

Abstract
The mechanisms by which growth factor-induced signals are propagated to the nucleus, leading to the activation of the transcription factor CREB, have been characterized. Nerve growth factor (NGF) was found to activate multiple signaling pathways that mediate the phosphorylation of CREB at the critical regulatory site, serine 133 (Ser-133). NGF activates the extracellular signal-regulated kinase (ERK) mitogen-activated protein kinases (MAPKs), which in turn activate the pp90 ribosomal S6 kinase (RSK) family of Ser/Thr kinases, all three members of which were found to catalyze CREB Ser-133 phosphorylation in vitro and in vivo. In addition to the ERK/RSK pathway, we found that NGF activated the p38 MAPK and its downstream effector, MAPK-activated protein kinase 2 (MAPKAP kinase 2), resulting in phosphorylation of CREB at Ser-133. Inhibition of either the ERK/RSK or the p38/MAPKAP kinase 2 pathway only partially blocked NGF-induced CREB Ser-133 phosphorylation, suggesting that either pathway alone is sufficient for coupling the NGF signal to CREB activation. However, inhibition of both the ERK/RSK and the p38/MAPKAP kinase 2 pathways completely abolished NGF-induced CREB Ser-133 phosphorylation. These findings indicate that NGF activates two distinct MAPK pathways, both of which contribute to the phosphorylation of the transcription factor CREB and the activation of immediate-early genes.
The neurotrophins are a family of secreted peptide growth factors that regulate the proliferation, differentiation, and survival of neurons and their precursors (44). The neurotrophin nerve growth factor (NGF) was the first growth factor to be identified and has served as a model for studying the mechanisms of action of neurotrophins and growth factors. The mechanisms by which neurotrophins generate diverse cellular responses have been studied extensively with the rat pheochromocytoma cell line PC12 (28). When exposed to NGF, PC12 cells exit the cell cycle and differentiate into sympathetic neuron-like cells (29). The effects of NGF on PC12 cells are mediated by the NGF receptor TrkA (40, 41). TrkA, like other growth factor receptors, is a tyrosine kinase that is activated upon ligand binding and receptor dimerization (37, 39). The activated TrkA tyrosine kinase triggers a signaling cascade that includes the activation of the small guanine nucleotide binding protein Ras (56) followed by the sequential phosphorylation and activation of Raf, MEK (mitogen-activated protein kinase [MAPK] or extracellular signal-regulated kinase [ERK] kinase), the ERKs, a subgroup of the MAPK superfamily, and RSKs (pp90 ribosomal S6 kinases) (67, 74). An important function of this kinase cascade is to induce the phosphorylation and activation of transcription factors in the nucleus to initiate new programs of gene expression (31).
Activation of the Ras/ERK pathway in NGF-treated PC12 cells has been shown to lead to the induction of a class of genes termed immediate-early genes (IEGs) (61, 64). IEGs are activated in a rapid, robust, and transient manner independently of new protein synthesis (26, 27). Many of the IEGs encode transcription factors whose expression is required for the activation of a class of delayed-response genes, which encode proteins that may contribute to the differentiation of NGF-treated PC12 cells (31, 43). Of the 50 to 100 IEGs that are activated by NGF and other growth factors, the c-fos proto-oncogene has been most extensively studied. One critical regulatory element within the c-fos promoter is the serum response element (SRE) (63, 71). The SRE is a binding site for a ternary transcription complex composed of a serum response factor (SRF) dimer and a single molecule of ternary complex factor (TCF), a family of Ets domain-containing transcription factors (53, 62, 68). Upon exposure of PC12 cells to NGF, the TCF component of the ternary complex becomes newly phosphorylated on several key amino acid residues; phosphorylation of these sites is critical for the ability of TCF to stimulate IEG transcription (22, 23, 34, 35, 47, 50, 55). A variety of experiments suggest that in NGF-treated PC12 cells, ERKs are the enzymes that catalyze the phosphorylation of TCF (50), although other members of the MAPK family may have a similar function.
In addition to the TCF-SRF complex, the cyclic AMP response element binding protein (CREB), which binds to three separate sequences within the c-fos promoter distinct from the SRE, appears to be required for NGF induction of c-fos transcription (5, 24). Mutations within the CREB binding sites in the c-fos promoter effectively abolish NGF induction of c-fos transcription (5, 24). NGF treatment triggers the phosphorylation of CREB at a critical regulatory site, Ser-133 (24). Once phosphorylated at this site, CREB induces c-fos transcription by cooperating with factors at the SRE, possibly SRF and TCF (5). This cooperation may be mediated by the transcriptional coactivator CREB binding protein (CBP), which specifically binds to Ser-133-phosphorylated CREB (8) and also binds to TCF (36) and SRF (57).
The identities of the growth factor-regulated CREB kinases are currently a subject of some controversy. A growth factor-inducible, Ras-dependent CREB Ser-133 kinase was identified (24), and its purification and sequencing revealed it to be identical to the Ser/Thr kinase RSK2 (76). NGF activation of the Ras/ERK pathway leads to the phosphorylation and activation of RSK2, which then phosphorylates CREB at Ser-133. In contrast to the findings with NGF, the p38 MAPK and its downstream kinase MAPK-activated protein kinase 2 (MAPKAP kinase 2), but not the ERK/RSK pathway, were recently shown to mediate CREB Ser-133 phosphorylation in cells exposed to basic fibroblast growth factor (bFGF) (66). In addition to RSK2 and MAPKAP kinase 2, the pp70 ribosomal S6 kinase (pp70S6K) was suggested to mediate serum-induced phosphorylation of CREM, a transcription factor closely related to CREB, at a site (Ser-117) which is equivalent to CREB Ser-133 (14). Thus, a number of different kinases may be capable of mediating growth factor induction of CREB phosphorylation under different circumstances, although the relative contributions of particular kinases in cells treated with a specific growth factor have not been investigated.
To characterize further the mechanisms by which NGF stimulates CREB Ser-133 phosphorylation, the roles played by the RSK family members RSK1, RSK2, and RSK3, pp70S6K, and the p38 MAPK in this process were examined. All three members of the RSK family were found to be activated by NGF and to be capable of phosphorylating CREB at Ser-133 in vitro and in vivo. In addition, NGF was found to activate p38 MAPK and its downstream effector MAPKAP kinase 2, which may then catalyze CREB phosphorylation at Ser-133. In NGF-treated PC12 cells, inhibition of both the ERK/RSK pathway and the p38/MAPKAP kinase 2 pathway was required to abolish CREB Ser-133 phosphorylation completely, indicating that both of these pathways contribute to CREB phosphorylation. Thus, a variety of signaling pathways have evolved that can trigger CREB phosphorylation at the critical amino acid residue Ser-133 and thereby activate IEG transcription.
MATERIALS AND METHODS
Materials.
NGF was prepared from mouse salivary glands by a previously described procedure (51). Human recombinant EGF was from Collaborative Biomedical Research. Platelet-derived growth factor (PDGF-BB) was from Upstate Biotechnology. The MEK inhibitor PD 098059 was from New England Biolabs, and the p38 MAPK inhibitor SB 203580 was from Calbiochem. Both compounds were dissolved in dimethyl sulfoxide. The expression vector pMT2-HA-RSK2 has been described previously (76). pMT2-HA-RSK1 and pMT2-HA-RSK3 were provided by J. Avruch and D. E. Moller, respectively. The p38 MAPK and MKK6 expression vectors were provided by R. Davis. The plasmid CMV-GAL4-CREB, encoding a hybrid protein that has the NH2-terminal 147 amino acids of the yeast transcription factor GAL4 fused to full-length CREB, has been described previously (4). The phosphospecific antibodies to ERK, p38 MAPK, and JNK were purchased from New England Biolabs. Antibodies to RSK1, RSK2, and MAPKAP kinase 2 were from Upstate Biotechnology. Antibodies to RSK3 were from Santa Cruz Biotechnology.
Cell culture and treatment.
Parental PC12 cells were grown under 10% CO2 on collagen-coated plates in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 5% fetal bovine serum and 10% horse serum. NIH 3T3 cells and COS cells were maintained under 5% CO2 in DMEM supplemented with 10% calf serum and 10% fetal bovine serum, respectively. HepG2 cell lines were maintained under 5% CO2 in DMEM containing 10% calf serum and 500 μg of G418 per ml. The cells were serum starved in DMEM plus 0.1% calf serum for 2 days before treatment and lysis.
Transfection of COS cells.
Transfection of COS cells was done by the DEAE-dextran method as previously described (76). Briefly, COS cells at about 70% confluence were used for transfection. Two days after transfection, the cells were treated as indicated, washed once in ice-cold phosphate-buffered saline, and lysed in boiling SDS sample buffer (62.5 mM Tris [pH 6.8], 1% sodium dodecyl sulfate [SDS], 10% glycerol, 5% β-mercaptoethanol) for SDS-polyacrylamide gel electrophoresis (PAGE) and immunoblot analysis. Alternatively, cells were lysed in Nonidet P-40 lysis buffer for immune complex kinase assays.
Immunoprecipitations.
The cells were washed once in ice-cold PBS and lysed on ice in Triton X-100 lysis buffer (20 mM Tris [pH 6.8], 137 mM NaCl, 50 mM β-glycerophosphate, 1 mM Na3VO4, 2 mM EDTA, 2 mM EGTA, 1 mM dithiothreitol [DTT], 1% Triton X-100, 10% glycerol, 1 mM benzamidine, leupeptin [20 μg/ml], pepstatin [10 μg/ml], aprotinin [20 μg/ml], 1 mM phenylmethylsulfonyl fluoride). Cell lysates were passed through a 22-gauge needle three times and centrifuged at 100,000 × g for 15 min at 4°C. Antibodies and protein A-Sepharose beads or protein G-Sepharose beads (washed in lysis buffer) were then added to the 100,000 × g supernatants. After a 1-h incubation at 4°C, the immune complex was collected by centrifugation and washed twice with lysis buffer and twice with kinase buffer [50 mM piperazine-N,N′-bis(2-ethanesulfonic acid) (PIPES; pH 7.3), 10 mM MgCl2, 1 mM DTT]. Washed immune complexes were then used for protein kinase assays. For MAPKAP kinase 2 assays, anti-MAPKAP kinase 2 antibodies were precoupled to protein A-Sepharose beads at 4°C for 1 h and washed once in lysis buffer. The protein A-antibody complex was then added to 100,000 × g supernatant which had been precleared with protein A-Sepharose beads. After a 1-h incubation at 4°C, the immune complexes were washed once in lysis buffer with 500 mM NaCl, once in lysis buffer, and twice in kinase buffer before being used for protein kinase assays.
Protein kinase assays.
For in vitro kinase assays with CREBtide (see below) as a substrate, immune complexes were added to a final kinase reaction mixture containing 50 mM PIPES (pH 7.2), 10 mM MgCl2, 50 μM ATP, 5 μCi of [γ-32P]ATP, 1 mM DTT, 1 μM protein kinase inhibitor, and 0.1 mM CREBtide in a final reaction volume of 50 μl. The reaction mixture was incubated at 30°C for 20 min. The reaction was terminated by spotting 25 μl of the reaction mixture onto 2- by 1-cm strips of p81 phosphocellulose filter paper. The filters were washed five times in 75 mM phosphoric acid and counted for 32P incorporation by liquid scintillation counting. JNK (c-Jun N-terminal kinase) and p38 MAPK activities were measured as described previously (75) with glutathione S-transferase (GST)–c-Jun (amino acid residues 1 to 79) and GST-ATF2 (amino acid residues 1 to 109), respectively, as substrates.
Immunoblot analysis.
The cells were treated with growth factors as indicated and then washed once in ice-cold phosphate-buffered saline. They were then lysed in boiling SDS sample buffer. The lysates were boiled for 5 min, separated by SDS-PAGE, and transferred to nitrocellulose membranes. The filters were incubated for 1 h at room temperature in TBST (20 mM Tris [pH 7.5], 145 mM NaCl, 0.05% Tween 20) containing 4% bovine serum albumin (BSA) (United States Biochemicals). The filters then were incubated for 2 to 3 h at room temperature with primary antibodies in TBST containing 4% BSA. The filters were washed four times for 5 min each in TBST containing 0.5% BSA and then incubated at room temperature for 1 h with horseradish peroxidase-conjugated secondary antibodies in TBST containing 4% BSA. The filters were washed four more times for 5 min each in TBST with 0.5% BSA, and bands were detected with an enhanced chemiluminescence system (Amersham).
RESULTS
All three members of the RSK family are activated in response to NGF and can phosphorylate CREB Ser-133 in vitro and in vivo.
We have previously reported that RSK2, a member of the pp90RSK family of Ser/Thr kinases, is activated in response to NGF treatment of PC12 cells and phosphorylates CREB at the critical regulatory site Ser-133 (76). pp90RSK was originally isolated from unfertilized Xenopus eggs, where it may phosphorylate the S6 protein of the 40S ribosomal subunit (19, 38). However, it has since been demonstrated with mammalian cells that pp70S6K, not pp90RSK, is the S6 kinase in vivo (3, 10). Three members of the pp90RSK family of serine/threonine kinases have been identified in mammalian cells (1, 30, 52). Members of the RSK family are activated by the ERK subfamily of MAPKs and translocate to the nucleus, where they presumably phosphorylate their nuclear targets including transcription factors (6, 7, 24, 58, 76, 79).
The finding that RSK2 is an NGF-inducible CREB Ser-133 kinase (76) raised the possibility that the other RSKs also contribute to NGF-induced CREB Ser-133 phosphorylation. Therefore, we investigated whether RSK1 and RSK3, like RSK2, are activated in response to NGF treatment of PC12 cells. RSK1, RSK2, and RSK3 were immunoprecipitated from PC12 extracts with antibodies specific to RSK1, RSK2, and RSK3, respectively, and their kinase activities were measured in an vitro kinase assay with CREBtide (a peptide which corresponds to amino acids 123 to 136 of CREB and contains the CREB Ser-133 phosphorylation site) (24) as a substrate. As shown in Fig. 1, all three RSKs were induced by NGF and phosphorylated CREBtide in vitro.
FIG. 1.

All three RSK family members are activated by NGF. PC12 cells were either untreated or treated with NGF (30 ng/ml) for 10 min. RSK1, RSK2, and RSK3 were immunoprecipitated from extracts of untreated or NGF-treated PC12 cells with antibodies specific for RSK1, RSK2, or RSK3 respectively. RSK kinase activities in the immune complexes were determined by an in vitro kinase assay with CREBtide as a substrate. The fold activation is the ratio of kinase activity from NGF-treated cells to that from untreated cells. Data are from three separate experiments. Mean values are plotted; error bars represent standard errors of the mean (SEM).
To determine which of the three RSKs was capable of phosphorylating CREB Ser-133 in vivo in response to growth factor stimulation, we introduced into COS cells hemagglutinin (HA) epitope-tagged versions of RSK1, RSK2, and RSK3. Expression vectors for HA-RSK1, HA-RSK2, or HA-RSK3 were transfected into COS cells together with Gal4-CREB, a hybrid protein containing the N-terminal 147 amino acids of the yeast transcription factor Gal4 fused to full-length CREB. The difference in mobility between Gal4-CREB and CREB on SDS-PAGE made it possible to examine the phosphorylation status of Ser-133 of the transfected Gal4-CREB without interference from the endogenous CREB. Transfected COS cells were stimulated with epidermal growth factor (EGF) rather than NGF since COS cells express the EGF receptor but do not express TrkA. After EGF treatment, the kinase activities of the HA-RSKs toward CREBtide were measured in an immune complex kinase assay using the monoclonal anti-HA antibody (12CA5) to precipitate the HA-RSKs (Fig. 2A). All three RSKs expressed in COS cells were activated upon EGF treatment to phosphorylate CREBtide as measured by an in vitro kinase assay. To determine whether the expression and activation of a RSK enhanced CREB phosphorylation within cells upon EGF treatment, the phosphorylation of CREB Ser-133 was monitored with an antibody that specifically recognizes the Ser-133-phosphorylated form of CREB (anti-PCREB) (24a). The expression of RSK1, RSK2, or RSK3 in COS cells substantially increased the level of EGF-induced Gal4CREB Ser-133 phosphorylation (Fig. 2B). Thus, all three RSK family members appear to be activated and able to phosphorylate CREB Ser-133 in cells upon growth factor stimulation.
FIG. 2.
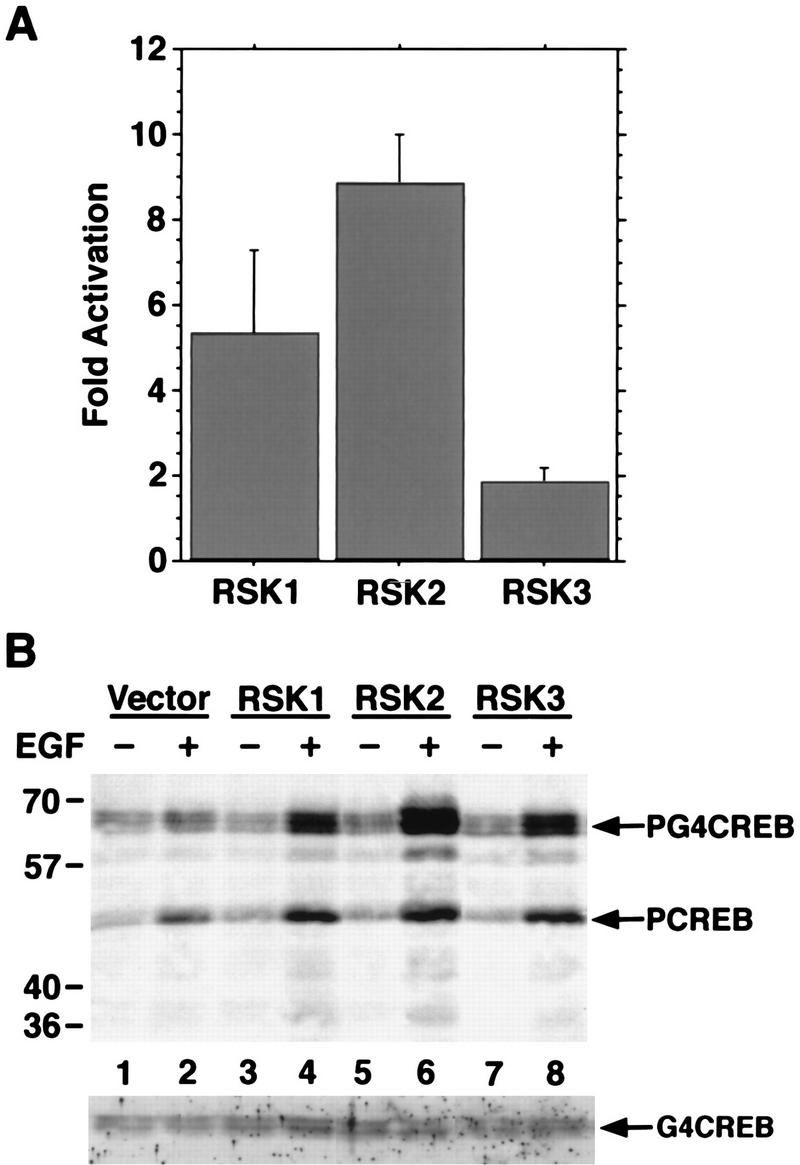
All three RSK family members phosphorylate CREB Ser-133 in response to growth factor stimulation. (A) COS cells were transfected with expression constructs for either HA-RSK1, HA-RSK2, or HA-RSK3. Cells were either left untreated or treated with EGF for 10 min. HA-tagged RSKs were immunoprecipitated from lysates of untreated or EGF-treated transfected COS cells with a monoclonal anti-HA antibody (12CA5). Kinase activities were determined by an in vitro kinase assay with CREBtide as a substrate. Fold activation indicates the ratio of kinase activity from EGF-treated cells to that from untreated cells. Data are from three separate experiments. Error bars represent SEM. (B) COS cells were transfected with 1 μg of CMV-Gal4-CREB together with 10 μg of pMT vector (lanes 1 and 2), pMT2-HA-RSK1 (lanes 3 and 4), pMT2-HA-RSK2 (lanes 5 and 6), or pMT-HA-RSK3 (lanes 7 and 8). Transfected COS cells were left untreated (lanes 1, 3, 5, and 7) or treated for 10 min with EGF (30 ng/ml) (lanes 2, 4, 6, and 8). Lysates of transfected COS cells were separated by SDS-PAGE and immunoblotted with anti-PCREB (upper panel) or anti-Gal4 (lower panel) antibodies.
To examine the contribution of the RSKs to CREB Ser-133 phosphorylation in NGF-treated PC12 cells, we used a synthetic compound, PD 098059, that specifically inhibits the Ras/MEK/ERK pathway by binding to the inactive form of MEK and preventing its activation (2, 17). Incubation with PD 098059 blocks the activation of MEK, ERK, and the RSKs in EGF-treated NIH 3T3 cells or NGF-treated PC12 cells but does not significantly affect the activation of a variety of other kinases, including the MEK-related kinase MKK4, p38 MAPK, JNK (the c-Jun N-terminal kinase), and pp70S6K, by various stimuli (2, 17, 54, 76a, 78) (see Fig. 8). Pretreatment of PC12 cells with PD 098059 almost completely blocked NGF activation of ERKs, as assayed with a phosphospecific ERK antibody that recognizes the dually phosphorylated forms of ERKs (p42 and p44 MAPK). In addition, PD 098059 effectively blocked NGF-induced RSK2 phosphorylation and activation of RSK2 kinase activity (Fig. 3A). PD 098059 pretreatment was also found to partially but not completely block NGF-induced CREB Ser-133 phosphorylation (Fig. 3B), suggesting that the MEK/ERK/RSK pathway contributes to NGF-induced CREB Ser-133 phosphorylation. It is possible that the ERK/RSK pathway was not completely blocked by PD 098059, since a low level of RSK2 activity was detected in the presence of the inhibitor (Fig. 3A). However, as demonstrated below, this low RSK2 activity is not likely to account for the significant level of CREB Ser-133 phosphorylation that is still detected when PC12 cells are exposed to NGF in the presence of PD 098059 (Fig. 3B). The finding that PD 098059 almost completely inhibited ERK and RSK activation but only partially blocked the NGF induction of CREB Ser-133 phosphorylation suggested that a signaling pathway(s) in addition to the MEK/ERK/RSK pathway might contribute to NGF-induced CREB Ser-133 phosphorylation.
FIG. 8.

MAPKAP kinase 2 is activated by NGF. PC12 cells were not pretreated, pretreated for 1 h with PD 098059 (100 μM), or pretreated for 1 h with SB 203580 (5 μM). The cells were then left untreated, treated for 10 min with NGF (30 ng/ml), or treated with UV irradiation. MAPKAP kinase 2 was immunoprecipitated from cell lysates, and its kinase activity was determined by an in vitro kinase assay with CREBtide as a substrate. Data are means from three independent experiments. Error bars represent SEM.
FIG. 3.
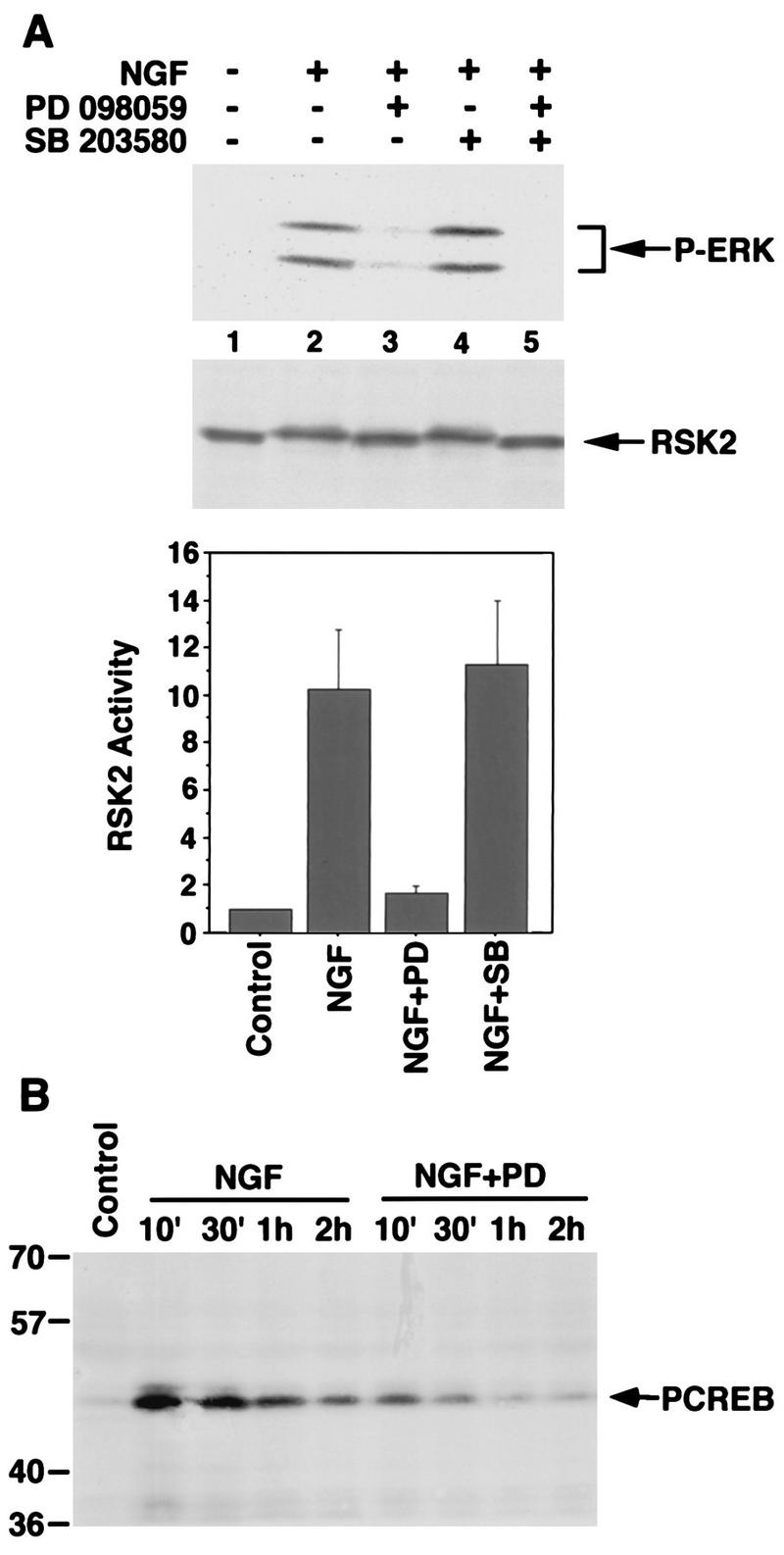
Inhibition of the MEK/ERK/RSK pathway leads to decreased NGF-stimulated CREB Ser-133 phosphorylation. (A) PC12 cells were either not pretreated, preincubated for 1 h with PD 098059 (100 μM), or preincubated for 1 h with SB 203580 (5 μM). The cells were then either lysed directly or treated with NGF (30 ng/ml) for 10 min before lysis. Cell lysates were separated by SDS-PAGE and immunoblotted with anti-phospho-ERK (top) or anti-RSK2 antibodies (middle). Cells were either left untreated, or treated for 10 min with NGF (30 ng/ml). RSK2 was immunoprecipitated from cell lysates, and kinase activity was determined by an in vitro kinase assay with CREBtide as the substrate (bottom). Data are means from three independent experiments. Error bars represent SEM. (B) PC12 cells were either not pretreated or pretreated for 1 h with PD 098059 (100 μM). The cells were then either untreated or treated with NGF (30 ng/ml) for the indicated periods. Cell lysates were separated by SDS-PAGE and immunoblotted with anti-PCREB antibodies.
pp70S6K is not involved in mediating NGF-induced CREB Ser-133 phosphorylation.
The observation that inhibition of the MEK/ERK/RSK pathway only partially blocked NGF-induced CREB Ser-133 phosphorylation prompted us to investigate the possibility that additional pathways also signal to CREB. The cyclic AMP-dependent protein kinase A and the multifunctional Ca2+-calmodulin-dependent kinases (CaMKs) mediate CREB Ser-133 phosphorylation in cells treated with agents that increase the level of intracellular cAMP or Ca2+, respectively (25, 65). However, these kinases were previously found not to mediate CREB Ser-133 phosphorylation in NGF-treated PC12 cells (24).
It has been suggested that pp70S6K plays a role in CREB Ser-133 phosphorylation in serum-stimulated cells (14). pp70S6K, which is activated in response to growth factor or serum stimulation, mediates the phosphorylation of the CREB-related transcription factor CREM at Ser-117 (14), a site that corresponds to Ser-133 within CREB. This finding raised the possibility that pp70S6K also catalyzes the phosphorylation of CREB at Ser-133 in growth factor-stimulated cells. However, treatment of PC12 cells with the immunosuppressant rapamycin effectively blocks NGF activation of pp70S6K but has no effect on NGF-induced CREB Ser-133 phosphorylation, indicating that pp70S6K activation is not required for NGF-induced CREB Ser-133 phosphorylation (24). Nevertheless, it remained possible that while pp70S6K might not be a critical mediator of CREB Ser-133 phosphorylation in NGF-treated PC12 cells, it was still capable of mediating this event within these or other cells.
To further examine if activation of pp70S6K contributes to growth factor-induced CREB Ser-133 phosphorylation, we used a series of HepG2 cell lines that stably express wild-type or mutant versions of the PDGF receptor (PDGFR) (70). Phosphorylation of specific tyrosine residues within the PDGFR is required for the recruitment and activation of various signaling molecules in response to PDGF (11). Phosphorylation of Y740 and Y751 leads to the activation of phosphatidylinositol 3-kinase (PI3K), while phosphorylation of Y771, Y1009, and Y1021 activates the GTPase-activating protein of Ras (RasGAP), the protein tyrosine phosphatase (SHP-2), and phospholipase Cγ1, respectively (11). Activation of PI3K is known to be necessary and sufficient for pp70S6K activation in response to PDGF treatment; thus, Y740 and Y751 within the PDGFR are required for PDGF induction of pp70S6K (9). Consistent with these observations, PDGF treatment activates PI3K and pp70S6K in HepG2 cells expressing the wild-type PDGFR or a mutant PDGFR in which Y771, Y1009, and Y1021 are mutated to phenylalanines, whereas PDGF treatment does not activate PI3K and pp70S6K in HepG2 lines expressing the F5 PDGFR, in which all five tyrosines are replaced by phenylalanine (9, 70).
We found that PDGF-stimulated CREB Ser-133 phosphorylation in HepG2 cells did not correlate with pp70S6K activation (Fig. 4). Although pp70S6K activation occurs in the F3 PDGFR mutant line (reference 9 and data not shown), CREB Ser-133 phosphorylation was not stimulated by PDGF treatment of these cells. In contrast, although pp70S6K was only minimally activated in cells expressing a mutant PDGFR (F740/751) in which the tyrosines required for PI3K activation (Y740 and Y751) are mutated to phenylalanine (reference 9 and data not shown), PDGF induced CREB Ser-133 phosphorylation to levels similar to those observed in the wild-type PDGFR-expressing line. These results suggest that pp70S6K activity is neither necessary nor sufficient for CREB Ser-133 phosphorylation in PDGF-treated cells. Thus, the available evidence suggests that pp70S6K does not mediate growth factor induction of CREB Ser-133 phosphorylation.
FIG. 4.

Growth factor-induced pp70S6K activation is not sufficient to stimulate CREB Ser-133 phosphorylation. HepG2 cell lines stably expressing wild-type and mutant versions of PDGFR were serum starved for 2 days and either left untreated (lanes 1, 3, 5, 7, and 9) or treated for 10 min with PDGF (20 ng/ml) (lanes 2, 4, 6, 8, and 10). Cell lysates were separated by SDS-PAGE and immunoblotted with anti-PCREB (top) or anti-CREB (bottom) antibodies. The HepG2 lines were stably transfected with the empty pLXSN vector (N), the wild-type PDGFR (wt PDGFR), a mutant PDGFR in which Y740, Y751, Y771, Y1009, and Y1021 were replaced by phenylalanine (F5), the F5 mutant in which F740 and F751 were changed back to tyrosines (Y740/Y751), and a mutant PDGFR in which Y740 and Y751 were changed to phenylalanine (F740/F751).
NGF activates the p38 MAPK pathway but not the JNK pathway.
In contrast to the ERK subgroup of the MAPK superfamily, which is activated by mitogens and growth factors (59), the JNK and p38 subgroups of the MAPK family are believed to be stimulated primarily by proinflammatory cytokines and cellular stress (15, 32, 42, 60). However, certain growth factors have also recently been reported to activate JNK and p38 MAPK (15, 20). In addition, a recent report demonstrated that exposure of SK-N-MC cells to FGF induces the p38 MAPK pathway, which then leads to CREB Ser-133 phosphorylation (66).
To address the possibility that NGF induction of the p38 MAPK or JNK signaling pathway stimulates CREB Ser-133 phosphorylation, we first examined whether NGF treatment of PC12 cells leads to p38 MAPK or JNK activation. p38 MAPK activation was monitored initially with a phosphospecific antibody that specifically recognizes p38 MAPK when it is phosphorylated at Thr-180 and Tyr-182, two sites that become newly phosphorylated as p38 MAPK becomes activated. The anti-phospho-p38 MAPK antibody detected the phosphorylated forms of p38 MAPK in extracts of NGF-treated PC12 cells but not in extracts of untreated PC12 cells (Fig. 5A). The level of p38 MAPK phosphorylation in NGF-treated PC12 cells was similar to the level of p38 MAPK phosphorylation detected when the cells were exposed to a chemical stress such as the protein synthesis inhibitor anisomycin. These findings suggest that NGF, like chemical stress, effectively triggers the phosphorylation and activation of p38 MAPK. In contrast to p38 MAPK, ERK was activated by NGF but not by anisomycin, since a phosphospecific ERK antibody recognized the phosphorylated forms of ERKs (p42 and p44 MAPK) in NGF-treated cells but not in anisomycin-treated cells (Fig. 5A). In addition to NGF, other growth factors were found to induce the phosphorylation of p38 MAPK. In NIH 3T3 cells, p38 MAPK phosphorylation at Thr-180 and Tyr-182 was stimulated by hyperosmotic concentrations of sorbitol and by EGF whereas ERK phosphorylation was induced strongly by EGF and only weakly by sorbitol (Fig. 5A). We conclude that in addition to being phosphorylated in response to stress, p38 MAPK becomes newly phosphorylated in response to a variety of growth factors that signal through receptor tyrosine kinases.
FIG. 5.
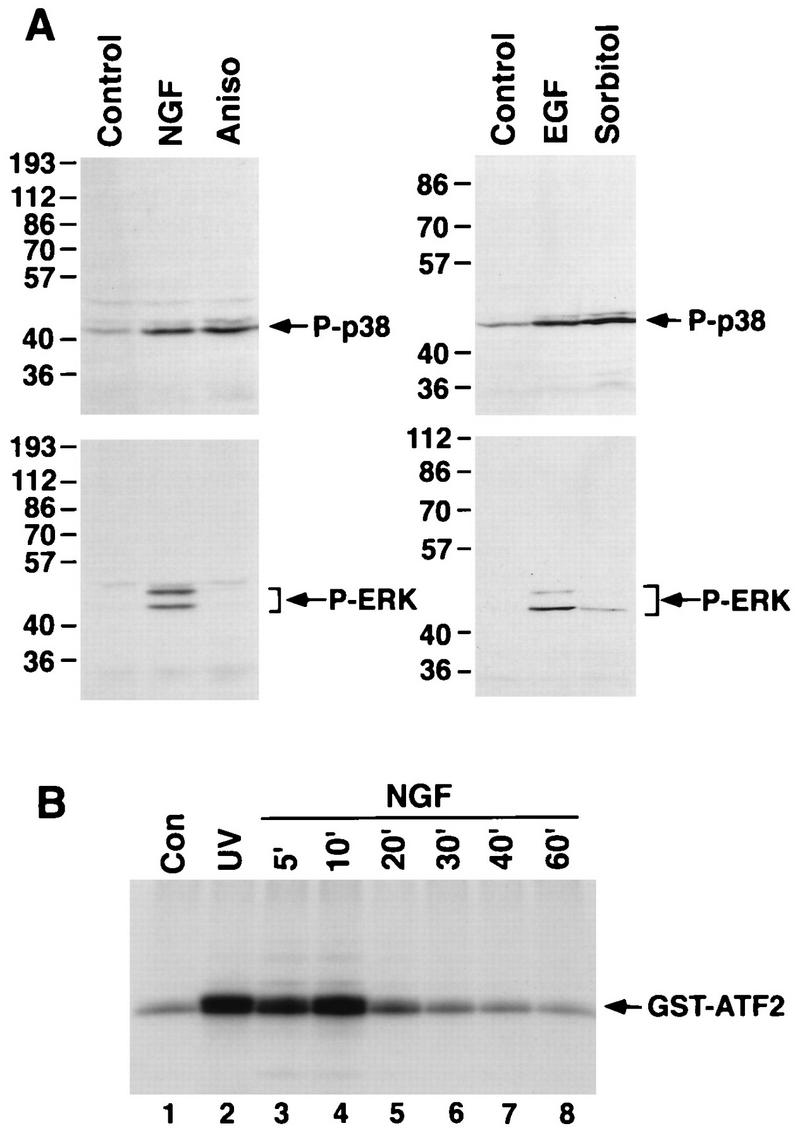
p38 MAPK is activated in response to growth factor stimulation. (A) (Left) Lysates of PC12 cells that either were untreated or had been treated for 10 min with NGF (30 ng/ml) or for 30 min with anisomycin (20 μg/ml) were separated by SDS-PAGE and immunoblotted with anti-phospho-p38 MAPK (top) or anti-phospho-ERK antibodies (bottom). (Right) Lysates of NIH 3T3 cells that were untreated or treated for 10 min with EGF (30 ng/ml), or for 15 min with sorbitol (0.3 M) were separated by SDS-PAGE and immunoblotted with anti-phospho-p38 MAPK (top) or anti-phospho-ERK (bottom) antibodies. (B) p38 MAPK was immunoprecipitated from lysates of PC12 cells that were untreated (lane 1), treated with UV irradiation (lane 2), or treated with NGF (30 ng/ml) for various periods (lanes 3 to 8). p38 MAPK activity in the immune complex was measured by an in vitro kinase assay with GST-ATF2 protein as a substrate. The reaction products were examined by SDS-PAGE and autoradiography.
NGF was also found to stimulate p38 MAPK activity, as measured in an immune complex kinase assay (Fig. 5B). p38 MAPK was immunoprecipitated from extracts of PC12 cells treated with UV irradiation or with NGF for various periods. The activity of immunoprecipitated p38 MAPK was measured in an vitro kinase assay with a recombinant form of ATF-2 (GST-ATF2) as the substrate. NGF treatment stimulated p38 MAPK activity to levels comparable to those stimulated by UV irradiation. Furthermore, NGF-induced p38 MAPK activation was transient, being maximal at 10 min and having declined to near control levels by 30 min.
In contrast, NGF treatment of PC12 cells failed to activate JNK. Extracts of PC12 cells that were treated with NGF for various periods and immunoblotted with an anti-phospho-JNK antibody showed no detectable induction of JNK phosphorylation. However, UV irradiation strongly induced JNK phosphorylation at two sites, Thr-183 and Tyr-185, whose phosphorylation correlates with and is required for JNK activation (Fig. 6A). JNK activity in immune complexes was also measured directly by an in vitro kinase assay with GST-Jun as a substrate. JNK activity was not significantly enhanced by NGF treatment, although it was strongly stimulated by UV irradiation (Fig. 6B).
FIG. 6.

NGF does not stimulate JNK activity. (A) PC12 cells were untreated, treated with NGF (30 ng/ml) for various periods as indicated, or treated with UV irradiation. Cell lysates were separated by SDS-PAGE and immunoblotted with P-JNK antibodies. (B) JNK was immunoprecipitated from lysates of PC12 cells that were untreated, treated with NGF for various periods as indicated, or treated with UV irradiation. JNK activity was measured by an in vitro kinase assay with GST-c-Jun as a substrate. The reaction products were separated by SDS-PAGE and subjected to autoradiography (top). Kinase activities were quantified with a PhosphorImager in arbitrary units (bottom).
The p38/MAPKAP kinase 2 pathway contributes to NGF induction of CREB Ser-133 phosphorylation.
Our finding that p38 MAPK was activated by NGF treatment of PC12 cells raised the possibility that the p38 MAPK pathway contributes to CREB Ser-133 phosphorylation in NGF-treated PC12 cells. To determine if activation of the p38 MAPK pathway is sufficient to trigger CREB Ser-133 phosphorylation in cells, COS cells were transfected with an expression vector for Gal4-CREB either alone or together with expression vectors for p38 MAPK and constitutively active or dominant negative forms of the p38 MAPK activator MKK6. Cotransfection of p38 MAPK and a constitutively active form of MKK6 led to high levels of Gal4-CREB phosphorylation at CREB Ser-133 (Fig. 7A), suggesting that activation of the p38 MAPK pathway is sufficient to induce CREB Ser-133 phosphorylation in intact cells.
FIG. 7.
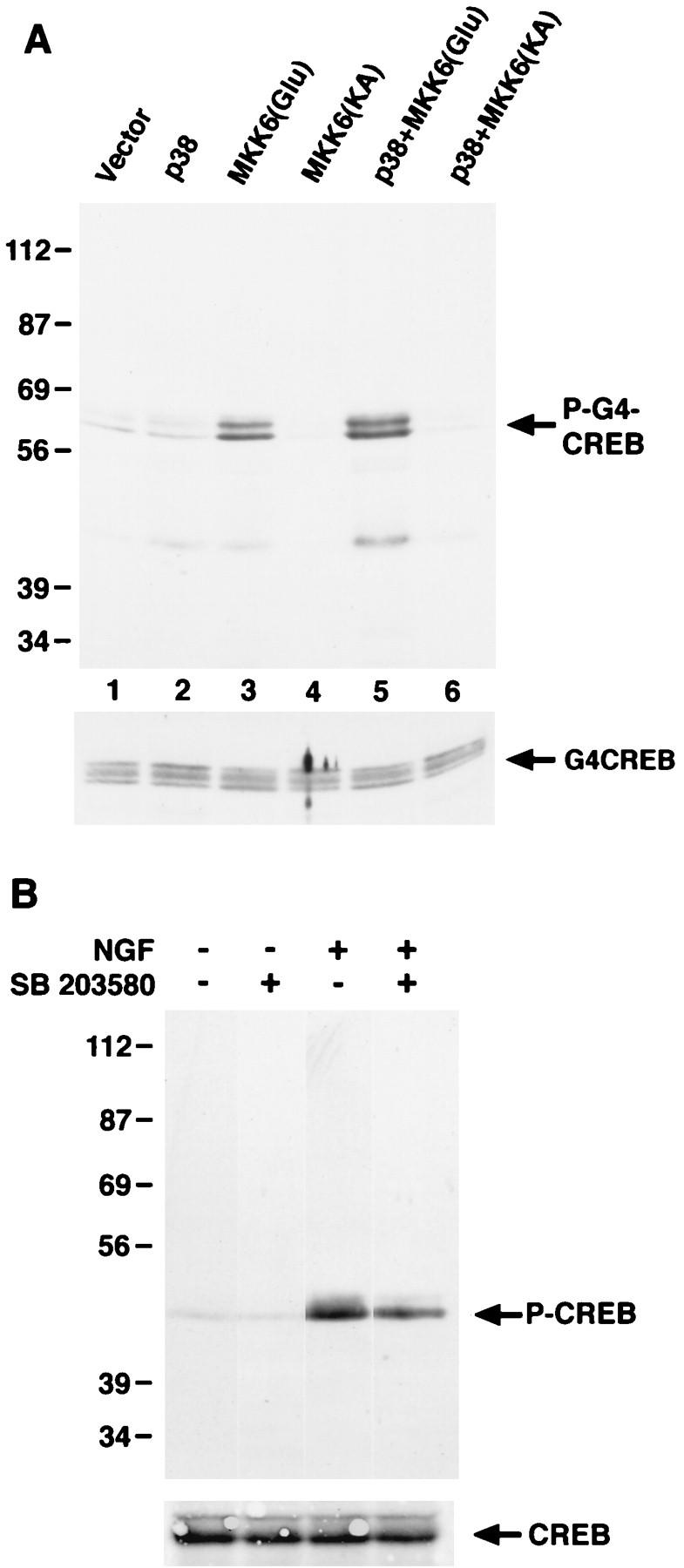
Activation of the p38 MAP kinase pathway leads to CREB Ser-133 phosphorylation in vivo. (A) COS cells were transfected with 1 μg of CMV-Gal4-CREB, together with either the empty vectors (pCMV6 and pcDNA3) or pCMV6-p38 and pcDNA3-MKK6(Glu) (constitutively active) or pcDNA3-MKK6(KA) (dominant negative). Two days after transfection, cell lysates were prepared, separated by SDS-PAGE, and immunoblotted with anti-PCREB (top) or anti-Gal4 (bottom) antibodies. (B) PC12 cells were either not pretreated or pretreated for 1 h with SB 203580 (5 μM) and were then either left untreated or treated for 10 min with NGF (30 ng/ml). Cell lysates were separated by SDS-PAGE and immunoblotted with an anti-PCREB antibody.
We also tested the role of the p38 MAPK pathway in CREB Ser-133 phosphorylation in PC12 cells by using a specific inhibitor of the p38 MAPK, SB 203580. Pretreatment of PC12 cells with a concentration of SB 203580 that had no effect on ERK and RSK activity (Fig. 3A) caused a partial inhibition of CREB Ser-133 phosphorylation in response to NGF (Fig. 7B). This result indicates that while p38 MAPK contributes to CREB phosphorylation induced by NGF, other signaling pathways must also be important. This is consistent with our findings that the MEK/ERK/RSK pathway also contributes to NGF-induced CREB phosphorylation but that inhibition of MEK only partially blocks CREB Ser-133 phosphorylation in response to NGF.
p38 MAPK is not likely to phosphorylate CREB directly, since the amino acids surrounding CREB Ser-133 do not form a typical consensus site for phosphorylation by proline-directed kinases of the MAPK superfamily. We therefore sought to identify kinases induced by p38 MAPK that might subsequently catalyze CREB phosphorylation. p38 MAPK is known to activate the Ser/Thr protein kinase MAPKAP kinase 2 (60) and a closely related kinase, MAPKAP kinase 3 (48), through phosphorylation at multiple sites (18). MAPKAP kinase 2 is activated by FGF and arsenite in SK-N-MC cells and phosphorylates CREB at Ser-133 (66). We investigated whether MAPKAP kinase 2 is activated by NGF in PC12 cells and contributes to NGF-induced CREB phosphorylation. MAPKAP kinase 2 was immunoprecipitated from extracts of PC12 cells treated with NGF or UV, and its activity was determined by an in vitro kinase assay with CREBtide as a substrate. We found that NGF treatment of PC12 cells stimulated the ability of MAPKAP kinase 2 to phosphorylate CREBtide (Fig. 8). The p38 MAPK inhibitor SB 203580 (13) but not the MEK inhibitor PD 098059 blocked MAPKAP kinase 2 activation in response to either NGF treatment or UV irradiation, suggesting that MAPKAP kinase 2 activation is mediated by the p38 MAPK pathway. The findings that NGF activates p38 MAPK and MAPKAP kinase 2 and that MAPKAP kinase 2 activation results in phosphorylation of CREB Ser-133 suggest that in addition to the ERK/RSK pathway, the p38/MAPKAP kinase 2 pathway contributes to NGF induction of CREB Ser-133 phosphorylation.
NGF-induced CREB Ser-133 phosphorylation is mediated by ERK/RSK and p38/MAPKAP kinase 2 pathways.
We next investigated whether the ERK/RSK and p38/MAPKAP kinase 2 pathways together were the major pathways that triggered CREB Ser-133 phosphorylation in NGF-treated PC12 cells. The ERK and p38 MAPK pathways were inhibited with PD 098059 and SB 203580, respectively, and the effects on CREB Ser-133 phosphorylation were examined (Fig. 9). NGF induction of CREB Ser-133 phosphorylation was reduced but not blocked completely by either PD 098059 or SB 203580, indicating that multiple pathways contribute to NGF-induced CREB Ser-133 phosphorylation. In contrast, PD 098059 and SB 203580 together inhibited NGF-induced CREB Ser-133 phosphorylation completely, indicating that the ERK and p38 pathways are the primary pathways responsible for NGF-stimulated CREB Ser-133 phosphorylation. We found that in addition to NGF, UV irradiation induced CREB Ser-133 phosphorylation in PC12 cells. UV-stimulated CREB Ser-133 phosphorylation was not affected by PD 098059 but was completely inhibited by SB 203580. This indicates that the p38 pathway is activated by UV stimulation and mediates CREB Ser-133 phosphorylation in response to UV irradiation whereas the ERK pathway does not contribute to UV-induced CREB Ser-133 phosphorylation, consistent with previous results showing that ERKs are not effectively activated by UV irradiation (15).
FIG. 9.

NGF-induced CREB Ser-133 phosphorylation is mediated by the ERK/RSK and p38/MAPKAP kinase 2 pathways. PC12 cells were not pretreated, preincubated for 1 h with PD 098059 (100 μM), or preincubated for 1 h with SB 203580 (5 μM). The cells were then lysed directly, treated with NGF (30 ng/ml), or treated with UV irradiation before lysis. Cell lysates were separated by SDS-PAGE and immunoblotted with an anti-PCREB antibody.
DISCUSSION
We have previously reported the identification of an NGF-inducible, Ras-dependent protein kinase that phosphorylates CREB at Ser-133 (24), a phosphorylation event that is necessary for CREB-dependent transcriptional activation. Purification, sequencing, and biochemical characterization of this CREB kinase revealed that it is identical to a member of the pp90RSK family, RSK2 (76). Identification of the NGF-inducible CREB kinase as RSK2 raised the question of the role of the other two members of the RSK family in mediating NGF-induced CREB Ser-133 phosphorylation. The three members of the RSK family are more than 80% identical in overall amino acid sequence and are even more highly conserved within their two catalytic kinase domains (1, 30, 52, 79). They also have similar substrate specificity and are known to translocate to the nucleus upon activation (6, 7, 30, 76, 78, 79), where they would have access to the potential substrate CREB. In this study, we show that all three members of the RSK family are activated in response to NGF and other growth factors and that they phosphorylate CREB Ser-133 both in vitro and in vivo. Our results indicate that RSK1, RSK2, and RSK3 function as a family of growth factor-inducible CREB Ser-133 kinases.
Several lines of evidence suggest that the RSK family members are not totally redundant in their in vivo function. First, the RSK family members display different patterns of tissue-specific gene expression (1, 52, 79). The expression of RSK2 appears to be widespread, whereas the expression of RSK1 and RSK3 is restricted to specific tissues (52). Second, mutations within the RSK2 gene have recently been found to be associated with Coffin-Lowry syndrome (CLS), an X-linked disorder characterized by mental retardation, facial and digital dysmorphisms, and skeletal deformations (69). Since RSK1 and RSK3 are expressed at normal levels in CLS patients, the presence of RSK1 and RSK3 does not appear to fully compensate for the loss of RSK2 function in CLS patients. We show here that a physiological role of the RSK family members is the phosphorylation of CREB Ser-133 in response to growth factor stimulation. It is possible that the contribution of individual RSK family members to growth factor-induced CREB Ser-133 phosphorylation in vivo varies depending on the tissue type or developmental stage.
Several factors could have influenced our previous identification of RSK2, instead of RSK1 or RSK3, as an NGF-inducible CREB Ser-133 kinase in PC12 cells. First, available evidence suggests that RSK2 is expressed in PC12 cells at higher levels than RSK1 (data not shown). Consistent with the idea that individual members of the RSK family are expressed at different levels in a given cell type, we failed to detect RSK1 protein or kinase activity in NIH 3T3 cells, either before or after growth factor stimulation (data not shown). Second, we cannot exclude the possibility that our methods for preparation and fractionation of cell extracts favored the extraction and preservation of activities of certain RSK family members over others. It is worth noting that most of the HA-RSK3 expressed in COS cells could be extracted only under harsh denaturing conditions (reference 78 and data not shown). In addition, RSK3 immunoprecipitated from PC12 cells exhibited a higher basal activity (in the absence of NGF) than did RSK1 and RSK2, which accounts, at least partially, for the lower fold activation of RSK3 observed with NGF stimulation (see Fig. 1).
Although the RSK family of Ser/Thr kinases is activated by NGF and other growth factors, and although RSK1, RSK2, and RSK3 phosphorylate CREB at Ser-133, inhibition of the ERK/RSK pathway did not completely block NGF induction of CREB Ser-133 phosphorylation. This finding suggested that there are other pathways in addition to the ERK/RSK pathway that mediate NGF induction of CREB Ser-133 phosphorylation. We have excluded the possible involvement of protein kinase A, CaMK, or pp70S6K in NGF-induced phosphorylation of CREB Ser-133 (24; see above). However, our results indicate that another member of the MAPK superfamily, the p38 MAPK, is activated along with ERK MAPKs in NGF-treated PC12 cells. The p38 MAPK is known to be strongly activated by the proinflammatory cytokines tumor necrosis factor alpha and interleukin-1 and by environmental stresses such as UV irradiation, heat, and osmotic stress (32, 60). However, recent evidence suggests that certain growth factors activate p38 MAPK as well. For example, FGF activates the p38 MAPK pathway in SK-N-MC cells (66), and p38 MAPK is also activated by hematopoietic growth factors including colony-stimulating factor type 1, granulocyte-macrophage colony-stimulating factor, and interleukin-3 in hematopoietic cells (20). In addition to NGF, we found that EGF activates p38 MAPK in 3T3 cells. These results suggest the possibility that activation of the p38 MAPK signaling pathway is a general mechanism by which growth factors elicit cellular responses and that the p38 MAPK might play a general role in mediating growth factor-induced CREB Ser-133 phosphorylation as well as CREB-dependent transcription (41a, 66). We have found, in preliminary studies, that inhibition of p38 MAPK with SB 203580 partially blocks NGF-induced CREB-dependent IEG transcription.
It is likely that p38 MAPK activation leads to CREB Ser-133 phosphorylation through the activation of MAPKAP kinase 2 or the closely related kinase MAPKAP kinase 3. It has been previously shown that MAPKAP kinase 2 can phosphorylate CREB Ser-133 in vitro (66). Taken together with our findings that NGF treatment of PC12 cells stimulates the ability of MAPKAP kinase 2 to phosphorylate CREBtide and that NGF activation of MAPKAP kinase 2 is blocked by the p38 MAPK inhibitor SB 203580, these results suggest that NGF activates a pathway in which the p38 MAPK phosphorylates and activates MAPKAP kinase 2, which then directly phosphorylates CREB Ser-133. Another recently identified kinase termed MNK1 (MAP kinase-interacting kinase 1) is activated by both ERKs and p38 MAPK (21, 73); however, the potential ability of this kinase to phosphorylate CREB Ser-133 has not yet been determined. It is possible that in addition to MAPKAP kinase 2, MNK1 or other unknown kinases activated by p38 MAPK also contribute to CREB phosphorylation in response to NGF.
The signaling pathway(s) which leads to p38 MAPK activation in response to growth factor stimulation is still not completely understood. An important mediator of NGF signaling is the guanine nucleotide-binding protein Ras (67, 74). NGF induction of CREB Ser-133 phosphorylation is completely blocked by a dominant interfering form of Ras (24), and CREB Ser-133 phosphorylation is potentiated by the expression of a constitutively active form of Ras (19a). These results imply that the signaling pathways that lead to CREB Ser-133 phosphorylation in NGF-stimulated PC12 cells function downstream of Ras activity. It has been well established that activation of the ERK-RSK pathway is dependent on Ras activity. We have found in preliminary experiments that NGF induction of p38 MAPK in PC12 cells may also be dependent on the activity of Ras, since expression of a dominant interfering form of Ras blocked NGF-induced p38 MAPK activation (data not shown). The suggestion that the two signaling pathways that mediate growth factor-induced CREB Ser-133 phosphorylation, the ERK/RSK pathway and the p38/MAPKAP kinase 2 pathway, may both be Ras dependent is consistent with the requirement of Ras activity for NGF-induced CREB Ser-133 phosphorylation.
The mechanism by which Ras might trigger p38 MAPK activation in response to growth factor stimulation is not clear. One possibility is that Ras regulates p38 MAPK by activating members of the Rho family of guanine nucleotide binding proteins (72). Rho proteins, including Rho, Rac1, and Cdc42, regulate the activation of p38 MAPK (12, 49). The active, GTP-bound form of Rho family members binds and activates the Pak (p21-activated kinase) family of Ser/Thr kinases (46, 77). The activation of Pak by Rho family members may involve membrane localization of Pak, a process that has been suggested to be mediated by the SH2/SH3 domain-containing adapter protein Nck (45). Paks have been proposed to be upstream activators of the MAPK kinase kinases (MKKKs) or MEK kinases (MEKKs) (72), which in turn phosphorylate and activate the MAPK kinases. Several MAPK kinases, including MKK3, MKK4, and MKK6, directly phosphorylate and activate p38 MAPK (16, 33). Although MKK4 also activates JNK, MKK3 and MKK6 appear to activate p38 MAPK exclusively.
Our finding that both the ERK pathway and the p38 pathway contribute to NGF-induced CREB Ser-133 phosphorylation raises the question of why there are two separate MAPK pathways that lead to CREB phosphorylation. One possibility is that both the ERK and p38 pathways are required to produce the maximal amount of CREB Ser-133 phosphorylation in response to NGF. Consistent with this idea, preincubation of PC12 cells with either PD 098059 to inhibit the ERK pathway or SB 203580 to inhibit the p38 MAPK pathway reduced the level of CREB Ser-133 phosphorylation in NGF-stimulated PC12 cells. Another possibility is that differences in the kinetics of ERK and p38 MAP kinase activation in NGF-treated PC12 cells allow for temporal changes in the extent of CREB phosphorylation. Support for this possibility comes from the finding that NGF activates the ERK/RSK pathway with prolonged kinetics while activation of the p38 MAPK pathway is transient. Thus, at early time points after NGF treatment, both ERK and p38 MAPK pathways contribute to CREB Ser-133 phosphorylation, but at later time points, the ERK/RSK pathway is likely to be solely responsible for CREB phosphorylation. Consistent with this idea, blocking the ERK/RSK pathway with PD 098059 changes the kinetics of NGF-induced CREB Ser-133 phosphorylation from prolonged to transient (data not shown). Therefore, at early times after growth factor stimulation, activation of both the ERK and p38 MAPK pathways may be necessary for maximal CREB Ser-133 phosphorylation and the full induction of IEGs that have CREs within their promoters. However, at later times after NGF addition, the ERK pathway alone may be sufficient to maintain CREB Ser-133 phosphorylation at a somewhat lower level that may be sufficient for the transcription of delayed response genes that contain CREB binding sites.
To conclude, CREB Ser-133 phosphorylation is a critical event for transcriptional activation that is induced by a variety of growth factors and neurotrophins. Despite the critical importance for CREB Ser-133 phosphorylation in growth factor signaling, the mechanisms by which CREB phosphorylation is induced by growth factors were not clear. Previous reports suggested the involvement of the ERK/RSK2, p38/MAPKAP kinase 2, and pp70S6K pathways in mediating CREB Ser-133 phosphorylation in NGF-treated PC12 cells, bFGF-treated SK-N-MC cells, and serum-treated NIH 3T3 cells, respectively (14, 66, 76). In this study, we have shown that both the ERK/RSK and the p38/MAPKAP kinase 2 pathways contribute to CREB Ser-133 phosphorylation in NGF-treated PC12 cells and that inhibition of both of these pathways is necessary to completely abolish NGF-induced CREB Ser-133 phosphorylation in these cells. In addition to their role in NGF-treated PC12 cells, the ERK and p38 MAPK pathways may function to mediate CREB Ser-133 phosphorylation in neurons and in nonneuronal cell lines. Our preliminary results show that p38 MAPK is activated in NGF-treated superior cervical ganglion neurons. In addition, p38 MAPK is activated by growth factors such as EGF and FGF in PC12 cells and NIH 3T3 cells. Thus, the ERK/RSK and p38/MAPKAP kinase 2 pathways are likely to play general roles in mediating CREB Ser-133 phosphorylation and IEG induction in response to a variety of neurotrophins and growth factors in a wide range of cell types.
ACKNOWLEDGMENTS
We thank R. J. Davis, J. Avruch, and D. E. Moller for gifts of plasmids. We are also most grateful to Yukiko Gotoh and Azad Bonni for critical reading of the manuscript and to other members of the Greenberg laboratory for useful discussions.
This work was supported by NIH grant CA43855 to M.E.G. and by NIH Mental Retardation Research Center grant P30-HD18655.
REFERENCES
- 1.Alcorta D A, Crews C M, Sweet L J, Bankston L, Jones S W, Erikson R L. Sequence and expression of chicken and mouse rsk: homologs of Xenopus laevis ribosomal S6 kinase. Mol Cell Biol. 1989;9:3850–3859. doi: 10.1128/mcb.9.9.3850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alessi D R, Cuenda A, Cohen P, Dudley D T, Saltiel A R. PD 098059 is a specific inhibitor of the activation of mitogen-activated protein kinase kinase in vitro and in vivo. J Biol Chem. 1995;270:27489–27494. doi: 10.1074/jbc.270.46.27489. [DOI] [PubMed] [Google Scholar]
- 3.Ballou L M, Luther H, Thomas G. MAP2 kinase and 70K S6 kinase lie on distinct signalling pathways. Nature. 1991;349:348–350. doi: 10.1038/349348a0. [DOI] [PubMed] [Google Scholar]
- 4.Berkowitz L A, Gilman M Z. Two distinct forms of active transcription factor CREB (cAMP response element binding protein) Proc Natl Acad Sci USA. 1990;87:5258–5262. doi: 10.1073/pnas.87.14.5258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bonni A, Ginty D D, Dudek H, Greenberg M E. Serine 133-phosphorylated CREB induces transcription via a cooperative mechanism that may confer specificity to neurotrophin signals. Mol Cell Neurosci. 1995;6:168–183. doi: 10.1006/mcne.1995.1015. [DOI] [PubMed] [Google Scholar]
- 6.Chen R-H, Abate C, Blenis J. Phosphorylation of the c-Fos transrepression domain by mitogen-activated protein kinase and 90-kDa ribosomal S6 kinase. Proc Natl Acad Sci USA. 1993;90:10952–10956. doi: 10.1073/pnas.90.23.10952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen R-H, Sarnecki C, Blenis J. Nuclear localization and regulation of erk- and rsk-encoded protein kinases. Mol Cell Biol. 1992;12:915–927. doi: 10.1128/mcb.12.3.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chrivia J C, Kwok R P, Lamb N, Hagiwara M, Montminy M R, Goodman R H. Phosphorylated CREB binds specifically to the nuclear protein CBP. Nature. 1993;365:855–859. doi: 10.1038/365855a0. [DOI] [PubMed] [Google Scholar]
- 9.Chung J, Grammer T C, Lemon K P, Kazlauskas A, Blenis J. PDGF- and insulin-dependent pp70S6K activation mediated by phosphatidylinositol-3-OH kinase. Nature. 1994;370:71–75. doi: 10.1038/370071a0. [DOI] [PubMed] [Google Scholar]
- 10.Chung J, Kuo C J, Crabtree G R, Blenis J. Rapamycin-FKBP specifically blocks growth-dependent activation of and signaling by the 70 kd S6 protein kinases. Cell. 1992;69:1227–1236. doi: 10.1016/0092-8674(92)90643-q. [DOI] [PubMed] [Google Scholar]
- 11.Claesson-Welsh L. Platelet-derived growth factor receptor signals. J Biol Chem. 1994;269:32023–32026. [PubMed] [Google Scholar]
- 12.Coso O A, Chiariello M, Yu J-C, Teramoto H, Crespo P, Xu N, Miki T, Gutkind J S. The small GTP-binding proteins Rac1 and Cdc42 regulate the activity of the JNK/SAPK signaling pathway. Cell. 1995;81:1137–1146. doi: 10.1016/s0092-8674(05)80018-2. [DOI] [PubMed] [Google Scholar]
- 13.Cuenda A, Rouse J, Doza Y N, Meier R, Cohen P, Gallagher T F, Young P R, Lee J C. SB 203580 is a specific inhibitor of a MAP kinase homologue which is stimulated by cellular stresses and interleukin-1. FEBS Lett. 1995;364:229–233. doi: 10.1016/0014-5793(95)00357-f. [DOI] [PubMed] [Google Scholar]
- 14.de Groot R P, Ballou L M, Sassone-Corsi P. Positive regulation of the cAMP-responsive activator CREM by the p70 S6 kinase: an alternative route to mitogen-induced gene expression. Cell. 1994;79:81–91. doi: 10.1016/0092-8674(94)90402-2. [DOI] [PubMed] [Google Scholar]
- 15.Dérijard B, Hibi M, Wu I-H, Barett T, Su B, Deng T, Karin M, Davis R J. JNK1: a protein kinase stimulated by UV light and Ha-Ras that binds and phosphorylates the c-Jun activation domain. Cell. 1994;76:1025–1037. doi: 10.1016/0092-8674(94)90380-8. [DOI] [PubMed] [Google Scholar]
- 16.Dérijard B, Raingeaud J, Barrett T, Wu I-H, Han J, Ulevitch R J, Davis R J. Independent human MAP kinase signal transduction pathways defined by MEK and MKK isoforms. Science. 1995;267:682–685. doi: 10.1126/science.7839144. [DOI] [PubMed] [Google Scholar]
- 17.Dudley D T, Pang L, Decker S J, Bridges A J, Saltiel A R. A synthetic inhibitor of the mitogen-activated protein kinase cascade. Proc Natl Acad Sci USA. 1995;92:7686–7689. doi: 10.1073/pnas.92.17.7686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Engel K, Schultz H, Martin F, Kotlyarov A, Plath K, Hahn M, Heinemann U, Gaestel M. Constitutive activation of mitogen-activated protein kinase-activated protein kinase 2 by mutation of phosphorylation sites and an A-helix motif. J Biol Chem. 1995;270:27213–27221. doi: 10.1074/jbc.270.45.27213. [DOI] [PubMed] [Google Scholar]
- 19.Erikson E, Maller J L. A protein kinase from Xenopus eggs specific for ribosomal protein S6. Proc Natl Acad Sci USA. 1985;82:742–746. doi: 10.1073/pnas.82.3.742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19a.Finkbeiner, S. M., and M. E. Greenberg. Unpublished data.
- 20.Foltz I N, Lee J C, Young P R, Schrader J W. Hemopoietic growth factors with the exception of interleukin-4 activate the p38 mitogen-activated protein kinase pathway. J Biol Chem. 1997;272:3296–3301. doi: 10.1074/jbc.272.6.3296. [DOI] [PubMed] [Google Scholar]
- 21.Fukunaga R, Hunter T. MNK1, a new MAP kinase-activated protein kinase, isolated by a novel expression screening method for identifying protein kinase substrates. EMBO J. 1997;16:1921–1933. doi: 10.1093/emboj/16.8.1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gille R, Kortenjann M, Thomae O, Moomaw C, Slaughter C, Cobb M, Shaw P. ERK phosphorylation potentiates Elk-1-mediated ternary complex formation and transactivation. EMBO J. 1995;14:951–962. doi: 10.1002/j.1460-2075.1995.tb07076.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gille R, Sharrocks A D, Shaw P E. Phosphorylation of transcription factor p62TCF by MAP kinase stimulates ternary complex formation at the c-fos promoter. Nature. 1992;358:414–417. doi: 10.1038/358414a0. [DOI] [PubMed] [Google Scholar]
- 24.Ginty D D, Bonni A, Greenberg M E. NGF activates a novel Ras-dependent protein kinase that stimulates c-fos transcription via phosphorylation of CREB. Cell. 1994;77:713–725. doi: 10.1016/0092-8674(94)90055-8. [DOI] [PubMed] [Google Scholar]
- 24a.Ginty D D, Kornhauser J M, Thompson M A, Bading H, Mayo K E, Takahashi J S, Greenberg M E. Regulation of CREB phosphorylation in the suprachiasmatic nucleus by light and a circadian clock. Science. 1993;260:238–241. doi: 10.1126/science.8097062. [DOI] [PubMed] [Google Scholar]
- 25.Gonzalez G A, Montminy M R. Cyclic AMP stimulates somatostatin gene transcription by phosphorylation of CREB at serine 133. Cell. 1989;59:675–680. doi: 10.1016/0092-8674(89)90013-5. [DOI] [PubMed] [Google Scholar]
- 26.Greenberg M E, Greene L A, Ziff E B. Nerve growth factor and epidermal growth factor induce rapid transient changes in proto-oncogene transcription in PC12 cells. J Biol Chem. 1985;260:14101–14110. [PubMed] [Google Scholar]
- 27.Greenberg M E, Hermanowski A L, Ziff E B. Effect of protein synthesis inhibitors on growth factor activation of c-fos, c-myc, and actin gene transcription. Mol Cell Biol. 1986;6:1050–1057. doi: 10.1128/mcb.6.4.1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Greene L A, Tischler A S. Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc Natl Acad Sci USA. 1976;73:2424–2428. doi: 10.1073/pnas.73.7.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Greene L A, Tischler A S. PC12 pheochromocytoma cultures in neurobiological research. Adv Cell Neurobiol. 1982;3:373–414. [Google Scholar]
- 30.Grove J R, Price D J, Banerjee P, Balasubramanyam A, Ahmad M F, Avruch J. Regulation of an epitope-tagged recombinant Rsk-1 S6 kinase by phorbol ester and erk/MAP kinase. Biochemistry. 1993;32:7727–7738. doi: 10.1021/bi00081a018. [DOI] [PubMed] [Google Scholar]
- 31.Halegoua S, Armstrong R C, Kremer N E. Dissecting the mode of action of a neuronal growth factor. Curr Top Microbiol Immunol. 1991;165:119–170. doi: 10.1007/978-3-642-75747-1_7. [DOI] [PubMed] [Google Scholar]
- 32.Han J, Lee J-D, Bibbs L, Ulevitch R J. A MAP kinase targeted by endotoxin and hyperosmolarity in mammalian cells. Science. 1994;265:808–811. doi: 10.1126/science.7914033. [DOI] [PubMed] [Google Scholar]
- 33.Han J, Lee J-D, Jiang Y, Li Z, Feng L, Ulevitch R J. Characterization of the structure and function of a novel MAP kinase kinase (MKK6) J Biol Chem. 1996;271:2886–2891. doi: 10.1074/jbc.271.6.2886. [DOI] [PubMed] [Google Scholar]
- 34.Hipskind R A, Büscher D, Nordheim A, Baccarini M. Ras/MAP kinase-dependent and -independent signaling pathways target distinct ternary complex factors. Genes Dev. 1994;8:1803–1816. doi: 10.1101/gad.8.15.1803. [DOI] [PubMed] [Google Scholar]
- 35.Janknecht R, Ernst W H, Pingoud V, Nordheim A. Activation of ternary complex factor Elk-1 by MAP kinases. EMBO J. 1993;12:5097–5104. doi: 10.1002/j.1460-2075.1993.tb06204.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Janknecht R, Nordheim A. Regulation of the c-fos promoter by the ternary complex factor Sap-1a and its coactivator CBP. Oncogene. 1996;12:1961–1969. [PubMed] [Google Scholar]
- 37.Jing S, Tapley P, Barbacid M. Nerve growth factor mediates signal transduction through trk homodimer receptors. Neuron. 1992;9:1067–1079. doi: 10.1016/0896-6273(92)90066-m. [DOI] [PubMed] [Google Scholar]
- 38.Jones S W, Erikson E, Blenis J, Maller J L, Erikson R L. A Xenopus ribosomal protein S6 kinase has two apparent kinase domains that are each similar to distinct protein kinases. Prot Natl Acad Sci USA. 1988;85:3377–3381. doi: 10.1073/pnas.85.10.3377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kaplan D R, Hempstead B L, Martin-Zanca D, Chao M V, Parada L F. The trk proto-oncogene product: a signal transducing receptor for nerve growth factor. Science. 1991;252:554–558. doi: 10.1126/science.1850549. [DOI] [PubMed] [Google Scholar]
- 40.Kaplan D R, Martin-Zanca D, Parada L F. Tyrosine phosphorylation and tyrosine kinase activity of the trk proto-oncogene product induced by NGF. Nature. 1991;350:158–160. doi: 10.1038/350158a0. [DOI] [PubMed] [Google Scholar]
- 41.Klein R, Jing S Q, Nanduri V, O’Rourke E, Barbacid M. The trk proto-oncogene encodes a receptor for nerve growth factor. Cell. 1991;65:189–197. doi: 10.1016/0092-8674(91)90419-y. [DOI] [PubMed] [Google Scholar]
- 41a.Kornhauser, J. M., and M. E. Greenberg. Unpublished data.
- 42.Kyriakis J M, Banerjee P, Nikolakaki E, Dai T, Rubie E A, Ahmed M F, Avruch J, Woodgett J R. The stress-activated protein kinase subfamily of c-Jun kinases. Nature. 1994;369:156–160. doi: 10.1038/369156a0. [DOI] [PubMed] [Google Scholar]
- 43.Leonard D G, Ziff E B, Greene L A. Identification and characterization of mRNAs regulated by nerve growth factor in PC12 cells. Mol Cell Biol. 1987;7:3156–3167. doi: 10.1128/mcb.7.9.3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lewin G R, Barde Y-A. Physiology of the neurotrophins. Annu Rev Neurosci. 1996;19:289–317. doi: 10.1146/annurev.ne.19.030196.001445. [DOI] [PubMed] [Google Scholar]
- 45.Lu W, Katz S, Gupta R, Mayer B J. Activation of Pak by membrane localization mediated by an SH3 domain from the adaptor protein Nck. Curr Biol. 1997;7:85–94. doi: 10.1016/s0960-9822(06)00052-2. [DOI] [PubMed] [Google Scholar]
- 46.Manser E, Leung T, Salihuddin H, Zhao Z S, Lim L. A brain serine/threonine protein kinase activated by Cdc42 and Rac1. Nature. 1994;367:40–46. doi: 10.1038/367040a0. [DOI] [PubMed] [Google Scholar]
- 47.Marais R, Wynne J, Treisman R. The SRF accessory protein Elk-1 contains a growth factor-regulated transcriptional activation domain. Cell. 1993;73:381–393. doi: 10.1016/0092-8674(93)90237-k. [DOI] [PubMed] [Google Scholar]
- 48.McLaughlin M M, Kumar S, McDonnell P C, Van Horn S, Lee J C, Livi G P, Young P R. Identification of mitogen-activated protein (MAP) kinase-activated protein kinase-3, a novel substrate of CSBP p38 MAP kinase. J Biol Chem. 1996;271:8488–8492. doi: 10.1074/jbc.271.14.8488. [DOI] [PubMed] [Google Scholar]
- 49.Minden A, Lin A, Claret F-X, Abo A, Karin M. Selective activation of the JNK signaling cascade and c-Jun transcriptional activity by the small GTPases Rac and Cdc42Hs. Cell. 1995;81:1147–1157. doi: 10.1016/s0092-8674(05)80019-4. [DOI] [PubMed] [Google Scholar]
- 50.Miranti C K, Ginty D D, Huang G, Chatila T, Greenberg M E. Calcium activates serum response factor-dependent transcription by a Ras- and Elk-1-independent mechanism that involves a Ca2+/calmodulin-dependent kinase. Mol Cell Biol. 1995;15:3672–3684. doi: 10.1128/mcb.15.7.3672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mobley W C, Schenker A, Shooter E M. Characterization and isolation of proteolytically modified nerve growth factor. Biochemistry. 1976;15:5543–5551. doi: 10.1021/bi00670a019. [DOI] [PubMed] [Google Scholar]
- 52.Moller D E, Xia C H, Tang W, Zhu A X, Jakubowski M. Human rsk isoforms: cloning and characterization of tissue-specific expression. Am J Physiol. 1994;266:C351–C359. doi: 10.1152/ajpcell.1994.266.2.C351. [DOI] [PubMed] [Google Scholar]
- 53.Mueller C G F, Nordheim A. A protein domain conserved between yeast MCM1 and human SRF directs ternary complex formation. EMBO J. 1991;10:4219–4229. doi: 10.1002/j.1460-2075.1991.tb05000.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pang L, Sawada T, Decker S J, Saltiel A R. Inhibition of MAP kinase kinase blocks the differentiation of PC-12 cells induced by nerve growth factor. J Biol Chem. 1995;270:13585–13588. doi: 10.1074/jbc.270.23.13585. [DOI] [PubMed] [Google Scholar]
- 55.Price M A, Rogers A E, Treisman R. Comparative analysis of the ternary complex factors Elk-1, SAP-1a and SAP-2(ERP/NET) EMBO J. 1995;14:2589–2601. doi: 10.1002/j.1460-2075.1995.tb07257.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Qiu M-S, Green S H. NGF and EGF rapidly activate p21ras in PC12 cells by distinct, convergent pathways including tyrosine phosphorylation. Neuron. 1991;7:937–946. doi: 10.1016/0896-6273(91)90339-2. [DOI] [PubMed] [Google Scholar]
- 57.Ramirez S, Ali S A S, Robin P, Trouche D, Harel-Bellan A. The CREB-binding protein (CBP) cooperates with the serum response factor for transactivation of the c-fos serum response element. J Biol Chem. 1997;272:31016–31021. doi: 10.1074/jbc.272.49.31016. [DOI] [PubMed] [Google Scholar]
- 58.Rivera V M, Miranti C K, Misra R P, Ginty D D, Chen R-H, Blenis J, Greenberg M E. A growth factor-induced kinase phosphorylates the serum response factor at a site that regulates its DNA-binding activity. Mol Cell Biol. 1993;13:6260–6273. doi: 10.1128/mcb.13.10.6260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Robinson M J, Cobb M H. Mitogen-activated protein kinase pathways. Curr Opin Cell Biol. 1997;9:180–186. doi: 10.1016/s0955-0674(97)80061-0. [DOI] [PubMed] [Google Scholar]
- 60.Rouse J, Cohen P, Trigon S, Morange M, Alonso-Llamazares A, Zamanillo D, Hunt T, Nebreda A R. A novel kinase cascade triggered by stress and heat shock that stimulates MAPKAP kinase-2 and phosphorylation of the small heat shock proteins. Cell. 1994;78:1027–1037. doi: 10.1016/0092-8674(94)90277-1. [DOI] [PubMed] [Google Scholar]
- 61.Segal R A, Greenberg M E. Intracellular signaling pathways activated by neurotrophic factors. Annu Rev Neurosci. 1996;19:463–489. doi: 10.1146/annurev.ne.19.030196.002335. [DOI] [PubMed] [Google Scholar]
- 62.Shaw P E. Ternary complex formation over the c-fos serum response element: p62TCF exhibits dual component specificity with contacts to DNA and an extended structure in the DNA-binding domain of p67SRF. EMBO J. 1992;11:3011–3019. doi: 10.1002/j.1460-2075.1992.tb05371.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sheng M, Dougan S T, McFadden G, Greenberg M E. Calcium and growth factor pathways of c-fos transcriptional activation require distinct upstream regulatory sequences. Mol Cell Biol. 1988;8:2787–2796. doi: 10.1128/mcb.8.7.2787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sheng M, Greenberg M E. The regulation and function of c-fos and other immediate early genes in the nervous system. Neuron. 1990;4:477–485. doi: 10.1016/0896-6273(90)90106-p. [DOI] [PubMed] [Google Scholar]
- 65.Sheng M, Thompson M A, Greenberg M E. CREB: a Ca2+-regulated transcription factor phosphorylated by calmodulin-dependent kinases. Science. 1991;252:1427–1430. doi: 10.1126/science.1646483. [DOI] [PubMed] [Google Scholar]
- 66.Tan Y, Rouse J, Zhang A, Cariati S, Cohen P, Comb M J. FGF and stress regulate CREB and ATF-1 via a pathway involving p38 MAP kinase and MAPKAP kinase-2. EMBO J. 1996;15:4629–4642. [PMC free article] [PubMed] [Google Scholar]
- 67.Thomas S M, DeMarco M, D’Arcangelo G, Halegoua S, Brugge J S. Ras is essential for nerve growth factor- and phorbol ester-induced tyrosine phosphorylation of MAP kinases. Cell. 1992;68:1031–1040. doi: 10.1016/0092-8674(92)90075-n. [DOI] [PubMed] [Google Scholar]
- 68.Treisman R. Identification and purification of a polypeptide that binds to the c-fos serum response element. EMBO J. 1987;6:2711–2717. doi: 10.1002/j.1460-2075.1987.tb02564.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Trivier E, De Cesare D, Jacquot S, Pannetier S, Zackai E, Young I, Mandel J-L, Sassone-Corsi P, Hanauer A. Mutations in the kinase Rsk-2 associated with Coffin-Lowry syndrome. Nature. 1996;384:567–570. doi: 10.1038/384567a0. [DOI] [PubMed] [Google Scholar]
- 70.Valius M, Kazlauskas A. Phospholipase C-γ1 and phosphatidylinositol 3 kinase are the downstream mediators of the PDGF receptor’s mitogenic signal. Cell. 1993;73:321–334. doi: 10.1016/0092-8674(93)90232-f. [DOI] [PubMed] [Google Scholar]
- 71.Visvader J, Sassone-Corsi P, Verma I M. Two adjacent promoter elements mediate nerve growth factor activation of the c-fos gene and bind distinct nuclear complexes. Proc Natl Acad Sci USA. 1988;85:9474–9478. doi: 10.1073/pnas.85.24.9474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Vojtek A B, Cooper J A. Rho family members: activators of MAP kinase cascades. Cell. 1995;82:527–529. doi: 10.1016/0092-8674(95)90023-3. [DOI] [PubMed] [Google Scholar]
- 73.Waskiewicz A J, Flynn A, Proud C G, Cooper J A. Mitogen-activated protein kinases activate the serine/threonine kinases Mnk1 and Mnk2. EMBO J. 1997;16:1909–1920. doi: 10.1093/emboj/16.8.1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wood K W, Sarnecki C, Roberts T M, Blenis J. ras mediates nerve growth factor modulation of three signal-transducing protein kinases: MAP kinase, Raf-1, and RSK. Cell. 1992;68:1041–1050. doi: 10.1016/0092-8674(92)90076-o. [DOI] [PubMed] [Google Scholar]
- 75.Xia Z, Dickens M, Raingeaud J, Davis R J, Greenberg M E. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- 76.Xing J, Ginty D D, Greenberg M E. Coupling of the Ras-MAPK pathway to gene activation by RSK2, a growth factor-regulated CREB kinase. Science. 1996;273:959–963. doi: 10.1126/science.273.5277.959. [DOI] [PubMed] [Google Scholar]
- 76a.Xing, J., and M. E. Greenberg. Unpublished observations.
- 77.Zhang S, Han J, Sells M A, Chernoff J, Knaus U G, Ulevitch R J, Bokoch G M. Rho family GTPases regulate p38 mitogen-activated protein kinase through the downstream mediator Pak1. J Biol Chem. 1995;270:23934–23936. doi: 10.1074/jbc.270.41.23934. [DOI] [PubMed] [Google Scholar]
- 78.Zhao Y, Bjørbæk C, Moller D E. Regulation and interaction of pp90rsk isoforms with mitogen-activated protein kinases. J Biol Chem. 1996;271:29773–29779. doi: 10.1074/jbc.271.47.29773. [DOI] [PubMed] [Google Scholar]
- 79.Zhao Y, Bjørbæk C, Weremowicz S, Morton C C, Moller D E. RSK3 encodes a novel pp90rsk isoform with a unique N-terminal sequence: growth factor-stimulated kinase function and nuclear translocation. Mol Cell Biol. 1995;15:4353–4363. doi: 10.1128/mcb.15.8.4353. [DOI] [PMC free article] [PubMed] [Google Scholar]