

Abstract
RNA editing at adenosine 1012 (amber/W site) in the antigenomic RNA of hepatitis delta virus (HDV) allows two essential forms of the viral protein, hepatitis delta antigen (HDAg), to be synthesized from a single open reading frame. Editing at the amber/W site is thought to be catalyzed by one of the cellular enzymes known as adenosine deaminases that act on RNA (ADARs). In vitro, the enzymes ADAR1 and ADAR2 deaminate adenosines within many different sequences of base-paired RNA. Since promiscuous deamination could compromise the viability of HDV, we wondered if additional deamination events occurred within the highly base paired HDV RNA. By sequencing cDNAs derived from HDV RNA from transfected Huh-7 cells, we determined that the RNA was not extensively modified at other adenosines. Approximately 0.16 to 0.32 adenosines were modified per antigenome during 6 to 13 days posttransfection. Interestingly, all observed non-amber/W adenosine modifications, which occurred mostly at positions that are highly conserved among naturally occurring HDV isolates, were found in RNAs that were also modified at the amber/W site. Such coordinate modification likely limits potential deleterious effects of promiscuous editing. Neither viral replication nor HDAg was required for the highly specific editing observed in cells. However, HDAg was found to suppress editing at the amber/W site when expressed at levels similar to those found during HDV replication. These data suggest HDAg may regulate amber/W site editing during virus replication.
Hepatitis delta virus (HDV) is a subviral human pathogen that increases the risk of severe liver disease in those infected with its helper, hepatitis B virus (34). The HDV genome is an ∼1,680-nucleotide (nt) circular RNA that replicates through a circular RNA intermediate, the antigenome (21). Both the genome and antigenome possess extensive intramolecular complementarity and are predicted to form rod-shaped structures in which about 70% of the nucleotides are base paired (39).
HDV produces two forms of the sole viral protein, hepatitis delta antigen (HDAg) (4), and both are translated from a single mRNA that is transcribed from the genomic RNA (16, 18, 40). The shorter form, HDAg-S, is required for RNA replication, while the longer form, HDAg-L, inhibits replication but is required for packaging the viral RNA with the envelope of hepatitis B surface antigen (19, 35, 42). The virus uses adenosine-to-inosine RNA editing activity of the host cell to produce HDAg-S and HDAg-L from the same open reading frame (6, 25). The editing does not occur on the mRNA directly but on adenosine 1012 of the antigenome (9, 32). The nucleotide change is subsequently passed to the genome during replication and to the mRNA during transcription. The ultimate effect is the conversion of the UAG amber termination codon of HDAg-S to a UGG tryptophan codon required to synthesize the slightly longer HDAg-L; because of the codon change produced by this editing event, the editing site is called the amber/W site (32).
We previously showed that double-stranded RNA (dsRNA) adenosine deaminase (ADAR1 [2]), purified from Xenopus laevis, can edit the amber/W site of HDV RNA in vitro (32). This enzyme converts adenosine to inosine in dsRNA (or RNA that is largely double stranded) by deamination (reviewed in reference 1). Editing at the HDV RNA amber/W site is affected identically by site-directed mutations in the base-paired structure around the amber/W site whether analyzed in cells or in vitro with purified Xenopus ADAR1 (32). Thus, ADAR1, or a closely related enzyme such as ADAR2 (formerly called RED-1 [26]), is thought to catalyze HDV RNA editing in vivo.
In vitro studies using synthetic substrates of ADAR1 (i.e., dsRNA) show that as many as 50% of the adenosines in a single RNA can be deaminated (1). However, the enzyme does not deaminate adenosine targets entirely randomly but rather exhibits deamination specificity, which is described by using the terms “preferences” and “selectivity” (31). ADAR1 shows preferences for certain adenosines, and the total number of deamination events in a single RNA molecule, or selectivity, varies for different substrates. The preference for editing at the HDV amber/W site and the selectivity that occurs on the HDV antigenome are likely to have important consequences for virus viability, particularly because sequence changes are passed to the genome. Excessive editing at the amber/W site would result in reduced levels of RNA replication and reduced production of viable virions because edited antigenomes encode HDAg-L, which inhibits RNA replication. Similarly, the amber/W adenosine would need to be a highly preferred deamination site compared to other adenosines, and the reaction would need to be very selective since promiscuous deamination could change the coding sequence in deleterious ways or alter nucleotide sequences required for other viral functions such as replication (3) or ribozyme activity (30).
Here we investigate the specificity of HDV editing as it occurs in transfected cells expressing HDV RNAs. We found that editing was very specific for the amber/W site and that neither virus replication nor HDAg was required for the high specificity. However, importantly, we observed that HDAg was able to suppress the extent of editing that occurred at the amber/W site and thus could play a role in regulating the extent of editing during HDV replication.
MATERIALS AND METHODS
Plasmids.
Construct pHDVx1.2-R, used for expression of replicating HDV RNA in transfected cells, was previously described as pCMV3-DC1x1.2 (9). Plasmid pHDVΔx1-NR, used for expression of nonreplicating antigenomic HDV RNA in transfected cells, was created by excising the 1,173-bp XbaI fragment containing a monomeric unit of HDV, less the deleted ApaI region, from pCMV3-DC-ΔApax1.2 (9). This fragment was inserted in the XbaI site of the expression vector pCMV-MCS3 (9) to generate pCMV-ΔApax1 (A), in which the cytomegalovirus (CMV) promoter is oriented to produce antigenomic-sense HDV RNA in transfected cells. The polyadenylation signal sequence was changed from AATAAA to AATAG by PCR site-directed mutagenesis; the 181-bp SalI-XbaI fragment containing this mutation replaced the corresponding fragment of pCMV-ΔApax1(A), which had been cut with SalI and partially digested with XbaI, to yield pHDVΔx1-NR. The disruption of the polyadenylation signal sequence was found to increase amber/W editing about twofold (data not shown). The sequence of the cloned fragment in pHDVΔx1-NR was obtained by cycle sequencing (Life Technologies, Grand Island, N.Y.) and verified both the presence of the desired site-directed mutation and the absence of additional mutations.
The HDAg-S expression plasmid pCMV-AgS was created as follows. The 785-bp HindIII-XbaI fragment from pGDC-1, which contains the HDAg coding sequences and the polyadenylation site, was inserted between the HindIII and XbaI sites of the expression vector pcDNAIneo (Invitrogen, San Diego, Calif.) to yield pcDNAneoAgS. From this plasmid, the 1,557-bp MluI-XbaI fragment containing the CMV promoter and HDV sequences was inserted between the MluI and XbaI sites of the vector pGEM-7Zf(+) (Promega, Madison, Wis.) to yield pCMV-AgS.
The expression plasmid pCMV-AgS(fs) is the same as pCMV-AgS except that it contains a stop codon and frameshift at codon 7 in the HDAg coding region (5). It was created by exchanging the HindIII-PstI fragment of pHDV · I(+)Ag(−) (5) for that of pCMV-AgS. The expression plasmid pCMV-AgSΔStuSma contains an in-frame deletion of nt 1110 to 1334 within the HDAg coding region. It was created by StuI and SmaI digestion of pCMV-AgS, followed by ligation.
Transfections.
Human Huh7 hepatoma cells were cultured and transfected by the calcium phosphate method as described previously (6). Transfections were done in duplicate or triplicate, as indicated. Where appropriate, the total amount of DNA in the calcium phosphate precipitate was adjusted to 5.5 μg by addition of the plasmid vector pCMV-MCS3. RNAs were prepared from cells harvested at indicated times as described previously (6, 9) except that proteinase K (1 mg/ml) was included in the lysis buffer for the transfection experiments shown in Fig. 3 to 5.
FIG. 3.

Effect of HDAg expression on editing at the amber/W site. Human Huh-7 hepatoma cells were transfected with the nonreplicating HDV RNA expression construct pHDVΔx1-NR plus equivalent amounts of the following constructs: pCMV-MCS3, the expression vector alone; pCMV-AgS, which expresses HDAg-S; pCMV-AgS(fs), which expresses HDAg mRNA with a stop codon and frameshift at codon 7; and pCMV-AgSΔStuSma, which expresses HDAg containing an internal deletion. All cells were harvested 5 days posttransfection and analyzed for editing at the amber/W site by the appearance of a StyI restriction site (9, 32). The autoradiogram shows 32P-labeled RT-PCR products, amplified with primers 7646 and 7647, uncut (−) or cut (+) with StyI. PCR products contained an additional StyI site that was not affected by editing (Fig. 1).
FIG. 5.

Changes in editing associated with altered HDAg levels do not correlate with altered RNA levels. Cells were cotransfected with various amounts of the HDAg expression construct pCMV-AgS and either 5 μg (columns A) or 0.5 μg (columns B) of the nonreplicating HDV RNA expression construct pHDVΔx1-NR. Numbers 1 to 6 refer to the same amounts of pCMV-AgS transfected as for Fig. 3. For each cotransfection, levels of amber/W editing were determined (bar height). In addition, cellular levels of HDV RNA were determined by radioanalytic imaging with a Packard InstantImager of Northern blots hybridized with genomic-sense HDV RNA; the number above each bar indicates the amount of HDV antigenomic RNA relative to that in column 1A. Cell transfection, RNA harvesting, and editing analysis were as for Fig. 3.
Analysis of RNA editing.
Editing assays were performed as described previously (9, 32). Briefly, RNA samples were treated with DNase and then subjected to reverse transcription-PCR (RT-PCR) amplification. Primers were 5414 and 5415 (9) or 7646 (5′-GGAGGTTGGGCCCGAAC-3′) and 7647 (5′-TGTGAGTGGAAACCCGCCTA-3′), as indicated. PCR products were analyzed for amber/W editing by single-cycle labeling with [α-32P]dCTP followed by NcoI or StyI restriction digestion as described previously (9, 32). Amber/W editing was indicated by the appearance of an NcoI or StyI restriction site in the amplification product. The effectiveness of the DNase treatment was verified by the absence of PCR products after amplification without prior reverse transcription. PCR products obtained without prior DNase treatment and without a prior reverse transcription reaction did not yield StyI digestion fragments related to editing. Editing was quantified by electrophoresis followed by radioanalytic imaging (Ambis [San Diego, Calif.] imager or InstantImager [Packard Instruments, Meriden, Conn.]).
Cloning and sequencing of PCR products.
RNA was reverse transcribed with Superscript II (100 U; Life Technologies) or Moloney murine leukemia virus reverse transcriptase (100 U; Life Technologies) in 10-μl reactions containing 1× forward reaction buffer (supplied by the manufacturer), 2 nmol of random hexamer, 1 mM deoxynucleoside triphosphates, and 10 U of RNasin (Promega). Reaction mixtures were incubated at 37°C for 15 min and then 42°C for 15 min. cDNA products were then amplified with Pfu polymerase (Stratagene, La Jolla, Calif.). Forty microliters of a PCR master mix, containing 1.25 U of Pfu DNA polymerase (Stratagene), 1× Pfu polymerase buffer (supplied by the manufacturer), and 25 pmol of primers, was added to the 10 μl of reverse transcription reaction mixture. cDNA was amplified for 25 or 30 cycles of 1 min at 94°C, 1 min at 55°C, and 1 min at 72°C. Primers were 5414 and 5415 (9). PCR products were cloned with a pCR-Script kit (Stratagene). Barrier pipette tips were used to set up all reactions, and standard precautions were taken to minimize potential contamination of samples prior to PCR analysis (20). Control reactions lacking reverse transcriptase or RNA were performed to ensure that the reactions were not contaminated.
For each experiment, multiple clones were obtained from independent amplification reactions from different RNA samples. Both strands of cloned PCR products were sequenced by the dye-terminator sequencing system on Applied Biosystems 373A DNA sequencers at the University of Utah Health Sciences Sequencing Facility. Sequence changes were considered bona fide only if observed on both strands.
Northern blot analysis of HDV RNA.
RNA was electrophoresed through 1.5% agarose gels containing 2.2 M formaldehyde, transferred to positively charge nylon membranes, and hybridized with a genomic-sense 32P-labeled probe as described previously (10); the hybridization temperature was 60°C, and the washing temperature was 70°C. The integrity of the RNA samples and equivalent loading were assessed by visualization of RNA bands after staining gels with ethidium bromide. Relative levels of HDV antigenomic RNA were determined by radioanalytic scanning of blots with a Packard InstantImager.
Immunoblot analysis.
Cell lysates were obtained by treatment with 2% sodium dodecyl sulfate (SDS)–0.2 M Tris-HCl (pH 7.5)–1 mM EDTA and analyzed for HDAg by SDS-polyacrylamide gel electrophoresis (PAGE) in 12% acrylamide gels and immunoblotting with human anti-HDAg as described previously (4). Relative HDAg levels were assessed by radioanalytic scanning with a Packard InstantImager.
RESULTS
We previously showed that the amber/W site in HDV antigenomic RNA can be edited in vitro by purified Xenopus ADAR1 (32). Editing was highly specific for the amber/W site: under conditions that produced 16% ± 1% editing at amber/W, the total amount of adenosine conversion was 0.81% among all 340 adenosines in the RNA (32). This value is much lower than the 50 to 60% deamination of adenosines typically observed in dsRNA substrates under similar reaction conditions. Nevertheless, 0.81% of 340 adenosines amounts to 2.7 A-to-I conversions per antigenome (32); this level of deamination could compromise the viability of the virus. We therefore investigated the specificity of editing in cells transfected with either replicating or nonreplicating HDV cDNA constructs.
Editing specificity in replicating HDV RNA in cells.
To examine the specificity of HDV RNA editing occurring in cells, human Huh-7 hepatoma cells were transfected in triplicate with the construct pHDVx1.2-R, which produces replicating HDV RNA (Fig. 1 and reference 6). Cells transfected with replication-competent HDV cDNA constructs produce replicating HDV RNA in which editing levels increase to a maximum of 20 to 35% by 12 to 15 days posttransfection (5, 6, 43). RNA was harvested 13 days posttransfection and analyzed for editing at the amber/W site by StyI restriction (editing at the amber/W site of the HDV antigenomic RNA creates a StyI restriction site in the corresponding cDNA [6, 32]) and by sequencing 84 cDNA clones from three separate amplification PCRs from different RNA samples. The sequenced region was a 358-nt segment corresponding to the C-terminal half of the HDAg-coding sequence (Fig. 1, positions 907 to 1264, numbered according to the genomic strand [39]) that includes the amber/W site (1012) and that has been used extensively in phylogenetic analyses of HDV isolates (7, 28, 37). This region accounts for 21% of the entire antigenome and contains 77 adenosines.
FIG. 1.
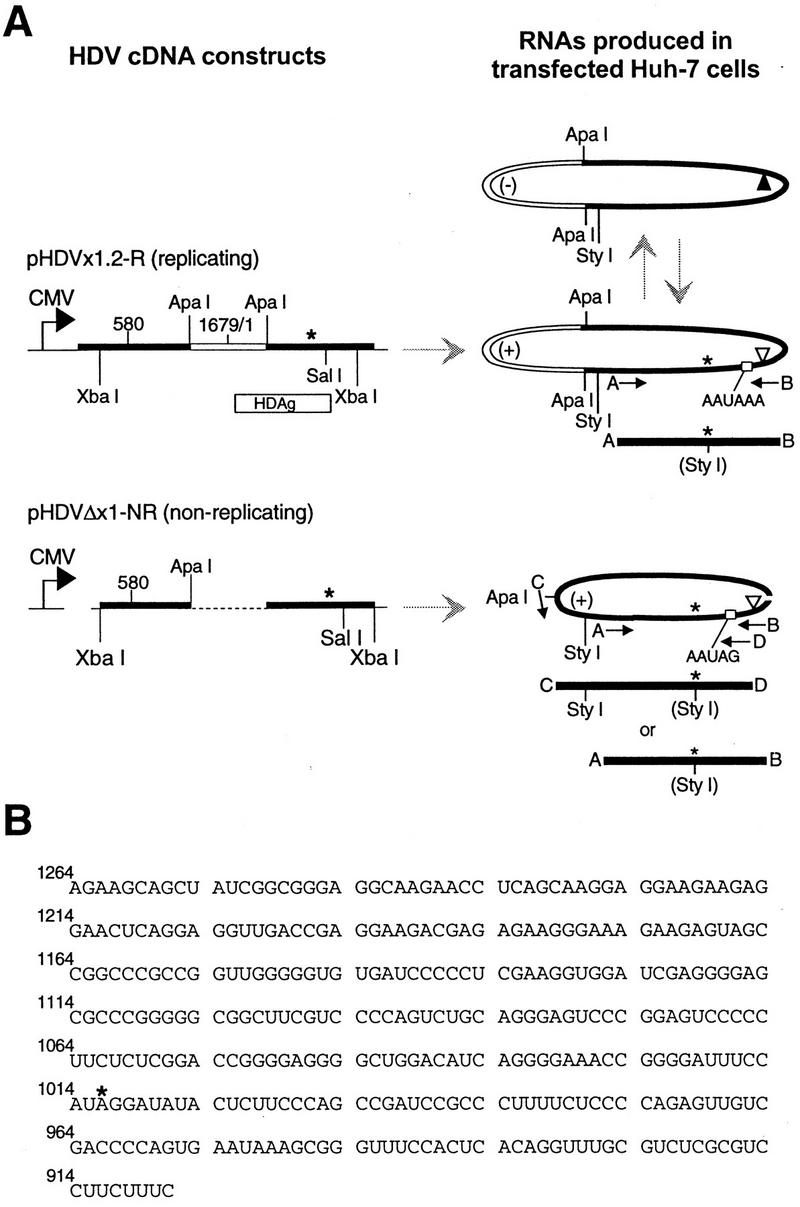
(A) Schematic diagram of HDV cDNA constructs, RNAs, and regions amplified by the PCR. Left: thick straight bar, HDV cDNA; thin line, plasmid sequences; dashed line pHDVΔx1-NR, sequences deleted between ApaI sites (9). Direction of transcription initiated by the CMV promoter is indicated by arrows. Right: oval shapes with heavy lines, expected HDV RNA species; (+), antigenomic sense; (−), genomic sense. The location of the amber/W site is indicated by an asterisk; the genomic and antigenomic ribozyme cleavage sites (38, 41) are indicated by solid and open triangles, respectively. Small open boxes indicate locations of wild-type (pHDVx1.2-R) and mutated (pHDVΔx1-NR) polyadenylation sites. Solid bars indicate expected PCR products; arrows mark primers A (5415), B (5414), C (7646), and D (7647). Primers 7646 and 7647 correspond to sequences present only in the RNA derived from the plasmid pHDVΔx1-NR. There is a single StyI site (indicated in parentheses) in cDNAs amplified with primers 5414 and 5415 and derived from edited RNA. Primers 7646 and 7647 yield cDNAs with the same editing-sensitive site, plus an existing site that is unaffected by editing. Nonreplicating RNA is produced in cells transfected with the deletion construct pHDVΔx1-NR, which contains an ∼514-nt internal deletion and a site-directed mutation at the polyadenylation signal site. The region to be deleted is indicated by an open segment for construct pHDVx1.2-R and its derived RNAs and by a dashed line for the construct pHDVΔx1-NR. Drawings are not necessarily to scale. Horizontal gray arrows indicate transcription of RNAs from plasmid DNA templates; vertical gray arrows indicate RNA template-driven transcription occurring during HDV RNA replication. (B) Sequence of the 358-nt region analyzed by cloning and sequencing after amplification with primers 5414 and 5415. Sequence shown is antigenomic sense; numbering corresponds to the genomic RNA (39). The amber/W site is indicated by an asterisk.
Sequence analysis indicated that editing of replicating RNAs in cells was highly specific. In good agreement with the amount of editing (25%) determined by restriction digestion assays on the RNA, 24 of 84 clones (29%) contained G at the amber/W site. However, of the 6,384 non-amber/W adenosines sequenced, only three A→G transitions (0.05%) were observed (Table 1, replicating; Fig. 2A). Amber/W editing accounted for at least 89% of all A→G changes on the replicating RNA in the 358-nt region analyzed (Table 1). Extrapolation of the average number of non-amber/W A→G changes over the entire antigenome indicated that, on average, only 0.16 such modifications occurred per molecule (Table 1). Thus, editing of replicating HDV RNA in cells exhibits considerably more specificity than that observed upon incubation of HDV RNA with purified Xenopus ADAR1 in vitro (between 1.6 and 2.7 non-amber/W changes per antigenome [32, 33]).
TABLE 1.
Specificity of HDV editing in transfected Huh-7 cells
| RNA (no. of clones analyzed) | No. (%) with:
|
A→G changes at amber/W as % of all A→G changesa | Non-amber/W A→G changes/RNA | ||
|---|---|---|---|---|---|
| Modifications at amber/W | Non-amber/W A→G changes | Non-A→G changes | |||
| Replicating (84) | 24 (29) | 3 (0.05b) | 9 (0.03)c | 89 | 0.16 |
| Nonreplicating (97) | 35 (36) | 7 (0.09) | 1 (.003) | 83 | 0.32 |
(No. of clones edited at the amber/W site)/(total no. of A→G changes)] × 100.
% = [(no. of changes)/(no. of adenosines × no. of clones)] × 100.
For replicating RNA, includes four U→C changes.
FIG. 2.

The percentage of HDV cDNA clones with A→G transitions at specific sites within the region from nt 907 to 1264 is plotted for cDNA populations derived from replicating or nonreplicating HDV RNAs. cDNA populations were derived from replicating HDV RNA harvested 13 days after Huh-7 cells were transfected with plasmid pHDVx1.2-R (84 clones) (A) and nonreplicating HDV RNA harvested 6 days after Huh-7 cells were transfected with the nonreplicating HDV RNA expression plasmid pHDVΔx1-NR (97 clones) (B). Sequence numbering refers to the genomic strand (39). Numbers in italics indicate sequence positions that exhibited A→G transitions.
Clones from replicating HDV RNA also exhibited nine additional changes other than A→G, but no insertions or deletions. Because the frequency of these non-A→G changes (0.03%) was not much greater than the misincorporation rate of Pfu polymerase, it is not certain whether these occurred in the cells during HDV replication or during the PCR amplification, particularly as none of these changes appeared in more than one clone. However, it is worth noting that among these nine additional modifications were four U→C transitions (in the antigenomic sequence) that could result from adenosine deaminations that occurred on the genomic RNA, because cells transfected with the replicating construct pHDVx1.2-R contained both genomic and antigenomic HDV RNA.
Highly specific editing observed in cells does not require HDAg or viral replication.
We considered the possibility that HDV itself was responsible for the high specificity of editing observed in cells. For example, it seemed possible that spurious modification was prevented in cells by the viral protein, HDAg, or that some aspect of the replication process could select against non-amber/W modifications. To determine whether HDV RNA replication or the presence of the viral protein was required for the highly specific amber/W editing observed in replicating RNAs in cells, Huh-7 cells were transfected in triplicate with the nonreplicating HDV antigenomic RNA expression construct pHDVΔx1-NR. This construct lacks a large portion of the genome that is essential for replication and HDAg expression (Fig. 1) (22); thus, transfected cells expressed only nonreplicating antigenomic HDV RNA (data not shown), and no HDAg was detectable (see Fig. 4B, lane 6).
FIG. 4.

Concentration-dependent effect of HDAg expression on editing at the amber/W site. Human Huh-7 hepatoma cells were transfected with 5 μg of the nonreplicating HDV RNA expression construct pHDVΔx1-NR plus different amounts of the HDAg expression construct pCMV-AgS (NR; lanes 1 to 6) or 5 μg of the replicating HDV RNA expression construct pHDVx1.2-R (R; lanes 7). The amounts of pCMV-AgS cotransfected were as follows: lanes 1, 0.2 μg; lanes 2, 0.05 μg; lanes 3, 0.01 μg; lanes 4, 0.002 μg; and lanes 5, 0.0005 μg. All cells were harvested 5 days posttransfection and analyzed for editing at the amber/W site as described for Fig. 3 (A) as well as for HDAg expression levels (B). (A) 32P-labeled RT-PCR products, uncut (−) or cut (+) with StyI. Lanes 1 to 6, PCR amplification with primers 7646 and 7647; lanes 7, primers 5414 and 5415. PCR products shown in lanes 1 to 6 contained an additional StyI site that was not affected by editing (Fig. 1 and 3). (B) SDS-PAGE-immunoblot analysis of HDAg expression (see Materials and Methods). The relative HDAg expression levels were determined by radioanalytic imaging with a Packard InstantImager. The numerical value indicated for lane 6 was obtained from a duplicate lane in the same gel that was not next to the strong signal in lane 7.
HDV RNA was harvested from Huh-7 cells 6 days after transfection with the nonreplicating HDV RNA expression construct pHDVΔx1-NR. RNAs were harvested earlier than in the comparable experiment with replicating RNAs because we sought to compare editing specificity in replicating and nonreplicating RNAs under conditions in which the amber/W site was edited to similar extents, and previous experiments had indicated that editing occurs more rapidly in the nonreplicating RNA expressed from pHDVΔx1-NR (5). Sequence analysis indicated that 35 of 97 cDNA clones (36%) were from RNAs that had been edited at the amber/W site. This value agreed with the amount of editing (35%) determined by the restriction enzyme digestion assay and was only slightly higher than the amount of editing observed 13 days posttransfection in cells transfected with the replicating HDV RNA expression construct pHDVΔx1.2-R. In the entire population of 97 clones, only seven A→G changes were detected at sites other than amber/W, nearly as few as in the replicating RNA (Fig. 2B; Table 1, nonreplicating). Editing at the amber/W site accounted for 83% of all A→G transitions. Extrapolating the average number of A→G changes observed outside of the amber/W site over the entire antigenome showed an average of 0.32 such modifications per molecule (Table 1). Thus, in cells, the specificity of editing was very similar for the nonreplicating and replicating HDV RNAs. This result suggests that neither HDV RNA replication nor the presence of HDAg nor selective pressure against genomes deaminated at adenosines essential for RNA replication was responsible for the high specificity of editing that occurred on replicating RNA in cells.
Possibly, some of the non-amber/W A→G transitions observed in cDNAs derived from HDV RNA in cells were due to processes other than deamination reactions; for example, some changes could have been due to transcription errors that occurred during viral replication or to misincorporation errors that occurred during PCR amplification. However, the number of non-amber/W A→G changes in both transfection experiments combined (10) is greater than the number of non A→G changes (6, not including the 4 U→C transitions), which encompass 10 different nucleotide substitutions and were likely due to such misincorporation errors (Table 1). Moreover, many of the sites modified in the transfected cells were found to be modified under more than one set of experimental conditions. For example, position 973 was modified in a single clone from both replicating and nonreplicating RNAs in cells (Fig. 2); this site was also a preferred site for deamination in vitro by ADAR1 purified from Xenopus (32, 33). In addition, the nonreplicating RNA yielded one modification each at positions 1131 and 1175 (Fig. 2), both of which were preferred sites for modification in vitro (32, 33). Three of the seven A→G transitions found in the 97 clones derived from nonreplicating RNA transcribed in cells occurred at position 1005 (Fig. 2); although this site was not modified in other clones analyzed in this study, it was modified in cDNAs derived from RNA of woodchucks infected with HDV (27).
Although the total number of non-amber/W transitions introduced per molecule is relatively low in transfected cells, comparison of these changes with naturally occurring sequence variations suggests that many of these changes could be deleterious. Of the seven non-amber/W transitions observed in RNAs from transfected cells, four produced nonconservative amino acid changes at positions that are more than 95% conserved among naturally occurring HDV isolates (positions 973, 1034, 1175, and 1177 [28, 37]), one produced a silent codon change in a fully conserved nucleotide position (position 1134 [28, 37]), and one introduced a naturally occurring variation (position 1228 [28, 37]). Thus, it seems likely that some of these changes would have negative effects on the ability of the virus to produce viable progeny. Remarkably, however, all cDNA clones modified at non-amber/W adenosines were also modified at amber/W (Table 2). This association was statistically significant for both the replicating RNA (P = 0.02) and the nonreplicating RNA (P = 0.005). Because genomes modified at amber/W are not viable (they encode HDAg-L, which inhibits replication), coordinate modification of non-amber/W adenosines with amber/W may limit the potentially deleterious effects of spurious editing on virus viability.
TABLE 2.
Comodification of non-amber/W sites with amber/W sites
| RNA | No. of clones with indicated combination of A→G changes
|
||||
|---|---|---|---|---|---|
| None | Amber/W only | Non-amber/W only | Amber/W plus nonamber/W | Pa | |
| Replicating | 60 | 21 | 0 | 3 | 0.02 |
| Nonreplicating | 62 | 30 | 0 | 5 | 0.005 |
| Nonreplicating and replicating | 122 | 51 | 0 | 8 | 0.0001 |
Fisher’s exact test.
HDAg inhibits amber/W editing.
Comparison of amber/W editing in replicating and nonreplicating RNAs suggested that editing occurred more efficiently in the absence of replication (Table 1). Not only was the level of editing slightly higher for the nonreplicating RNA, but the higher level was attained in less than half the time (6 days versus 13 days; see the legend to Fig. 2). Since HDAg was expressed in cells transfected with the replication-competent HDV construct pHDVx1.2-R but not in those transfected with the nonreplicating construct pHDVΔx1-NR (see Fig. 4B, lanes 6 and 7), it seemed possible that the lower amounts of editing in the replicating cells was due to the presence of HDAg. This was a particularly attractive hypothesis since HDAg has been shown to bind HDV RNA (11, 12).
To determine whether HDAg could suppress editing, Huh-7 cells were cotransfected with the nonreplicating RNA expression construct pHDVΔx1-NR and the HDAg expression construct pCMV-AgS, which directs expression of HDAg-S. In separate transfections, we included either pCMV-AgS(fs), which produces no HDAg due to a frameshift-stop codon mutation introduced at codon 7 (8), or pCMV-AgSΔStuSma, which produces HDAg from which 75 amino acids (aa), including the RNA binding domain (23, 24), have been deleted. Immunoblot analysis of lysates from transfected cells indicated that the 75-aa deletion does not affect the level of HDAg expression (data not shown). Although HDAg-S can support replication of some HDV RNAs defective for HDAg synthesis, it does not support replication of the construct used here because RNA elements essential for replication have been deleted (22). To avoid potential PCR amplification of unedited HDAg-encoding mRNA produced from the HDAg expression vector, the primers used were specific for the nonreplicating RNA derived from pHDVΔx1-NR (Fig. 1) and did not amplify any detectable species from cells transfected with the HDAg expression vector alone (data not shown).
Coexpression of HDAg with nonreplicating HDV antigenomic RNA strongly suppressed editing at the amber/W site (Fig. 3). Cotransfection of the expression plasmid for HDAg mRNA with a frameshift-stop codon mutation did not suppress editing (Fig. 3). Thus, suppression required the expression of HDAg and was not due to the mRNA alone, which could conceivably interfere with the editing reaction by base pairing with the substrate. No suppression of editing was observed when the construct pCMV-AgSΔStuSma was cotransfected (Fig. 3), suggesting that the RNA binding domain of HDAg was required for suppression.
To determine whether the suppression of amber/W editing by HDAg might be biologically significant, Huh-7 cells were cotransfected with the nonreplicating RNA expression construct pHDVΔx1-NR along with various amounts of the HDAg expression construct pCMV-AgS. For comparison, Huh-7 cells were also transfected with the replication-competent construct pHDVx1.2-R. The transfections were repeated three times and produced essentially identical results each time; the data shown in Fig. 4 & 5 are representative.
Analysis of amber/W editing 5 days posttransfection indicated that editing levels were about eightfold lower in replicating RNAs than in nonreplicating RNAs produced in the absence of HDAg (Fig. 4A, lanes 6 and 7). However, coexpression of HDAg with nonreplicating HDV RNA strongly suppressed editing at amber/W, and the amount of suppression increased with higher levels of HDAg expression (Fig. 4). At the highest level of HDAg expression, amber/W editing was 10-fold lower than that observed in the absence of HDAg. For an amount of HDAg expression similar to that in cells with replicating HDV RNA, the levels of editing were similar for the replicating and nonreplicating RNAs (Fig. 4A and B; compare lanes 1, 2, and 7), suggesting that the observed inhibition of editing by HDAg is biologically important.
HDAg has been shown to stabilize nonreplicating HDV RNA in transfected cells (23). Indeed, for cells transfected with the nonreplicating construct pHDVΔx1-NR, HDV RNA levels were about 15-fold higher in the presence of the highest amount of HDAg (Fig. 5). Because ADAR1 activity is inhibited in vitro by high levels of substrate (15), we considered the possibility that the observed inhibition of editing by HDAg was an indirect effect of the stabilization of HDV RNA. To address the effect of RNA levels on editing, cells were also cotransfected, in duplicate, with 10-fold less of the nonreplicating HDV RNA expression plasmid pHDVΔx1-NR than was used in the studies presented in Fig. 4. Analysis of amber/W editing by RT-PCR and StyI digestion, and of RNA levels by blot hybridization, indicated that inhibition of editing by HDAg was not related to high RNA levels (Fig. 5). Dramatically different levels of amber/W editing were observed in cells with very similar amounts of HDV RNA but different amounts of HDAg (Fig. 5, columns 1B and 6A). Indeed, in the absence of HDAg, cells expressing lower levels of HDV RNA actually exhibited a slightly decreased level of amber/W editing (28% versus 41% [Fig. 5, column 6]).
DISCUSSION
We have investigated the specificity of HDV RNA editing by sequencing cDNAs derived from transfected cells harboring either replicating or nonreplicating HDV RNA. Consistent with previous studies performed in vitro with purified ADAR1 from Xenopus (32), we found that the biologically significant amber/W site was the preferred modification site. Editing at this site accounted for 89% of all A→G transitions observed in cDNA populations derived from HDV RNA replicating in cells. Editing was highly selective overall: very few A→G transitions occurred at non-amber/W adenosines, and the majority of cDNA clones contained none. For example, under conditions where 29% of the cDNAs from replicating RNA showed an A→G change at the amber/W site, only 0.05% of all other adenosines showed A→G transitions. By extrapolating data obtained from the 358-nt region sequenced to the entire 1,679-nt antigenome, we estimate that 0.16 of the 340 non-amber/W adenosines in each antigenomic RNA were deaminated during 13 days posttransfection. The high specificity of editing observed in vivo did not require viral replication or the viral protein, HDAg, because editing of nonreplicating HDV antigenomes was similarly selective.
The minimal number of A→G changes, or high selectivity, observed on single HDV cDNAs, both in vitro and in cells, is remarkable for several reasons. First, data from in vitro studies of ADAR1, using completely base paired, synthetic substrates, indicate that ∼50% of the adenosines can be deaminated in a molecule with a length comparable to that of HDV RNA (29). Of course, the HDV antigenome differs from the artificial substrates in that it is not completely base paired. As proposed to explain the high selectivity observed for editing of gluR-B mRNAs (17), it seems likely that the numerous mismatches, bulges, and internal loops found in the HDV structure are in part responsible for the high selectivity. Indeed, editing of HDV antigenomic RNA with purified ADAR1 from Xenopus was also found to be highly selective (0.48% non-amber/W adenosines converted versus 16% of amber/W [32, 33]). Thus, the selectivity may be due in large measure to interactions between the RNA and the deaminase.
The 358-nt region analyzed in this study is well conserved among HDV genotype I isolates (28, 37). Of the seven adenosines (excluding amber/W) that were found modified among the 181 clones analyzed (Fig. 2), five are more than 95% conserved among over 100 HDV genotype I isolates. The conservation suggests adenosines at these positions could be necessary for virus propagation and that their deamination could interfere with the viral life cycle. Alternatively, it is also possible that low-level modification at some sites serves an important function in the viral life cycle, similar to what occurs at the amber/W site. The lack of substantial correlation between positions observed to be modified in this study and those seen to vary among different isolates, or among those observed to change during high-dose passage in infected woodchucks (27), could indicate that adenosine deamination is not a predominant mechanism for genetic drift of HDV. Given the high degree of sequence conservation of many of the deamination sites, it is significant that our data showed that modification of non-amber/W sites in vivo was linked to modification at amber/W. All 10 adenosine conversions at other positions occurred in RNAs that were also modified at amber/W (Table 2). A similar, though weaker, linkage was observed for modification of sites neighboring the Q/R site in gluR6 pre-mRNAs (14).
When put in the context of the viral life cycle, the high degree of specificity associated with HDV RNA editing, as well as the coordinate modification of non-amber/W adenosines on RNAs also modified at amber/W, makes biological sense. Sequence changes at non-amber/W adenosines will be passed to the genome during HDV replication. Promiscuous deamination of non-amber/W adenosines within the HDAg coding region could cause deleterious effects on protein expression and function, or deamination within noncoding sequences could alter RNA secondary structures essential for replication and/or packaging. Perhaps during the evolution of HDV there has been selective pressure toward sequences and structures that are not optimal for deamination. The linkage of non-amber/W modifications with amber/W site changes is significant because genomes edited at amber/W produce only the long form of HDAg, which inhibits RNA replication, and would not be able to subsequently infect other cells. Thus, the accumulation of additional modifications on genomes edited at amber/W would not further limit the viability of viral progeny.
One of the most interesting outcomes of our study is the observation that HDAg can alter the extent of editing at the amber/W site or, using the previously defined term (1, 31), the preference for this site. Although there has been considerable speculation that ADAR1 activity is regulated by accessory factors in vivo and some evidence to support such a view (13, 36), our result is the first example in which a specific factor has been identified. Clearly, editing at the amber/W site must be regulated in some manner since complete editing at this site would result in viral genomes that could no longer produce HDAg-S, which is absolutely required for replication (19). The observed ability of HDAg to inhibit amber/W editing in a concentration-dependent manner suggests a role for HDAg in this regulation. Because HDAg can bind HDV RNA (11, 12), and a 75-aa segment including the RNA binding domain was required for the inhibitory effect (Fig. 3), it seems possible that the inhibition could occur by direct steric interference through binding at or near the amber/W site. Certainly our future experiments will address this and related issues.
ACKNOWLEDGMENTS
This work was supported by funds to J.L.C. from NIAID (contract NO1-AI-45179) and funds to B.L.B. from the National Institutes of Health (grant GM 44073) and the David and Lucile Packard Foundation. Oligodeoxyoligonucleotides were synthesized by the Utah Cancer Center Protein/DNA core facility, supported by the National Cancer Institute (grant 5 P30 CA42014). B.L.B. is a Howard Hughes Medical Institute associate investigator.
We thank Thomas L. Brown for excellent technical assistance, R. Hough and S. Hurst for providing purified ADAR1, and M. Robertson and E. Lawrence (Health Sciences Center Sequencing Facility) for their extensive sequencing of numerous cDNA clones. We also appreciate the helpful suggestions and discussions from the members of our research groups.
REFERENCES
- 1.Bass B L. RNA editing and hypermutation by adenosine deamination. Trends Biochem Sci. 1997;22:157–162. doi: 10.1016/s0968-0004(97)01035-9. [DOI] [PubMed] [Google Scholar]
- 2.Bass B L, Nishikura K, Keller W, Seeburg P H, Emeson R B, Ma O C, Samuel C E, Herbert A. A standardized nomenclature for adenosine deaminases that act on RNA. RNA. 1997;3:947–949. [PMC free article] [PubMed] [Google Scholar]
- 3.Beard M R, MacNaughton T B, Gowans E J. Identification and characterization of a hepatitis delta virus RNA transcriptional promoter. J Virol. 1996;70:4986–4995. doi: 10.1128/jvi.70.8.4986-4995.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bergmann K F, Gerin J L. Antigens of hepatitis delta virus in the liver and serum of humans and animals. J Infect Dis. 1986;154:702–706. doi: 10.1093/infdis/154.4.702. [DOI] [PubMed] [Google Scholar]
- 5.Casey, J. L. Unpublished observations.
- 6.Casey J L, Bergmann K F, Brown T L, Gerin J L. Structural requirements for RNA editing in hepatitis delta virus: evidence for a uridine-to-cytidine editing mechanism. Proc Natl Acad Sci USA. 1992;89:7149–7153. doi: 10.1073/pnas.89.15.7149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Casey J L, Brown T L, Colan E J, Wignall F S, Gerin J L. A genotype of hepatitis D virus that occurs in northern South America. Proc Natl Acad Sci USA. 1993;90:9016–9020. doi: 10.1073/pnas.90.19.9016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Casey, J. L., and J. L. Gerin. Genotype-specific complementation of hepatitis delta virus RNA replication by hepatitis delta antigen. J. Virol., in press. [DOI] [PMC free article] [PubMed]
- 9.Casey J L, Gerin J L. Hepatitis D virus RNA editing: specific modification of adenosine in the antigenomic RNA. J Virol. 1995;69:7593–7600. doi: 10.1128/jvi.69.12.7593-7600.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Casey J L, Koeller D M, Ramin V C, Klausner R D, Harford J B. Iron regulation of transferrin receptor mRNA levels requires iron-responsive elements and a rapid turnover determinant in the 3′ untranslated region of the mRNA. EMBO J. 1989;8:3693–3699. doi: 10.1002/j.1460-2075.1989.tb08544.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chang M F, Baker S C, Soe L H, Kamahora T, Keck J G, Makino S, Govindarajan S, Lai M M. Human hepatitis delta antigen is a nuclear phosphoprotein with RNA-binding activity. J Virol. 1988;62:2403–2410. doi: 10.1128/jvi.62.7.2403-2410.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chao M, Hsieh S Y, Taylor J. The antigen of hepatitis delta virus: examination of in vitro RNA-binding specificity. J Virol. 1991;65:4057–4062. doi: 10.1128/jvi.65.8.4057-4062.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dabiri G A, Lai F, Drakas R A, Nishikura K. Editing of the GLuR-B ion channel RNA in vitro by recombinant double-stranded RNA adenosine deaminase. EMBO J. 1996;15:34–45. [PMC free article] [PubMed] [Google Scholar]
- 14.Herb A, Higuchi M, Sprengel R, Seeburg P H. Q/R site editing in kainate receptor GluR5 and GluR6 pre-mRNAs requires distant intronic sequences. Proc Natl Acad Sci USA. 1996;93:1875–1880. doi: 10.1073/pnas.93.5.1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hough R F, Bass B L. Purification of the Xenopus laevis double-stranded RNA adenosine deaminase. J Biol Chem. 1994;269:9933–9939. [PubMed] [Google Scholar]
- 16.Hsieh S Y, Chao M, Coates L, Taylor J. Hepatitis delta virus genome replication: a polyadenylated mRNA for delta antigen. J Virol. 1990;64:3192–3198. doi: 10.1128/jvi.64.7.3192-3198.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hurst S R, Hough R F, Aruscavage P J, Bass B L. Deamination of mammalian glutamate receptor RNA by Xenopus dsRNA adenosine deaminase: similarities to in vivo RNA editing. RNA. 1995;1:1051–1060. [PMC free article] [PubMed] [Google Scholar]
- 18.Jilbert A R, Gowans E J, Macnaughton T B, Burrell C J. Characterization of a subgenomic HDV-specific poly(A)+ RNA species isolated from human hepatoma cells supporting HDV RNA replication. Prog Clin Biol Res. 1991;364:309–314. [PubMed] [Google Scholar]
- 19.Kuo M Y, Chao M, Taylor J. Initiation of replication of the human hepatitis delta virus genome from cloned DNA: role of delta antigen. J Virol. 1989;63:1945–1950. doi: 10.1128/jvi.63.5.1945-1950.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kwok S, Higuchi R. Avoiding false positives with PCR. Nature. 1989;339:237–238. doi: 10.1038/339237a0. [DOI] [PubMed] [Google Scholar]
- 21.Lai M M. The molecular biology of hepatitis delta virus. Annu Rev Biochem. 1985;64:259–286. doi: 10.1146/annurev.bi.64.070195.001355. [DOI] [PubMed] [Google Scholar]
- 22.Lazinski D W, Taylor J M. Expression of hepatitis delta virus RNA deletions: cis and trans requirements for self-cleavage, ligation, and RNA packaging. J Virol. 1994;68:2879–2888. doi: 10.1128/jvi.68.5.2879-2888.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lazinski D W, Taylor J M. Relating structure to function in the hepatitis delta virus antigen. J Virol. 1993;67:2672–2680. doi: 10.1128/jvi.67.5.2672-2680.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee C Z, Lin J H, Chao M, McKnight K, Lai M M. RNA-binding activity of hepatitis delta antigen involves two arginine-rich motifs and is required for hepatitis delta virus RNA replication. J Virol. 1993;67:2221–2227. doi: 10.1128/jvi.67.4.2221-2227.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Luo G X, Chao M, Hsieh S Y, Sureau C, Nishikura K, Taylor J. A specific base transition occurs on replicating hepatitis delta virus RNA. J Virol. 1990;64:1021–1027. doi: 10.1128/jvi.64.3.1021-1027.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Melcher T, Maas S, Higuchi M, Keller W, Seeburg P H. Editing of alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptor gluR-B pre-mRNA in vitro reveals site-selective adenosine to inosine conversion. J Biol Chem. 1995;270:8566–8570. doi: 10.1074/jbc.270.15.8566. [DOI] [PubMed] [Google Scholar]
- 27.Netter H J, Wu T T, Bockol M, Cywinski A, Ryu W S, Tennant B C, Taylor J M. Nucleotide sequence stability of the genome of hepatitis delta virus. J Virol. 1995;69:1687–1692. doi: 10.1128/jvi.69.3.1687-1692.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Niro G A, Smedile A, Andriulli A, Rizzetto M, Gerin J L, Casey J L. Predominance of hepatitis delta virus genotype I among chronically infected Italian patients. Hepatology. 1997;25:728–734. doi: 10.1002/hep.510250339. [DOI] [PubMed] [Google Scholar]
- 29.Nishikura K, Yoo C, Kim U, Murray J M, Estes P A, Cash F E, Liebhaber S A. Substrate specificity of the dsRNA unwinding/modifying activity. EMBO J. 1991;10:3523–3532. doi: 10.1002/j.1460-2075.1991.tb04916.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Perrotta A T, Been M D. A pseudoknot-like structure required for efficient self-cleavage of hepatitis delta virus RNA. Nature. 1991;350:434–436. doi: 10.1038/350434a0. [DOI] [PubMed] [Google Scholar]
- 31.Polson A G, Bass B L. Preferential selection of adenosines for modification by double-stranded RNA adenosine deaminase. EMBO J. 1994;13:5701–5711. doi: 10.1002/j.1460-2075.1994.tb06908.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Polson A G, Bass B L, Casey J L. RNA editing of hepatitis delta virus antigenome by dsRNA-adenosine deaminase. Nature. 1996;380:454–456. doi: 10.1038/380454a0. [DOI] [PubMed] [Google Scholar]
- 33.Polson, A. G., H. L. Ley, B. L. Bass, and J. L. Casey. Unpublished results.
- 34.Rizzetto M. The delta agent. Hepatology. 1983;3:729–737. doi: 10.1002/hep.1840030518. [DOI] [PubMed] [Google Scholar]
- 35.Ryu W S, Bayer M, Taylor J. Assembly of hepatitis delta virus particles. J Virol. 1992;66:2310–2315. doi: 10.1128/jvi.66.4.2310-2315.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Saccomanno L, Bass B L. The cytoplasm of Xenopus oocytes contains a factor that protects double-stranded RNA from adenosine-to-inosine modification. Mol Cell Biol. 1994;14:5425–5432. doi: 10.1128/mcb.14.8.5425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shakil A O, Hadziyannis S, Hoofnagle J H, DiBisceglie A M, Gerin J L, Casey J L. Geographic distribution and genetic variability of hepatitis delta virus genotype I. Virology. 1997;234:160–167. doi: 10.1006/viro.1997.8644. [DOI] [PubMed] [Google Scholar]
- 38.Sharmeen L, Kuo M Y, Taylor J. Self-ligating RNA sequences on the antigenome of human hepatitis delta virus. J Virol. 1989;663:1428–1430. doi: 10.1128/jvi.63.3.1428-1430.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang K S, Choo Q L, Weiner A J, Ou J H, Najarian R C, Thayer R M, Mullenbach G T, Denniston K J, Gerin J L, Houghton M. Structure, sequence and expression of the hepatitis delta viral genome. Nature. 1986;323:508–514. doi: 10.1038/323508a0. [DOI] [PubMed] [Google Scholar]
- 40.Weiner A J, Choo Q L, Wang K S, Govindarajan S, Redeker A G, Gerin J L, Houghton M. A single antigenomic open reading frame of the hepatitis delta virus encodes the epitope(s) of both hepatitis delta antigen polypeptides p24 delta and p27 delta. J Virol. 1988;62:594–599. doi: 10.1128/jvi.62.2.594-599.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu H N, Lai M M. Reversible cleavage and ligation of hepatitis delta virus. Science. 1989;234:652–654. doi: 10.1126/science.2492677. [DOI] [PubMed] [Google Scholar]
- 42.Wu J C, Chen P J, Kuo M Y, Lee S D, Chen D S, Ting L P. Production of hepatitis delta virus and suppression of helper hepatitis B virus in a human hepatoma cell line. J Virol. 1991;65:1099–1104. doi: 10.1128/jvi.65.3.1099-1104.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zheng H, Fu T B, Lazinski D, Taylor J. Editing on the genomic RNA of human hepatitis delta virus. J Virol. 1992;66:4693–4697. doi: 10.1128/jvi.66.8.4693-4697.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]