

Abstract
RAD52 and RAD9 are required for the repair of double-strand breaks (DSBs) induced by physical and chemical DNA-damaging agents in Saccharomyces cerevisiae. Analysis of EcoRI endonuclease expression in vivo revealed that, in contrast to DSBs containing damaged or modified termini, chromosomal DSBs retaining complementary ends could be repaired in rad52 mutants and in G1-phase Rad+ cells. Continuous EcoRI-induced scission of chromosomal DNA blocked the growth of rad52 mutants, with most cells arrested in G2 phase. Surprisingly, rad52 mutants were not more sensitive to EcoRI-induced cell killing than wild-type strains. In contrast, endonuclease expression was lethal in cells deficient in Ku-mediated end joining. Checkpoint-defective rad9 mutants did not arrest cell cycling and lost viability rapidly when EcoRI was expressed. Synthesis of the endonuclease produced extensive breakage of nuclear DNA and stimulated interchromosomal recombination. These results and those of additional experiments indicate that cohesive ended DSBs in chromosomal DNA can be accurately repaired by RAD52-mediated recombination and by recombination-independent complementary end joining in yeast cells.
DNA double-strand breaks (DSBs) occur spontaneously, after exposure to ionizing radiation or various chemical clastogens, and as part of normal cellular development. Developmental processes which are initiated by enzymatically induced DSBs include V(D)J recombination in mammalian cells and mating-type switching, intron homing, and meiotic recombination in yeast cells (17, 25, 37, 56, 58, 61). Repair of DSBs in eukaryotic DNA involves complex interactions between proteins associated with chromatin structure, enzymatic repair of broken DNA ends, and cell cycling. In mammalian cells, the primary mechanism of repair of DSBs is through nonhomologous end-joining processes (25, 61, 70). In Saccharomyces cerevisiae, the predominant pathway of DSB repair is through homologous recombination, although other, less common pathways have been described (discussed below). Models for the recombinational repair of DSBs in yeast DNA which involve pairing of broken DNA ends with complementary strands of an unbroken homolog have been proposed (63, 75, 77; see references in reference 56).
Recombinational repair of damage-induced DSBs occurring during mitotic growth of yeast cells requires genes within the RAD52 epistasis group, which includes RAD50-59, MRE11, and XRS2 (2, 16, 19, 56, 65). RAD52 group mutants are hypersensitive to ionizing radiation and methyl methanesulfonate but display only a slight sensitivity to UV light. Physical analysis of DNA from rad51, rad52, and rad54 mutants demonstrated that these strains are specifically deficient in the rejoining of radiation-induced DSBs (15, 16, 64). Recent studies have suggested that Rad51, Rad52, Rad54, Rad55, and Rad57 associate to form a multiprotein complex (2, 20, 70). Furthermore, both Rad52 and Rad51, which are conserved in lower and higher eukaryotes, display intrinsic strand-annealing activity (49, 70). Although RAD52 group-mediated homologous recombination is the primary pathway of repair of DSBs in yeast cells, additional pathways have been described. These include nonconservative repair processes which occur in DNA containing direct repeats (e.g., single-strand annealing [30, 56]) and nonrecombinational end-joining pathways, which may be either precise or error prone (9, 13, 39, 44, 45, 47, 68, 72, 78). Genes which have been associated with end-joining pathways include HDF1/YKU70, HDF2/YKU80, RAD50, XRS2, and MRE11. End-joining mechanisms in yeast share several characteristics of DSB repair observed in higher eukaryotes and involve two genes (HDF1/YKU70 and HDF2/YKU80) which are homologs of genes required for DSB repair and V(D)J recombination in mammalian cells (Ku70 and Ku80 [25]). Recent experiments have established that the yeast Ku70 homolog is also essential for maintenance of normal telomere lengths (59).
Eukaryotic cells possess checkpoint mechanisms which interrupt cell cycling when chromosomal DNA is damaged. This arrest ensures that DNA repair is completed before downstream cycling events are initiated. Exposure of yeast cells to ionizing radiation or methyl methanesulfonate causes growing cells to arrest in G2. Arrested cells retain large buds and a single undivided nucleus (55, 71, 80, 82). Initial experiments established that rad9 strains were deficient in DNA damage-induced cell cycle arrest (80), and additional genes were subsequently identified (71). Direct evidence that DSBs can initiate the arrest of yeast cells has been obtained from analysis of the consequences of HO endonuclease expression (described below). DSB-induced cell cycle arrest has also been observed in mammalian cells and is dependent upon p53 (50). Recent studies have demonstrated that additional damage-responsive checkpoints in G1 and S phase are present in yeast cells (71).
Analysis of DSB repair in yeast has been facilitated by the development of systems for studying the genetic and cytological consequences of a single DSB induced by HO endonuclease. This enzyme stimulates mating-type switching, a gene conversion process, by producing a single DSB at the MAT locus (17, 26, 51). The ends of breaks produced by HO retain complementary 3′ extensions which are 4 nucleotides in length (51). Several groups have used plasmid and chromosomal fusions containing the HO gene under the control of a galactose-regulated promoter to examine effects of DSBs on viability, chromosome metabolism, and cell division. Processing of breaks occurring at MAT has been studied (see, e.g., references 12, 26, and 47), as well as at synthetic HO target sites located on other chromosomes (see, e.g., references 6, 7, 43, 52, 62, and 66). In addition, the consequences of HO expression in radiation-sensitive mutants have been investigated (14, 18, 28, 40, 42, 74, 79). These experiments revealed that HO-induced strand breaks have several of the characteristics of damage produced by ionizing radiation treatment of cells. Such DSBs are recombinogenic, increase aneuploidy and mutagenesis, can arrest cell cycling in G2, and inhibit the growth of radiation-sensitive RAD52 group mutants.
High-level expression of two other endonucleases, the restriction enzyme EcoRI and mitochondrial endonuclease I-SceI, has also been examined in yeast cells. Expression of EcoRI in vivo resulted in the formation of DSBs at its target sequence (G∧AATTC) and inhibition of the growth of Rad+ and rad52 cells (3). A single DSB produced by I-SceI efficiently induced homologous recombination in a plasmid containing its recognition sequence (58).
While investigating the genetic consequences of enzymatically induced DSBs containing cohesive ends (EcoRI and HO) or blunt ends (PvuII), we confirmed that expression of EcoRI inhibits the growth of rad52 cells. However, most Rad+ strains which were tested grew after the induction of plasmid or chromosomal GAL1::EcoRI fusions. EcoRI-induced DSBs inhibited the growth of rad52 strains and arrested cell division in the G2 phase. Remarkably, rad52 mutants were not more sensitive to killing than wild-type cells. Thus, RAD52 was required for the growth of cells continuously expressing the endonuclease but not for precise repair of induced DSBs after EcoRI synthesis was repressed. In contrast, strains deficient in Ku70-mediated end joining or RAD9-dependent cell cycle checkpoint responses were hypersensitive to killing by EcoRI.
MATERIALS AND METHODS
Yeast strains and plasmids.
Strain designations, genotypes, and sources of the yeast strains used are listed in Table 1. The reg1-501 mutant strain T334, used for most of the experiments described here, is a derivative of 334 (24) in which the TRP1 gene has been deleted. The GAL1::EcoRI cassette was integrated by PCR fragment-mediated gene disruption (5). Briefly, primers containing 50 bases of HIS3 or LYS2 gene sequence and 26 bases corresponding to the vector pRS314 (73) were used to amplify sequences within pLKL31. pLKL31 was constructed by inserting a GAL1::EcoRI cassette into pRS314 (TRP1) between the EcoRI and BamHI sites within this vector. Fragments generated with primer pairs prslysA and prslysC or prshisA and prshisC were transformed into T334 to create strains YLKL340 [Δlys2::(GAL1::EcoRI TRP1)] and YLKL350 [Δhis3::(GAL1::EcoRI TRP1)]. EcoRI synthesis in these strains is regulated by GAL1, but transcription of the associated TRP1 gene is controlled by its natural promoter. All the oligonucleotides were obtained from BioServe Biotechnologies. The plasmid p316Gal was created by inserting a GAL1-10 promoter fragment (27) into the EcoRI and BamHI sites of pRS316. Plasmid pGALHO has been described previously (22), and YCpGal:Rlb (URA3) was a gift from J. Rine (3). Disruption/deletion of RAD52 was accomplished with pΔ52Blast (Δrad52::hisG-URA3-hisG) or pΔ52Leu (Δrad52::LEU2), provided by Ed Perkins. The RAD9 gene was deleted with pRR330 (67), and Δhdf1::HIS3 strains were created by PCR as described previously (5).
TABLE 1.
Yeast strains used in this study
| Strain | Genotype | Source or reference |
|---|---|---|
| T334 | MATα ura3-52 leu2-3,112 Δtrp1::hisG reg1-501 gal1 pep4-3 prb1-1122 | E. Perkins |
| T334ΔR52T | T334, Δrad52::TRP1 | E. Perkins |
| YLKL292 | T334, Δrad9::hisG | This study |
| YLKL340 | T334, Δlys2::(GAL1::EcoRI TRP1) Δhis3::hisG | This study |
| YLKL341 | YLKL340, Δrad52::LEU2 | This study |
| YLKL355 | YLKL340, Δrad9::hisG-URA3-hisG | This study |
| YLKL350 | T334, Δhis3::(GAL1::EcoRI TRP1) | This study |
| YLKL351 | YLKL350, Δrad52::LEU2 | This study |
| YLKL366 | YLKL350, Δrad9::hisG-URA3-hisG | This study |
| YLKL389 | YLKL350, Δhdf1::HIS3 | This study |
| RM10-32D | MATa ura3-1 leu1-c trp5-c lys2-2 tyr1-2 his7-1 ade2-1 ade6 met13c cyh2r | 41 |
| RM26-26C | MATα ura3-1 leu1-12 trp5-2 lys2-1 his1 ade2-1 ade5 canr | 41 |
| EPY214-1B | MATa ura3-52 leu2-Δ1 Δtrp1::hisG his1-7 lys2 | 32 |
| MAR1530 | MATα ura3-52 leu2-Δ1 ilv1-92 cha1 | 60 |
| VL6α | MATα ura3-52 trp1-Δ63 lys2 his3 met14 ade1 | 33 |
| SSL231 | MATα ura3-52 leu2 trp1 ade2-101 can1::BYA112 (his3-Δ200) | 28 |
| LS20 | matΔ his3 ade2 can1 trp1 ura3 leu2 lys5 cyhrade3::GALHO | 66 |
| GRY1060 | matα-inc::(trp1-089 his3-621) ura3-52 trp1-Δ1 his3-Δ200 tyr7-1 (leu2-Δ1) | 43 |
| GRY1078 | mata-inc::(trp1-488 his3-192) ura3-52 trp1-Δ1 his3-Δ200 leu2-Δ1 ade2-101 lys2-801 cry1 | 43 |
| S1InsE4A | MATα ura3-52 leu2-3,112 trp1-289 his7-2 ade5-1 lys2::lnsE-4A | 48 |
| BGW1-7a | MATa ura3-52 leu2-3,112 his4-519 ade1-100 | 81 |
| tNR85T1 | mat::LEU2 hmr-Δ3 ura3-52 trp1 leu2 mal2 thr4 | 6 |
Yeast growth media.
High-purity d-(+)-galactose from Pfanstiehl (no. G106) was used for experiments in which galactose was needed as the sole carbon source. For growth of reg1-501 strains in glucose-plus-galactose media, 98% pure galactose (no. G105) was used. Synthetic and YP media were prepared as described previously (69).
Growth curves, plating efficiencies, and cell cycle analysis.
The growth of various published haploid strains containing YCpGal:Rlb or the control vector pRS316 was analyzed by aliquoting or replica plating strains grown in synthetic 2% glucose medium without uracil (Glu − Ura) to 2% galactose-minus-uracil plates (Gal − Ura). The plating efficiencies of plasmid-containing T334 cells were assessed after log-phase cultures were spread to Glu − Ura plates or Glu − Ura plates supplemented with 2% galactose (Gal + Glu − Ura). Strains containing integrated (GAL1::EcoRI TRP1) constructions were spread to YPD and YPD+2%Gal plates. Gal+ colonies observed on six plates were counted after 3 days, and the results were averaged. Plating efficiency is defined here as the viable cell titer calculated from observed CFU on plates divided by the cell titer determined by hemacytometer counts.
For growth curves and viability tests, plasmid-containing T334 cells (including Δrad52 and Δrad9 derivatives) were grown to a density of approximately 7 × 106 to 10 × 106 cells per ml in Glu − Ura and shifted to 2% Gal + Glu − Ura at ∼5 × 105/ml. All the cultures (5 ml) were shaken vigorously at 30°C. At each time point, aliquots of cells were sonicated, counted with a hemacytometer, diluted, and spread to glucose-complete (Glu-Com) and Glu − Ura plates. Three or four independent cultures were assayed for each strain. Plasmid loss was analyzed by comparing plating efficiency on Glu − Ura plates to that on Glu-Com plates. Strains containing integrated GAL1::EcoRI cassettes were grown to 1 × 107 to 5 × 107/ml in YPD and split into separate YPD or YPD + 2% Gal cultures at 2 × 105 to 3 × 105/ml. Aliquots were subsequently sonicated, counted, and spread to YPD plates, and the results were averaged. The fraction of cells which were unbudded, small-budded, or large-budded was analyzed with a hemacytometer after sonication. A total of 100 to 300 cells were counted at each time point, and large-budded cells were defined as those in which the bud was >50% of the size of the mother cell.
Purification and analysis of yeast genomic DNA.
DNA was purified from cells containing plasmid and chromosomal GAL1::EcoRI fusions by three distinct methods. DNA prepared from galactose-induced cultures by alkali lysis (34), by lysis in the presence of Triton X-100, sodium dodecyl sulfate, and phenol-chloroform (23), or after vortexing with glass beads in the presence of high levels of EDTA (see below) exhibited EcoRI-specific chromosomal DNA repeat bands on agarose gels (described in reference 57). Using a fourth purification protocol, Barnes and Rine (3) also observed extensive cleavage of chromosomal DNA when EcoRI was expressed in vivo. The analysis presented in Fig. 8 used DNA prepared from wild-type and rad52 cells by a procedure which requires only a few minutes of extraction in high-EDTA buffer. Between 25 and 200 ml of cultured cells was precipitated and resuspended in 400 μl of 10 mM Tris (pH 8.0)–100 mM EDTA. Approximately 400 μl of acid-washed glass beads was added, and the tube was vortexed hard for 90 s and microcentrifuged for 1 min at room temperature. The resulting supernatant was recentrifuged for 4 min and transferred, and 200 μl of 10 mM Tris (pH 8.0)–1 mM EDTA (TE) was added. After gentle extraction with phenol, 50:50 phenol-chloroform, and chloroform, the DNA preparation was treated with RNase A, precipitated with isopropanol, washed with 70% ethanol, and resuspended in TE. The DNA concentrations were determined by fluorometry. The purified DNA was heated at 68°C for 5 min and cooled on ice before 0.8 or 1.0 μg was loaded onto 0.6% agarose gels. Varying the initial steps of this procedure by using 50, 100, or 200 mM EDTA, by adding 40 U of commercial EcoRI to the cells before lysis, or by resuspending cells at 4°C rather than at room temperature did not alter the results (data not shown).
FIG. 8.
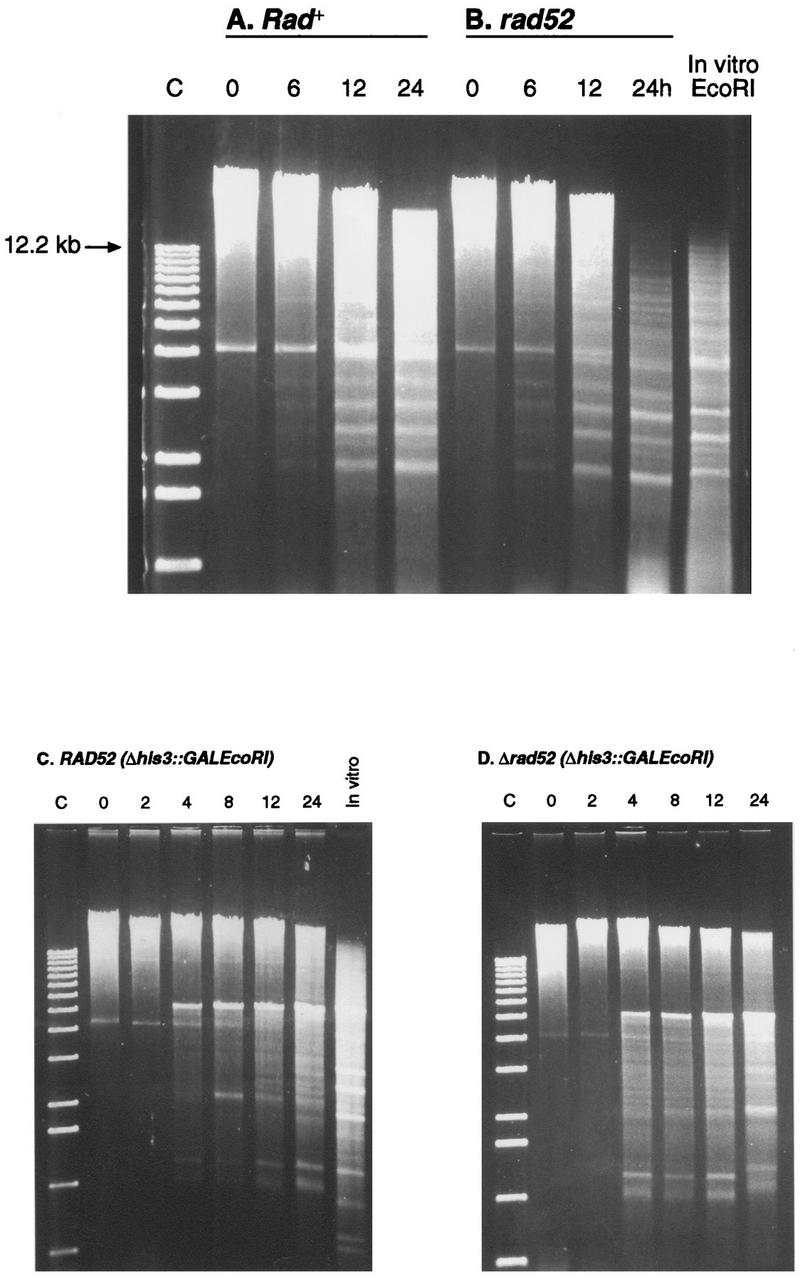
Expression of EcoRI in Rad+ and rad52 cells produces extensive breakage of chromosomal DNA. (A and B) DNA was purified from cells containing YCpGal:RIb at the indicated times after induction in galactose. (C and D) Analysis of DNA purified from cells containing chromosomal Δhis3::(GAL1::EcoRI) fusions (YLKL350 and YLKL351).
EcoRI-induced interchromosomal recombination rates.
Diploid strains created by crossing RM10-32D and RM26-26C (Table 1) containing p316Gal or YCpGal:Rlb were used in the assay. Cells grown in Glu − Ura were spread at low density to Glu − Ura and Gal − Ura plates. After 3 or 4 days of growth at 30°C, 11 colonies from each strain were harvested, diluted, and spread to Glu − Ura plates to determine total cells and to Glu − Leu and Glu − Trp plates to detect LEU1 and TRP5 recombinants.
RESULTS
Growth of rad52 but not Rad+ cells is inhibited by EcoRI.
Expression of EcoRI in vivo was previously shown to inhibit the growth of both Rad+ and rad52 haploid yeast cells (3). These experiments were performed with YCpGal:Rlb (URA3), a centromeric plasmid containing a fusion of the EcoRI R gene with the GAL1 promoter (with no EcoRI sites within the plasmid). During a study of the effects of EcoRI-induced DSBs on chromosome stability, we observed that the growth rates of most Rad+ strains containing YCpGal:Rlb were only slightly reduced on plates containing 2% galactose (Gal − Ura plates). To investigate this discrepancy further, haploid Rad+ strains obtained from several sources were transformed with the GAL1::EcoRI plasmid and with a control vector (pRS316). Transformants of each strain were replica plated as patches or pronged cells from Glu − Ura plates to Gal − Ura plates and incubated at 30°C for 3 days. The strains which were tested (including A364a, S288c, and SK1 strain backgrounds) are listed and referenced in Table 1. Most Rad+ background strains tested (RM10-32D, RM26-26C, EPY214-1B, MAR1530, VL6α, SSL231, LS20, GRY1060, S1InsE4A [CG379], and T334) Table 1) grew at near-normal rates in galactose media. Another strain, GRY1078 (43), grew slowly and only two strains, tNR85T1 and BGW1-7a, were unable to grow when the endonuclease was expressed. tNR85T1 also exhibits other strain-specific phenotypes in response to enzymatically induced DSBs (6) (see Discussion). The growth of rad52 mutants was inhibited by EcoRI expression in all strains examined, although some variation in the degree of inhibition was observed. Thus, the consequences of EcoRI-generated DSBs on the growth of most Rad+ and rad52 strains were comparable to those of a single HO-induced break at MAT (14, 28, 31, 45).
The effects of EcoRI-induced DSBs on cell growth have been examined quantitatively with strain T334. This strain contains the mutant alleles reg1-501, alleviating glucose repression of the GAL1 and GAL10 promoters, and gal1, blocking the metabolism of galactose (24, 53). Addition of galactose to T334 cells cultured in glucose media induces the GAL1 promoter while permitting the cells to continue growth on glucose. T334 also contains mutations within the protease-encoding genes PEP4 and PRB1, potentially increasing the stability of heterologous proteins. The plating efficiencies of wild-type and rad52 cells containing YCpGal:Rlb on synthetic Gal + Glu − Ura and Glu − Ura plates were determined (Table 2). The relative plating efficiency of rad52 strains containing the plasmid (on galactose versus glucose plates) was approximately 600-fold lower than that of wild-type cells expressing the endonuclease. T334 strains containing a chromosomal GAL1::EcoRI cassette at LYS2 (YLKL340) or HIS3 (YLKL350) were also tested. Δrad52::LEU2 derivatives of each strain exhibited poor plating efficiencies on YPD+2%Gal plates (Table 2).
TABLE 2.
Plating efficiencies of cells expressing EcoRI or HOa
| Strain | PE on Gal/ PE on Glu | Fold decrease (rad52 vs RAD52) | Refer- ence |
|---|---|---|---|
| RAD52 (pRS316) | 1.06 | ||
| rad52 (pRS316) | 0.89 | 1.2 | |
| RAD52 (YCpGal:Rlb) | 0.32 | ||
| rad52 (YCpGal:Rlb) | 5.1 × 10−4 | 627.5 | |
| RAD52 (Δlys2::GALEcoRI) | 0.71 | ||
| rad52 (Δlys2::GALEcoRI) | 18 × 10−4 | 394.4 | |
| RAD52 (Δhis3::GALEcoRI) | 0.52 | ||
| rad52 (Δhis3::GALEcoRI) | 14 × 10−4 | 371.4 | |
| RAD52 (pGALHO) | 0.46 | ||
| rad52 (pGALHO) | 3.3 × 10−4 | 1,394 | |
| Previous resultsb | |||
| rad52 (pGALHO) | ∼1 × 10−4 | 14 | |
| rad52 (pGALHO) | (1–4) × 10−4 | 31 | |
| rad52 (pGALHO) | (0.1–1) × 10−4 | 28c | |
| rad52 (pGALHO) | 16 × 10−4 | 45 |
Plating efficiencies (PE) (plating CFU/hemacytometer counts) were determined at 30°C with synthetic Glu − Ura and Gal + Glu − Ura plates for plasmid-containing strains and YPD and YPD+2%Gal plates for cells containing integrated fusions. T334 (reg1-501) and derivatives used in the assay are described in the text.
Previously published plating efficiencies for REG1 rad52 strains obtained by similar methods.
Gal/Glu plating efficiencies were determined at 25 and 33°C.
The plating efficiencies of cells containing pGALHO were also assessed to compare growth-inhibitory effects in T334 to results obtained previously with REG1 strains. Several studies have indicated that rad52 mutants containing a GAL10::HO fusion on a plasmid are unable to grow on selective galactose media. The relative plating efficiencies (on Gal versus Glu plates) have ranged from 10−3 to 10−4 (14, 28, 31, 45). As shown in Table 2, a similar result was obtained with T334ΔR52T cells (Δrad52::TRP1) containing pGALHO (relative plating efficiency, 3.3 × 10−4).
Effect of plasmid-mediated EcoRI and HO expression on viability and cell cycling in Rad+, rad52, and rad9 cells.
The reduced plating efficiencies on galactose media relative to glucose media (Table 2) demonstrated that growth of rad52 cells is inhibited by EcoRI. To examine changes in cell survival and cycling produced by plasmid-based synthesis of the endonuclease, a liquid culture assay system was developed (Fig. 1). pGALHO was again included as a control. Logarithmically growing Rad+ cells expressing EcoRI or HO progressed at near-normal growth rates to stationary phase (∼4 × 107 to 8 × 107 cells/ml in synthetic Gal + Glu − Ura media). In contrast, both rad52 and rad9 cells halted growth at mid-logarithmic densities.
FIG. 1.

Expression of EcoRI and HO from centromeric plasmids inhibits the growth of rad52 and rad9 strains. Late-log-phase cells were inoculated into Gal + Glu − Ura liquid medium at time zero and counted with a hemacytometer as described in Materials and Methods. Abbreviations: pRS, pRS316; pHO, pGALHO; pERI, YCpGal:RIb.
Microscopic examination of wild-type and mutant cells at various times after galactose induction revealed striking differences (Fig. 2). Rad+ cells expressing EcoRI or HO progressed through log phase (a mixture of unbudded, small-budded, and large-budded cells after 12 h) to early stationary phase (mostly unbudded G1 cells [Fig. 2A to C]). This was also true for rad52 cells containing the control vector pRS316 (Fig. 2D). However, rad52 cells expressing EcoRI or HO accumulated as large-budded cells (Fig. 2E and F). Staining with 4′,6-diamidino-2-phenylindole (DAPI) revealed that most of these cells contain a single nucleus located near the neck of the new bud (data not shown). Furthermore, the mother and daughter portions of many of the large-budded cells appeared greatly enlarged. A similar G2 arrest phenotype has been reported for yeast cells treated with X rays and MMS (55, 71, 80). Induction of G2 arrest by HO endonuclease has been observed previously in strains containing HO recognition sites on chromosomes or plasmids (6, 18, 66).
FIG. 2.

rad52 cells synthesizing EcoRI and HO are chronically arrested as large-budded cells. The distribution of unbudded (▩), small-budded (░⃞), and large-budded (▪) cells during growth of T334 (Rad+) and T334ΔR52T (rad52) strains in galactose media is shown (see Materials and Methods).
The results presented in Fig. 2B and C suggest that elongation of G2 phase occurred in the wild-type strain when EcoRI and HO were expressed (note the increase in the number of large-budded cells at 12 h; the cells were at mid-logarithmic-phase growth densities at this point [Fig. 1]). This transient arrest and its dependence on RAD9 were examined by using shorter time intervals, and the results are shown in Fig. 3. Rad+ cells synthesizing EcoRI contained 50 to 60% large-budded cells at 9, 12, and 15 h after induction. The control population (pRS316) achieved a maximum of ∼30% large-budded cells in this medium. The lengthening of G2 phase generated by EcoRI and HO expression was largely RAD9 dependent; Δrad9 strains expressing EcoRI or HO did not exceed 35% G2/M cells (Fig. 3).
FIG. 3.

Endonuclease-induced G2 arrest is largely dependent on rad9. The accumulation of large-budded cells in Rad+ and rad9 strains after induction of EcoRI and HO endonuclease expression is shown.
The effects of EcoRI- and HO-generated DSBs on cell viability were assessed by growing cells in liquid Gal + Glu − Ura medium (as for Fig. 1), counting them with a hemacytometer, and rescuing them onto Glu-Com plates. Cell survival at 0, 12, 24 and 48 h after induction of HO or EcoRI in selective medium is presented in Fig. 4. Rad+ cells expressing each endonuclease exhibited consistent plating efficiencies (50 to 70%) when rescued onto Glu-Com plates. Surprisingly, rad52 cells also retained high viability throughout the time course (Fig. 4B). Thus, rad52 cells displayed growth inhibition and G2 arrest when EcoRI and HO were synthesized but did not exhibit appreciable killing. In contrast, both EcoRI and HO induced extensive cell killing in rad9 mutants. The percentage of viable cells decreased from 60 to 1–2% after EcoRI and HO were induced (Fig. 4C).
FIG. 4.

Plasmid-based synthesis of EcoRI and HO causes cell killing in rad9 mutants but not in Rad+ or rad52 cells. Cells grown in Gal + Glu − Ura medium (maintaining selection for the plasmid) were rescued onto synthetic Glu-Com plates as described in Materials and Methods. Symbols: □, pRS316; ◊, pGALHO; ○, YCpGal:Rlb.
Although selection was maintained for each plasmid throughout the time course (Gal + Glu − Ura medium), the possible effects of plasmid instability were addressed by determining the percentage of cells retaining each plasmid at 0, 12, and 24 h after galactose induction. A comparison of colonies arising on Glu-Com and Glu − Ura plates is presented in Table 3. pRS316 was stably maintained in wild-type and mutant cells. Loss of the GAL1::EcoRI plasmid was similar in all three strains (ca. five- to sevenfold reduction after 24 h). pGALHO was stably propagated in Rad+ and rad9 cells but was very unstable in rad52 cells expressing the endonuclease (∼13-fold loss). The uniform instability of the GAL1::EcoRI plasmid in all backgrounds and the nearly identical survival results obtained with integrated fusions (described below) suggest that the higher survival of Rad+ and rad52 strains than of rad9 mutants is not attributable to plasmid loss. It is not possible to assess the extent of HO-induced cell killing from these experiments because of the instability of pGALHO in induced rad52 cells. However, the observation that rad52 mutants were chronically arrested as large-budded cells (Fig. 1 and 2E) suggests that cleavage at MAT occurred in most cells (also see Discussion).
TABLE 3.
Stabilities of HO and EcoRI expression plasmidsa
| Vector | Strain | % of plasmid-containing
cells at time (h):
|
||
|---|---|---|---|---|
| 0 | 12 | 24 | ||
| pRS316 | Rad+ | 67.5 | 60.5 | 88.9 |
| Δrad52 | 47.2 | 66.7 | 67.8 | |
| Δrad9 | 52.9 | 45.7 | 81.0 | |
| pGALHO | Rad+ | 55.4 | 48.2 | 52.1 |
| Δrad52 | 45.7 | 27.0 | 3.8 | |
| Δrad9 | 61.9 | 60.2 | 51.2 | |
| YCpGal:RIb | Rad+ | 72.9 | 45.7 | 18.1 |
| Δrad52 | 55.5 | 41.5 | 7.6 | |
| Δrad9 | 75.0 | 45.3 | 15.8 | |
The mean percentage of cells retaining the plasmid is shown. Aliquots of cells grown in Gal + Glu − Ura liquid medium were removed and spread directly to synthetic Glu-Com and Glu − Ura plates.
Consequences of chromosome-based GAL1::EcoRI synthesis on growth, cell cycling, and viability.
To measure the effects of EcoRI-induced cleavage under conditions which ensured that endonuclease expression occurred in all cells, GAL1::EcoRI cassettes were integrated into T334 strains at LYS2 and HIS3, yielding strains YLKL340 [Δlys2::(GAL1::EcoRI TRP1)] and YLKL350 [Δhis3::(GAL1::EcoRI TRP1)] (Table 1). The growth of Rad+ and rad52-derived strains with and without inducer is shown in Fig. 5. Cells were grown in YPD without galactose and then shifted to YPD or YPD+2%Gal. The timescale is shortened in Fig. 5 compared to Fig. 1 because of the faster induction of the GAL1 promoter and the shorter doubling time of cells in YPD+2%Gal than in synthetic Gal + Glu medium (24). As observed with the plasmid-based expression systems, the growth of Rad+ cells was only modestly affected by EcoRI synthesis. The growth of rad52 mutants was impaired, especially in cells containing EcoRI integrated at HIS3; these cells achieved only one doubling between 4 and 24 h after induction (Fig. 5).
FIG. 5.

Growth of rad52 cells containing integrated GAL1::EcoRI is inhibited in 2% galactose medium. Growth of cells containing GAL1::EcoRI integrated at LYS2 or HIS3 in glucose (YPD) or Glu + Gal (YPD+Gal) medium.
EcoRI-generated DSBs caused a modest increase in the number of large-budded cells in wild-type yeast and a much larger increase (typically 75 to 85% of total cells) in rad52 mutants (Fig. 6A and B). Data obtained with the Δlys2::(GAL1::EcoRI) construct is shown in the figure. Similar results were obtained with strains containing GAL1::EcoRI integrated at HIS3 (data not shown). Arrest of cell cycling was not observed in rad9 mutants (Fig. 6C). In Fig. 6, data are presented for cells cultured in YPD+0.5%Gal and YPD+2.0%Gal. The kinetics of induction of the GAL1 promoter in reg1-501 cells is dependent upon the concentration of the inducer (24). As shown in Fig. 6, 0.5% galactose was nearly as effective as 2.0% galactose at inducing cell cycle arrest.
FIG. 6.

Effects of chromosome-based expression of EcoRI on cell cycling and survival. (A to C) Analysis of cell cycling after induction of endonuclease synthesis in Δlys2::(GAL1::EcoRI) fusion strains. (D to F) Survival of Rad+, rad52, and rad9 strains containing the Δlys2::(GAL1::EcoRI) construction after growth in YPD or YPD + Gal medium.
Analysis of the effects of endonuclease expression on cell survival is presented in Fig. 6D to F. While the viability of Rad+ and rad52 cells was only modestly reduced (ca. two- to threefold for both the lys2 and his3 constructs [Fig. 6 and data not shown]), rad9 strains lost viability rapidly after galactose induction (from 60% viable cells to ∼1% at 24 h [Fig. 6F]). These results are similar to the survival curves obtained with the GAL1::EcoRI plasmid (Fig. 4). One difference observed with strains containing integrated fusions was that rad9 mutants had reduced viability after approximately 24 h of growth in YPD medium without galactose. This result suggests that glucose depletion in stationary-phase YPD cultures resulted in derepression of GAL1 promoter activity. In reg1-501 strains, GAL promoters are repressed in 2% Glu, are derepressed in the absence of Glu and Gal (resulting in a modest elevation of transcription), and are induced in Gal or Gal + Glu media (increasing transcription 1,000 to 2,000-fold relative to repressed levels) (24, 53). The decrease in survival demonstrates that even low levels of EcoRI expression are lethal to rad9 strains.
A possible explanation for the high viability of rad52 strains containing integrated GAL1::EcoRI is that they rapidly produce EcoRI-defective and/or EcoRI-insensitive mutants during galactose induction. Continual production and growth of such mutants might then contribute to the observed slow growth and apparent high survival of the total population. To address this possibility, colonies formed on YPD plates as part of the survival tests for lys2::(GAL1::EcoRI) and his3::(GAL1::EcoRI) strains were replica plated to YPD+2%Gal plates. rad52 strains containing a functional GAL1::EcoRI cassette cannot grow on these plates. rad52 cells grown in YPD or YPD+2%Gal did not accumulate a significant percentage of mutants capable of growing on galactose (≤1%) after 4, 8, or 12 h of growth (data not shown). After 24 h of continuous EcoRI expression, however, rad52 cells capable of growing on galactose were observed [average, 14.6% for lys2::(GAL1::EcoRI) and 34.4% for his3::(GAL1::EcoRI) strains]. Growth inhibition, G2 arrest, and EcoRI-specific DSBs were detectable approximately 4 h after induction. These results indicate that the high viability of rad52 cells during the first 12 h (Fig. 6E) is not due to the appearance of mutants.
Previous studies have demonstrated that linearized plasmid DNA containing complementary overhangs is efficiently recircularized after transformation into wild-type and rad52 cells but not in strains lacking components of the DNA end-binding Ku complex (Hdf1/Yku70 and Hdf2/Yku80 [9, 44, 45]). A recent report by Barnes and Rio (4) indicated that hdf1 cells containing a GAL1::EcoRI expression plasmid could not grow on selective galactose plates. We have confirmed that plasmid- and chromosome-based expression of the endonuclease blocks the growth of Δhdf1 strains (Fig. 7A and data not shown). Analysis of cell killing after induction of a chromosomal fusion [Δhis3::(GAL1::EcoRI)] demonstrated that Ku70 is required for survival (Fig. 7B). The percentage of viable cells decreased from 70% to 2–3% after 12 h. The slight increase in the percentage of surviving hdf1 cells after 24 h correlates with an increase in the number of mutant cells able to grow on galactose plates (data not shown). Accumulation of such mutants was also observed after prolonged expression of EcoRI in rad52 cells (described above).
FIG. 7.

Expression of EcoRI causes growth inhibition and cell killing in hdf1 mutants. Strains YLKL350, YLKL351, and YLKL389 (Δhis3::GALEcoRI) were used for the assays. (A) Growth of YLKL389 (Δhdf1::HIS3) in YPD and YPD+2%Gal. (B) Survival of Rad+, Δrad52, and Δhdf1 strains in YPD+2%Gal.
EcoRI expression produces extensive breakage of chromosomal DNA in Rad+ and rad52.
Barnes and Rine (3) previously used ethidium bromide-stained agarose gels and Southern blot experiments to show that EcoRI expressed in yeast cuts specifically at its 6-bp recognition site (G∧AATTC). To investigate the extent of EcoRI-induced DNA cleavage in T334 cells under the conditions used in these experiments, three distinct DNA purification methods were used (see Materials and Methods for descriptions of the methods and control experiments). Galactose induction of plasmid-based and integrated GAL1::EcoRI fusions resulted in extensive digestion of chromosomal DNA in both Rad+ and rad52-deleted T334 cells (Fig. 8). Genomic DNA prepared from T334 cells and cut with commercial EcoRI is included for comparison. EcoRI treatment of purified yeast DNA produces several characteristic bands (57). These bands are derived from ribosomal DNA repeats, Ty elements, and telomeric Y′ repeats. As shown in Fig. 8, many repeat bands produced after complete digestion in vitro were present after 12 or 24 h of digestion in vivo. Differences in band intensities between DNA cleaved in vitro and in vivo may be due to differential access of the enzyme to sites in cellular DNA. The single low-molecular-weight band present at time zero in Fig. 8 may represent double-stranded RNA (57). The kinetics of break formation in wild-type and mutant cells appeared similar, although increased relative amounts of low-molecular-weight DNA were clearly visible in rad52 cells. In particular, DNA purified from rad52 cells after 24 h of plasmid-based expression was consistently very low in molecular weight (compare Fig. 8A and B). DNA breakage was not as extensive after 24 h of expression in rad52 strains containing the GAL1::EcoRI fusion integrated at HIS3 or LYS2 (Fig. 8D and data not shown). The gel electrophoresis data suggest that a larger number of unrepaired breaks were present in rad52 cells. However, characterization of differences in the efficiency of rejoining of DNA ends will require further studies.
Expression of EcoRI induces recombination between homologous chromosomes.
As discussed above, a single DSB produced in vivo by HO or I-SceI endonuclease is recombinogenic in yeast cells. The effects of EcoRI-induced DSBs on mitotic interchromosomal recombination were assayed in the diploid strain RM10-32D × RM26-26C (Table 1) containing YCpGal:Rlb. This strain contains heteroalleles of LEU1 and TRP5 whose recombination has been studied previously (41). There are two recognition sites for EcoRI in the coding sequence of LEU1 and three sites within TRP5. Recombination rates in cells grown under inducing conditions (on Gal − Ura plates) were 23 times (leu1) and 33 times (trp5) higher than in cells grown on glucose plates (Table 4). Thus, as previously observed for DSBs produced by treatment with ionizing radiation or expression of HO and I-SceI (see the introduction), EcoRI-induced DSBs stimulated homologous interchromosomal recombination.
TABLE 4.
Recombination rates in cells expressing EcoRI
| Alleles | Plasmid | Recombination
ratea in cells grown on:
|
Fold increase | |
|---|---|---|---|---|
| Glucose | Galactose | |||
| leu1-12/leu1-c | p316Gal | 4.92 × 10−6 | 5.18 × 10−6 | |
| YCpGal:RIb | 5.36 × 10−6 | 121.00 × 10−6 | 22.6 | |
| trp5-2/trp5-c | p316Gal | 2.70 × 10−6 | 1.75 × 10−6 | |
| YCpGal:RIb | 2.39 × 10−6 | 79.50 × 10−6 | 33.3 | |
Number of events per cell per generation.
DISCUSSION
We have examined the effects of EcoRI-induced DSBs on viability and cell cycle progression in wild-type, repair-deficient, and checkpoint-defective yeast cells. Several of the responses observed in cells expressing EcoRI have also been found in cells containing DSBs generated by ionizing radiation or after induction of a single DSB by HO endonuclease. Expression of EcoRI in logarithmically growing Rad+ cells caused slightly decreased growth rates and elongation of G2 phase, but cells were able to progress to stationary phase. This result suggests that most breaks were efficiently repaired in all stages of the cell cycle. Previous assays of GAL promoter activity have demonstrated that galactose-induced transcription occurs throughout the cell cycle (12, 26). Synthesis of EcoRI or HO in rad52 mutants inhibited cell growth and caused cells to arrest in G2. Induction of G2 arrest by HO has been observed previously in strains containing either chromosomal or plasmid recognition sites for the enzyme (6, 18, 66). Other examples of DSB-induced cell cycle arrest have been reported for cells containing a dicentric chromosome (10) and for cells expressing a mutant topoisomerase I enzyme (34).
EcoRI-induced DSBs can be repaired in rad52 mutants.
Surprisingly, Rad+ and rad52 cells expressing EcoRI in liquid galactose media could be rescued onto glucose plates with approximately equal efficiency (Fig. 4 and 6). This result indicates that there is an alternative pathway for the repair of enzymatically induced DSBs in chromosomal DNA which is RAD52 independent. Expression of EcoRI in rad52 derivatives of several other strains (EPY214, MAR1530, VL6α, and SSL231 [Table 1]) also did not result in cell killing (data not shown). Although the protein was not essential for the maintenance of cell viability, some function(s) of Rad52 was clearly required for efficient progression of cells past the checkpoint at G2. This implies that a significant fraction of broken DNA ends in logarithmically growing Rad+ cells were repaired through a RAD52-dependent repair process, allowing passage beyond G2, but that this pathway did not provide a survival advantage over rad52 cells. Furthermore, growing and stationary-phase Rad+ cells in G1 lacked sister chromatids and were unable to participate in RAD52-mediated recombination but remained viable when EcoRI was expressed. The high viability of wild-type and rad52 cells was observed even though continuous expression of EcoRI produced numerous DSBs within cellular DNA (Fig. 8) (3). This result suggests that the broken ends produced after endonuclease cleavage remain in proximity, probably due to constraints imposed by chromatin structure (including components of the nucleosome and possibly end-joining proteins such as Hdf1 and Hdf2). It is possible that the 4-bp overhangs produced by EcoRI are efficiently reannealed in vivo, creating DNA segments containing two staggered nicks which are repairable by RAD52-independent mechanisms.
Several previous studies have established that rad52 cells are highly susceptible to killing by ionizing radiation and are specifically unable to rejoin radiation-induced DSBs (15, 16, 64). The broken ends of irradiated DNA usually have associated base or sugar damage and are often missing one nucleotide base (54). It is therefore likely that such ends require extensive processing and/or recombinational repair to restore the intact, unmutagenized strand. RAD52 is also required for the repair of DSBs containing modified termini such as those generated by various chemical clastogens, e.g., bleomycin (29, 46). Furthermore, we have recently observed that high-level expression of PvuII, which generates blunt termini, produces much greater killing in rad52 cells than in Rad+ strains (36). These results suggest that repair of DSBs containing complementary overhangs can be RAD52 independent but that repair of DSBs with damaged or blunt termini requires homologous recombination.
The possibility that EcoRI-induced DSBs in yeast chromosomes can be repaired by nonrecombination-independent pathways has gained considerable additional support from previous studies: (i) recircularization of linear, cohesive-ended plasmid DNA after transformation into yeast cells is not greatly reduced in rad52 mutants (9, 45) (see below); (ii) Schiestl et al. (68) observed that restriction enzyme-mediated integration of linear DNA into yeast chromosomes in vivo is RAD52 independent, but RAD50, which has been associated with end-joining pathways of plasmid DSB repair (described below), was required for this process; (iii) a study by Heitman et al. (21) found that Escherichia coli cells deficient in the recombinase enzyme RecA, which plays a central role in radiation resistance, recombination, and mutagenesis (76), were not more sensitive to EcoRI-induced killing than wild-type cells; and (iiii) expression of restriction endonucleases in mammalian cells produces many of the same effects as treatment with X rays, e.g., decreased viability and an increase chromosomal aberrations, mutations and cellular transformation. However, several studies have indicated that DSBs with complementary overhangs produce such effects less efficiently than do DSBs with blunt termini (11, 54). These data are consistent with the hypothesis that prokaryotic and eukaryotic cells have repair mechanisms for cohesive-ended DSBs which are not available for damaged or blunt termini.
Plasmid-based expression of HO endonuclease, which produces a single cohesive-ended DSB at the MAT locus, caused prolonged G2 arrest of rad52 mutants but did not produce detectable killing (although rad9 mutants were killed [Fig. 4 and see below]). However, the centromeric plasmid used in these studies (pGALHO) was unstable in rad52 cells in galactose medium despite selection for the vector (Table 3). Under the same conditions, pGALHO was stably maintained in Rad+ and rad9 cells. Thus, the effects of high-level expression of HO on viability could not be determined from these experiments. The instability of pGALHO in rad52 mutants may be due to the chronic disruption of the chromosome and cell cycles in these cells.
A number of studies have shown that the expression of HO from its natural promoter or from a GAL promoter blocks the growth of rad52 mutants (see the introduction). This effect has generally been interpreted as evidence of lethality, but this conclusion has not been tested rigorously. Using reg1-501 strains containing integrated GAL10::HO cassettes, we have recently observed that high levels of expression of HO (in 0.2 to 2% galactose) produced greater killing in rad52 cells than in Rad+ cells. However, synthesis of low levels of HO (with 0.005% galactose) resulted in high viabilities for both strains (36). The growth of rad52 cells was inhibited at this lower galactose concentration, with >70% of cells arrested in G2, indicating that breakage occurred in most cells. Thus, when the level of endonuclease activity was reduced (presumably corresponding to a reduction in repeated cleavage, repair, and recleavage at MAT), RAD52 was required for cell growth but not for survival. These results are analogous to the effects of EcoRI expression in rad52 mutants and suggest that DSBs induced by both HO and EcoRI can be repaired by recombination-dependent and -independent mechanisms.
How are DSBs repaired in rad52 mutants?
The growth and survival of haploid cells synthesizing the endonuclease required a functional HDF1 (YKU70) gene (Fig. 7). Past studies have demonstrated that repair of damage-induced DSBs in S. cerevisiae occurs primarily by conservative, homologous recombination mechanisms (16, 56, 70). However, recombination-independent end-joining processes, which may be either precise or error prone (discussed below), and nonconservative forms of repair within long direct repeat sequences have been described. Repair of DSBs in DNA with direct repeats often results in formation of deletions and may be RAD52 dependent or independent (30).
Genes which have been associated with end-joining pathways include HDF1/YKU70, HDF2/YKU80, RAD50, XRS2 and MRE11 (9, 13, 39, 44, 45, 47, 68, 72, 78). HDF1 and HDF2 encode subunits of a DNA end-binding complex which is found in yeast and in higher eukaryotes (referred to as Ku70 and Ku80, respectively). This heterodimeric complex participates in the repair of radiation-induced DSBs and V(D)J recombination in mammalian cells (25). Recent experiments have demonstrated that the repair of plasmid DNA containing a DSB with complementary overhangs is highly precise in yeast cells (9, 45). In these studies, linearized plasmids containing cohesive ends (produced by a single restriction enzyme) were efficiently recircularized after transformation into Rad+ cells. This end-joining process was accurate and was reduced only two- to threefold in rad52 mutants. However, in yeast Ku70- and/or Ku80-deficient strains, transformation efficiencies were reduced 10- to 14-fold (45) and 25- to 400-fold (9). In addition, the proportion of recircularized plasmids that contained precisely joined termini was greatly reduced in Ku-defective mutants. In contrast to the results obtained for DNA with cohesive ends, DNA molecules containing blunt ends could not be repaired by Ku-dependent end joining (9).
The results described above suggest that pathways of repair are operative for DSBs containing enzymatically induced complementary overhangs that are not available for DSBs produced by physical and chemical DNA-damaging agents (Fig. 9). It is likely that wild-type cells are able to cycle and survive when EcoRI is expressed because at least three principal pathways of DSB repair are available: (i) RAD52-dependent homologous recombinational repair; (ii) Ku-mediated complementary end joining; and (iii) nonconservative repair of breaks occurring in dispensable direct-repeat DNA (e.g., within ribosomal DNA) which has variable dependency on RAD52 (30). In rad52 strains, only pathways (ii) and (iii) are operative. These repair systems are insufficient to permit cells to progress past the G2 checkpoint when EcoRI is continuously expressed. However, after transfer to glucose medium (inhibiting the synthesis of EcoRI), these two pathways are sufficient for repair of the broken chromosomes, and cycling resumes without cell death.
FIG. 9.

Alternative pathways of repair for DSBs containing damaged ends (∗) or ends with complementary overhangs. RAD52-mediated homologous recombination is required for repair of DSBs with damaged termini produced by physical and chemical DNA-damaging agents (e.g., ionizing radiation) but is not essential for repair of DSBs containing complementary ends (see the text for details).
Requirement for a functional cell cycle checkpoint.
Induction of G2 arrest by EcoRI was largely dependent on RAD9. The growth rate of rad9 cells was strongly reduced by plasmid-based expression of EcoRI and was modestly inhibited in cells containing GAL1::EcoRI fusions integrated at LYS2 and HIS3. In contrast to results obtained with rad52 strains, rad9 mutants died rapidly after galactose induction of the endonuclease (Fig. 4 and 6). Killing was also seen in rad9 strains after HO endonuclease-induced breakage at MAT (using pGALHO). In addition, we have recently observed that expression of EcoRI and HO produces growth inhibition and cell killing in rad17 mutants, which are also defective in damage-induced cell cycle arrest (36).
RAD9 plays a critical role in the cell-cycling response of yeast cells to DNA damage and is required for cell cycle arrest responses in G1 and G2 but not S (1, 67, 71, 80). Damage-induced G2 arrest requires RAD17, RAD24, and MEC3 in addition to RAD9. Interestingly, analysis of UV light and methyl methanesulfonate sensitivity in single and double mutants has demonstrated that RAD9 acts in a different repair pathway(s) from the other three genes (38). Recent experiments have indicated that RAD9 is also required for DNA damage-induced transcription of several repair genes, including RAD51 and RAD54 (1). EcoRI-induced DSBs caused transient arrest in Rad+ cells and prolonged G2 arrest in rad52 mutants, but most rad9 cells continued cycling. This suggests that endonuclease-induced killing of rad9 mutants was a result of defective arrest mechanisms. However, impaired transcriptional induction of genes required for DSB repair may also be involved (1).
Rad+ strains continue cycling when EcoRI is expressed.
Most previously published Rad+ strains containing the GAL1::EcoRI plasmid formed colonies on selective galactose medium at near normal growth rates (10 of 12 strain backgrounds tested, including A364a, S288c, and SK1 strains). Two strains, tNR85T1 (6) and BGW1-7a (81), were unable to grow when EcoRI was expressed. These strains are radiation resistant and it remains unclear whether the strain disparities are attributable to differences in galactose induction kinetics, EcoRI stability and/or transport, chromatin structure, or some other factor. We note, however, that induction of GAL1-10 promoters and growth in galactose are much faster in tNR85T1 and BGW1-7a than in T334 and several other strains we have tested (Table 1 and data not shown). Growth of tNR85T1 was previously reported to be hypersensitive to induction of a single DSB on a plasmid by HO endonuclease (6). It seems likely that HO-mediated killing and EcoRI-induced blockage of growth of tNR85T1 (and BGW1-7a) are related phenomena. Recent experiments with diploid strains created by mating BGW1-7a (EcoRIs) with MAR1530 and VL6α (EcoRIr) (Table 1) or with a MATα version of BGW1-7a have established that the sensitivity phenotype is recessive (8).
Summary.
We have shown that EcoRI endonuclease expression is lethal in Ku-deficient end-joining mutants and checkpoint-defective rad9 mutants but not in Rad+ or rad52 cells. These results suggest that DSBs containing complementary ends produced by EcoRI (or by low-level synthesis of HO) can be repaired via recombination-dependent and -independent mechanisms. The data also suggests that developmentally programmed DSBs, e.g., endonuclease-induced DSBs initiating meiotic recombination, intron homing, or mating-type switching, might in principle be accurately repaired by two separate pathways. In the first pathway, processing of the ends (e.g., 5′→3′ exonuclease digestion of one strand [17]) precedes strand exchange and subsequent resolution of recombination intermediates. The second pathway may serve as a conservative restitution mechanism which rejoins the broken DNA molecules without deletion or strand exchange.
ACKNOWLEDGMENTS
We thank James Haber, Vladimir Larionov, Dennis Livingston, Robert Malone, Ed Perkins, Jeff Strathern, Akio Sugino, and Hiep Tran for providing yeast strains. We also thank Craig Bennett, Dmitri Gordenin, and Amy Greene for critical reviews of the manuscript and Philip Erhlich and Mark Roberts for expert technical assistance.
REFERENCES
- 1.Aboussekhra A, Vialard J E, Morrison D E, de la Torre-Ruiz M A, Cernakova L, Fabre F, Lowndes N F. A novel role for the budding yeast RAD9checkpoint gene in DNA damage-dependent transcription. EMBO J. 1996;15:3912–3922. [PMC free article] [PubMed] [Google Scholar]
- 2.Bai Y, Symington L S. A RAD52 homolog is required for RAD51-independent mitotic recombination in Saccharomyces cerevisiae. Genes Dev. 1996;10:2025–2037. doi: 10.1101/gad.10.16.2025. [DOI] [PubMed] [Google Scholar]
- 3.Barnes G, Rine J. Regulated expression of endonuclease EcoRI in Saccharomyces cerevisiae: nuclear entry and biological consequences. Proc Natl Acad Sci USA. 1985;82:1354–1358. doi: 10.1073/pnas.82.5.1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barnes G, Rio D. DNA double-strand-break sensitivity, DNA replication, and cell cycle arrest phenotypes of Ku-deficient Saccharomyces cerevisiae. Proc Natl Acad Sci USA. 1997;94:867–872. doi: 10.1073/pnas.94.3.867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baudin A, Ozier-Kalogeropoulos O, Denouel A, Lacroute F, Cullin C. A simple and efficient method for direct gene deletion in Saccharomyces cerevisiae. Nucleic Acids Res. 1993;21:3329–3330. doi: 10.1093/nar/21.14.3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bennett C B, Lewis A L, Baldwin K K, Resnick M A. Lethality induced by a single site-specific double-strand break in a dispensable yeast plasmid. Proc Natl Acad Sci USA. 1993;90:5613–5617. doi: 10.1073/pnas.90.12.5613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bennett C B, Westmoreland T J, Snipe J R, Resnick M A. A double-strand break within a yeast artificial chromosome (YAC) containing human DNA can result in YAC loss, deletion, or cell lethality. Mol Cell Biol. 1996;16:4414–4425. doi: 10.1128/mcb.16.8.4414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bennett, C. B., J. R. Snipe, and M. A. Resnick. Unpublished data.
- 9.Boulton S J, Jackson S P. Saccharomyces cerevisiaeKu70 potentiates illegitimate DNA double-strand break repair and serves as a barrier to error-prone DNA repair pathways. EMBO J. 1996;15:5093–5103. [PMC free article] [PubMed] [Google Scholar]
- 10.Brock J K, Bloom K. A chromosome breakage assay to monitor mitotic forces in budding yeast. J Cell Sci. 1994;107:891–902. doi: 10.1242/jcs.107.4.891. [DOI] [PubMed] [Google Scholar]
- 11.Bryant P E, Johnston P J. Restriction-endonuclease-induced DNA double-strand breaks and chromosomal aberrations in mammalian cells. Mutat Res. 1993;299:289–296. doi: 10.1016/0165-1218(93)90105-m. [DOI] [PubMed] [Google Scholar]
- 12.Connolly B, White C I, Haber J E. Physical monitoring of mating type switching in Saccharomyces cerevisiae. Mol Cell Biol. 1988;8:2342–2349. doi: 10.1128/mcb.8.6.2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Feldmann H, Driller L, Meier B, Mages G, Kellermann J, Winnacker E. HDF2, the second subunit of the Ku homologue from Saccharomyces cerevisiae. J Biol Chem. 1996;271:27765–27769. doi: 10.1074/jbc.271.44.27765. [DOI] [PubMed] [Google Scholar]
- 14.Firmenich A A, Elias-Arnanz M, Berg P. A novel allele of Saccharomyces cerevisiae RFA1 that is deficient in recombination and repair and suppressible by RAD52. Mol Cell Biol. 1995;15:1620–1631. doi: 10.1128/mcb.15.3.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Frankenberg-Schwager M, Frankenberg D. DNA double-strand breaks: their repair and relationship to cell killing in yeast. Int J Radiat Biol. 1990;58:569–575. doi: 10.1080/09553009014551931. [DOI] [PubMed] [Google Scholar]
- 16.Game J C. DNA double-strand breaks and the RAD50–RAD57 genes in Saccharomyces. Cancer Biol. 1993;4:73–83. [PubMed] [Google Scholar]
- 17.Haber J E. Mating-type gene switching in Saccharomyces cerevisiae. Trends Genet. 1992;8:446–452. doi: 10.1016/0168-9525(92)90329-3. [DOI] [PubMed] [Google Scholar]
- 18.Halbrook J, Hoekstra M F. Mutations in the Saccharomyces cerevisiae CDC1gene affect double-strand-break-induced intrachromosomal recombination. Mol Cell Biol. 1994;14:8037–8050. doi: 10.1128/mcb.14.12.8037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Haynes R H, Kunz B A. DNA repair and mutagenesis in yeast. In: Strathern J N, Jones E W, Broach J R, editors. The molecular biology of the yeast Saccharomyces: life cycle and inheritance. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory; 1981. pp. 371–414. [Google Scholar]
- 20.Hays S L, Firmenich A A, Berg P. Complex formation in yeast double-strand break repair: participation of Rad51, Rad52, Rad55, and Rad57 proteins. Proc Natl Acad Sci USA. 1995;92:6925–6929. doi: 10.1073/pnas.92.15.6925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Heitman J, Zinder N D, Model P. Repair of the Escherichia coli chromosome after in vivo scission by the EcoRI endonuclease. Proc Natl Acad Sci USA. 1989;86:2281–2285. doi: 10.1073/pnas.86.7.2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Herskowitz I, Jensen R E. Putting the HOgene to work: practical uses for mating-type switching. Methods Enzymol. 1991;194:132–146. doi: 10.1016/0076-6879(91)94011-z. [DOI] [PubMed] [Google Scholar]
- 23.Hoffman C S, Winston F. A ten-minute DNA preparation from yeast efficiently releases autonomous plasmids for transformation of E. coli. Gene. 1987;57:267–272. doi: 10.1016/0378-1119(87)90131-4. [DOI] [PubMed] [Google Scholar]
- 24.Hovland P, Flick J, Johnston M, Sclafani R A. Galactose as a gratuitous inducer of GALgene expression in yeasts growing on glucose. Gene. 1989;83:57–64. doi: 10.1016/0378-1119(89)90403-4. [DOI] [PubMed] [Google Scholar]
- 25.Jeggo P A, Taccioli G E, Jackson S P. Menage a trois: double strand break repair, V(D)J recombination and DNA-PK. Bioessays. 1995;17:949–957. doi: 10.1002/bies.950171108. [DOI] [PubMed] [Google Scholar]
- 26.Jensen R E, Herskowitz I. Directionality and regulation of cassette substitution in yeast. Cold Spring Harbor Symp Quant Biol. 1984;49:97–104. doi: 10.1101/sqb.1984.049.01.013. [DOI] [PubMed] [Google Scholar]
- 27.Johnston M, Davis R W. Sequences that regulate the divergent GAL1 GAL10 promoter in Saccharomyces cerevisiae. Mol Cell Biol. 1984;4:1440–1448. doi: 10.1128/mcb.4.8.1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kaytor M D, Livingston D M. Saccharomyces cerevisiae RAD52alleles temperature-sensitive for the repair of DNA double-strand breaks. Genetics. 1994;137:933–944. doi: 10.1093/genetics/137.4.933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Keszenman D J, Salvo V A, Nunes E. Effects of bleomycin on growth kinetics and survival of Saccharomyces cerevisiae: a model of repair pathways. J Bacteriol. 1992;174:3125–3132. doi: 10.1128/jb.174.10.3125-3132.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Klein H L. Genetic control of intrachromosomal recombination. Bioessays. 1995;17:147–159. doi: 10.1002/bies.950170210. [DOI] [PubMed] [Google Scholar]
- 31.Kramer K M, Brock J A, Bloom K, Moore J K, Haber J E. Two different types of double-strand breaks in Saccharomyces cerevisiae are repaired by similar RAD52-independent, nonhomologous recombination events. Mol Cell Biol. 1994;14:1293–1301. doi: 10.1128/mcb.14.2.1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Larionov V, Kouprina N, Eldarov M, Perkins E, Porter G, Resnick M A. Transformation-associated recombination between diverged and homologous DNA repeats is induced by strand breaks. Yeast. 1994;10:93–104. doi: 10.1002/yea.320100109. [DOI] [PubMed] [Google Scholar]
- 33.Larionov V, Kouprina N, Nikolaishvili N, Resnick M A. Recombination during transformation as a source of chimeric mammalian artificial chromosomes in yeast (YACs) Nucleic Acids Res. 1994;22:4154–4162. doi: 10.1093/nar/22.20.4154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee F S. Modified protocol for yeast DNA mini-preparation. BioTechniques. 1992;12:677. [PubMed] [Google Scholar]
- 35.Levin N A, Bjornsti M, Fink G R. A novel mutation in DNA topoisomerase I of yeast causes DNA damage and RAD9-dependent cell cycle arrest. Genetics. 1993;133:799–814. doi: 10.1093/genetics/133.4.799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lewis, L. K., J. W. Westmoreland, and M. A. Resnick. Unpublished data.
- 37.Liu J, Wu T, Lichten M. The location and structure of double-strand DNA breaks induced during yeast meiosis: evidence for a covalently linked DNA-protein intermediate. EMBO J. 1995;14:4599–4608. doi: 10.1002/j.1460-2075.1995.tb00139.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lydall D, Weinert T. Yeast checkpoint genes in DNA damage processing: implications for repair and arrest. Science. 1995;270:1488–1491. doi: 10.1126/science.270.5241.1488. [DOI] [PubMed] [Google Scholar]
- 39.Mages G J, Feldmann G M, Winnacker E. Involvement of the Saccharomyces cerevisiae HDF1gene in DNA double-strand break repair and recombination. J Biol Chem. 1996;271:7910–7915. doi: 10.1074/jbc.271.14.7910. [DOI] [PubMed] [Google Scholar]
- 40.Malkova A, Ivanov E L, Haber J E. Double-strand break repair in the absence of RAD51in yeast: a possible role for break induced DNA replication. Proc Natl Acad Sci USA. 1996;93:7131–7136. doi: 10.1073/pnas.93.14.7131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Malone R E. Multiple mutant analysis of recombination in yeast. Mol Gen Genet. 1983;189:405–412. doi: 10.1007/BF00325902. [DOI] [PubMed] [Google Scholar]
- 42.Malone R E, Esposito R E. The RAD52gene is required for homothallic interconversion of mating types and spontaneous mitotic recombination in yeast. Proc Natl Acad Sci USA. 1980;77:503–507. doi: 10.1073/pnas.77.1.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McGill C B, Shafer B K, Derr L K, Strathern J N. Recombination initiated by double-strand breaks. Curr Genet. 1993;223:305–314. doi: 10.1007/BF00310891. [DOI] [PubMed] [Google Scholar]
- 44.Mezard C, Nicolas A. Homologous, homeologous, and illegitimate repair of double-strand breaks during transformation of a wild-type strain and a rad52 mutant strain of Saccharomyces cerevisiae. Mol Cell Biol. 1994;14:1278–1292. doi: 10.1128/mcb.14.2.1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Milne G T, Jin S, Shannon K B, Weaver D T. Mutations in two Ku homologs define a DNA end-joining repair pathway in Saccharomyces cerevisiae. Mol Cell Biol. 1996;16:4189–4198. doi: 10.1128/mcb.16.8.4189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Moore C W. Control of in vivo (cellular) phleomycin sensitivity by nuclear genotype, growth phase, and metal ions. Cancer Res. 1982;42:929–933. [PubMed] [Google Scholar]
- 47.Moore J K, Haber J E. Cell cycle and genetic requirements of two pathways of nonhomologous end-joining repair of double-strand breaks in Saccharomyces cerevisiae. Mol Cell Biol. 1996;16:2164–2173. doi: 10.1128/mcb.16.5.2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Morrison A, Johnson A L, Johnson L H, Sugino A. Pathway correcting DNA replication errors in Saccharomyces cerevisiae. EMBO J. 1993;12:1467–1473. doi: 10.1002/j.1460-2075.1993.tb05790.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mortensen U H, Bendixen C, Sunjevaric I, Rothstein R. DNA strand annealing is promoted by the yeast Rad52 protein. Proc Natl Acad Sci USA. 1996;93:10729–10734. doi: 10.1073/pnas.93.20.10729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nelson W G, Kastan M B. DNA strand breaks: the DNA template alterations that trigger p53-dependent DNA damage response pathways. Mol Cell Biol. 1994;14:1815–1823. doi: 10.1128/mcb.14.3.1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nickoloff J A, Chen E Y, Heffron F. A 24-base-pair DNA sequence from the MATlocus stimulates intergenic recombination in yeast. Proc Natl Acad Sci USA. 1986;83:7831–7835. doi: 10.1073/pnas.83.20.7831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nickoloff J A, Singer F D, Hoekstra M F, Heffron F. Double strand breaks stimulate alternative mechanisms of recombination repair. J Mol Biol. 1989;207:527–541. doi: 10.1016/0022-2836(89)90462-2. [DOI] [PubMed] [Google Scholar]
- 53.Niederacher D, Entian K. Characterization of Hex2 protein, a negative regulatory element necessary for glucose repression in yeast. Eur J Biochem. 1991;200:311–319. doi: 10.1111/j.1432-1033.1991.tb16187.x. [DOI] [PubMed] [Google Scholar]
- 54.Obe G, Johannes C, Schulte-Frohlinde D. DNA double-strand breaks induced by sparsely ionizing radiation and endonucleases as critical lesions for cell death, chromosomal aberrations, mutations and oncogenic transformation. Mutagenesis. 1992;7:3–12. doi: 10.1093/mutage/7.1.3. [DOI] [PubMed] [Google Scholar]
- 55.Paulovich A G, Hartwell L H. A checkpoint regulates the rate of progression through S phase in S. cerevisiaein response to DNA damage. Cell. 1995;82:841–847. doi: 10.1016/0092-8674(95)90481-6. [DOI] [PubMed] [Google Scholar]
- 56.Petes T D, Malone R E, Symington L S. Recombination in yeast. In: Broach J R, Pringle J R, Jones E W, editors. The molecular biology of the yeast Saccharomyces: genome dynamics, protein synthesis, and energetics. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory; 1991. pp. 407–521. [Google Scholar]
- 57.Philippsen P, Stotz A, Scherf C. DNA of Saccharomyces cerevisiae. Methods Enzymol. 1991;194:169–182. doi: 10.1016/0076-6879(91)94014-4. [DOI] [PubMed] [Google Scholar]
- 58.Plessis A, Perrin A, Haber J E, Dujon B. Site-specific recombination determined by I-SceI, a mitochondrial group I intron-encoded endonuclease expressed in the yeast nucleus. Genetics. 1992;130:451–460. doi: 10.1093/genetics/130.3.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Porter S E, Greenwell P W, Ritchie K B, Petes T. The DNA-binding protein Hdf1p (a putative Ku homologue) is required for maintaining normal telomere length in Saccharomyces cerevisiae. Nucleic Acids Res. 1996;24:582–585. doi: 10.1093/nar/24.4.582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Priebe S D, Westmoreland J, Nilsson-Tillgren T, Resnick M A. Induction of recombination between homologous and diverged DNAs by double-strand gaps and breaks and role of mismatch repair. Mol Cell Biol. 1994;14:4802–4814. doi: 10.1128/mcb.14.7.4802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ramsden D A, Gellert M. Formation and resolution of double-strand break intermediates in V(D)J rearrangement. Genes Dev. 1995;9:2409–2420. doi: 10.1101/gad.9.19.2409. [DOI] [PubMed] [Google Scholar]
- 62.Ray A, Siddiqi I, Kolodkin A L, Stahl F W. Intra-chromosomal gene conversion induced by a DNA double-strand break in Saccharomyces cerevisiae. J Mol Biol. 1988;201:247–260. doi: 10.1016/0022-2836(88)90136-2. [DOI] [PubMed] [Google Scholar]
- 63.Resnick M A. The repair of double-strand breaks in DNA: a model involving recombination. J Theor Biol. 1976;59:97–106. doi: 10.1016/s0022-5193(76)80025-2. [DOI] [PubMed] [Google Scholar]
- 64.Resnick M A, Martin P. The repair of double-strand breaks in the nuclear DNA of Saccharomyces cerevisiaeand its genetic control. Mol Gen Genet. 1976;143:119–129. doi: 10.1007/BF00266917. [DOI] [PubMed] [Google Scholar]
- 65.Saeki T, Machida I, Nakai S. Genetic control of diploid recovery after γ irradiation in the yeast Saccharomyces cerevisiae. Mutat Res. 1980;73:251–265. doi: 10.1016/0027-5107(80)90192-x. [DOI] [PubMed] [Google Scholar]
- 66.Sandell L L, Zakian V A. Loss of a yeast telomere: arrest, recovery, and chromosome loss. Cell. 1993;75:729–739. doi: 10.1016/0092-8674(93)90493-a. [DOI] [PubMed] [Google Scholar]
- 67.Schiestl R H, Reynolds P, Prakash S, Prakash L. Cloning and sequence analysis of the Saccharomyces cerevisiae RAD9gene and further evidence that its product is required for cell cycle arrest induced by DNA damage. Mol Cell Biol. 1989;9:1882–1896. doi: 10.1128/mcb.9.5.1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schiestl R H, Zhu J, Petes T D. Effect of mutations in genes affecting homologous recombination on restriction enzyme-mediated and illegitimate recombination in Saccharomyces cerevisiae. Mol Cell Biol. 1994;14:4493–4500. doi: 10.1128/mcb.14.7.4493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sherman F. Getting started with yeast. Methods Enzymol. 1991;194:3–21. doi: 10.1016/0076-6879(91)94004-v. [DOI] [PubMed] [Google Scholar]
- 70.Shinohara A, Ogawa T. Homologous recombination and the roles of double-strand breaks. Trends Biochem Sci. 1995;20:387–391. doi: 10.1016/s0968-0004(00)89085-4. [DOI] [PubMed] [Google Scholar]
- 71.Siede W. Cell cycle arrest in response to DNA damage: lessons from yeast. Mutat Res. 1995;337:73–84. doi: 10.1016/0921-8777(95)00023-d. [DOI] [PubMed] [Google Scholar]
- 72.Siede W, Friedl A A, Dianova I, Eckardt-Schupp F, Friedberg E C. The Saccharomyces cerevisiaeKu autoantigen homologue affects radiosensitivity only in the absence of homologous recombination. Genetics. 1996;142:91–102. doi: 10.1093/genetics/142.1.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sikorski R S, Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sugawara N, Haber J E. Characterization of double-strand break-induced recombination: homology requirements and single-stranded DNA formation. Mol Cell Biol. 1992;12:563–575. doi: 10.1128/mcb.12.2.563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Szostak J W, Orr-Weaver T L, Rothstein R J, Stahl F W. The double-strand break repair model for recombination. Cell. 1983;33:25–35. doi: 10.1016/0092-8674(83)90331-8. [DOI] [PubMed] [Google Scholar]
- 76.Takahashi M, Maraboeuf F, Norden B. Locations of functional domains in the RecA protein. Overlap of domains and regulation of activities. Eur J Biochem. 1996;242:20–28. doi: 10.1111/j.1432-1033.1996.0020r.x. [DOI] [PubMed] [Google Scholar]
- 77.Thaler D S, Stahl F W. DNA double-chain breaks in recombination of phage lambda and of yeast. Annu Rev Genet. 1988;22:169–197. doi: 10.1146/annurev.ge.22.120188.001125. [DOI] [PubMed] [Google Scholar]
- 78.Tsukamoto Y, Kato J, Ikeda H. Hdf1, a yeast Ku-protein homologue, is involved in illegitimate recombination, but not in homologous recombination. Nucleic Acids Res. 1996;24:2067–2072. doi: 10.1093/nar/24.11.2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Weiffenbach B, Haber J E. Homothallic mating-type switching generates lethal chromosome breaks in rad52 strains of Saccharomyces cerevisiae. Mol Cell Biol. 1981;1:522–534. doi: 10.1128/mcb.1.6.522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Weinert T A, Hartwell L H. The RAD9 gene controls the cell cycle response to DNA damage in Saccharomyces cerevisiae. Science. 1988;241:317–322. doi: 10.1126/science.3291120. [DOI] [PubMed] [Google Scholar]
- 81.White C I, Haber J E. Intermediates of recombination during mating type switching in Saccharomyces cerevisiae. EMBO J. 1990;9:663–673. doi: 10.1002/j.1460-2075.1990.tb08158.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wintersberger U, Karwan A. Retardation of cell cycle progression in yeast cells recovering from DNA damage: a study at the single cell level. Mol Gen Genet. 1987;207:320–327. doi: 10.1007/BF00331596. [DOI] [PubMed] [Google Scholar]