expression in other cell lines (PC12 and NIH 3T3) results in a similar partial inhibition of 3′-phosphorylated PI lipid formation in vivo. In addition, it has been shown by us (10) and others (35) that expression of RasAsn17 does not block p85-induced PI 3-kinase activity as measured by an in vitro kinase assay on antiphosphotyrosine immunoprecipitates, indicating that association of p85 with tyrosine-phosphorylated proteins occurs independently of Ras. Data obtained by Klinghoffer et al. (25) show that for mutant receptors, recruitment of p85 to the membrane by receptor binding is not sufficient to stimulate PI 3-kinase activity. This is in apparent contrast to our data showing that inhibition of Ras still leads to PI 3-kinase activation but also to the increased ligand-independent activation observed with artificially membrane-targeted p110 (p110CAAX [19, 26]). The whole of these data suggests that proper positioning of p110 at the plasma membrane is crucial to its activation. If so, it could be that there is a critical difference between recruitment of p85 to mutant receptors and p85 binding to wild-type endogenous receptors.
It is reasonable to assume that the relative contribution of Ras and p85 to PI 3-kinase activation may vary between cell lines and receptors. Although not many cell types have been tested, RasAsn17 expression clearly has a more profound effect on PKB activation in some cells (NIH 3T3 cells) (17) than in other cells (NIH 3T3 cells overexpressing the insulin receptor and Rat1 cells) (3). It is, however, surprising that in the cells used in this study, the contribution of endogenous Ras to PI 3-kinase activation was very restricted, compared to the introduction of constitutively active Ras. Even high levels of active endogenous Ras only marginally induce PI 3-kinase-mediated events, and lower levels of endogenous Ras activation, as induced by, for instance, LPA, are not even sufficient to induce detectable PKB activation (3). It could be that only a small fraction of endogenous Ras is available for the activation and that the majority of endogenous Ras is involved in the activation of other effectors, such as the members of the Raf1 and the RalGEF family. Alternatively, it has recently been reported that besides Ras, ectopic expression of constitutively active R-Ras results in PI 3-kinase-dependent PKB activation (27). Therefore, it could be that R-Ras rather than Ras is involved in the regulation of growth factor-induced PI 3-kinase activation. Also, in cases in which RasAsn17 has a clear effect, this could be due to inhibition of R-Ras rather than Ras. However, using a similar type of assay to that described here for Ras (Fig. 5B), we have not found evidence that R-Ras might be activated by receptor tyrosine kinases. Also, we have thus far not observed any effect of expression of dominant negative R-RasAsn43 on PKB activation (8).
Finally, with respect to tumorigenesis, it is important to note that our results show that the expression of constitutively active Ras clearly affects signaling in a different way from regular activation of Ras by growth factors. Thus, in tumors containing mutant Ras, deregulation of signaling pathways may go beyond the deregulation of signaling in which endogenous Ras is actually involved.
PKB and lamellipodia represent separate PI 3-kinase-dependent pathways.
Differential regulation of PI 3-kinase by bFGF does not explain why bFGF induces PKB activation and not lamellipodium formation. However, we show here that PKB and lamellipodia are not on the same linear pathway but represent a bifurcation in PI 3-kinase-dependent signaling. Constitutively active or dominant negative PKB does not interfere with lamellipodium formation (which is Rac dependent); conversely, constitutively active or dominant negative Rac does not interfere with PKB activation. The latter result is consistent with recent studies by others that also do not indicate a role for Rac in PKB activation (5, 26).
Since PKB and lamellipodia formation lie on separate pathways, the observation that bFGF induces a small increase in PI-3,4P2 and PI-3,4,5P3 levels and concomitantly a small increase in PKB activation with no lamellipodium formation suggests that different threshold levels of 3′-phosphorylated PI lipids, either PI-3,4P2, which is the major regulator of PKB (16), or PI-3,4,5P3, regulate downstream signaling. We note that the activation of PKB by EGF(Ret) in the presence of RasAsn17 is similar to the activation of PKB by bFGF alone. However, EGF(Ret) in the presence of RasAsn17 induces lamellipodium formation and bFGF does not. This additionally argues that PKB and lamellipodia represent separate pathways, but if PKB activation is a direct reflection of 3′-phosphorylated PI lipid levels, this would also argue against a role for threshold levels of 3′-phosphorylated PI lipids. Alternatively, it could be that the cellular location of 3′-phosphorylated PI lipid formation determines the outcome of PI 3-kinase-dependent signaling. Indeed, most receptors are localized at specific plasma membrane sites by their interaction with cytoskeleton elements, and one can envision that within a tripartite complex (receptor–PI 3-kinase–Ras), 3′-phosphorylated PI lipid production will occur at these specific sites in the vicinity of other downstream molecules. If activation occurs within the bipartite (Ras-p110) complex, 3′-phosphorylated PI lipid formation may be more diffuse and may therefore not touch target molecules present at specific locations. Unfortunately, a rigorous analysis of the latter hypothesis is at present not possible since it requires antibodies to PI-3P lipids applicable in immunohistochemistry.
Irrespective of these interesting possibilities, our results show that growth factor-specific differences in the use of Ras and p85 in the activation of PI 3-kinase translates into different biological responses toward these growth factors.
ACKNOWLEDGMENTS
This work was supported by grants from the Netherlands Foundation for Chemical Research (SON) with financial support from the Netherlands Organization for Scientific Research (NWO) (to D.H.J.W.), the Dutch Cancer Society (KWF) (to B.M.T.B.), and the Foundation for Life Sciences (SLW) (to J.R.).
REFERENCES
- 1.Alessi D R, Andjelkovich M, Caudwell B, Cron P, Morrice N, Cohen P, Hemmings B A. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 1996;15:6541–6551. [PMC free article] [PubMed] [Google Scholar]
- 2.Alessi D R, James S R, Downes C P, Holmes A B, Gaffney P R, Reese C B, Cohen P. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase B-alpha. Curr Biol. 1997;7:261–269. doi: 10.1016/s0960-9822(06)00122-9. [DOI] [PubMed] [Google Scholar]
- 2a.Burgering, B. M. T., et al. Unpublished data.
- 3.Burgering B M T, Coffer P J. Protein kinase B (c-Akt) in phosphatidylinositol-3-OH kinase signal transduction. Nature. 1995;376:599–602. doi: 10.1038/376599a0. [DOI] [PubMed] [Google Scholar]
- 4.Carpenter C L, Auger K R, Duckworth B C, Hou W M, Schaffhausen B, Cantley L C. A tightly associated serine/threonine protein kinase regulates phosphoinositide 3-kinase activity. Mol Cell Biol. 1993;13:1657–1665. doi: 10.1128/mcb.13.3.1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chou M M, Blenis J. The 70 kDa S6 kinase complexes with and is activated by the Rho family G proteins Cdc42 and Rac1. Cell. 1996;85:573. doi: 10.1016/s0092-8674(00)81257-x. [DOI] [PubMed] [Google Scholar]
- 6.Cross D A, Alessi D R, Cohen P, Andjelkovich M, Hemmings B A. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378:785–789. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- 7.Datta S R, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg M E. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- 8.De Rooij, J., and B. M. T. Burgering. Unpublished results.
- 9.De Rooij J, Bos J L. Minimal Ras-binding domain of Raf1 can be used as an activation-specific probe for Ras. Oncogene. 1997;14:623–625. doi: 10.1038/sj.onc.1201005. [DOI] [PubMed] [Google Scholar]
- 10.de Vries-Smits A M M, Burgering B M T, Leevers S J, Marshall C J, Bos J L. Involvement of p21ras in activation of extracellular signal-regulated kinase 2. Nature. 1992;357:602–604. doi: 10.1038/357602a0. [DOI] [PubMed] [Google Scholar]
- 11.Dhand R, Hiles I, Panayotou G, Roche S, Fry M J, Gout I, Totty N F, Truong O, Vicendo P, Yonezawa K. PI 3-kinase is a dual specificity enzyme: autoregulation by an intrinsic protein-serine kinase activity. EMBO J. 1994;13:522–533. doi: 10.1002/j.1460-2075.1994.tb06290.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Diaz-Meco M T, Lozano J, Municio M M, Berra E, Frutos S, Sanz L, Moscat J. Evidence for the in vitro and in vivo interaction of Ras with protein kinase C-zeta. J Biol Chem. 1994;269:31706–31710. [PubMed] [Google Scholar]
- 13.Dudek H, Datta S R, Franke T F, Birnbaum M J, Yao R, Cooper G M, Segal R A, Kaplan D R, Greenberg M E. Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science. 1997;275:661–665. doi: 10.1126/science.275.5300.661. [DOI] [PubMed] [Google Scholar]
- 14.Fantl W J, Escobedo J A, Martin G A, Turck C W, del Rosario M, McCormick F. Distinct phosphotyrosines on a growth factor receptor bind to specific molecules that mediate different signaling pathways. Cell. 1992;69:413–423. doi: 10.1016/0092-8674(92)90444-h. [DOI] [PubMed] [Google Scholar]
- 15.Feig L A, Urano T, Cantor S. Evidence for a Ras/Ral signaling cascade. Trends Biochem Sci. 1996;21:438–441. doi: 10.1016/s0968-0004(96)10058-x. [DOI] [PubMed] [Google Scholar]
- 16.Franke T F, Kaplan D R, Cantley L C, Toker A. Direct regulation of the Akt proto-oncogene product by phosphatidylinositol-3,4-bisphosphate. Science. 1997;275:665–668. doi: 10.1126/science.275.5300.665. [DOI] [PubMed] [Google Scholar]
- 17.Franke T F, Yang S-I, Chan T O, Datta K, Kazlauskas A, Morrison D K, Kaplan D R, Tsichlis P N. The protein kinase encoded by the Akt proto-oncogene is a target of the PDGF-activated phosphatidylinositol 3-kinase. Cell. 1995;81:727–736. doi: 10.1016/0092-8674(95)90534-0. [DOI] [PubMed] [Google Scholar]
- 18.Hawkins P T, Eguinoa A, Qiu R-G, Stokoe D, Cooke F T, Walters R, Wennström S, Claesson-Welsh L, Evans T, Symons M, Stephens L. PDGF stimulates an increase in GTP-Rac via activation of phosphoinositide 3-kinase. Curr Biol. 1995;5:393–403. doi: 10.1016/s0960-9822(95)00080-7. [DOI] [PubMed] [Google Scholar]
- 19.Hu Q, Klippel A, Muslin A J, Fantl W J, Williams L T. Ras-dependent induction of cellular responses by constitutively active phosphatidylinositol-3 kinase. Science. 1995;268:100–102. doi: 10.1126/science.7701328. [DOI] [PubMed] [Google Scholar]
- 20.Isakoff S J, Taha C, Rose E, Marcusohn J, Klip A, Skolnik E Y. The inability of phosphatidylinositol 3-kinase activation to stimulate GLUT4 translocation indicates additional signaling pathways are required for insulin-stimulated glucose uptake. Proc Natl Acad Sci USA. 1995;92:10247–10251. doi: 10.1073/pnas.92.22.10247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jackson T R, Stephens L R, Hawkins P T. Receptor specificity of growth factor-stimulated synthesis of 3-phosphorylated inositol lipids in Swiss 3T3 cells. J Biol Chem. 1992;267:16627–16636. [PubMed] [Google Scholar]
- 22.James S R, Downes C P, Gigg R, Grove S J, Holmes A B, Alessi D R. Specific binding of the Akt-1 protein kinase to phosphatidylinositol 3,4,5-triphosphate without subsequent activation. Biochem J. 1996;315:709–713. doi: 10.1042/bj3150709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kauffmann-Zeh A, Rodriguez-Viciana P, Ulrich E, Gilbert C, Coffer P J, Downward J, Evan G. Suppression of c-Myc-induced apoptosis by Ras signalling through PI(3)K and PKB. Nature. 1997;385:544–548. doi: 10.1038/385544a0. [DOI] [PubMed] [Google Scholar]
- 24.Kazlauskas A, Cooper J A. Autophosphorylation of the PDGF receptor in the kinase insert region regulates interactions with cell proteins. Cell. 1989;58:1121–1133. doi: 10.1016/0092-8674(89)90510-2. [DOI] [PubMed] [Google Scholar]
- 25.Klinghoffer R A, Duckworth B, Valius M, Cantley L, Kazlauskas A. Platelet-derived growth factor-dependent activation of phosphatidylinositol 3-kinase is regulated by receptor binding of SH2-domain-containing proteins which influence Ras activity. Mol Cell Biol. 1996;16:5905–5914. doi: 10.1128/mcb.16.10.5905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Klippel A, Reinhard C, Kavanaugh W M, Apell G, Escobedo M A, Williams L T. Membrane localization of phosphatidylinositol 3-kinase is sufficient to activate multiple signal-transducing kinase pathways. Mol Cell Biol. 1996;16:4117–4127. doi: 10.1128/mcb.16.8.4117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marte B M, Rodriguez-Viciana P, Wennström S, Warne P H, Downward J. R-Ras can activate the phosphoinositide 3-kinase but not the MAP kinase arm of the Ras effector pathways. Curr Biol. 1996;7:63–70. doi: 10.1016/s0960-9822(06)00028-5. [DOI] [PubMed] [Google Scholar]
- 28.Ming X-F, Burgering B M T, Claesson-Welsh L, Wennström S, Heldin C-H, Bos J L, Kozma S C, Thomas G. Activation of p70/p85 S6 kinase by a pathway independent of p21ras. Nature. 1994;371:426–429. doi: 10.1038/371426a0. [DOI] [PubMed] [Google Scholar]
- 29.Moodie S A, Wolfman A. The 3Rs of life: Ras, Raf and growth regulation. Trends Genet. 1994;10:44–48. doi: 10.1016/0168-9525(94)90147-3. [DOI] [PubMed] [Google Scholar]
- 30.Nishiyama T, Sasaki T, Takaishi K, Kato M, Yaku H, Araki K, Matsuura Y, Takai Y. Rac p21 is involved in insulin-induced membrane ruffling and Rho p21 is involved in hepatocyte growth factor- and 12-O-tetradecanoylphorbol-13-acetate (TPA)-induced membrane ruffling in KB cells. Mol Cell Biol. 1994;14:2447–2456. doi: 10.1128/mcb.14.4.2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nobes C D, Hawkins P, Stephens L, Hall A. Activation of the small GTP-binding proteins Rho and Rac by growth factor receptors. J Cell Sci. 1995;108:225–233. doi: 10.1242/jcs.108.1.225. [DOI] [PubMed] [Google Scholar]
- 32.Nori M, L’Allemain G, Weber M J. Regulation of tetradecanoyl phorbol acetate-induced responses in NIH 3T3 cells by GAP, the GTPase-activating protein associated with p21c-Ras. Mol Cell Biol. 1992;12:936–945. doi: 10.1128/mcb.12.3.936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Panayotou G, Bax B, Gout I, Ferderwisch M, Wroblowski B, Dhand R, Fry M J, Blundell T L, Wollmer A, Waterfield M D. Interaction of the p85 subunit of PI 3-kinase and its N-terminal SH2 domain with a PDGF receptor phosphorylation site: structural features and analysis of conformational changes. EMBO J. 1992;11:4261–4272. doi: 10.1002/j.1460-2075.1992.tb05524.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ridley A J, Paterson H F, Johnston C L, Diekmann D, Hall A. The small GTP-binding protein Rac regulates growth factor-induced membrane ruffling. Cell. 1992;70:401–410. doi: 10.1016/0092-8674(92)90164-8. [DOI] [PubMed] [Google Scholar]
- 35.Rodriguez-Viciana P, Warne P H, Dhand R, Vanhaesebroeck B, Gout I, Fry M J, Waterfield M D, Downward J. Phosphatidylinositol-3-OH kinase as a direct target of Ras. Nature. 1994;370:527–532. doi: 10.1038/370527a0. [DOI] [PubMed] [Google Scholar]
- 36.Rodriguez-Viciana P, Warne P H, Khwaja A, Marte B M, Pappin D J, Das P, Waterfield M D, Ridley A J, Downward J. Role of phosphoinositide 3-OH kinase in cell transformation and control of the actin cytoskeleton by Ras. Cell. 1997;87:457–467. doi: 10.1016/s0092-8674(00)80226-3. [DOI] [PubMed] [Google Scholar]
- 37.Rodriguez-Viciana P, Warne P H, Vanhaesebroeck B, Waterfield M D, Downward J. Activation of phosphoinositide 3-kinase by interaction with Ras and by point mutation. EMBO J. 1996;15:2442–2451. [PMC free article] [PubMed] [Google Scholar]
- 38.Rordorf-Nikolic T, Van Horn D J, Chen D, White M F, Backer J M. Regulation of phosphatidylinositol 3′-kinase by tyrosyl phosphoproteins. Full activation requires occupancy of both SH2 domains in the 85-kDa regulatory subunit. J Biol Chem. 1995;270:3662–3666. doi: 10.1074/jbc.270.8.3662. [DOI] [PubMed] [Google Scholar]
- 39.Santoro M, Wong W T, Aroca P, Santos E, Matoskova B, Grieco M, Fusco A, Di Fiore P P. An epidermal growth factor receptor/ret chimera generates mitogenic and transforming signals: evidence for a ret-specific signaling pathway. Mol Cell Biol. 1994;14:663–675. doi: 10.1128/mcb.14.1.663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schaap D, Van der Wal J, Howe L R, Marshall C J, Blitterswijk W J. A dominant-negative mutant of Raf blocks mitogen-activated protein kinase activation by growth factors and oncogenic Ras. J Biol Chem. 1993;268:20232–20236. [PubMed] [Google Scholar]
- 41.Stephens L R, Jackson T R, Hawkins P T. Agonist-stimulated synthesis of phosphatidylinositol (3,4,5)-triphosphate: a new intracellular signalling system. Biochim Biophys Acta. 1993;1179:27–75. doi: 10.1016/0167-4889(93)90072-w. [DOI] [PubMed] [Google Scholar]
- 42.Tolias K F, Cantley L C, Carpenter C L. Rho family GTPases bind to phosphoinositide kinases. J Biol Chem. 1995;270:17656–17659. doi: 10.1074/jbc.270.30.17656. [DOI] [PubMed] [Google Scholar]
- 43.Ullrich A, Schlessinger J. Signal transduction by receptors with tyrosine kinase activity. Cell. 1990;61:203–212. doi: 10.1016/0092-8674(90)90801-k. [DOI] [PubMed] [Google Scholar]
- 44.Vanhaesebroeck B, Leevers S J, Panayotou G, Waterfield M D. Phosphoinositide 3-kinases: a conserved family of signal transducers. Trends Biochem Sci. 1997;22:267–272. doi: 10.1016/s0968-0004(97)01061-x. [DOI] [PubMed] [Google Scholar]
- 45.van Puijenbroek A A F L, van Weering D H J, van den Brink C E, Bos J L, van der Saag P T, de Laat S W, den Hertog J. Cell scattering of SK-N-MC neuroepithelioma cells in response to Ret and FGF receptor tyrosine kinase activation is correlated with sustained ERK2 activation. Oncogene. 1997;14:1147–1158. doi: 10.1038/sj.onc.1200911. [DOI] [PubMed] [Google Scholar]
- 46.van Weering D H J, Bos J L. Glial cell line-derived neurotrophic factor induces Ret-mediated lamellipodia formation. J Biol Chem. 1997;272:249–254. doi: 10.1074/jbc.272.1.249. [DOI] [PubMed] [Google Scholar]
- 46a.van Weering, D. H. J., C. J. Marshall, J. L. Bos, and B. T. M. Burgering. Unpublished data.
- 47.van Weering D H J, Medema J P, van Puijenbroek A, Burgering B M T, Baas P D, Bos J L. Ret receptor tyrosine kinase activates extracellular signal-regulated kinase 2 in SK-N-MC cells. Oncogene. 1995;11:2207–2214. [PubMed] [Google Scholar]
- 48.Zheng Y, Bagrodia S, Cerione R A. Activation of phosphoinositide 3-kinase activity by Cdc42Hs binding to p85. J Biol Chem. 1994;269:18727–18730. [PubMed] [Google Scholar]