

Abstract
The steroid hormone progesterone regulates proliferation and differentiation in the mammary gland and uterus by cell cycle phase-specific actions. In breast cancer cells the predominant effect of synthetic progestins is long-term growth inhibition and arrest in G1 phase. Progestin-mediated growth arrest of T-47D breast cancer cells was preceded by inhibition of cyclin D1-Cdk4, cyclin D3-Cdk4, and cyclin E-Cdk2 kinase activities in vitro and reduced phosphorylation of pRB and p107. This was accompanied by decreases in the expression of cyclins D1, D3, and E, decreased abundance of cyclin D1- and cyclin D3-Cdk4 complexes, increased association of the cyclin-dependent kinase (CDK) inhibitor p27 with the remaining Cdk4 complexes, and changes in the molecular masses and compositions of cyclin E complexes. In control cells cyclin E eluted from Superdex 200 as two peaks of ∼120 and ∼200 kDa, with the 120-kDa peak displaying greater cyclin E-associated kinase activity. Following progestin treatment, almost all of the cyclin E was in the 200-kDa, low-activity form, which was associated with the CDK inhibitors p21 and p27; this change preceded the inhibition of cell cycle progression. These data suggest preferential formation of this higher-molecular-weight, CDK inhibitor-bound form and a reduced number of cyclin E-Cdk2 complexes as mechanisms for the decreased cyclin E-associated kinase activity following progestin treatment. Ectopic expression of cyclin D1 in progestin-inhibited cells led to the reappearance of the 120-kDa active form of cyclin E-Cdk2 preceding the resumption of cell cycle progression. Thus, decreased cyclin expression and consequent increased CDK inhibitor association are likely to mediate the decreases in CDK activity accompanying progestin-mediated growth inhibition.
Steroid hormones regulate cellular proliferation and differentiation by cell cycle phase-specific actions (40). Estrogen, acting in concert with other hormones and growth factors, appears to be the main drive to proliferation in the female reproductive tract and mammary gland. In contrast with the proliferative effects of estrogen, progesterone acts as the differentiating female sex steroid. In this role it can either stimulate or inhibit proliferation in a cell type- and tissue-specific manner (5). For example, the primary function of progesterone in the uterus is to facilitate implantation, and in this organ progesterone acts synergistically with estrogen to stimulate proliferation of stromal cells but inhibits estrogen-induced mitosis in the epithelium. In the mammary gland progesterone stimulates proliferation and development of alveoli, a requirement for subsequent lactation. In breast cancer cells, a widely used model for studies of the effects of steroids on cell proliferation, treatment with synthetic progestins results in a biphasic change in the rate of cell cycle progression, consisting of an initial transient acceleration through G1 phase and a subsequent increase in the S phase fraction, followed by cell cycle arrest and growth inhibition accompanied by a decrease in the S phase fraction (23, 25, 38, 55, 61). Thus, two distinct, opposing effects of progestins on cell cycle progression can be observed within the one cell type, emphasizing the complexity of progestin effects on cell proliferation. Data from both breast cancer cells in tissue culture and in vivo studies of the uterus and mammary gland demonstrate that sensitivity to both stimulation and inhibition is present only during G1 phase (5, 38, 55).
Since endogenous hormones play a key role in the development of hormone-dependent cancers, exposure to exogenous steroid hormones through the use of oral contraceptives and hormone replacement therapies might influence the risk of developing such cancers. Combined oral contraceptives or hormone replacement therapies containing both an estrogen and progestin confer protection from endometrial cancer, while treatment with estrogen alone leads to an increase in risk (46). In contrast, while the effect of hormonal therapies on breast cancer risk has been controversial, there appears to be a slight increase in risk in recent or current users (7, 8), and in postmenopausal women the risk of breast cancer associated with estrogen use does not appear to be reduced by the addition of progestin (6). Thus, progestins appear to be protective against endometrial cancer but not breast cancer. Nevertheless, synthetic progestins have an established role in the therapy of both breast and endometrial cancers (46, 49, 60). The mechanism for the antitumor action of progestins is unknown, but inhibition of breast cancer cell proliferation is a likely contributor. Despite these issues and the role of progesterone in normal mammary development and differentiation, the effects of progesterone and synthetic progestins on cell proliferation have not been widely studied from a mechanistic viewpoint, and mechanisms for progestin inhibition of proliferation remain unidentified at a molecular level. However, the demonstration of steroidal control of cell cycle progression at defined points within G1 phase (40) suggests that these agents and their antagonists act via their respective receptors to regulate, directly or indirectly, the expression of genes with a central role in the control of cell cycle progression through G1 phase. Research aimed towards identification of these molecular targets has focused on the roles of c-Myc and cyclins and the associated cyclin-dependent kinases (CDKs) (1, 10, 11, 35, 37, 38, 44, 64).
The sequential activation of CDKs and consequent phosphorylation of specific substrates govern progress through the cell cycle. Key substrates of the CDKs with G1 phase-specific actions include the retinoblastoma gene product, pRB, and the related protein p107, although it is likely that other substrates remain to be identified: unlike cyclin D1, cyclin E is required for G1 progression in the absence of functional pRB (reviewed in references 24, 50, and 51). CDK activity is subject to multiple levels of regulation. Since CDKs are inactive in the absence of cyclin binding, cyclin abundance is a major determinant of cyclin-CDK activity (24, 32, 50, 51). Each cyclin is thus typically present for only a restricted portion of the cell cycle. Alteration of cyclin abundance is sufficient to alter the rate of cell cycle progression, since overexpression of either of the principal G1 cyclins, cyclins D and E, accelerates cells through G1 and, conversely, inhibition of their function by antibody microinjection prevents entry into S phase (24, 50, 51). CDK activity is also regulated by a network of kinases and phosphatases so that cyclin binding is sufficient only for partial activation (32). The CDK-activating kinase (CAK) phosphorylates a conserved threonine residue in the T-loop of the kinase, stabilizing the cyclin-CDK interaction and altering the conformation of the substrate binding site (33); this phosphorylation is necessary for full activity. Even in the presence of activating phosphorylation at this threonine residue and cyclin binding, CDKs can be inhibited by phosphorylation of threonine and tyrosine residues within the catalytic cleft. The dual-specificity Cdc25 phosphatases activate CDKs by dephosphorylating these inhibitory residues (32). In vertebrate cells three such phosphatases have been identified: Cdc25A, -B, and -C. Cdc25A appears to target CDKs active at the G1-S transition, since microinjection of anti-Cdc25A antibodies blocked progression from G1 into S phase (22, 27), while anti-Cdc25C antibodies led to arrest in G2 phase (31). Overexpression of dominant negative Cdc25B mutants indicates that Cdc25B regulates G2-M transit (13).
A further level of control results from the actions of two families of specific CDK-inhibitory proteins. One family, for which the prototype is p16INK4, specifically targets the kinases which associate with the D-type cyclins, Cdk4 and Cdk6. Their inhibitory activity appears to be largely due to competition with the cyclin for CDK binding, although they can also interact with cyclin D-Cdk4 and cyclin D-Cdk6 complexes (21, 45, 51). Members of the other family, of which p21 (WAF1, Cip1, sdi1) and p27 (Kip1) are the best studied, interact with a broader range of CDKs, including Cdk2 as well as Cdk4 and Cdk6. Recent structural studies of p27 bound to cyclin A-Cdk2 have indicated that p27 interacts with both cyclin A and Cdk2, occluding the catalytic cleft and causing multiple structural changes around it (47). Sequence conservation of the residues involved in cyclin and CDK interaction suggests that other members of this inhibitor family probably bind in a similar fashion (47). Both CAK and Cdc25 activation of the kinase are also prevented by inhibitor binding, probably by steric hindrance (2, 48). Despite these multiple modes of inhibition of CDK activity, not all cyclin-CDK complexes containing p21 or p27 are inactive. Histone H1 kinase activity, most likely due to Cdk2, is present in p21 immunoprecipitates (66), perhaps representing an alternate mode of interaction where either the cyclin or CDK, but not both, interacts with p21 (12). Histone H1 activity is not present in p27 immunoprecipitates, although they display pRB kinase activity, apparently due to Cdk4 and Cdk6 (53). Recent data indicate that p21 and p27 as well as a related inhibitor, p57Kip2, stabilize cyclin D-Cdk4 and cyclin D-Cdk6 complexes in vitro (30). High-stoichiometry p21 or p27 binding appears to be required for inhibition of Cdk4 activity (3, 30). Consequently, these molecules appear to have functions in addition to CDK inhibition, perhaps as adapters which not only promote assembly of the cyclin-CDK complexes but also target these complexes to specific intracellular compartments or substrates.
To identify the sequence of molecular events linking progestin action with inhibition of cell cycle progression, we have examined the effects of progestin treatment of T-47D human breast cancer cells on the abundance and activity of cyclin D1-Cdk4, cyclin D3-Cdk4, and cyclin E-Cdk2, the major G1 phase CDK complexes in these cells. These experiments demonstrate that progestins not only regulate cyclin abundance but also lead to increased association of the CDK inhibitor p27 with G1 cyclin-CDK complexes, providing a mechanism for their growth-inhibitory effects in breast cancer cells.
MATERIALS AND METHODS
Cell culture.
T-47D human breast cancer cells (obtained from the EG & G Mason Research Institute, Worcester, Mass.) were cultured in RPMI 1640 medium supplemented with 5% fetal calf serum, insulin (10 μg/ml), and gentamicin (20 μg/ml). ORG 2058 (16α-ethoxy-21-hydroxy-19-norpregn-4-en-3,20-dione; Amersham Australia, Castle Hill, New South Wales, Australia) was dissolved in ethanol at a 1,000- or 2,000-fold final concentration and added to cells in exponential growth. Control cultures received ethanol to the same final concentration. In some experiments untreated control cultures were harvested at the time of ORG 2058 or vehicle addition to the remainder of the replicate cultures.
A clonal derivative of T-47D cells expressing cyclin D1 under control of the metal-responsive metallothionein promoter, T-47D ΔMTcycD1-3, was used for the experiments presented in Fig. 12. The derivation and characteristics of this cell line have been previously described (36, 39); these cells retain progestin sensitivity similar to that of the parent cell line. T-47D ΔMTcycD1-3 cells were treated with 10 nM ORG 2058 or vehicle as described above, and 24 to 48 h later either zinc (50 to 75 μM ZnSO4) or water (vehicle) was added.
FIG. 12.

Resumption of cell cycle progression after cyclin D1 induction in progestin-pretreated cells. Exponentially proliferating T-47D ΔMTcycD1-3 breast cancer cells, expressing cyclin D1 under the control of a zinc-inducible promoter, were pretreated with the synthetic progestin ORG 2058 (10 nM) or vehicle (ethanol [EtOH]) and then treated with either zinc (ZnSO4) or vehicle (water). (A) Percentages of cells in S phase after 24 h of ORG 2058 or EtOH pretreatment followed by 15 h of zinc (50 μM) or water treatment. (B) Lysates harvested from cells pretreated for 30 h with either ORG 2058 or EtOH followed by 15 h of zinc (75 μM) or water treatment were Western blotted for the indicated proteins or immunoprecipitated with either Cdk4 or cyclin E antibodies for measurement of kinase activity as described in the legend to Fig. 3. (C) Whole-cell lysates from the experiment presented in panel B were fractionated on a Superdex 200 gel filtration column (void volume, 45 ml). Fractions (2 ml) corresponding to elution volumes of 57 to 82 ml were acetone precipitated and then Western blotted for the proteins indicated. The elution volumes of markers with known molecular masses (ferritin, 440 kDa; aldolase, 158 kDa) are indicated; ovalbumin (43 kDa) eluted at 85 ml, corresponding to fraction 15. (D) The Western blots presented in panel C were quantitated by densitometry and are presented as percentages of the total signal observed for each protein.
Generation of recombinant proteins.
The pRB fusion protein substrate for the Cdk4 activity assay was a glutathione S-transferase (GST)-pRB construct encoding amino acids 773 to 928 (supplied by Ed Harlow, Massachusetts General Hospital Cancer Center, Charlestown). A restriction fragment of pVL1392 His6-Cdk2 (44) encompassing the Cdk2 open reading frame was cloned into the expression vector pGEX-2T (Pharmacia, Uppsala, Sweden) to yield a construct encoding the GST-Cdk2 fusion protein used as a substrate for measurement of CAK activity. GST-Cdc25A was generated by using a construct provided by David Beach, Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y. Escherichia coli transformed with the appropriate expression vectors was induced by the addition of 0.2 to 0.4 mM isopropylthiogalactopyranoside and incubated for 3 h at room temperature. The bacterial pellets were lysed by sonication, and the fusion proteins were purified by affinity chromatography on glutathione-agarose beads and then eluted with 15 mM reduced glutathione.
Western blot analysis and immunoprecipitation.
Cells were lysed as follows. Cell monolayers were washed twice with ice-cold phosphate-buffered saline and then scraped into ice-cold lysis buffer (50 mM HEPES [pH 7.5], 150 mM NaCl, 10% [vol/vol] glycerol, 1% Triton X-100, 1.5 mM MgCl2, 1 mM EGTA, 10 μg of aprotinin per ml, 10 μg of leupeptin per ml, 1 mM phenylmethylsulfonyl fluoride, 200 μM sodium orthovanadate, 10 mM sodium pyrophosphate, 20 mM NaF) or ice-cold kinase lysis buffer (50 mM HEPES [pH 7.5], 1 mM dithiothreitol, 150 mM NaCl, 10% [vol/vol] glycerol, 0.1% Tween 20, 1 mM EDTA, 2.5 mM EGTA, 10 mM β-glycerophosphate, 10 μg of aprotinin per ml, 10 μg of leupeptin per ml, 0.1 mM phenylmethylsulfonyl fluoride, 0.1 mM sodium orthovanadate, 1 mM NaF). At selected time points an aliquot of this suspension was diluted in RPMI 1640–5% fetal calf serum and stained for later flow cytometric DNA analysis by addition of ethidium bromide (50 μg/ml) and Triton X-100 (0.2%). Lysates in lysis buffer were incubated for 5 min on ice, and the cellular debris was cleared by centrifugation (15,000 × g, 5 min, 4°C). Lysates in kinase lysis buffer were snap frozen in liquid nitrogen and later thawed on ice, vortexed every 10 min for 60 min, and finally cleared by centrifugation (15,000 × g, 5 min, 4°C). The cleared lysates were aliquoted and stored at −80°C. Similar results were obtained from Western blotting or immunoprecipitation by either lysis technique.
For immunoprecipitation, whole-cell lysates (typically 500 μg) or lysates fractionated by gel filtration chromatography were precleared by incubation with protein A-Sepharose beads (Zymed, San Francisco, Calif.) (1 h, 4°C) and then immunoprecipitated by incubation (1 to 2 h, 4°C) with either anti-cyclin E (C19) or anti-Cdk4 (H22) antibodies (Santa Cruz Biotechnology, Inc., Santa Cruz, Calif.) followed by incubation with protein A-Sepharose beads (0.5 to 1 h, 4°C). In some experiments the antibodies were chemically cross-linked to the protein A-Sepharose beads by incubation in 5 mg of dimethyl pimelimidate per ml–0.2 M sodium tetraborate (pH 9.0) for 30 min at room temperature, essentially as described previously (17). The immunoprecipitated proteins were washed with lysis buffer and resuspended in sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) sample buffer (63 mM Tris-HCl [pH 6.8], 10% [vol/vol] glycerol, 2% SDS, 5% β-mercaptoethanol).
Samples of immunoprecipitated or total protein (40 to 100 μg) in SDS-PAGE sample buffer were heated to 95°C for 3 min and then separated by SDS-PAGE and transferred to nitrocellulose. Following transfer, the nitrocellulose membrane was stained with 0.5% Ponceau S in 10% acetic acid to monitor protein loading; blots with more than one lane not displaying equivalent loading were discarded, and the procedure was repeated. The membranes were incubated (1 to 2 h at room temperature or overnight at 4°C) with the following primary antibodies: p107 (C-18), cyclin D3 (C-16), cyclin E (HE12), Cdk2 (M2), and Cdk4 (C-22) antibodies from Santa Cruz Biotechnology; cyclin D1 antibody (DCS6) from Novocastra, Newcastle-upon-Tyne, United Kingdom; pRB (14001A) antibody from Pharmingen, San Diego, Calif.; p21 (C24420) and p27 (K25020) antibodies from Transduction Laboratories, Lexington, Ky.; rabbit polyclonal antibody raised against a pRB peptide phosphorylated on the site corresponding to Ser780, a residue preferentially phosphorylated by cyclin D1-Cdk4 (29), generously provided by Yoichi Taya, National Cancer Center Research Institute, Tokyo, Japan; and affinity-purified rabbit polyclonal Cdc25A antibody, generously provided by Ingrid Hoffmann, Deutsches Krebforschungszentrum, Heidelberg, Germany. Following incubation (1 h at room temperature) with horseradish peroxidase-conjugated sheep anti-mouse or donkey anti-rabbit secondary antibody (Amersham Australia), specific proteins were visualized by chemiluminescence (Dupont NEN, Boston, Mass.). Where the proteins of interest were of sufficiently different mobilities, membranes were incubated either sequentially or simultaneously with several primary antibodies. Before incubation with primary antibodies requiring a different secondary antibody, the horseradish peroxidase on the initial secondary antibody was inactivated by incubation (overnight, 4°C) in 10 mM Tris (pH 7.4)–150 mM NaCl–0.05% Triton X-100–5% skim milk powder–0.2% sodium azide. Where proteins of similar mobility were examined by using the same membrane, it was stripped by incubation (20 min, 50°C) in 62.5 mM Tris-HCl (pH 6.7)–2% SDS–100 mM β-mercaptoethanol.
Relative abundances were quantitated with a Molecular Dynamics (Sunnyvale, Calif.) densitometer and IP LabGel analysis software (Signal Analytics, Vienna, Va.). Quantitation of protein levels by this method was linear over the range of intensities measured. Within each experiment data were available from multiple control samples (typically four or five vehicle-treated controls harvested in parallel with the ORG 2058-treated samples and, in some experiments, an additional untreated control harvested at the time of treatment). Protein abundances in treated cultures were calculated relative to the means for these controls, which typically had a standard error of the mean (SEM) of ∼10%.
Kinase assays.
The histone H1 kinase activity of cyclin E immunoprecipitates (100 μg of cell lysate) was measured as previously described (37, 44) with 10 μg of histone H1 as the substrate. There was little or no detectable phosphorylation in samples immunoprecipitated by using beads without antibody or following blocking of specific antibody binding with the appropriate antigenic peptide. For Cdk4 assays, cells were harvested and lysed by using kinase lysis buffer as described above. The kinase activity of Cdk4 immunoprecipitates of >400 μg of these lysates was measured by using 10 μg of GST-pRB773-928 fusion protein substrate as previously described (44). The degree of background phosphorylation in each pRB phosphorylation assay was estimated from parallel control samples immunoprecipitated either by using beads without antibody or following blocking of specific antibody binding with the appropriate antigenic peptide. The two controls yielded similar amounts of background phosphorylation.
Cdk7 (06-377; Upstate Biotechnology, Lake Placid, N.Y.) immunoprecipitates from whole-cell lysates were assayed for CAK activity by using GST-Cdk2 as the substrate. To determine the degree of background phosphorylation, control lysate was immunoprecipitated with antibody which had been preincubated in the presence of competing antigenic peptide at 30°C for 30 min. The kinase reaction was initiated by resuspending each immunoprecipitate in 30 μl of 50 mM HEPES (pH 7.5)–30 mM MgCl2–1 mM dithiothreitol–90 μM ATP containing 10 μCi of [γ-32P]ATP and 10 μg of GST-Cdk2. After incubation for 20 min at 30°C, the reaction was terminated by the addition of SDS-PAGE sample buffer.
Following termination of kinase reactions, samples were incubated at 95°C for 2 min in SDS sample buffer and separated by SDS–10% PAGE. Substrate phosphorylation was quantitated with a Molecular Dynamics PhosphorImager Scanner (model 445 SI) followed by analysis with IP LabGel analysis software (Signal Analytics), and relative activity was calculated as described above for Western blots.
RNA isolation and Northern analysis.
Total RNA was extracted (by a guanidinium isothiocyanate-cesium chloride procedure) and blotted as previously described, using 20 μg of total RNA/lane (4). Filters were hybridized (overnight, 50°C) with cDNA probes radioactively labelled with [α-32P]dCTP to a specific activity of approximately 109 cpm/μg of DNA by using the Prime-A-Gene labelling kit (Promega Australia, Annandale, New South Wales, Australia). Restriction enzyme digestion was used to provide the listed fragments from plasmids supplied by the following investigators: cyclin D1, a 1.3-kb EcoRI fragment encompassing the entire open reading frame (David Beach, Cold Spring Harbor Laboratory); cyclin E, a 2.5-kb EcoRI fragment encompassing the entire open reading frame (Steven Reed, Scripps Research Institute, La Jolla, Calif.); Cdc25A, a 2.3-kb EcoRI fragment encompassing the entire open reading frame (David Beach); c-myc, a 0.45-kb PstI fragment corresponding to the second exon (Geoff Symonds, RW Johnson Pharmaceutical Research Institute, Sydney, New South Wales, Australia); and 36B4, a 0.22-kb PstI fragment (Pierre Chambon, Institut de Genetique et de Biologie Moleculaire et Cellulaire, Strasbourg, France).
Cdc25A activation assay.
Cyclin E or Cdk4 immunoprecipitates were washed twice in lysis buffer or kinase lysis buffer, respectively, without phosphatase inhibitors and twice in phosphatase buffer containing 50 mM HEPES (pH 8.2), 15 mM MgCl2, 1 mM dithiothreitol, and 1 mM ATP. The immunoprecipitates were incubated with GST-Cdc25A in phosphatase buffer for at least 1 h at 30°C and then washed twice in lysis buffer and twice in 10 mM HEPES (pH 7.5)–1 mM dithiothreitol. Kinase activity was assayed as described above.
Gel filtration.
Cell lysates prepared in kinase lysis buffer were passed through a 0.22-μm-pore-size MILLEX-GV4 filter (Millipore, Lane Cove, New South Wales, Australia) and fractionated on a Hiload 16/60 Superdex 200 column-fast protein liquid chromatography system (Pharmacia Biotech, Uppsala, Sweden) with 20 mM HEPES (pH 7.5)–250 mM NaCl–1 mM EDTA–0.1% (vol/vol) β-mercaptoethanol–0.01% Tween 20 at a flow rate of 1.2 ml/min. The void volume of the column was 45 ml. Either 2- or 3-ml fractions were collected between elution volumes of 55 and 95 ml. Column calibration was performed under the same conditions with ferritin (440 kDa), aldolase (158 kDa), and ovalbumin (43 kDa). Protein was concentrated prior to Western blotting: 500 μl of each fraction was placed at −70°C for 3 h with 2.5 ml of acetone and 10 μg of carrier bovine serum albumin protein, and pellets were collected by centrifugation (15,000 × g, 10 min, 4°C) and then resuspended by boiling for 4 min in 30 μl of SDS sample buffer.
RESULTS
Previous experiments in this laboratory characterized the effects of synthetic progestins on the cell proliferation kinetics of human breast cancer cell lines, particularly T-47D, and defined a biphasic response consisting of initial acceleration of cell cycle progression and subsequent inhibition of proliferation (38, 55). The relative magnitudes of the two components of the response are dependent in part on the growth rate and hence on the culture conditions. To emphasize the growth-inhibitory response, for the present series of experiments cells were cultured under conditions leading to optimal growth rates, i.e., in medium supplemented with insulin and fetal calf serum. Consistent with previous data (38, 55), treatment with a maximally effective concentration of the progestin ORG 2058 (10 nM) led to an initial small increase in the S phase fraction, followed by arrest in G1 phase and consequent accumulation of G1 phase cells at the expense of the other phases of the cell cycle (Fig. 1). Inhibition of entry into S phase was first apparent after 18 h of treatment, as indicated by a relative lack of cells in early S phase (Fig. 1A), although the S phase fraction had not yet decreased significantly below that of control cells (Fig. 1B). The decrease in the S phase fraction was maximal at 24 h (Fig. 1B), and the G2-plus-M fraction subsequently decreased (Fig. 1A) such that by 30 h up to 86% of the cells were in G1 phase.
FIG. 1.

Progestin inhibition of cell cycle progression. Exponentially proliferating T-47D breast cancer cells were treated with the synthetic progestin ORG 2058 (10 nM) or vehicle (Control). At intervals thereafter, cells were lysed and an aliquot was stained for flow cytometric measurement of DNA content. (A) Representative DNA histograms. (B) Percentages of control cells (○) or ORG 2058-treated cells (•) in S phase. Data from a total of six experiments have been pooled and are shown as means ± SEMs, where the SEM exceeds the size of the symbol used.
Inhibition of G1 CDK activity and reduced phosphorylation of pRB and p107 following progestin treatment.
Initial experiments examined the phosphorylation of pRB and p107, in vivo substrates of G1 CDKs. In control exponentially proliferating cells both phosphorylated and underphosphorylated pRB were apparent, although the underphosphorylated form comprised only ∼20% of the total (Fig. 2A and B). Little change in the proportions of phosphorylated and underphosphorylated pRB was apparent at 6 or 12 h after progestin treatment (Fig. 2A and B), although there was a slight, <50%, increase in the total pRB abundance and a consequent increase in the absolute amount of underphosphorylated pRB at 12 h (Fig. 2C). By 18 h the proportion of pRB in the underphosphorylated form had begun to increase, and after 24 h or more, 70 to 80% of pRB was in this form (Fig. 2A and B). Although the total pRB abundance decreased by ∼50% at 24 to 30 h, the large increase in the proportion of underphosphorylated pRB resulted in an ∼2-fold increase in the abundance of the underphosphorylated form from 18 to 30 h (Fig. 2C). Phosphorylated pRB, detected by antibodies recognizing either the whole molecule or a site specifically phosphorylated by cyclin D1-Cdk4 (RB-P-Ser780 [29]), began to decrease in abundance by 18 h and was markedly reduced by 24 h (Fig. 2). The effects of progestin treatment on p107 phosphorylation and abundance paralleled those on pRB: at 18 h more underphosphorylated p107 was apparent, and after 24 h or more, p107 was largely unphosphorylated and reduced in total abundance (Fig. 2A). Comparison of the data in Fig. 1B and 2C indicated that the accumulation of underphosphorylated pRB reached its maximum by 18 h, a time when little change in the S phase fraction was detected, and thus accumulation of the inhibitory form of pRB preceded the inhibition of entry into S phase by up to 6 h.
FIG. 2.
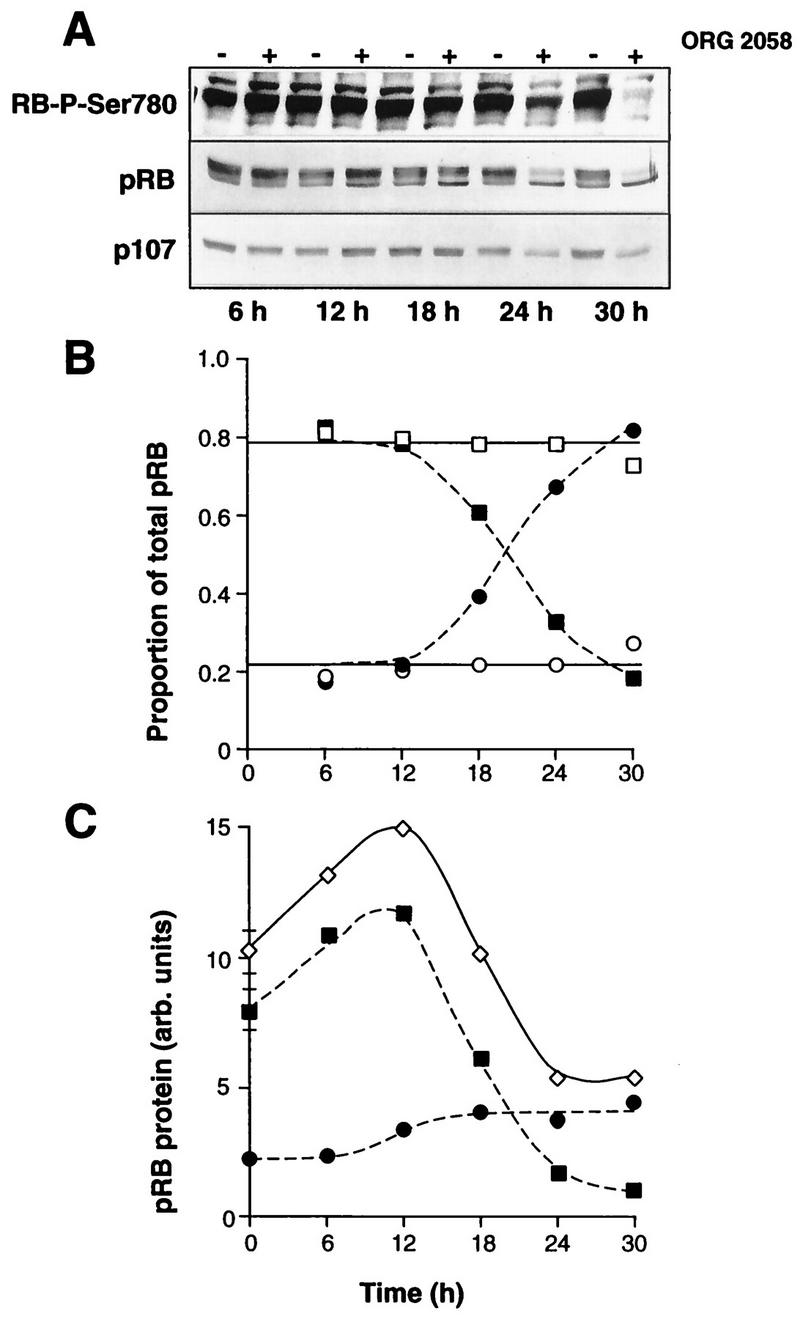
Progestin effects on pRB and p107. Exponentially proliferating T-47D breast cancer cells were treated with the synthetic progestin ORG 2058 (10 nM) or vehicle. At intervals thereafter whole-cell lysates were immunoblotted with antibodies raised against pRB, p107, or a pRB-derived phosphopeptide that contains a cyclin D1-Cdk4-specific target (RB-P-Ser780). (A) Representative blots from one of five experiments (pRB) or two experiments (p107 and RB-P-Ser780). (B) The slower-mobility (hyperphosphorylated) (□ and ▪) and faster-mobility (hypophosphorylated) (○ and •) forms of pRB in the blot shown in panel A were quantitated by densitometry and are presented as the proportion of total pRB for both control cells (open symbols) and ORG 2058-treated cells (closed symbols). (C) Raw densitometric measurements of pRB abundance after ORG 2058 treatment for the representative blot shown in panel A. For clarity, data from control lanes have been pooled and are presented (means ± SEMs) as the 0-h points. ◊, total; ▪, hyperphosphorylated; •, hypophosphorylated. arb., arbitrary.
Since decreased CDK activity was a probable mechanism for the decreased phosphorylation of pRB and p107, the activities of CDK complexes active in G1 phase were measured by using in vitro kinase assays. T-47D cells express both cyclin D1 and cyclin D3, but cyclin D2 is essentially undetectable (4, 56). Both Cdk4 and Cdk6 are expressed, but cyclin D1 immunoprecipitates contain little detectable Cdk6 under conditions where Cdk6 can be readily detected in parallel immunoprecipitates from other cell lines (56). Thus, measurement of the activities of Cdk4 immunoprecipitates (comprising both cyclin D1-Cdk4 and cyclin D3-Cdk4) and cyclin E immunoprecipitates allows estimation of activity of the major G1 phase CDK complexes in these cells. Cdk4 immunoprecipitates from control cells phosphorylated a truncated pRB fusion protein, and this kinase activity was substantially reduced in the presence of a competing peptide or in the absence of primary antibody (Fig. 3A and data not shown), confirming the specificity of the assay. No significant change in Cdk4 activity was apparent in the first 12 h following progestin treatment (Fig. 3). However, Cdk4 activity declined after ≥18 h of progestin treatment (Fig. 3A), such that the specific Cdk4 activity (after background subtraction) was reduced to 40% of the control value by 18 h and to 10% of the control value by 24 h (Fig. 3B). A slight increase in cyclin E-associated kinase activity was apparent at 6 h, followed by a reduction to ∼40% of the control value by 18 h (Fig. 3). The activity of this kinase reached a minimum of 23% at 24 h and remained below 50% of the control value at 30 h, although partial recovery of kinase activity was observed at 30 h (Fig. 3B). Since a marked decrease in the activities of both Cdk4 and cyclin E-associated kinases preceded a significant decrease in the S phase fraction by up to 6 h (compare Fig. 1 and 3), the decrease in CDK activity is implicated in the accompanying inhibition of pRB and p107 phosphorylation and hence arrest in G1 phase following progestin treatment.
FIG. 3.
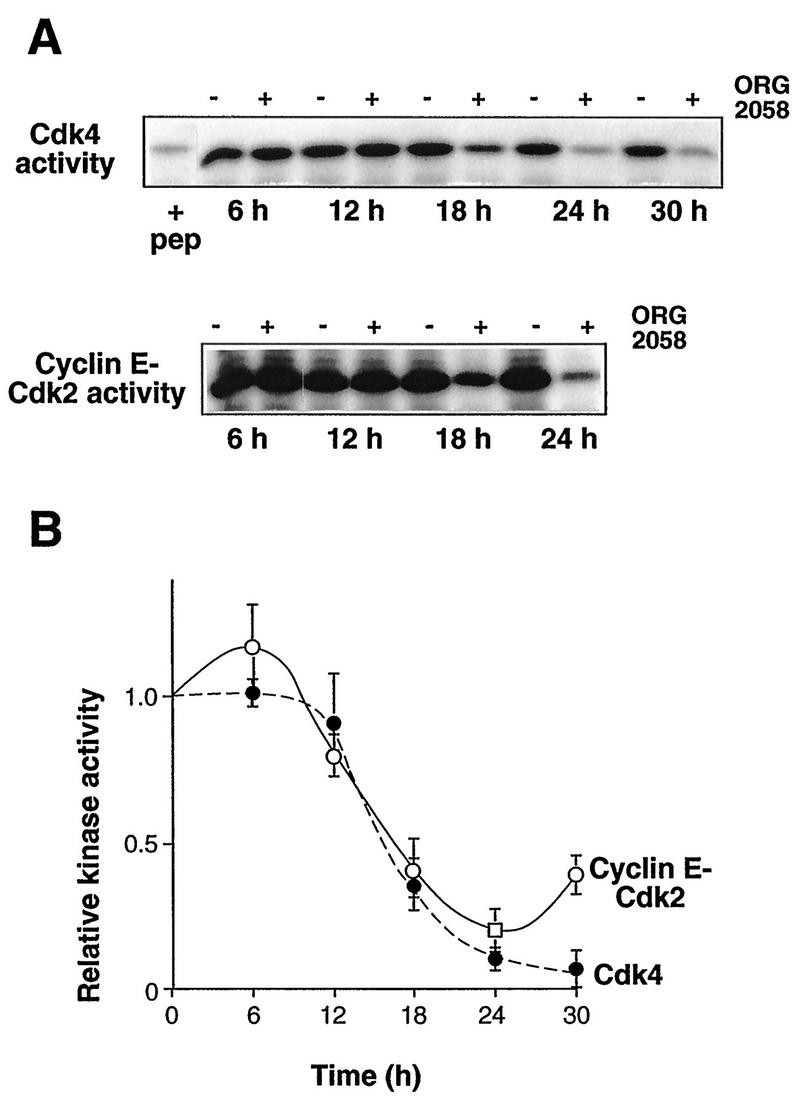
Progestin inhibition of CDK activity. Exponentially proliferating T-47D breast cancer cells were treated with the synthetic progestin ORG 2058 (10 nM) or vehicle. At intervals thereafter whole cell-lysates were prepared and immunoprecipitated with antibodies to Cdk4 or cyclin E. Either GST-pRB773-928 (Cdk4 activity) or histone H1 (cyclin E-Cdk2 activity) was used as a substrate. (A) The upper panel shows phosphorylated GST-pRB773-928 from a representative experiment, visualized by SDS-PAGE and autoradiography. The first lane (+ pep) contains a sample from untreated cells harvested at the time of ORG 2058 or vehicle addition, in which the immunoprecipitation was performed in the presence of competing antigenic peptide. The lower panel shows phosphorylated histone H1 from a representative experiment visualized by SDS-PAGE and autoradiography. (B) Phosphorylated substrates were detected with PhosphorImager. Points represent the means ± SEMs from two to four observations, relative to the average control value for each experiment. For Cdk4 activity the background in each assay was estimated from samples immunoprecipitated in the presence of competing peptide or in the absence of antibody and has been subtracted from the individual data points. •, Cdk4 activity; ○, cyclin E-Cdk2 activity.
Decreased expression of cyclins D1, D3, and E and increased expression of the CDK inhibitors p21 and p27 following progestin treatment.
To investigate possible causes of the decreased kinase activity, the expression of components of the G1 cyclin complexes was examined. Cyclin D1 protein initially slightly increased in abundance (Fig. 4). However, this was transient, and at 18 to 24 h cyclin D1 levels in progestin-treated cells were ∼50% of those in control cells (Fig. 4). Northern analysis showed that, similarly, cyclin D1 mRNA was initially induced but subsequently declined to ∼60% of control at 18 h, remaining below control levels at 24 h (Fig. 4C). A slight decrease in cyclin E protein expression was apparent as early as 12 h, and the decline continued until cyclin E reached a minimum of 25% of the control value at 24 h (Fig. 4). Consistent with the partial recovery of cyclin E-associated kinase activity observed at 30 h (Fig. 3), cyclin E expression partially recovered between 24 and 30 h to reach 50% of the control value (Fig. 4). The cyclin E mRNA abundance was reduced following 18 to 24 h of progestin treatment, to ∼50% of the control value or less (Fig. 4C). The changes in cyclin D1 and E mRNAs were similar in timing and magnitude to the changes in protein abundance, suggesting that progestin treatment regulated cyclin D1 and E expression in large part via transcriptional mechanisms. No significant changes in cyclin D3, Cdk2, or Cdk4 protein expression were apparent until 24 h after progestin treatment, when decreases of 40 to 50% were observed; the decreases were maintained at 30 h (Fig. 4). The abundance of p21 protein did not change until 18 h, when it increased ∼2-fold; the increase was maintained at 24 h but was of smaller magnitude thereafter (Fig. 5). Upon more extended progestin treatment, p21 levels fell below control values (see Fig. 12). The abundance of p27 began to increase slightly at 18 to 24 h and had increased approximately twofold at 30 h (Fig. 5). Thus, of the proteins examined, only cyclin D1, cyclin E, and p21 displayed alterations in abundance of ≥2-fold at 18 h, when marked decreases in Cdk4 activity and cyclin E-associated kinase activity were observed.
FIG. 4.
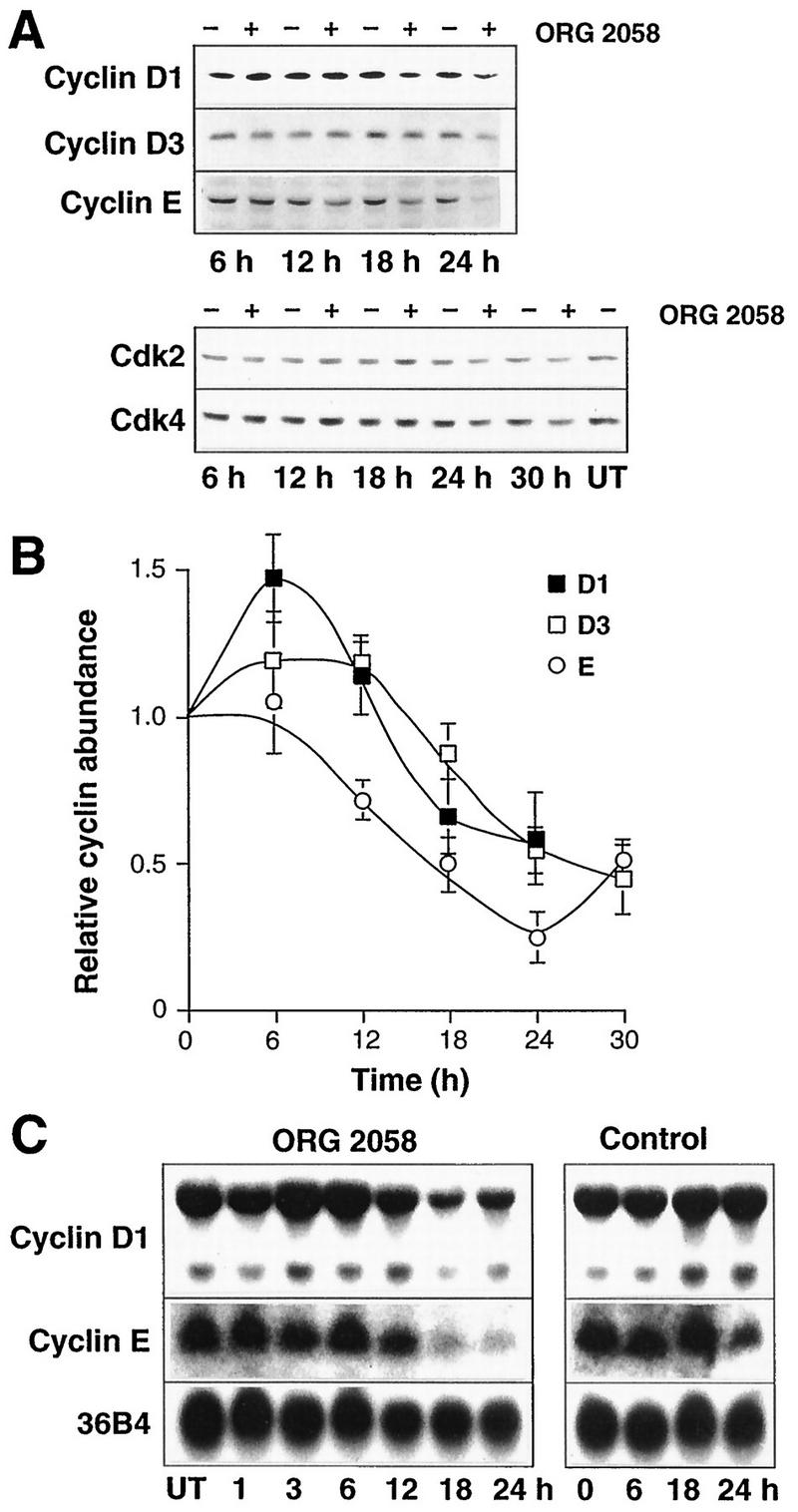
Cyclin and CDK expression following progestin treatment. Exponentially proliferating T-47D breast cancer cells were treated with the synthetic progestin ORG 2058 (10 nM) or vehicle, and at intervals thereafter whole-cell lysates or total cellular RNA were prepared. (A) Representative Western blots for cyclin D1, cyclin D3, cyclin E, Cdk2, and Cdk4. UT, untreated control harvested at the time of treatment. (B) Data representing the means ± SEMs from three to five observations (cyclin D1), three or four observations (cyclin D3), or two to four observations (cyclin E) are expressed relative to the average control value in each experiment. (C) Representative Northern blots for cyclin D1, cyclin E, and, as a control for RNA loading on the cyclin E blot, 36B4. The loading control for the cyclin D1 blot is shown in Fig. 6.
FIG. 5.

CDK inhibitor expression following progestin treatment. Exponentially proliferating T-47D breast cancer cells were treated with the synthetic progestin ORG 2058 (10 nM) or vehicle, and at intervals thereafter whole-cell lysates were prepared. (A) Representative Western blots for p21 and p27. (B) Data representing the means ± SEMs from three to six (p21) or four or five (p27) observations are expressed relative to the average control value in each experiment.
Changes in Cdc25 or CAK activity do not account for progestin-induced decreases in CDK activity.
Further experiments examined the possibility that altered CDK phosphorylation might contribute to the decrease in activity following progestin treatment. First, regulation of phosphorylation at inhibitory residues within the catalytic cleft was addressed, since such inhibition is dominant over activation resulting from either cyclin binding or CAK activation (see the introduction). Of the family of phosphatases responsible for relieving this inhibition, Cdc25A appears to have specificity for G1 CDKs in vivo, and therefore its expression was examined. Following ORG 2058 treatment a small increase of <50% was apparent at 6 h, but thereafter Cdc25A mRNA decreased in abundance to reach a minimum of ∼30% of the control value at 18 to 24 h (Fig. 6A). In view of evidence for c-myc regulation of Cdc25A (14), c-myc expression was also examined. Consistent with previous data (38, 65), progestin treatment increased c-myc expression after 1 h, but thereafter c-myc expression decreased to <50% of the control value (Fig. 6A). The decrease in c-myc expression was apparent within 6 h, thus preceding the decline in Cdc25A expression by >12 h. Measurement of Cdc25A protein levels revealed only an ∼50% decrease in abundance at 18 to 24 h (Fig. 6B).
FIG. 6.

Cdc25A and c-myc expression following progestin treatment. Exponentially proliferating T-47D breast cancer cells were treated with the synthetic progestin ORG 2058 (10 nM) or vehicle (Control), and at intervals thereafter total cellular RNA or whole-cell lysates were prepared. (A) Representative Northern blot sequentially probed with Cdc25A, c-myc, and 36B4 (as a control for RNA loading). (B) Representative Western blot for Cdc25A. UT, untreated cells harvested at the time of treatment.
To determine whether a lack of activating phosphatase activity might contribute to the reduction in CDK activity following progestin treatment, attempts were made to reactivate kinase complexes from ORG 2058-treated cells by using bacterially expressed recombinant GST-Cdc25A. Incubation of GST-Cdc25A with cyclin E immunoprecipitates from exponentially proliferating T-47D cells resulted in activation of the cyclin E-associated kinase (Fig. 7A). The activation was abolished in the presence of the phosphatase inhibitors sodium vanadate and sodium fluoride, and incubation with recombinant GST alone had no effect on cyclin E-associated kinase activity (Fig. 7A), demonstrating that the activation of the kinase resulted from Cdc25A phosphatase activity. The effect of Cdc25A on the CDK activity of cyclin E immunoprecipitates from control or progestin-treated cells was then examined. These experiments demonstrated activation of cyclin E-Cdk2 from both control and progestin-treated cells (Fig. 7B). However, since the degree of activation was similar in both cases, following Cdc25A activation the cyclin E-Cdk2 activity from progestin-treated lysates remained at ∼20% of that observed in Cdc25A-activated immunoprecipitates from control cells. This difference could not be accounted for by a lack of Cdk2 in the immunoprecipitates, since in parallel cyclin E immunoprecipitates the amount of Cdk2 was reduced by <40% (Fig. 7B). Thus, a lack of Cdc25A activity did not appear to contribute to the decreased cyclin E-Cdk2 activity following progestin treatment. Similar experiments failed to demonstrate Cdc25A activation of Cdk4 immunoprecipitates from either control or progestin-treated cells, despite 15-fold increases in the amount of added Cdc25A (Fig. 7C). These data argue against decreased Cdc25A expression contributing to the decrease in Cdk4 activity following progestin treatment. Since the presence of inhibitory phosphorylation did not contribute to the decrease in CDK activity following progestin treatment, the possibility that CAK activity might be regulated by progestins was next examined. Bacterially expressed GST-Cdk2 was used as a substrate for Cdk7 immunoprecipitates, since in eukaryotes complexes containing Cdk7 (MO15) appear to be responsible for CAK activity (32). However, these experiments did not demonstrate a decrease in CAK activity following progestin treatment (Fig. 7D).
FIG. 7.

Regulation of Cdc25 or CAK activity does not account for decreased CDK activity following progestin treatment. (A) Replicate cyclin E immunoprecipitates from 250 μg of lysate prepared from exponentially proliferating T-47D cells were incubated with increasing amounts of GST-Cdc25A (170, 340, 680, and 1,360 μg) or GST (360 and 1,440 μg) or without further addition (−) at 30°C for 1 h and washed, and then kinase activity was measured with a histone H1 substrate. One sample (Van) was incubated with 680 μg of GST-Cdc25A in the presence of the phosphatase inhibitors vanadate (10 mM) and NaF (50 mM). (B) T-47D breast cancer cells were treated with the synthetic progestin ORG 2058 (10 nM) or vehicle for 24 h. Cyclin E immunoprecipitates were incubated with or without Cdc25A as indicated, and cyclin E-Cdk2 activity was measured with a histone H1 substrate. Parallel immunoprecipitates were Western blotted with Cdk2 antibodies. (C) Cdk4 immunoprecipitates from control or ORG 2058-treated (10 nM, 30 h) cells were incubated with GST-Cdc25A, and Cdk4 activity was measured with GST-pRB773-928. (D) The kinase activity of Cdk7 immunoprecipitates from control or ORG 2058-treated T-47D cells was measured with a GST-Cdk2 substrate. The first lane (+ pep) contains a sample from untreated cells in which the immunoprecipitation was performed in the presence of competing antigenic peptide.
Progestin treatment alters the molecular mass of cyclin E complexes but not cyclin D complexes.
The data presented in Fig. 7 argued that changes in CDK phosphorylation were unlikely to contribute to the decreases in Cdk4 and cyclin E-associated kinase activities. This suggested that alterations in either the abundance or composition of the CDK complexes were probable mechanisms for the decreased kinase activity. Since recent data have indicated that CDK complexes with different compositions and activities can be resolved by gel filtration chromatography (for example, see references 44 and 54), lysates from control and progestin-treated cells were fractionated to determine whether progestin treatment affected the formation or composition of these complexes. Aliquots of protein eluted from a Superdex 200 column were acetone precipitated and Western blotted. In control lysates cyclin D1 typically eluted in a single major broad peak with an apparent molecular mass of ∼160 kDa (Fig. 8A), consistent with previous data from Superose 12 fractionation of Swiss 3T3 cells (42); in some experiments (e.g., that shown in Fig. 8), a proportion also eluted at a lower apparent molecular mass (Fig. 8A). The majority of cyclin D3 coeluted with cyclin D1 at ∼160 kDa, but a form with a lower apparent molecular mass was consistently observed. Since the lower-molecular-mass forms of cyclin D1 and D3 coeluted with cyclin D1 produced by in vitro transcription and translation in reticulocyte lysate (data not shown), it is likely that these represent free cyclin D. Cyclin E eluted over a broad range of apparent molecular mass, indicating the presence of two overlapping peaks with apparent molecular masses of ∼120 and ∼200 kDa (Fig. 8B and 9; see also Fig. 12).
FIG. 8.
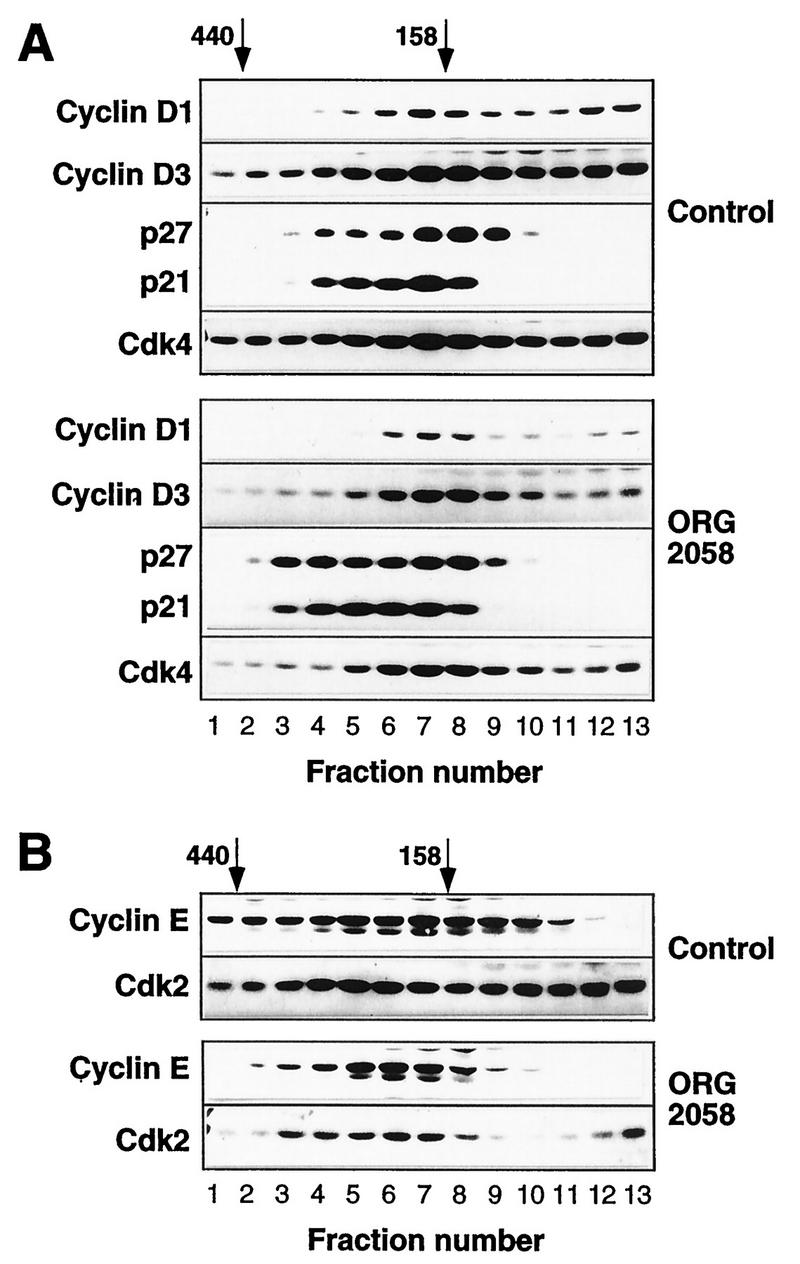
Elution profiles of cyclins, CDKs, and CDK inhibitors in control and progestin-treated lysates. Exponentially proliferating T-47D breast cancer cells were treated with the synthetic progestin ORG 2058 (10 nM) or vehicle (Control) for 24 h. Whole-cell lysates were fractionated on a Superdex 200 gel filtration column (void volume, 45 ml). The elution volumes of markers with known molecular masses (ferritin, 440 kDa; aldolase, 158 kDa) are indicated; ovalbumin (43 kDa) eluted at 85 ml, corresponding to fraction 15. Fractions (2 ml) corresponding to elution volumes of 57 to 82 ml were acetone precipitated and then Western blotted for the proteins indicated.
FIG. 9.

Time course of changes in elution profiles of cyclin E, p21, and p27 following progestin treatment. Exponentially proliferating T-47D breast cancer cells treated with the synthetic progestin ORG 2058 (10 nM) or vehicle (Control) in the same experiment as presented in Fig. 8 were harvested after 12 or 18 h. Whole-cell lysates were fractionated on a Superdex 200 gel filtration column (void volume, 45 ml). The elution volumes of markers with known molecular masses (ferritin, 440 kDa; aldolase, 158 kDa) are indicated; ovalbumin (43 kDa) eluted at 85 ml, corresponding to fraction 15. (A) Fractions (2 ml) corresponding to elution volumes of 57 to 80 ml were acetone precipitated and then Western blotted for the proteins indicated. (B) Western blots of 2-ml fractions were quantitated by densitometry and are presented as percentages of the total signal observed for each protein. Data from three control samples (12, 18, and 24 h of vehicle treatment) have been pooled and are presented as means ± SEMs, where the SEM exceeds the size of the symbol. ○, control; •, ORG 2058.
Both Cdk2 and Cdk4 eluted in two major peaks. One peak, representing about 50% of the Cdk4 and the majority of the Cdk2, eluted at an apparent molecular mass of <50 kDa (data not shown), close to the expected position of monomeric CDK. For Cdk4 the other peak coeluted with the major peaks of cyclins D1 and D3 at ∼160 kDa (Fig. 8A). The higher-apparent-molecular-mass form of Cdk2 encompassed the fractions containing both the 120- and 200-kDa forms of cyclin E. A small proportion of cyclins D1, D3, and E, Cdk2, and Cdk4 could be detected at very high apparent molecular masses, >400 kDa (Fig. 8), suggesting the existence of a small fraction of cyclin-CDK complexes associated with other intracellular proteins. Examination of the elution profiles of p21 and p27 suggested that these CDK inhibitors were all associated with cyclin-CDK complexes, since no p21 or p27 with an apparent mobility of below ∼120 kDa was detected and both coeluted with cyclin D1, cyclin D3, and cyclin E (Fig. 8). Overall, the mobilities of p27 and p21 were similar, although there was a tendency for more p21 to elute at a higher apparent molecular weight (Fig. 8A and 9).
In lysates prepared following 24 h of progestin treatment, several significant differences in the elution profiles of the cyclins, CDKs, and CDK inhibitors were apparent. The most marked of these was loss of the ∼120-kDa peak of cyclin E, such that essentially all the detectable cyclin E coeluted with the higher-molecular-weight form of cyclin E present in control cells (Fig. 8B and 9). There was a corresponding shift in the elution profile of Cdk2, such that less Cdk2 was present in fractions corresponding to the 120-kDa peak of cyclin E protein (Fig. 8B). In contrast with the marked changes in the elution profile of cyclin E, there was no apparent change in the position of the major peak of cyclin D1 or D3 (Fig. 8A). While a significant fraction of p21 and p27 coeluted with cyclins D1 and D3 in lysates from either control or progestin-treated cells, there was a clear increase in the amount of p27 and a less marked increase in the amount of p21 eluting at apparent molecular masses of ≥200 kDa following progestin treatment (Fig. 8A and 9). These higher-apparent-molecular-mass fractions contained cyclin E, suggesting increased association between cyclin E-Cdk2 complexes and these inhibitors following progestin treatment.
The changes in the elution profiles of cyclin E, p27, and p21 were further examined to determine the temporal relationship between these changes and the inhibition of proliferation. After 12 h of progestin treatment, before any inhibition of entry into S phase could be detected, no change in the elution profile of any of these proteins was detected (Fig. 9). However, after 18 h of progestin treatment, when inhibition of entry into S phase was just commencing (Fig. 1), the proportion of cyclin E in the 120-kDa form was markedly reduced, and there was a corresponding increase in the proportion in the 200-kDa form (Fig. 9). Similarly, after 18 h of progestin treatment, the amounts of p21 and p27 eluting at >200 kDa (fractions 3 to 5 in Figure 9) were increased compared with the control values. The changes in the elution profiles of cyclin E and p27 were more pronounced after 24 h than after 18 h of treatment (Fig. 9). Thus, these changes paralleled the decrease in cyclin E-Cdk2 activity and preceded the decrease in the S phase fraction by several hours, suggesting that they were a cause rather a consequence of the inhibition of proliferation.
Increased association of p27 but not p21 with cyclin D-Cdk4 complexes following progestin treatment.
Since the data presented in Fig. 8 indicated that the majority of cyclin-CDK complexes might also contain CDK inhibitors, in further experiments immunoprecipitates of fractionated lysates were used in in vitro kinase assays to determine which fractions contained active CDK. In view of the limited sensitivity of Cdk4 assays, Cdk4 activity was measured in 3-ml fractions rather than in 2-ml fractions. Cdk4 activity from control lysates displayed an elution profile similar to that of the high-molecular-mass form of Cdk4, reaching a maximum at 150 to 200 kDa (compare Fig. 8A and 10). However, consistent with data obtained with whole-cell lysates (Fig. 3), progestin treatment resulted in a marked decrease in Cdk4 activity (Fig. 10A). Western blotting of Cdk4 immunoprecipitates from fractionated control lysates indicated that the active fractions contained significant amounts of coimmunoprecipitating cyclin D1, cyclin D3, Cdk4, p21, and p27 (Fig. 10B). The same fractions from progestin-treated lysates contained less Cdk4 and significantly less cyclin D1 and cyclin D3, consistent with the decrease in total abundance of these proteins (Fig. 10B). However, no increase in p21 was observed, and in some experiments the abundance of p21 appeared to decrease relative to the abundance of other components of the complex following progestin treatment (Fig. 10B). In contrast, the decrease in the amount of cyclin D1 and cyclin D3 in the Cdk4 immunoprecipitates was more marked than the decrease in the amount of associated p27 (Fig. 10B). In fraction 3, the abundance of cyclin D3 decreased by ∼60% while the abundance of p27 changed little, such that the p27/cyclin D3 ratio increased 2.3-fold. Similarly, in fraction 5, the abundance of cyclin D1 decreased by >50% and there was a smaller decrease in the amount of p27, such that the p27/cyclin D1 ratio increased ∼2-fold. These data suggest that the decreased abundance of cyclins D1 and D3 is a major factor in the decrease in Cdk4 activity but that increased p27 association also likely contributes to the loss of activity.
FIG. 10.

Cdk4 activity and complex composition of fractionated T-47D lysates. Exponentially proliferating T-47D breast cancer cells were treated with the synthetic progestin ORG 2058 (10 nM) or vehicle (Control) for 24 h. Whole-cell lysates were fractionated on a Superdex 200 gel filtration column, and 3-ml fractions corresponding to elution volumes of 58 to 81 ml were then immunoprecipitated with a Cdk4 antibody. (A) Kinase activities of the immunoprecipitates were measured with a GST-pRB773-928 substrate. (B) Immunoprecipitates of the fractions indicated were Western blotted with the antibodies indicated.
The low-activity form of cyclin E is associated with CDK inhibitors.
Measurement of kinase activity in cyclin E immunoprecipitates from gel filtration fractions revealed a peak of activity associated with the 120-kDa form of cyclin E in control cells (fractions 8 to 10) but much less activity in fractions 4 to 7, associated with the ∼200-kDa form (Fig. 11A), although similar amounts of cyclin E were present in the two forms (Fig. 8B and 9). Some activity, comprising up to ∼20% of the total, was present in the fractions with very high apparent molecular masses (>400 kDa). Following progestin treatment, near-background levels of kinase activity were observed in fractions 8 to 11, consistent with the lack of cyclin E in these fractions, and little activity could be detected associated with the remaining major ∼200-kDa peak of cyclin E protein (Fig. 11A). The peak of activity in fraction 4 of progestin-treated lysate in Fig. 11A was not consistently observed. Thus, while kinase activity was reduced across the elution profile, this was most marked in the fractions corresponding to the 120-kDa form of cyclin E.
FIG. 11.
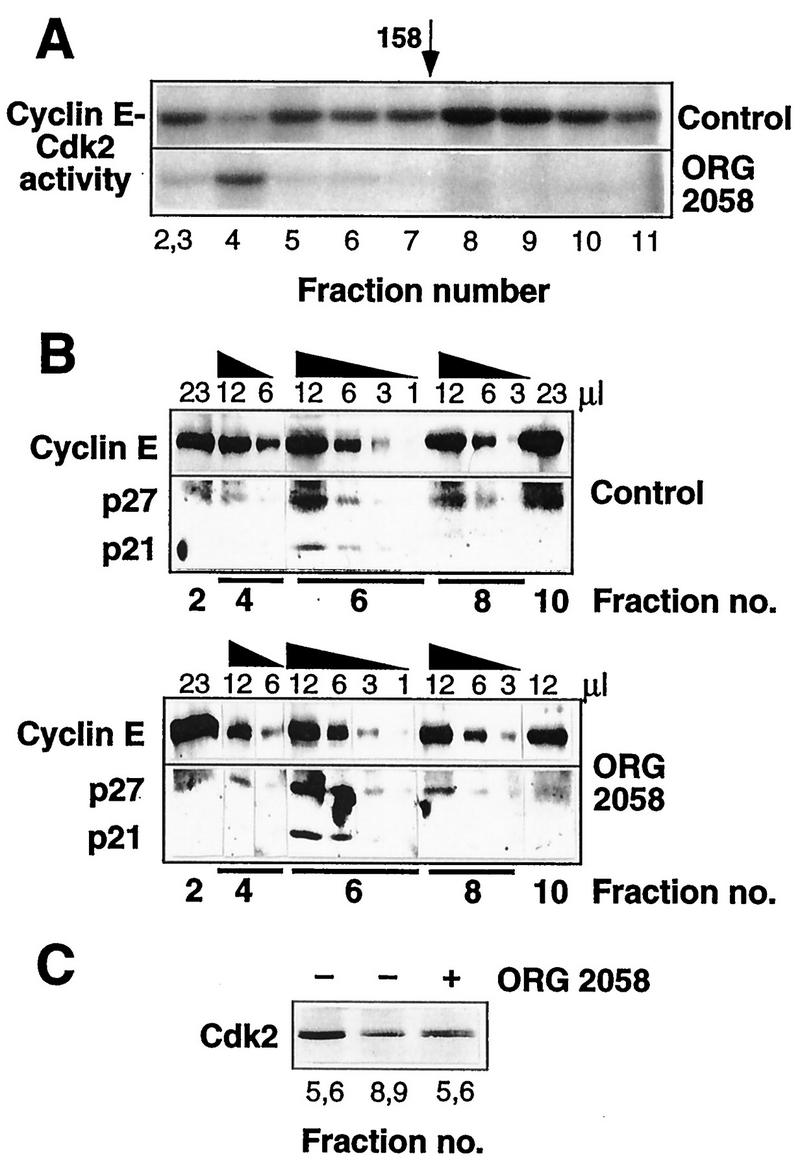
Cyclin E-Cdk2 activity and complex composition of fractionated T-47D lysates. Exponentially proliferating T-47D breast cancer cells were treated with the synthetic progestin ORG 2058 (10 nM) or vehicle (Control) for 24h. Whole-cell lysates were fractionated on a Superdex 200 gel filtration column, and fractions were then immunoprecipitated with a cyclin E antibody. (A) Kinase activities of immunoprecipitates corresponding to elution volumes of 59 to 61 (fractions 2 and 3), and 63 to 78 ml in 2-ml increments (fractions 4 to 11) were measured with a histone H1 substrate. (B) Immunoprecipitates from fractions encompassing the peak of cyclin E protein were resuspended in 25 μl of SDS sample buffer, and various volumes were Western blotted in parallel with the antibodies indicated. (C) Cyclin E immunoprecipitates corresponding to elution volumes of 65 to 68 ml (fractions 5 and 6) and 72 to 74 ml (fractions 8 and 9) were Western blotted with a Cdk2 antibody.
Fractions encompassing the major peaks of cyclin E protein were immunoprecipitated to examine whether differences in composition were associated with the differences in activity. To facilitate comparison between fractions containing different amounts of cyclin E, various amounts of each fraction were Western blotted in parallel. These experiments revealed clearly detectable p21 and p27 in fraction 6, corresponding to the ∼200-kDa form of cyclin E, in control cells but little in the remaining fractions (Fig. 11B). Thus, the ∼120-kDa form was relatively deficient in p21 and p27. Cdk2 displays a characteristic increase in electrophoretic mobility upon activation via CAK phosphorylation (15), and higher-resolution Western blotting of cyclin E immunoprecipitates from control cells demonstrated that all the detectable Cdk2 in the lower-molecular-weight complexes was apparently CAK phosphorylated, while the higher-molecular-weight complexes contained Cdk2 of both electrophoretic mobilities (Fig. 11C). Following progestin treatment, fractions encompassing the peak of cyclin E protein at 200 kDa (fractions 4, 6, and 8), contained a 2.0- to 2.5-fold increase in the relative abundance of cyclin E-associated p27 compared with the same fraction from control lysates (Fig. 11B). Although in the experiment presented in Fig. 11B the relative abundance of p21 also increased in fraction 6 following ORG 2058 treatment, this was not a consistent observation. In progestin-treated cells, a smaller proportion of cyclin E-associated Cdk2 was CAK phosphorylated (Fig. 11C). The mobility shift of the Cdk2 in these fractions following ORG 2058 treatment suggested that the reduced specific activity of the complexes might in part be due to a lack of CAK activation, perhaps resulting from increased p27 association.
Cyclin D1 induction in progestin-inhibited cells leads to a reinitiation of cell cycle progression and altered molecular mass of cyclin E complexes.
To determine to what degree reduced cyclin expression contributed to the reduction in CDK activity and inhibition of cell cycle progression following progestin treatment, a clonal derivative of T-47D cells expressing cyclin D1 under the control of the heavy-metal-responsive metallothionein promoter, T-47D ΔMTcycD1-3, was treated with progestin for 24 to 48 h to inhibit cell cycle progression and then with zinc to induce cyclin D1 expression. Induction of cyclin D1 was sufficient to stimulate a cohort of progestin-inhibited cells to resume cell cycle progression, such that the S phase fraction, 10% after progestin pretreatment, increased to a maximum of >40% after zinc treatment (Fig. 12A). Zinc treatment of cells pretreated with vehicle rather than progestin increased the proportion of cells in S phase to a similar maximum level, although the relative increase was smaller due to the higher S phase fraction in the absence of progestin pretreatment (Fig. 12A). Western blots of cells harvested after zinc treatment of either vehicle- or progestin-pretreated cells demonstrated marked induction of cyclin D1 in progestin-pretreated cells and more modest induction of cyclin D1 in vehicle-pretreated cells (Fig. 12B). In progestin-pretreated cells cyclin D1 induction restored cyclin E expression to a level similar to that observed in cells not treated with progestin, consistent with the observation that cyclin D1 induction of cell cycle progression in these cells leads to induction of cyclin E (39), but had little effect on the abundances of p21 and p27. Induction of cyclin D1 in progestin-pretreated cells also increased Cdk4 activity and cyclin E-Cdk2 activity to levels similar to that following cyclin D1 induction in vehicle-pretreated cells (Fig. 12B).
To determine whether the restoration of CDK activity and resumption of cell cycle progression following cyclin D1 induction was accompanied by alteration in the molecular mass of the cyclin-CDK complexes, lysates from zinc-treated cells were separated by gel filtration chromatography. Despite the partial synchronization following cyclin D1 induction in vehicle-pretreated cells, the elution profiles of cyclin D1, cyclin E, p27, and p21 were similar to those observed in the absence of zinc treatment (compare Fig. 12C and D with Fig. 8 and 9). As expected, the elution profiles of cyclins D1 and E, p21, and p27 in progestin-treated T-47D ΔMTcycD1-3 cells were similar to those observed in progestin-treated T-47D cells. Interestingly, some p27 eluted at a low molecular mass (∼70 to 90 kDa) following progestin treatment (ORG 2058/water in Fig. 12C and D); this likely represents monomeric p27 (42). However, following cyclin D1 induction in progestin-pretreated cells, the elution profiles of cyclin E and p27 were altered compared with those from cells treated with progestin but not zinc, such that they closely resembled the profiles obtained from control cells. Thus, the 120-kDa form of cyclin E was clearly apparent following cyclin D1 induction in progestin-pretreated cells, while the amount of p27 present in high-molecular-mass (>200-kDa) complexes decreased (Fig. 12C and D). These changes in elution profile preceded entry into S phase, since the lysates used for the gel filtration chromatography were harvested from cells just beginning to enter S phase following cyclin D1 induction; i.e., the S phase fraction in progestin-pretreated cells had not increased to more than 20%. These data suggest that altered cyclin abundance is a major determinant of the altered cyclin-CDK complex distribution following progestin treatment.
DISCUSSION
This study has focused on regulation of CDK activity accompanying growth inhibition by the progestin ORG 2058 as a step towards dissecting the mechanisms that mediate progestin effects on normal physiology and on breast cancer. Marked decreases in both Cdk4 activity and cyclin E-Cdk2 activity, with consequent decreases in the phosphorylation of pRB and p107, preceded inhibition of entry into S phase. Several different molecular events potentially contributing to the decrease in CDK activity were observed: initial alterations in the abundances of cyclin D1 and cyclin E, later changes in the abundances of cyclin D3 and p27, and increased relative p27 association with cyclin D-Cdk4 and cyclin E-Cdk2 complexes. These events were all preceded by an early decline in c-myc expression, first apparent within 3 to 6 h of progestin treatment.
Since both Cdk4 activity and cyclin E-Cdk2 activity are required for progress from G1 into S phase (24, 50, 51), the observed decrease in these activities (Fig. 3) could account for progestin-mediated inhibition of proliferation. Substrates for these CDKs, including pRB and p107, can thus be seen as the eventual targets for this aspect of progestin action. Both pRB and p107 decreased in total abundance as well as phosphorylation, such that very little of the phosphorylated form of either protein was present following 24 h or more of progestin treatment. However, despite the decrease in total abundance, the underphosphorylated form was increased in abundance, reaching a maximum by 18 h, which was maintained until at least 30 h. Thus, the accumulation of the growth-inhibitory forms of these proteins preceded the inhibition of entry into S phase, which was first apparent at 18 h. The partial recovery of cyclin E-associated kinase activity between 24 and 30 h was not accompanied by a detectable increase in pRB phosphorylation. Since at that time little Cdk4 activity was present, this observation is consistent with the view that cyclin E-Cdk2 phosphorylation of pRB is dependent on prior pRB phosphorylation by cyclin D-associated kinases (19). However, it is also possible that the recovery of cyclin E-Cdk2 activity is sufficient to overcome the lack of Cdk4 activity in a fraction of the cell population, thus contributing to the slow resumption of proliferation following extended progestin treatment (55). Induction of differentiation is a major physiological role of progestins (5), but the decrease in pRB abundance following progestin treatment contrasts with the frequent observation of increased pRB expression accompanying differentiation in diverse cell types (9, 52, 57). However, since the mammary gland differs from most other tissues in that differentiation is followed by involution, some differences in control of differentiation may be expected. In this context it is interesting that pRB deficiency is associated with increased apoptosis (63).
Examination of potential mechanisms for the decrease in CDK activity accompanying progestin inhibition of proliferation revealed decreased expression of mRNA encoding the CDK-activating phosphatase Cdc25A. Reduced Cdc25A expression and increased tyrosine phosphorylation of Cdk2 and Cdk4 have been implicated in growth arrest by different agents, including UV irradiation, alpha interferon, and transforming growth factor β (TGF-β) (26, 58, 59). In addition, since cyclin E-Cdk2 activates Cdc25A by phosphorylation (22), decreased Cdc25A expression in combination with decreased cyclin E-Cdk2 activity might result in marked decreases in Cdc25A activity. Thus, lack of Cdc25A activity provided an attractive mechanism contributing to reduced CDK activity following progestin treatment. However, the decrease in Cdc25A protein levels was less marked than the decrease in the corresponding mRNA, consistent with the suggestion that the protein may have a long half-life (26). Furthermore, treatment with recombinant Cdc25A failed to restore the activity of cyclin E-associated kinases to a level consistent with the level of Cdk2 present in the complexes, and no Cdc25A activation of Cdk4 from either control or progestin-treated lysates was observed. Although the possibility that the amount of Cdc25A used was insufficient to activate Cdk4 cannot be excluded, this result is consistent with the lack of tyrosine phosphorylation on Cdk4 and Cdk6 (but not Cdk2) in exponentially proliferating MCF10A mammary epithelial cells (26). These data indicate that regulation of Cdc25A does not make a major contribution to progestin inhibition of proliferation.
The initial progestin-mediated decreases in Cdk4 and cyclin E-Cdk2 activity were accompanied by alterations in the abundances of several of the components of these complexes, i.e., cyclin D1, cyclin E, and p21. Separation of whole-cell lysates by gel filtration chromatography indicated that cyclin E and Cdk2 displayed a distinct alteration in elution profile following progestin treatment. In control cells the major peak of cyclin E-associated kinase activity eluted at ∼120 kDa, although some activity, ∼20% of the total, was present in complexes of very high molecular mass. A significant fraction of the cyclin E in control cells eluted at ∼200 kDa and displayed relatively little associated kinase activity, likely due to p21 and p27 association. This observation is consistent with other data indicating that cyclin E is present in a largely inactive ∼250-kDa form and a more active ∼120-kDa form (54) and that a minority of cyclin E accounts for the majority of the associated kinase activity (41, 44). Following progestin treatment, almost all of the cyclin E was in a low-activity, p21- and p27-bound form eluting at ∼200 kDa. This peak of cyclin E also contained less CAK-activated Cdk2 than the corresponding fractions from a control lysate, consistent with data demonstrating that these CDK inhibitors interfere with activation by CAK (2). Thus, the reduced number of cyclin E-Cdk2 complexes present following progestin treatment preferentially assemble into the less active CDK-inhibitor bound form at the expense of the more active 120-kDa form. This redistribution preceded the inhibition of entry into S phase and coincided with the inhibition of cyclin E-Cdk2 activity, implicating it as a cause of the inhibition of kinase activity. Further evidence for a causative link between the redistribution of cyclin E-Cdk2 complexes and their activity is provided by the observation that following cyclin D1 induction in progestin-pretreated cells, the more active 120-kDa form of cyclin E-Cdk2 reappeared and kinase activity was restored at a time when the majority of the stimulated population had yet to reenter S phase. These data also suggest that alterations in cyclin abundance, especially cyclin D1 abundance, are sufficient to lead to changes in the compositions as well as the relative abundances of various cyclin-CDK complexes.
Cyclin D1 and cyclin D3 differed from cyclin E in that the cyclin-CDK complexes and the associated kinase activity displayed the same elution profile, peaking at ∼160 kDa, although this peak clearly contained p21- and p27-bound CDK (Fig. 10). This is consistent with data indicating that p21 and p27 association with Cdk4 does not necessarily inhibit kinase activity (3, 30, 53), and indeed, some pRB kinase activity was present in p27 immunoprecipitates from exponentially proliferating T-47D cells, equivalent to approximately a third of the Cdk4 activity (data not shown). Following progestin treatment, although major shifts in the elution profile of cyclin D1, cyclin D3, or Cdk4 were not observed, Cdk4 immunoprecipitates of fractionated lysates contained less cyclin D1 and cyclin D3 and had an increased p27/cyclin D ratio (Fig. 10). Overall, these data suggest that a major factor in the decrease in Cdk4 activity following progestin treatment is a decrease in the abundance of the cyclin D1-Cdk4 and cyclin D3-Cdk4 complexes and that increased p27 association with these complexes may contribute to their decreased kinase activity. The ability of ectopic cyclin D1 induction to reinitate cell cycle progression following progestin pretreatment is consistent with this conclusion.
The data presented above indicate a redistribution of p27 between different cyclin-CDK complexes at times when there was little alteration in p27 abundance. Furthermore, although the p21 abundance increased ∼2-fold following progestin treatment, there appeared to be no corresponding recruitment of p21 into either cyclin E-Cdk2 complexes or Cdk4 complexes. One explanation for the latter observation might be that following progestin treatment p21 was in excess over the number of available binding sites, but gel filtration indicated that neither p21 nor p27 was present in monomeric form except after extended progestin treatment. Furthermore, immunoprecipitation studies indicated that the majority of p21 and p27 is associated with cyclin D1 and Cdk4, in either the presence or absence of progestin treatment (reference 37 and data not shown). In contrast, while a significant proportion of cyclin E is associated with p21 and p27, this accounts for a small fraction (<10%) of the total pool of these CDK inhibitors.
One important factor in the formation of complexes between these proteins is the relative affinities of the CDK inhibitors for different cyclin-CDK complexes, and this, combined with the reduction in the abundance of several cyclin-CDK complexes, suggests a possible explanation for the redistribution of these CDK inhibitors. Experiments using purified components have indicated that the affinity of p27 for cyclin E-Cdk2 is ∼10-fold higher than its affinity for cyclin D2-Cdk4, while in contrast, p21 has a higher affinity for cyclin D2-Cdk4 than for cyclin E-Cdk2 (18). If cyclin D1-Cdk4 and cyclin D3-Cdk4 have affinities for p27 similar to that of cyclin D2-Cdk4, cyclin D-Cdk4 complexes are likely to act as high-capacity, low-affinity binding sites for p27, consistent with the idea that they represent a sink for p27 (24, 50, 51). Thus, the data are consistent with the interpretation that a relatively small decrease in the abundance of cyclin D-Cdk4 complexes makes sufficient p27 available for cyclin E-Cdk2 binding to account for its redistribution following progestin treatment, and conversely, induction of cyclin D1 sequesters p27 in cyclin D1-Cdk4 complexes, favoring the formation of the more active, p27-depleted form of cyclin E-Cdk2. Thus, increased p27 association with cyclin E-Cdk2 following progestin treatment may be a consequence of the decreased abundances of cyclins D1 and D3. In contrast with p27, the lower relative affinity of p21 for cyclin E-Cdk2 suggests that upon reduction in cyclin D-Cdk4 complex numbers, it would compete poorly with p27 for the available cyclin E-Cdk2 and in consequence associate with other cyclin-CDK complexes, perhaps cyclin A-Cdk2, for which it displays an affinity similar to its affinity for cyclin D2-Cdk4 (18). This suggestion is consistent with observations that following progestin treatment, an increased proportion of p21 was present in the fractions containing cyclin A-Cdk2 (∼300 kDa) and that Cdk2 immunoprecipitates contained increased amounts of p21 (data not shown).
While it is likely that the composition of cyclin-CDK complexes is largely determined by the relative abundances and affinities of the cyclins, CDKs, and CDK inhibitors involved, it is also possible that interactions with other intracellular proteins contribute to the availability or association of these molecules. For example, a Myc-regulated heat-labile inhibitor of p21 has been described (20). By an apparently similar mechanism, in cells which express abundant p27 but little p21, Myc induction inhibits the ability of p27 to bind cyclin E-Cdk2, most likely by causing the p27 to be sequestered (34, 41, 62). While cyclin D-Cdk4 or cyclin D-Cdk6 complexes provide an attractive possibility for the sequestration of p27 or p21, increased association between these proteins and p27 was not observed following c-Myc induction (62). Similarly, data from this laboratory suggest that following c-Myc induction in human breast cancer cells, p21 and p27 are not sequestered by cyclin D-Cdk4 (43). Recent data suggest that the sequestered p27 is targeted for degradation, raising the possibility that the sequestering proteins are involved in the degradation process (34). It is possible that the decrease in c-myc expression following progestin treatment results in decreased expression of p21- and p27-sequestering molecules, contributing to increased availability of p21 and p27 for cyclin-CDK binding. However, the ability of cyclin D1 induction to restore CDK activity and cell cycle progression in progestin-pretreated cells suggests a more major role for cyclin expression than for c-myc expression in the redistribution of cyclin E-Cdk2 complexes.
Decreased CDK activity preceded inhibition of entry into S phase and thus provides a mechanism for inhibition of proliferation by progestins. However, no decreases in cyclin expression or CDK activity were apparent after less than 12 h of progestin treatment, indicating that while these are unlikely to be consequences of inhibition of proliferation, they are likely to be indirect effects of progestin action. Cell cycle kinetic studies of progestin inhibition in T-47D cells led to the postulation of a delay of at least 5 h before the processes resulting in progestin inhibition began to take effect (55), consistent with the relatively late effects on cyclin expression and CDK activity. This delay does not result from a delay in progestin activation of its receptor, since some rapid progestin responses are apparent in these cells (for example, see reference 38). Rather, it may be that progestin inhibition of proliferation occurs as an indirect consequence of progestin action. The possibility that this may be due to induction of TGF-β is unlikely, since there is no correlation between growth inhibition by progestins and production of or sensitivity to TGF-β (28). Alternatively, it is possible that decreased c-myc expression, which precedes decreased cyclin expression by 6 h or more, may be involved in the later decreases in cyclin expression and cell cycle progression. However, targeted disruption of one c-myc allele in a fibroblast cell line delayed induction of cyclin E but not cyclin D1 or cyclin D3 following mitogen stimulation (16), suggesting that decreased c-myc expression is at most only a partial explanation for the later decrease in cyclin expression. An additional possibility suggested by the physiological role of progesterone is that the growth inhibition is the indirect result of induction of a differentiation program. Further dissection of the mechanisms underlying progestin inhibition of cell proliferation will involve defining the basis for their delayed action on CDK activity and the relative contributions of inhibition of Cdk4 and cyclin E-Cdk2 to the overall effect.
ACKNOWLEDGMENTS
This research was supported by research grants from the National Health and Medical Research Council of Australia and the New South Wales State Cancer Council.
We thank Owen Prall for advice on gel filtration and kinase assays, generous supply of reagents, and valuable discussions during the course of these experiments.
ADDENDUM IN PROOF
While this paper was under review, another study addressing the mechanisms for progestin regulation of proliferation was published (S. D. Groshong, G. I. Owen, B. Grimison, I. E. Schauer, M. C. Todd, T. A. Langan, R. A. Sclafani, C. A. Lange, and K. B. Horwitz, Mol. Endocrinol. 11:1593–1607, 1997). This independent study provides data on regulation of cyclin and CDK inhibitor expression and pRB phosphorylation essentially in agreement with those presented here (Fig. 2, 4, and 5).
REFERENCES
- 1.Altucci L, Addeo R, Cicatiello L, Davois S, Parker M G, Truss M, Beato M, Sica V, Bresciani F, Weisz A. 17β-Estradiol induces cyclin D1 gene transcription, p36D1-p34cdk4 complex activation and p105RB phosphorylation during mitogenic stimulation of G1-arrested human breast cancer cells. Oncogene. 1996;12:2315–2324. [PubMed] [Google Scholar]
- 2.Aprelikova O, Xiong Y, Liu E T. Both p16 and p21 families of cyclin-dependent kinase (CDK) inhibitors block the phosphorylation of cyclin-dependent kinases by the CDK-activating kinase. J Biol Chem. 1995;270:18195–18197. doi: 10.1074/jbc.270.31.18195. [DOI] [PubMed] [Google Scholar]
- 3.Blain S W, Montalvo E, Massagué J. Differential interaction of the cyclin-dependent kinase (Cdk) inhibitor p27Kip1 with cyclin A-Cdk2 and cyclin D2-Cdk4. J Biol Chem. 1997;272:25863–25872. doi: 10.1074/jbc.272.41.25863. [DOI] [PubMed] [Google Scholar]
- 4.Buckley M F, Sweeney K J E, Hamilton J A, Sini R L, Manning D L, Nicholson R I, deFazio A, Watts C K W, Musgrove E A, Sutherland R L. Expression and amplification of cyclin genes in human breast cancer. Oncogene. 1993;8:2127–2133. [PubMed] [Google Scholar]
- 5.Clarke C L, Sutherland R L. Progestin regulation of cellular proliferation. Endocr Rev. 1990;11:266–302. doi: 10.1210/edrv-11-2-266. [DOI] [PubMed] [Google Scholar]
- 6.Colditz G A, Hankinson S E, Hunter D J, Willett W C, Manson J E, Stampfer M J, Hennekens C, Rosner B, Speizer F E. The use of estrogens and progestins and the risk of breast cancer in postmenopausal women. N Engl J Med. 1995;332:1589–1593. doi: 10.1056/NEJM199506153322401. [DOI] [PubMed] [Google Scholar]
- 7.Collaborative Group on Hormonal Factors in Breast Cancer. Breast cancer and hormonal contraceptives: collaborative reanalysis of individual data on 53 297 women with breast cancer and 100 239 women without breast cancer from 54 epidemiological studies. Lancet. 1996;347:1713–1727. doi: 10.1016/s0140-6736(96)90806-5. [DOI] [PubMed] [Google Scholar]
- 8.Collaborative Group on Hormonal Factors in Breast Cancer. Breast cancer and hormone replacement therapy: collaborative reanalysis of data from 51 epidemiological studies of 52 705 women with breast cancer and 108 411 women without breast cancer. Lancet. 1997;350:1047–1059. [PubMed] [Google Scholar]
- 9.Coppola J A, Lewis B A, Cole M D. Increased retinoblastoma gene expression is associated with late stages of differentiation in many different cell types. Oncogene. 1990;5:1731–1733. [PubMed] [Google Scholar]
- 10.Dubik D, Dembinski T C, Shiu R P C. Stimulation of c-myc oncogene expression associated with estrogen-induced proliferation of human breast cancer cells. Cancer Res. 1987;47:6517–6521. [PubMed] [Google Scholar]
- 11.Foster J S, Wimalasena J. Estrogen regulated activity of cyclin-dependent kinases and retinoblastoma protein phosphorylation in breast cancer cells. Mol Endocrinol. 1996;10:488–498. doi: 10.1210/mend.10.5.8732680. [DOI] [PubMed] [Google Scholar]
- 12.Fotedar R, Fitzgerald P, Rouselle T, Cannella D, Dorée M, Messier H, Fotedar A. p21 contains independent binding sites for cyclin and cdk2: both sites are required to inhibit cdk2 kinase activity. Oncogene. 1996;12:2155–2164. [PubMed] [Google Scholar]
- 13.Gabrielli B G, De Souza C P C, Tonks I D, Clark J M, Hayward N K, Ellem K A O. Cytoplasmic accumulation of cdc25B phosphatase in mitosis triggers centrosomal microtubule nucleation in HeLa cells. J Cell Sci. 1996;109:1081–1093. doi: 10.1242/jcs.109.5.1081. [DOI] [PubMed] [Google Scholar]
- 14.Galaktionov K, Chen X, Beach D. Cdc25 cell-cycle phosphatase as a target of c-myc. Nature. 1996;382:511–517. doi: 10.1038/382511a0. [DOI] [PubMed] [Google Scholar]
- 15.Gu Y, Rosenblatt J, Morgan D O. Cell cycle regulation of CDK2 activity by phosphorylation of threonine 160 and tyrosine 15. EMBO J. 1992;11:3995–4005. doi: 10.1002/j.1460-2075.1992.tb05493.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hanson K D, Shichiri M, Follansbee M R, Sedivy J M. Effects of c-myc expression on cell cycle progression. Mol Cell Biol. 1994;14:5748–5755. doi: 10.1128/mcb.14.9.5748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harlow E, Lane D. Antibodies: a laboratory manual. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory; 1988. [Google Scholar]
- 18.Harper J W, Elledge S J, Keyomarsi K, Dynlacht B, Tsai L-H, Zhang P, Dobrowlski S, Bai C, Connell-Crowley L, Swindell E, Fox M P, Wei N. Inhibition of cyclin-dependent kinases by p21. Mol Biol Cell. 1995;6:387–400. doi: 10.1091/mbc.6.4.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hatakeyama M, Brill J A, Fink G R, Weinberg R A. Collaboration of G1 cyclins in the functional inactivation of the retinoblastoma protein. Genes Dev. 1994;8:1759–1771. doi: 10.1101/gad.8.15.1759. [DOI] [PubMed] [Google Scholar]
- 20.Hermeking H, Funk J O, Reichert M, Ellwart J W, Eick D. Abrogation of p53-induced cell cycle arrest by c-Myc: evidence for an inhibitor of p21WAF1/CIP1/SDI1. Oncogene. 1995;11:1409–1415. [PubMed] [Google Scholar]
- 21.Hirai H, Roussel M F, Kato J-Y, Ashmun R A, Sherr C J. Novel INK4 proteins, p19 and p18, are specific inhibitors of the cyclin D-dependent kinases CDK4 and CDK6. Mol Cell Biol. 1995;15:2672–2681. doi: 10.1128/mcb.15.5.2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hoffman I, Draetta G, Karsenti E. Activation of the phosphatase activity of human cdc25A by a cdk2-cyclin E dependent phosphorylation at the G1/S transition. EMBO J. 1994;13:4302–4310. doi: 10.1002/j.1460-2075.1994.tb06750.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Horwitz K B, Freidenberg G R. Growth inhibition and increase of insulin receptors in antiestrogen-resistant T47Dco human breast cancer cells by progestins: implications for endocrine therapies. Cancer Res. 1985;45:167–173. [PubMed] [Google Scholar]
- 24.Hunter T, Pines J. Cyclins and cancer. II. Cyclin D and CDK inhibitors come of age. Cell. 1994;79:573–582. doi: 10.1016/0092-8674(94)90543-6. [DOI] [PubMed] [Google Scholar]
- 25.Iacobelli S, Sica G, Natoli C, Gatti D. Inhibitory effects of medroxyprogesterone acetate on the proliferation of human breast cancer cells. In: Campio L, Robustelli della Cuna G, Taylor R W, editors. Role of medroxyprogesterone in endocrine-related tumors. New York, N.Y: Raven Press; 1983. pp. 1–6. [Google Scholar]
- 26.Iavarone A, Massagué J. Repression of the CDK activator Cdc25A and cell-cycle arrest by cytokine TGF-β in cells lacking the CDK inhibitor p15. Nature. 1997;387:417–422. doi: 10.1038/387417a0. [DOI] [PubMed] [Google Scholar]
- 27.Jinno S, Suto K, Nagata A, Igarashi M, Kanaoka Y, Nojima H, Okayama H. Cdc25A is a novel phosphatase functioning early in the cell cycle. EMBO J. 1994;13:1549–1556. doi: 10.1002/j.1460-2075.1994.tb06417.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kalkhoven E, Kwakkenbos-Isbrücker L, Mummery C L, de Laat S W, van den Eijnen-van Raij A J M, van der Saag P T, van der Burg B. The role of TGF-β production in growth inhibition of breast-tumor cells by progestins. Int J Cancer. 1995;61:80–86. doi: 10.1002/ijc.2910610114. [DOI] [PubMed] [Google Scholar]
- 29.Kitagawa M, Higashi H, Jung H-K, Suzuki-Takahashi I, Ikeda M, Tamai K, Kato J-Y, Segawa K, Yoshida E, Nishimura S, Taya Y. The consensus motif for phosphorylation by cyclin D1-Cdk4 is different from that for phosphorylation by cyclin A/E-Cdk2. EMBO J. 1996;15:7060–7069. [PMC free article] [PubMed] [Google Scholar]
- 30.LaBauer J, Garrett M D, Stevenson L F, Slingerland J M, Sandhu C, Chou H S, Fattaey A, Harlow E. New functional activities for the p21 family of CDK inhibitors. Genes Dev. 1997;11:847–862. doi: 10.1101/gad.11.7.847. [DOI] [PubMed] [Google Scholar]
- 31.Millar J B, Blevitt J, Gerace L, Sadhu K, Featherstone C, Russell P. p55CDC25 is a nuclear protein required for the initiation of mitosis in human cells. Proc Natl Acad Sci USA. 1991;88:10500–10504. doi: 10.1073/pnas.88.23.10500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morgan D O. Principles of CDK regulation. Nature. 1995;374:131–134. doi: 10.1038/374131a0. [DOI] [PubMed] [Google Scholar]
- 33.Morgan D O. The dynamics of cyclin dependent kinase structure. Curr Opin Cell Biol. 1996;8:767–772. doi: 10.1016/s0955-0674(96)80076-7. [DOI] [PubMed] [Google Scholar]
- 34.Müller D, Bouchard C, Rudolph B, Steiner P, Stuckmann I, Saffrich R, Ansorge W, Huttner W, Eilers M. Cdk2-dependent phosphorylation of p27 facilitates its Myc-induced release from cyclin E/cdk2 complexes. Oncogene. 1997;15:2561–2576. doi: 10.1038/sj.onc.1201440. [DOI] [PubMed] [Google Scholar]
- 35.Musgrove E A, Hamilton J A, Lee C S L, Sweeney K J E, Watts C K W, Sutherland R L. Growth factor, steroid, and steroid antagonist regulation of cyclin gene expression associated with changes in T-47D human breast cancer cell cycle progression. Mol Cell Biol. 1993;13:3577–3587. doi: 10.1128/mcb.13.6.3577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Musgrove E A, Lee C S L, Buckley M F, Sutherland R L. Cyclin D1 induction in breast cancer cells shortens G1 and is sufficient for cells arrested in G1 to complete the cell cycle. Proc Natl Acad Sci USA. 1994;91:8022–8026. doi: 10.1073/pnas.91.17.8022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Musgrove E A, Lee C S L, Cornish A L, Swarbrick A, Sutherland R L. Antiprogestin inhibition of cell cycle progression in T-47D breast cancer cells is accompanied by induction of the CDK inhibitor p21. Mol Endocrinol. 1997;11:54–66. doi: 10.1210/mend.11.1.9869. [DOI] [PubMed] [Google Scholar]
- 38.Musgrove E A, Lee C S L, Sutherland R L. Progestins both stimulate and inhibit breast cancer cell cycle progression while increasing expression of transforming growth factor α, epidermal growth factor receptor, c-fos, and c-myc genes. Mol Cell Biol. 1991;11:5032–5043. doi: 10.1128/mcb.11.10.5032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Musgrove E A, Sarcevic B, Sutherland R L. Inducible expression of cyclin D1 in T-47D human breast cancer cells is sufficient for CDK2 activation and pRB hyperphosphorylation. J Cell Biochem. 1996;60:362–377. doi: 10.1002/(SICI)1097-4644(19960301)60:3%3C363::AID-JCB8%3E3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 40.Musgrove E A, Sutherland R L. Cell cycle control by steroid hormones. Semin Cancer Biol. 1994;5:381–389. [PubMed] [Google Scholar]
- 41.Pérez-Roger I, Solomon D L C, Sewing A, Land H. Myc activation of cyclin E/Cdk2 kinase involves induction of cyclin E gene transcription and inhibition of p27Kip1 binding to newly formed complexes. Oncogene. 1997;14:2373–2381. doi: 10.1038/sj.onc.1201197. [DOI] [PubMed] [Google Scholar]
- 42.Poon R Y C, Toyoshima H, Hunter T. Redistribution of the CDK inhibitor p27 between different cyclin. CDK complexes in the mouse fibroblast cell cycle and in cells arrested with lovastatin or ultraviolet irradiation. Mol Biol Cell. 1995;6:1197–1213. doi: 10.1091/mbc.6.9.1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Prall, O. W. J., E. M. Rogan, E. A. Musgrove, C. K. W. Watts, and R. L. Sutherland. Submitted for publication.
- 44.Prall O W J, Sarcevic B, Musgrove E A, Watts C K W, Sutherland R L. Estrogen-induced activation of Cdk4 and Cdk2 during G1-S phase progression is accompanied by increased cyclin D1 expression and decreased cyclin-dependent kinase inhibitor association with cyclin E-Cdk2. J Biol Chem. 1997;272:10882–10894. doi: 10.1074/jbc.272.16.10882. [DOI] [PubMed] [Google Scholar]
- 45.Reynisdóttir I, Massagué J. The subcellular locations of p15Ink4b and p27Kip1 coordinate their inhibitory interactions with cdk4 and cdk2. Genes Dev. 1997;11:492–503. doi: 10.1101/gad.11.4.492. [DOI] [PubMed] [Google Scholar]
- 46.Rose P G. Endometrial cancer. N Engl J Med. 1996;335:640–649. doi: 10.1056/NEJM199608293350907. [DOI] [PubMed] [Google Scholar]
- 47.Russo A A, Jeffrey P D, Patten A K, Massagué J, Pavletich N. Crystal structure of the p27Kip1 cyclin-dependent-kinase inhibitor bound to the cyclin A-Cdk2 complex. Nature. 1996;382:325–331. doi: 10.1038/382325a0. [DOI] [PubMed] [Google Scholar]
- 48.Saha P, Eichbaum Q, Silberman E D, Mayer B J, Dutta A. p21CIP1 and Cdc25A: competition between an inhibitor and an activator of cyclin-dependent kinases. Mol Cell Biol. 1997;17:4338–4345. doi: 10.1128/mcb.17.8.4338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Santen R J, Manni A, Harvey H, Redmond C. Endocrine treatment of breast cancer in women. Endocr Rev. 1990;11:221–265. doi: 10.1210/edrv-11-2-221. [DOI] [PubMed] [Google Scholar]
- 50.Sherr C J. G1 phase progression: cycling on cue. Cell. 1994;79:551–555. doi: 10.1016/0092-8674(94)90540-1. [DOI] [PubMed] [Google Scholar]
- 51.Sherr C J, Roberts J M. Inhibitors of mammalian G1 cyclin-dependent kinases. Genes Dev. 1995;9:1149–1163. doi: 10.1101/gad.9.10.1149. [DOI] [PubMed] [Google Scholar]
- 52.Slack R S, Hamel P A, Bladon T S, Gill R M, McBurney M W. Regulated expression of the retinoblastoma gene in differentiating embryonal carcinoma cells. Oncogene. 1993;8:1585–1591. [PubMed] [Google Scholar]
- 53.Soos T J, Kiyokawa H, Yan J S, Rubin M S, Giordano A, DeBlasio A, Bottega S, Wong B, Mendelsohn J, Koff A. Formation of p27-CDK complexes during the human mitotic cell cycle. Cell Growth Differ. 1996;7:135–146. [PubMed] [Google Scholar]
- 54.Steiner P, Philipp A, Lukas J, Godden-Kent D, Pagano M, Mittnacht S, Bartek J, Eilers M. Identification of a Myc-dependent step during the formation of active G1 cyclin-cdk complexes. EMBO J. 1995;14:4814–4826. doi: 10.1002/j.1460-2075.1995.tb00163.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sutherland R L, Hall R E, Pang G Y N, Musgrove E A, Clarke C L. Effect of medroxyprogesterone acetate on proliferation and cell cycle kinetics of human mammary carcinoma cells. Cancer Res. 1988;48:5084–5091. [PubMed] [Google Scholar]
- 56.Sweeney, K. J., A. Swarbrick, R. L. Sutherland, and E. A. Musgrove. Lack of relationship between CDK activity and G1 cyclin expression in breast cancer cells. Oncogene, in press. [DOI] [PubMed]
- 57.Szekely L, Jiang W Q, Bulic-Jakus F, Rosen A, Ringertz N, Klein G, Wiman K G. Cell type and differentiation dependent heterogeneity in retinoblastoma protein expression in SCID mouse fetuses. Cell Growth Differ. 1992;3:149–156. [PubMed] [Google Scholar]
- 58.Terada Y, Tatsuka M, Jinno S, Okayama H. Requirement for tyrosine phosphorylation of Cdk4 in G1 arrest induced by ultraviolet irradiation. Nature. 1995;376:358–362. doi: 10.1038/376358a0. [DOI] [PubMed] [Google Scholar]
- 59.Tiefenbrun N, Melamed D, Levy N, Resnitzky D, Hoffman I, Reed S I, Kimchi A. Alpha interferon suppresses the cyclin D3 and cdc25A genes, leading to a reversible G0-like arrest. Mol Cell Biol. 1996;16:3934–3944. doi: 10.1128/mcb.16.7.3934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Veronesi U, Goldhirsch A, Yarnold J. Breast cancer. In: Peckham M, Pinedo H M, Veronesi U, editors. Oxford textbook of oncology. Oxford, United Kingdom: Oxford University Press; 1995. pp. 1243–1289. [Google Scholar]
- 61.Vignon F, Bardon S, Chalbos D, Rochefort H. Antiestrogenic effect of R5020, a synthetic progestin in human breast cancer cells in culture. J Clin Endocrinol Metab. 1983;56:1124–1130. doi: 10.1210/jcem-56-6-1124. [DOI] [PubMed] [Google Scholar]
- 62.Vlach J, Hennecke S, Alevizopoulos K, Conti D, Amati B. Growth arrest by the cyclin-dependent kinase inhibitor p27Kip1 is abrogated by c-Myc. EMBO J. 1996;15:6595–6604. [PMC free article] [PubMed] [Google Scholar]
- 63.Wang J Y J. Retinoblastoma protein in growth suppression and death protection. Curr Opin Genet Dev. 1997;7:39–45. doi: 10.1016/s0959-437x(97)80107-4. [DOI] [PubMed] [Google Scholar]
- 64.Watts C K W, Brady A, Sarcevic B, deFazio A, Musgrove E A, Sutherland R L. Antiestrogen inhibition of cell cycle progression in breast cancer cells is associated with inhibition of cyclin-dependent kinase activity and decreased retinoblastoma protein phosphorylation. Mol Endocrinol. 1995;9:1804–1813. doi: 10.1210/mend.9.12.8614416. [DOI] [PubMed] [Google Scholar]
- 65.Wong M S J, Murphy L C. Differential regulation of c-myc by progestins and antiestrogens in T-47D human breast cancer cells. J Steroid Biochem Mol Biol. 1991;39:39–44. doi: 10.1016/0960-0760(91)90010-3. [DOI] [PubMed] [Google Scholar]
- 66.Zhang H, Hannon G J, Beach D. p21-containing cyclin kinases exist in both active and inactive states. Genes Dev. 1994;8:1750–1758. doi: 10.1101/gad.8.15.1750. [DOI] [PubMed] [Google Scholar]