

Abstract
The estrogen receptor α (ER), a member of the steroid receptor superfamily, contains an N-terminal hormone-independent transcriptional activation function (AF-1) and a C-terminal hormone-dependent transcriptional activation function (AF-2). Here, we used in-gel kinase assays to determine that pp90rsk1 activated by either epidermal growth factor (EGF) or phorbol myristate acetate specifically phosphorylates Ser-167 within AF-1. In vitro kinase assays demonstrated that pp90rsk1 phosphorylates the N terminus of the wild-type ER but not of a mutant ER in which Ser-167 was replaced by Ala. In vivo, EGF stimulated phosphorylation of Ser-167 as well as Ser-118. Ectopic expression of active pp90rsk1 increased the level of phosphorylation of Ser-167 compared to that of either a mutant pp90rsk1, which is catalytically inactive in the N-terminal kinase domain, or to that of vector control. The ER formed a stable complex with the mutant pp90rsk1 in vivo. Transfection of the mutant pp90rsk1 depressed ER-dependent transcription of both a wild-type ER and a mutant ER that had a defective AF-2 domain (ER TAF-1). Furthermore, replacing either Ser-118 or Ser-167 with Ala in ER TAF-1 showed similar decreases in transcription levels. A double mutant in which both Ser-118 and Ser-167 were replaced with Ala demonstrated a further decrease in transcription compared to either of the single mutations. Taken together, our results strongly suggest that pp90rsk1 phosphorylates Ser-167 of the human ER in vivo and that Ser-167 aids in regulating the transcriptional activity of AF-1 in the ER.
The estrogen receptor α (ER) belongs to a superfamily of ligand-activated transcription factors whose transcriptional activities are influenced by various second messenger signaling pathways. In common with other steroid hormone receptors, the ER has an N-terminal domain with a hormone-independent transcriptional activation function (AF-1), a central DNA-binding domain, and a C-terminal ligand-binding domain with a hormone-dependent transcriptional activation function (AF-2) (18).
Like other members of the steroid receptor superfamily, the ER becomes hyperphosphorylated in the presence of its ligand and several nonsteroidal agents including epidermal growth factor (EGF) (32). Transcriptional activation by EGF has been shown to require AF-1 but not AF-2 (5, 15). EGF stimulates the mitogen-activated protein kinase (MAPK) pathway and causes an increase in the level of phosphorylation of Ser-118 in the human ER (5, 15). Ser-118 appears to be a critical residue in EGF-dependent transcriptional activation of the ER, because mutation of Ser-118 to Ala causes a reduction in the level of transcription (5). However, EGF has a role in addition to phosphorylating Ser-118, since EGF could still stimulate transcription from a mutant ER in which Ser-118 was replaced by Glu (5).
EGF also leads to the activation of the 90-kDa ribosomal S6 kinase (pp90rsk1), a Ser/Thr protein kinase. In response to growth factors, pp90rsk1 is phosphorylated by MAPK and is transported into the nucleus (4), where it forms a stable complex with CBP, a transcriptional coactivator (20). The pp90rsk1-CBP complex has been shown to regulate transcription (20). CBP has also been found to associate in a complex that contains the ER (26). Ectopic expression of CBP enhances ER transcriptional activity (26).
Three mammalian isoforms of pp90rsk1 have been previously identified (19). Each of the isoforms contains two nonidentical kinase domains (10). The amino-terminal kinase domain is responsible for phosphorylating exogenous substrates (3, 10). pp90rsk1 has been shown in vitro to phosphorylate Nur 77 (an orphan nuclear receptor), c-Fos, serum response factor (4), and glycogen synthase kinase-3 (9) and in vivo to phosphorylate IκBα (25).
There is evidence to suggest that in the human ER, Ser-167, like Ser-118, becomes significantly phosphorylated in response to estradiol (1). Ser-167 is contained within the sequence RLASTND. In vitro evidence suggests that casein kinase II phosphorylates Ser-167 (1) and that this residue may be important in DNA binding (1, 29). However, the sequence RXXS is identical to the site in c-Fos which is phosphorylated by pp90rsk1 (6). Therefore, based on the sequence match to a known pp90rsk1 phosphorylation site and the fact that both pp90rsk1 and the ER are known to interact with CBP, we investigated the possibility that pp90rsk1 plays a role in ER-mediated transcription.
Taken together, our results strongly suggest that pp90rsk1 phosphorylates Ser-167 of the human ER in vivo and that Ser-167 aids in regulating the transcriptional activity of AF-1 in the ER. These results suggest that phosphorylation of the ER by pp90rsk1 is involved in ER-mediated transcription.
MATERIALS AND METHODS
Materials.
[32P]orthophosphate (8,500 to 9,120 Ci/mmol) and [γ-32P]ATP (3,000 Ci/mmol) were obtained from DuPont NEN, Boston, Mass.
Expression vectors and receptor mutants.
P. Chambon, D. McDonnell, N. Ahn, and M. Cobb kindly provided HEGO (27), ER-TAF-1 (30), MAPKK constructs (11), and Erk2 (16), respectively. pp90rskα, Xenopus homolog of pp90rsk1 (31), and H-Ras, SrcY527F, and Raf-1 (22) were generously provided in baculoviral vectors by T. Vik and D. Morrison. pp90rsk1 constructs have been described elsewhere (10). The mutants S104/106/118A-HEGO, S167A-HEGO, and S118/167A-HEGO were created by site-directed mutagenesis of pSG5-HEGO using the megaprimer method (2). Each of these mutants was then subcloned into ER TAF-1. Glutathione-S transferase (GST) fusion proteins contained amino acids 2 to 184 of either wild-type or mutant ERs and were created by subcloning into pGEX-2T (Pharmacia Biotech, Inc.). GST fusion proteins were prepared by batch purification as described by the instructions of the manufacturer. Where indicated, GST was cleaved from the GST fusion protein by incubation with thrombin (24).
Cell culture.
COS-1, MCF-7, and BHK cell lines were maintained in phenol-red free medium with charcoal-dextran-treated fetal bovine serum as previously described (12, 13).
In-gel kinase assays.
For the in-gel kinase assay, MCF-7 cells were washed with phosphate-buffered saline and lysed (as shown in Fig. 1A) with ice-cold extraction buffer (20 mM Tris-Cl [pH 7.5], 5 mM EGTA, 0.5% Triton X-100, 6 mM dithiothreitol, 50 mM β-glycerophosphate, 1 mM phenylmethylsulfonyl fluoride [PMSF], 1 mM Na3VO4, 10-μg/ml aprotinin and leupeptin, and 2-μg/ml pepstatin). The supernatant was cleared by centrifugation, and sodium dodecyl sulfate (SDS) sample buffer added. In the case of Fig. 1C, MCF-7 cells were lysed with ice-cold lysis buffer (50 mM Tris-HCl [pH 7.4], 1% Nonidet P-40 [NP-40], 150 mM NaCl, 5 mM EDTA, 1 mM PMSF, 2-μg/ml aprotonin, leupeptin, and pepstatin, 1 mM Na3VO4, 20 mM NaF, 20 mM β-glycerophosphate, 200 nM okadaic acid). pp90rsk1 was immunoprecipitated with 2 μg of anti-pp90rsk1 antibody (Santa Cruz) and 3 μg of protein G Sepharose. The immunoprecipitates were washed four times with a buffer identical to the lysis buffer, except that only 0.1% NP-40 was used. The immunoprecipitates were eluted from the protein G-Sepharose and electrophoresed on an SDS-7% polyacrylamide gel electrophoresis (PAGE) gel with either 0.05 mg of NER or 0.07 mg of S104/106/118A-NER or S167A-NER per ml polymerized in the gel. Following electrophoresis, the proteins were denatured and renatured as described elsewhere (14). The gels were incubated for 30 min at 22°C in 40 mM HEPES-NaOH [pH 8.0]–2 mM DTT–0.1 mM EGTA–5 mM MgCl2–25 μM ATP (25-mCi/ml [γ-32P]ATP). The gels were washed as described elsewhere (14), dried, and autoradiographed.
FIG. 1.

pp90rsk1 phosphorylates Ser-167 of the ER in vitro. (A) Extracts made from MCF-7 cells that were serum starved for 24 h and then treated for 10 or 30 min with 100 nM PMA, 100-ng/ml EGF, or vehicle (lanes C). These extracts were used in an in-gel kinase assay containing NER in the gel. Lane 1 contained 14C-labelled protein molecular weight standards. (B) Western blot of the extracts used in panel A with anti-pp90rsk1 antibody (1:5,000) (Transduction Laboratories). (C) Serum-starved MCF-7 cells treated for 10 min with EGF, PMA, or vehicle. pp90rsk1 was immunoprecipitated with an anti-pp90rsk1 antibody. The immunoprecipitates were used in an in-gel kinase assay with gels containing either NER, S104/106/118A-NER, or S167A-NER. The locations of the molecular weight standards are shown. (D) In vitro kinase reactions performed with either pp90rskα, a Xenopus homolog of pp90rsk1, or Erk2 with GST-NER, GST-S104/106/118A-NER, or GST-S167A-NER as substrate. The proteins were resolved on an SDS-PAGE gel and analyzed. The experiment shown represents three independent trials. Panels C and D were obtained by scanning the autoradiograms with a Molecular Dynamics Personal Densitometer with ImageQuant software. The resultant images were cropped in Photoshop and labelled by using Freehand. Stds, standards.
In vitro kinase assays.
The in vitro kinase assay mixtures contained 2 μg of GST-NER, GST-S104/106/118A-NER, or GST-S167A-NER, 20 mM HEPES (pH 7.4), 50 mM β-glycerophosphate, 0.5 mM Na3VO4, 10 mM MgCl2, 50 μM ATP (2.5 μCi of [γ-32P]ATP), 0.5 mM DTT, 20 mM protein kinase C inhibitor peptide, 2 mM cyclic AMP-dependent kinase inhibitor peptide, 20 mM R24571 (calmodulin kinase inhibitor), 0.1 mg of bovine serum albumin per ml, and either purified activated pp90rskα or purified activated Erk2 in 30 μl. Samples were incubated for 15 min at 30°C. The reaction was stopped by the addition of SDS-PAGE sample buffer and boiling. Samples were electrophoresed on an SDS–10% PAGE gel. Following electrophoresis, the gels were stained with Coomassie Blue, dried, and autoradiographed.
Active Erk2 was purified to apparent homogeneity from Escherichia coli cells engineered to coexpress the enzyme with its activator, essentially as described elsewhere (16). Active pp90rskα, a Xenopus homolog of pp90rsk1, was extensively purified from Sf9 cells by modifications of a previously published procedure (31). pp90rskα was partially activated in situ by coexpression together with H-Ras, SrcY527F, and Raf-1. Sf9 cells were infected at a multiplicity of infection (MOI) of 5 for pp90rskα and an MOI of 1 for the upstream activators, and the cells were lysed after 60 h. Purification was achieved by sequential chromatographies of the 100,000 × g supernatant: Fast Flow S-Sepharose, phosphocellulose (Whatman P11), Superose 6, and MonoQ. The purity of the final preparation was estimated as 80% by silver staining.
In vivo [32P]orthophosphate cell labelling.
COS-1 cells on 6-cm plates were transfected by the diethylaminoethyl-dextran method as modified by Reese and Katzenellenbogen (23) with 3 μg of HEGO, S104/106/118A-HEGO, or S167A-HEGO (see Fig. 2) or 1 μg of S104/106/118A-HEGO plus 3 μg of either pMT2-RSK (avian) or pMT2-K112R-RSK (avian) or the empty vector pGL2 (Promega) (see Fig. 3). Empty pMT2 inhibits the synthesis of S104/106/118A-ER; therefore, pGL2 which does not inhibit the expression of the mutant ER, was used for the control. Thirty-two hours after transfection, the cells were labelled with 0.5 mCi of [32P]orthophosphate in 1 ml of minimal essential medium with no phosphate, no serum, and no phenol red for 4 h. EGF (100 ng/ml) or vehicle was added for either the last 30 min (see Fig. 2) or 1 h (see Fig. 3) of labelling. The cells were lysed in buffer A (with DNase, MgCl2, and 0.2 μM okadaic acid), and the ER was immunoprecipitated with monoclonal antibody α78 (28) as described elsewhere (12). Immunoprecipitated proteins were eluted from protein A-Sepharose, resolved on an SDS-7% PAGE gel, and electrotransferred to nitrocellulose. The phosphorylated ER was detected by autoradiography. Quantification of 32P label was done either by Cerenkov counting of excised bands (located by Ponceau S staining) (see Fig. 2) or by using the Molecular Dynamics PhosphorImager with ImageQuant software (see Fig. 3).
FIG. 2.
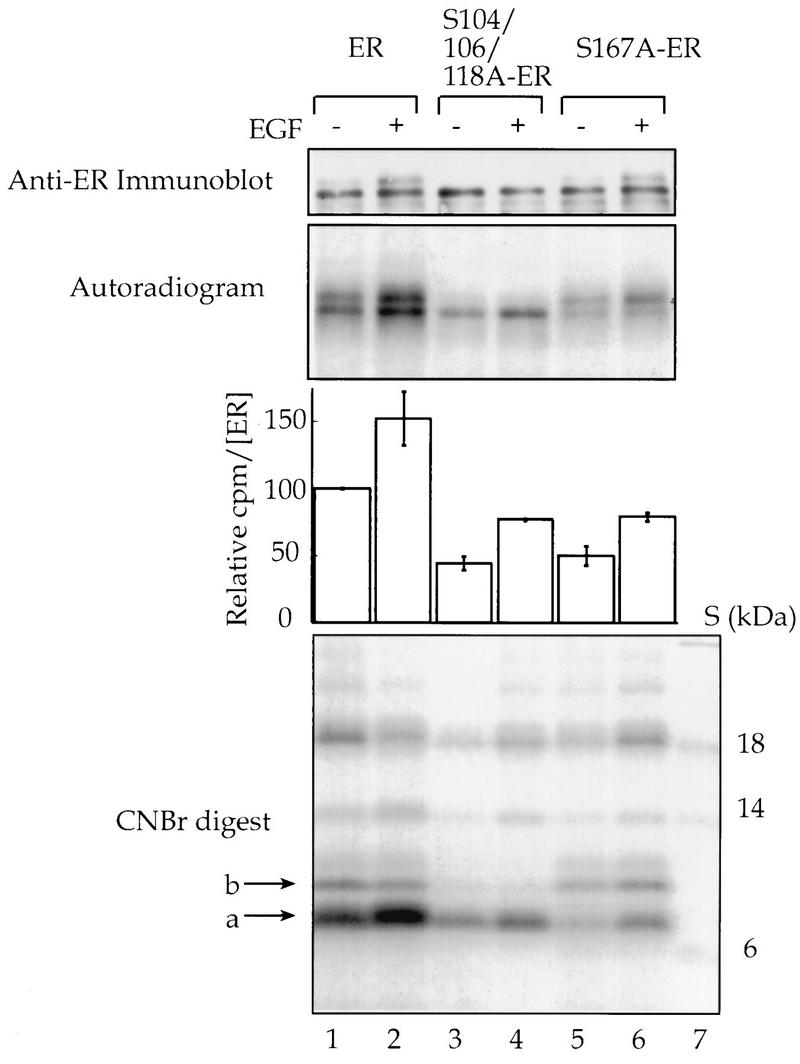
EGF stimulates phosphorylation of Ser-167 in vivo. COS-1 cells were transfected with expression vectors for wild-type human ER or for the mutants S104/106/118A-ER and S167A-ER. Cells were incubated with [32P]orthophosphate, and EGF (100 ng/ml) or vehicle was added for the last 30 min of labelling. The ER was immunoprecipitated, and an aliquot of the immunoprecipitates was resolved on an SDS-PAGE gel, transferred to nitrocellulose, and immunoblotted with monoclonal antibody EVG F9 (1:4,000), and the ER was quantitated (28). The remainder of the immunoprecipitated ER was resolved by SDS-PAGE, transferred to nitrocellulose, and autoradiographed. The ER-containing bands were excised and counted, and counts per minute were normalized for the amount of ER present. The average counts per minute ± the range as for duplicates are shown. The ER was digested with CNBr, and the peptides were resolved by Tricine-SDS-PAGE. The arrows on the autoradiogram of the CNBr digests indicate the 7-kDa peptide band that contained Ser-118 and Ser-167 (a) and the 9-kDa peptide band that contained Ser-104 and Ser-106 (b). Lane 7, 14C-labelled protein molecular mass standards. The images were obtained as described in the legend to Fig. 1.
FIG. 3.
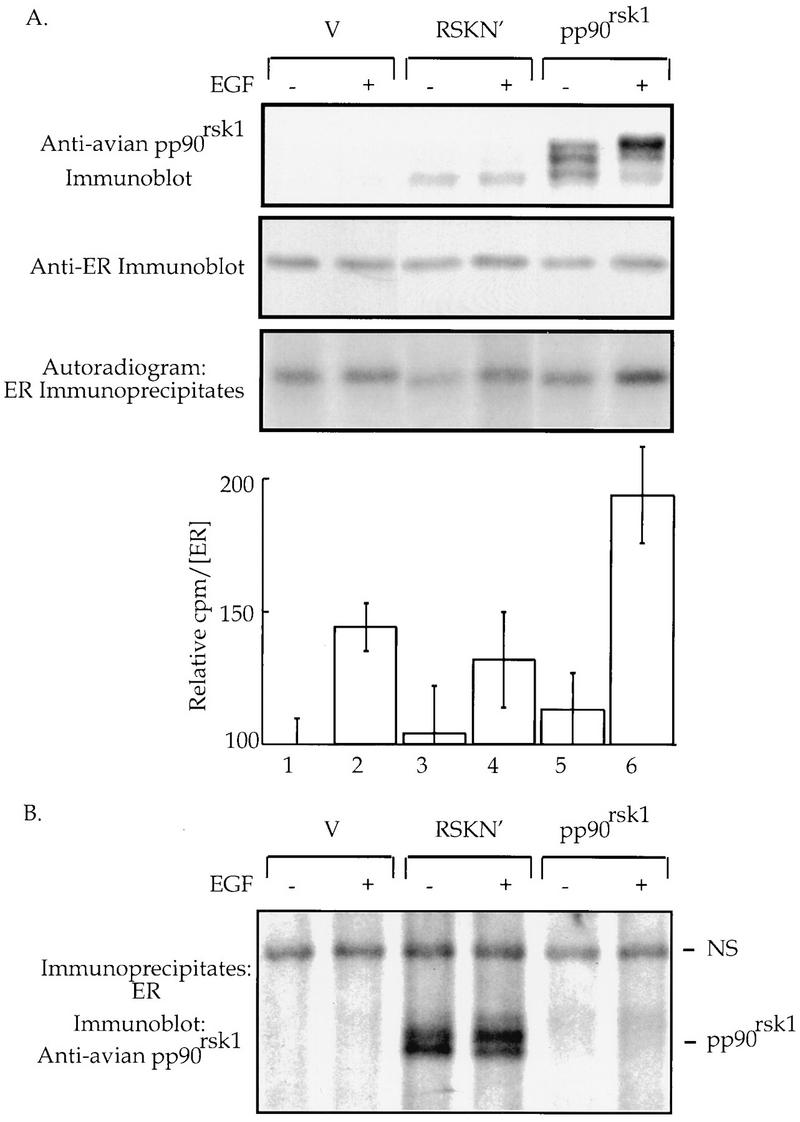
pp90rsk1 phosphorylates Ser-167 in vivo. (A) COS-1 cells cotransfected with expression vectors for the mutant S104/106/118A-ER and either wild-type pp90rsk1 or a mutant (RSK N′) or vector control. The cells were incubated with [32P]orthophosphate, and EGF (100 ng/ml) or vehicle was added for the last hour of labelling. An aliquot of cell extracts was resolved by SDS-PAGE, transferred to nitrocellulose, and immunoblotted with anti-avian pp90rsk1 antibodies (1:2,000) (10). The mutant ER was immunoprecipitated. The immunoprecipitates were resolved by SDS-PAGE, transferred to nitrocellulose, immunoblotted with the monoclonal ER AB 10 (Neo Markers) (1:400), and analyzed, and the ER was quantitated. The relative counts per minute in the ER-containing bands were determined with the Molecular Dynamics PhosphorImager by using ImageQuant software. The data were normalized for the amount of ER present. The means ± standard deviations for triplicate samples are shown. (B) The blot of ER immunoprecipitates shown in panel A stripped and reprobed with anti-avian pp90rsk1 antibodies (1:2,000). NS, nonspecific band indicating that equal amounts of protein were loaded into each gel well.
ER bands excised from the nitrocellulose membrane were digested with CNBr, and the peptides were resolved on a Tricine-SDS-PAGE gel as described elsewhere (12). The phosphorylated bands on the dried gel were detected by autoradiography and quantitated on a Molecular Dynamics PhosphorImager with ImageQuant software.
Immunoblots.
All immunoblots used anti-mouse immunoglobulin G conjugated to horseradish peroxidase as a secondary antibody. Antigen-antibody interactions were detected with the DuPont NEN Western chemiluminescence reagent. Various exposures of the film were taken. A Molecular Dynamics Personal Densitometer with ImageQuant software was used to determine the relative amounts of ER in the immunoblot bands. To ensure that a linear response over the range of ER concentrations obtained from the experiments was achieved, a standard curve relating the amount of ER to the densitometric reading was performed. The intensities of the ER bands were quantitated from the various exposures of the film. The relative intensities among the ER bands were not found to change over a wide range of exposures, since the amounts of ER were relatively similar for samples within an experiment.
Transcriptional activation analysis.
MCF-7 cells in 60-mm dishes were transfected with Lipofectamine with 500 ng of reporter plasmid (2ERE-MpGL2 containing the human metallothionien promoter [−69 to +37] [12]), 750 ng of the plasmid containing β-galactosidase (12), and 2.25 μg of the pp90rsk1 expression vectors. EGF (100 ng/ml) with estradiol (E2) (1 nM), ICI 182,780 (1 μM), or vehicle as a control were added for 5.5 h before luciferase and β-galactosidase activities were assayed. BHK cells were transfected with calcium phosphate in 24-well plates with 2.5 ng of plasmid containing the indicated receptors, 250 ng of reporter plasmid, 125 ng of the plasmid containing β-galactosidase, 125 ng of MAPK kinase expression vectors, and, when indicated, 6.25 ng of either wild-type or mutant pp90rsk1 constructs or vector alone. The total amount of DNA transfected was increased to 812.5 ng with empty vector DNA. E2 or vehicle was added 36 h before luciferase and β-galactosidase activities were assayed. The luciferase and β-galactosidase assays were performed as described elsewhere (12).
RESULTS
In vitro evidence suggests that Ser-167 of the human ER is phosphorylated by pp90rsk1.
Phosphorylation of the ER is induced by phorbol esters (phorbol myristate acetate [PMA]) and EGF (5, 12, 15, 17). To identify the kinase(s) responsible for this phosphorylation, an in-gel kinase assay was performed with extracts from MCF-7 cells that had been treated with PMA, EGF, or vehicle as a control. The substrate embedded in the gel was the N-terminal domain of the ER (NER) comprising amino acids 2 to 184. MCF-7 cells were used in these studies as a physiologically relevant model system since they are a breast cancer cell line that contains endogenous ER. Both PMA and EGF stimulated a protein kinase with a molecular mass of ∼90 kDa (Fig. 1A, lanes 3, 4, 6, and 7), suggesting that PMA and EGF activated a protein kinase that phosphorylates the ER. The band seen in all lanes migrating slightly ahead of the 90-kDa kinase is an autophosphorylated protein seen in an in-gel kinase assay performed with no substrate in the gel (13a). It is of interest to note that MAPK was unable to phosphorylate the NER in an in-gel kinase reaction. However, in an in-gel kinase assay in which myelin basic protein was used as a substrate, a band at approximately 42 kDa was detected in cell extracts that had been obtained from cells treated with either EGF or PMA (13). This result indicates that MAPK is active in the in-gel kinase assay. Therefore, NER appears to be a poor MAPK substrate in an in-gel kinase assay but is effectively phosphorylated by MAPK in solution (Fig. 1D). Presumably, in the in-gel kinase assay NER does not refold sufficiently to be a good MAPK substrate.
To determine if the 90-kDa kinase could be pp90rsk1, the extracts used in the in-gel kinase assay were screened with pp90rsk1 antibodies on an immunoblot. The extracts from cells treated with PMA and EGF showed proteins with reduced mobilities, which is characteristic of activated pp90rsk1 (7) at the same location as that of the 90-kDa kinase seen in the in-gel kinase assay (Fig. 1B, lanes 3, 4, 6, and 7).
To confirm that pp90rsk1 was the 90-kDa kinase observed in the in-gel kinase assay, pp90rsk1 was immunoprecipitated from MCF-7 cells that had been treated with PMA, EGF, or vehicle as a control. To ensure that we were able to immunoprecipitate pp90rsk1 in an activated form, an immunoblot of the pp90rsk1 immunoprecipitates was performed (13a). This immunoblot demonstrated that in response to PMA and EGF, pp90rsk1 showed a reduced gel mobility compared to that of the vehicle control. These immunoprecipitates were used in an in-gel kinase assay with NER as the substrate (Fig. 1C, lanes 1 to 3). Only activated pp90rsk1 is able to phosphorylate NER (Fig. 1C, lane 1 versus lanes 2 and 3). Ser-167 of the human ER is contained within the sequence RXXS and, therefore, lies within a potential pp90rsk1 site of phosphorylation. Ser-104, -106, and -118 are also sites of phosphorylation in the ER (12, 17). To compare Ser-167 and the Ser-Pro sites at Ser-104, -106, and -118 as substrates for pp90rsk1, in-gel kinase assays were performed with two mutated NER constructs, S104/106/118A-NER and S167A-NER. S104/106/118A-NER contains three point mutations in which the indicated Ser residues were mutated to Ala. S167A-NER contains a single point mutation in which the indicated Ser is mutated to Ala. pp90rsk1 that was activated by either EGF or PMA could phosphorylate S104/106/118A-NER (Fig. 1C, lanes 5 and 6) but could not phosphorylate S167A-NER (Fig. 1C, lanes 8 and 9). These results indicate that Ser-167 is a specific substrate for pp90rsk1.
To further determine if Ser-167 of NER can be phosphorylated by pp90rsk1, an enzyme preparation containing purified activated pp90rskα, the Xenopus homolog of pp90rsk1, was used in an in vitro kinase assay with GST-NER, GST-S104/106/118A-NER, and GST-S167A-NER as substrates. Both GST-NER and GST-S104/106/118A-NER were highly phosphorylated (Fig. 1D, lanes 1 and 2), but GST-S167A-NER was not phosphorylated by the enzyme preparation (Fig. 1D, lane 3). A Coomassie Blue stain of the same gel indicated that approximately equal amounts of the three substrates were present in the kinase assay mixtures (13a). These data provide additional evidence that Ser-167 can be specifically phosphorylated by pp90rsk1. In a parallel experiment in which activated ERK2 instead of activated pp90rsk1 was used, both GST-NER and GST-S167A-NER were phosphorylated but GST-S104/106/118A-NER was not phosphorylated (Fig. 1D, lanes 4 to 6). Thus, GST-S167A-NER is still a substrate for MAPK. Moreover, this result confirms that NER is a possible physiological substrate for MAPK.
EGF stimulates phosphorylation of Ser-167 in vivo.
Ser-167 of the human ER has been shown to be phosphorylated in MCF-7 cells treated with estradiol (1), but no effect of PMA or EGF on the phosphorylation of Ser-167 in vivo has been reported. To determine whether EGF does increase the phosphorylation of Ser-167 in vivo, COS-1 cells were transfected with expression vectors for either the wild type or the mutant S104/106/118A-ER or S167A-ER. COS-1 cells were used in these studies because sufficient quantities of ER can be obtained to perform biochemical analysis. COS-1 cells contain the large T antigen, and, thus, transfected plasmids that contain the simian virus 40 (SV40) origin of replication will be efficiently replicated. Thus, approximately 1 μg of ER can be obtained from a 100-mm dish of transfected COS-1 cells. The cells were incubated with [32P]orthophosphate and treated for 30 min with EGF or vehicle, and the ERs were then immunoprecipitated. We have shown previously that the ER runs as a doublet and that the phosphorylation of Ser-118 causes the receptor to upshift (12). The basal incorporation of [32P]orthophosphate into the mutant ERs lacking either Ser-167 or Ser-104, -106, and -118 was reduced by approximately 50% compared to that for the wild-type ER (Fig. 2, lane 1 versus lanes 3 and 5). EGF increased the incorporation of [32P]orthophosphate into the wild-type and mutant receptors by ≥50% (Fig. 2, lanes 2, 4, and 6 versus lanes 1, 3, and 5).
To determine the site of [32P]orthophosphate incorporation into the ER, the ER bands in the experiment described above were excised from the nitrocellulose and treated with CNBr, and the peptides were resolved (Fig. 2). All bands greater than 10 kDa represent incomplete scission by the CNBr. The band at 7 kDa (labelled a) has been sequenced previously and has been shown to contain only the peptide with amino acids 110 to 174 (12). Thus, this band contains both Ser-118 and Ser-167. The band at 9 kDa (labelled b) has also been sequenced and contains amino acids 13 to 109. Thus, this band contains Ser-104 and Ser-106 and was not labelled in samples with Ser-104 and Ser-106 mutated to Ala (Fig. 2, lanes 3 and 4). The majority of the 32P label was in the 7-kDa band for all samples. This band also showed the largest difference in intensities among the wild-type and mutant ERs, in agreement with our assumption that most of the 32P label is in Ser-118 and/or Ser-167. The label in the 7-kDa band was increased by EGF for both the wild-type and the mutant receptors. This result indicates that EGF causes an increase in the level of phosphorylation of Ser-167 as well as Ser-118 in vivo.
pp90rsk1 increases the phosphorylation of Ser-167 in vivo.
To determine if pp90rsk1 phosphorylates Ser-167 in vivo, COS-1 cells were cotransfected with expression vectors for S104/106/118A-ER and either the wild-type or a mutant avian pp90rsk1 (RSK N′) or vector alone. RSK N′ contains the point mutation K112R, which makes the N-terminal kinase domain catalytically inactive (10). The mutant S104/106/118A-ER was chosen to eliminate phosphorylation into serines 104, 106, and 118 and thus to aid in interpretation of the results. Following transfection, the COS-1 cells were incubated with [32P]orthophosphate and treated with EGF or vehicle, and the ER was immunoprecipitated (Fig. 3). Overexpression of wild-type pp90rsk1 increased the level of phosphorylation of the mutant ER by 50% in EGF-stimulated cells compared to that for the vector control (Fig. 3, lane 6 versus lane 2). This result suggests that Ser-167 of the ER is a physiological substrate for pp90rsk1. Expression of RSK N′ did not enhance ER phosphorylation compared to that for the vector control (lane 4 versus lane 2). This result demonstrates that the N-terminal region of pp90rsk1 is most likely responsible for phosphorylating the ER. This observation is in agreement with previous studies, which demonstrated that the N-terminal kinase domain of pp90rsk1 appears to be responsible for phosphorylating exogenous substrates (3, 10).
To further demonstrate that the ER is a physiological substrate for pp90rsk1, we determined whether the ER and pp90rsk1 could physically associate in vivo. The nitrocellulose containing the immunoprecipitated ER shown in Fig. 3A was stripped and reprobed with anti-avian pp90rsk1 antibody. Interestingly, even though RSK N′ is expressed at considerably lower levels than those of the wild-type pp90rsk1 (Fig. 3A), the mutant RSK N′ associates with the ER, whereas no visible association was observed between either activated or unactivated wild-type pp90rsk1 and the ER (Fig. 3B). Thus, the ER and RSK N′ coimmunoprecipitate. Overexposure of the autoradiogram shown in Fig. 3B demonstrated that activated pp90rsk1 weakly associated with the ER (13a).
pp90rsk1 is involved in transcriptional activation by endogenous ER in MCF-7 cells.
To determine if pp90rsk1 has any physiological role in ER action, the ability of pp90rsk1 to influence ER-mediated transcription was analyzed. MCF-7 cells were cotransfected with a reporter construct that contained a luciferase gene regulated by two estrogen response elements (EREs) and a plasmid that permitted constitutive expression of β-galactosidase. β-Galactosidase expression allows the luciferase data to be normalized for transfection efficiency. In addition to the reporter and β-galactosidase construct, the MCF-7 cells were transfected with expression vectors for pp90rsk1 or RSK N′ or with vector alone. As shown in Fig. 4A, the most striking effect on ligand-stimulated transcription was the addition of the mutant RSK N′, which appeared to depress transcriptional activation by the ER. Exogenously added pp90rsk1 had no effect on ligand-induced transcription (Fig. 4A). Treatment of MCF-7 cells with ICI 182,780, a pure antagonist for the ER, decreased transcription to the same extent for all conditions (Fig. 4A). ICI 182,780 causes rapid degradation of the ER (8, 13). This result shows that the inhibitory effect of the mutant RSK N′ is specific for ER-mediated transcription, since basal transcription, which was observed in the presence of ICI 182,780, was not affected.
FIG. 4.
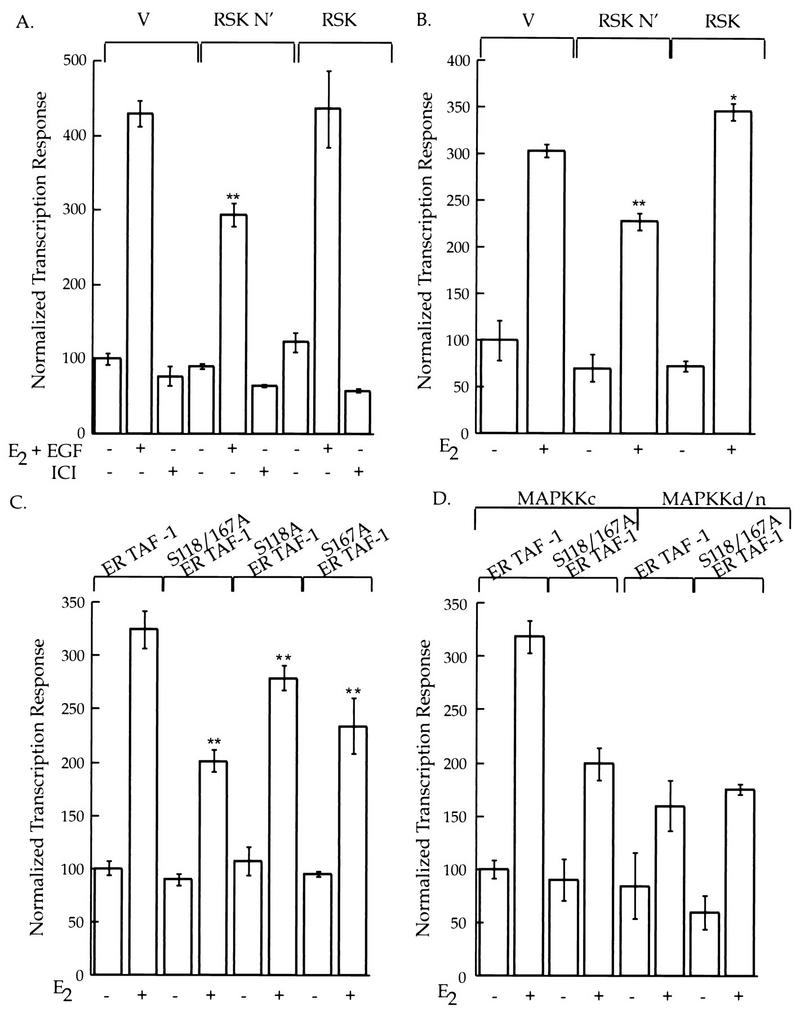
pp90rsk1 and Ser-167 regulate ER-mediated transcription. (A) MCF-7 cells cotransfected with a reporter construct that contained a luciferase gene regulated by two EREs and a plasmid that permitted constitutive expression of β-galactosidase. Additionally, the cells were transfected with expression vectors for either wild-type pp90rsk1 or the mutant RSK N′ or vector (V) alone. EGF (100 ng/ml) and estradiol (E2) (1 nM), ICI 182,780 (1 μM), or vehicle was added. Luciferase activity was normalized to β-galactosidase activity. The normalized luciferase values were divided by the value obtained in the absence of exogenous agents and multiplied by 100. The means ± standard errors of the means are shown. (B) BHK cells transfected as described for panel A, except that constructs containing a MAPKKc and ER TAF-1 were also cotransfected. E2 or vehicle was added, and the assay and analysis were performed as described for panel A. The means ± standard errors of the means are shown. (C) BHK cells transfected with the reporter and β-galactosidase constructs as described for panel A and the MAPKKc construct as described for panel B. Additionally, constructs that contained either ER TAF-1, ER S118/167A TAF-1, ER S118A TAF-1, or ER S167A TAF-1 were transfected. E2 or vehicle was added, and the assay and analysis were performed as described for panel A. (D) BHK cells were cotransfected with the reporter and β-galactosidase constructs as described for panel A as well as with constructs containing either ER TAF-1 or ER S118/167A TAF-1. Additionally, constructs that contained either MAPKKc or MAPKKd/n were transfected. E2 or vehicle was added, and the assay and analysis were performed as described for panel A. The means ± standard errors of the means are shown. ∗∗, P of <0.001; ∗, P of <0.05.
Ser-167 regulates AF-1 activation of transcription.
The above data suggest that pp90rsk1 regulates transcriptional activation by the ER through its ability to phosphorylate Ser-167. Furthermore, since Ser-167 is contained within the AF-1 region, it presumably is involved in regulating the transcriptional activity of AF-1. Mutant ERs were constructed in which either Ser-118 or Ser-167 or both were replaced with Ala, and each was then subcloned into ER TAF-1. ER TAF-1 contains three point mutations in the AF-2 domain which make the AF-2 transcriptional activation domain nonfunctional, but nonetheless ER TAF-1 still binds estradiol with wild-type affinity (30). By eliminating the transcriptional activation produced by AF-2, we can directly address the role of Ser-167 in controlling AF-1 function.
Neither MCF-7 cells nor COS-1 cells were suitable for transcriptional analysis of the mutant ERs. MCF-7 cells contain endogenous wild-type ER. COS-1 cells were not suitable, since all plasmids used in this study contained the SV40 origin of replication. BHK cells were chosen for the study of the mutant ERs since they do not contain ER or the large T antigen and have a transfection efficiency of greater than 50%. To determine whether the activity of ER TAF-1 could be regulated by pp90rsk1, BHK cells were cotransfected with the ER TAF-1 construct together with the reporter, the β-galactosidase expression vector, and one of the pp90rsk1 plasmids described above. Additionally, a construct that allowed the expression of a constitutively active MAPK kinase (MAPKKc) was cotransfected. MAPKKc was used to bypass any rate-limiting steps that might exist before the activation of MAPK and the resulting activation of pp90rsk1. The mutant RSK N′ suppressed transcription, and wild-type pp90rsk1 slightly stimulated transcription (Fig. 4B). Thus, the effect of the pp90rsk1 constructs on the transcriptional activity induced by ER TAF-1 is essentially the same as that produced by the wild-type ER. Therefore, pp90rsk1 predominately influences the ability of AF-1 to activate transcription. These results would be predicted from the ability of pp90rsk1 to phosphorylate Ser-167 in AF-1.
Since the transcriptional activity of ER TAF-1 was found to be regulated by pp90rsk1, the effect of mutating either Ser-118, Ser-167, or both on transcription mediated by ER TAF-1 was tested. Figure 4C shows that replacing either Ser-118 or Ser-167 with Ala decreases transcription to the same extent, i.e., ∼15%, whereas the double mutant decreases transcription by ∼40%. It is not possible to determine the expression level of the wild-type and mutant ERs under the conditions used for the transcriptional assays. However, immunoblot analysis of COS cells transfected with wild-type or mutant ERs demonstrated that mutation of Ser-118 and Ser-167 does not effect the relative expression levels compared to that for the wild-type ER (13a). In other experiments, EGF instead of MAPKKc was used to activate MAPK and pp90rsk1. The results were similar to those shown in Fig. 4C (13a). These results demonstrate that mutating Ser-118 and Ser-167 results in a receptor with a diminished capability to activate transcription. We also created a mutant ER in which Ser-118 and Ser-167 were replaced with Ala (S118/167A-ER). In response to estradiol and EGF, the mutant S118/167A-ER showed approximately 56% of the transcriptional response shown by the wild-type receptor (13a). Thus, Ser-118 and Ser-167 are also important in the context of a receptor with a functional AF-2 domain.
Others have shown that MAPK phosphorylates Ser-118 (5, 15); we have demonstrated that pp90rsk1 phosphorylates Ser-167. In our transcription experiments, the MAPK pathway is partially active because of the serum present during the transfection. Therefore, we used a dominant-negative MAPKK (MAPKKd/n) to inhibit this basal activity of the MAPK pathway. If Ser-118 and Ser-167 are the only residues that are significantly regulated by the MAPK pathway, then the wild-type receptor in the absence of an activated MAPK pathway should have essentially the same level of transcriptional activity as that of the ER double mutant S118/167A in the presence of an activated MAPK pathway. Figure 4D shows that Ser-118 and Ser-167 are apparently the key residues in the ER under the regulation of the MAPK pathway.
DISCUSSION
Four different lines of evidence support the conclusion that Ser-167 of the human ER is directly phosphorylated by pp90rsk1 in vivo. First, in-gel kinase assays demonstrated that pp90rsk1, stimulated by either PMA or EGF, specifically phosphorylates Ser-167. Second, pp90rskα, a Xenopus homolog of pp90rsk1, specifically phosphorylates Ser-167 in vitro. Third, COS-1 cells transfected with pp90rsk1 demonstrated enhanced levels of Ser-167 phosphorylation compared to COS-1 cells transfected with either a mutant pp90rsk1 that was mutated in the N-terminal kinase domain (RSK N′) or a vector control. Fourth, the ER and RSK N′ were found to physically associate in vivo.
A physiological role for phosphorylation of Ser-167 is provided by the observations that RSK N′ can suppress ER- mediated transcription and that a mutant ER in which Ser-167 is replaced with Ala cannot maximally activate transcription. pp90rsk1 regulates the transcriptional activity of the ER through its effect on AF-1, since RSK N′ depressed transcriptional activation of the mutant ER TAF-1 to the same extent as the wild-type receptor.
Ser-118 and Ser-167 are important residues in regulating ER-mediated transcription either by the mutant ER TAF-1 or the wild-type ER. Estradiol was able to activate the double mutant, although the transcriptional response was impaired compared to that of the wild type. This result indicates that residues in addition to Ser-118 and Ser-167 are also important in regulating ER-mediated transcription. These additional residues could be other phosphorylation sites or residues that have been exposed in response to estradiol binding and which are now in a more favorable position to interact with coactivators.
A possible mechanism for the ability of RSK N′ to inhibit the transcriptional activity of the ER is suggested by our observations. pp90rsk1 associates with the ER and phosphorylates Ser-167. Presumably, phosphorylation of Ser-167 allows the ER to be released from pp90rsk1. This hypothesis is supported by our observations that only a weak association was observed between the ER and activated pp90rsk1. RSK N′ also associates with the ER, but because of the mutation in the N-terminal kinase domain, RSK N′ cannot phosphorylate Ser-167. The ER and RSK N′ form a stable complex which prevents endogenous pp90rsk1 from phosphorylating Ser-167. Therefore, RSK N′ suppresses ER-mediated transcription. The reason that we did not observe a decrease in S104/106/118A-ER phosphorylation with RSK N′ in COS-1 cells may be that not all of the ER was complexed with RSK N′. Thus, the ER that was not bound to RSK N′ was free to associate with the endogenous activated pp90rsk1. However, a more complicated mechanism may account for the ability of RSK N′ to inhibit the transcriptional activity of the ER. It is possible that the stable interaction between RSK N′ and the ER sterically prevents the ER from interacting with either its specific DNA binding sequence or with other proteins that are involved in ER-mediated transcription. Further experiments are needed to determine whether RSK N′ inhibits ER-mediated transcription either by preventing phosphorylation of Ser-167 or by steric inhibition or a combination of both mechanisms.
In vitro evidence suggests that c-fos is also a substrate for MAPK and pp90rsk1 (6). The significance of two Ser residues in either c-fos or the ER being phosphorylated by different kinases on the MAPK pathway is not understood. MAPK and pp90rsk1 are known to physically associate with each other (21). It may be that a complex consisting of MAPK and pp90rsk1 interacts with the ER and that the kinases then coordinately phosphorylate Ser-118 and Ser-167. The presence or absence of coordinate phosphorylation of these residues may provide the ER with a mechanism of differentiating signals from various second messenger pathways.
Approximately one-third of breast cancers are hormone dependent and will regress upon treatment with antiestrogens or inhibitors of aromatase, a rate-limiting enzyme for estrogen biosynthesis in postmenopausal women (33). It is well known that some patients with breast cancer experience further tumor regression upon secondary endocrine-based therapies, even though circulating estrogen levels have been lowered. It is possible that inhibition of ligand-independent activation of the ER is responsible for further tumor regression. It appears that Ser-118 and Ser-167 are important residues in regulating ER-induced transcription through the MAPK pathway. It may be that specific inhibition of the MAPK pathway as a secondary treatment for hormone-dependent breast cancer may provide further tumor regression. The MAPK pathway may also be of importance in some hormone-independent breast cancers in which mutated forms of the ER are present.
ACKNOWLEDGMENTS
We especially thank Abdulmaged Traish (Boston University) for providing the anti-ER antibodies used in this study. We also thank Ian Macara and David Brautigan (University of Virginia) for their helpful discussions and critical reading of the manuscript.
This work was supported by Public Health Service grant CA-55887 from the National Cancer Institute.
REFERENCES
- 1.Arnold S F, Obourn J D, Jaffe H, Notides A C. Serine 167 is the major estradiol-induced phosphorylation site on the human estrogen receptor. Mol Endocrinol. 1994;8:1208–1214. doi: 10.1210/mend.8.9.7838153. [DOI] [PubMed] [Google Scholar]
- 2.Barik S. Site-directed mutagenesis by double polymerase chain reaction. Methods Mol Biol. 1993;15:277–286. doi: 10.1385/0-89603-244-2:277. [DOI] [PubMed] [Google Scholar]
- 3.Bjorbaek C, Zhao Y, Moller D E. Divergent functional roles for p90rsk kinase domains. J Biol Chem. 1995;270:18848–18852. doi: 10.1074/jbc.270.32.18848. [DOI] [PubMed] [Google Scholar]
- 4.Blenis J. Signal transduction via the MAP kinases: proceed at your own RSK. Proc Natl Acad Sci USA. 1993;90:5889–5892. doi: 10.1073/pnas.90.13.5889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bunone G, Briand P-A, Miksicek R J, Picard D. Activation of the unliganded estrogen receptor by EGF involves the MAP kinase pathway and direct phosphorylation. EMBO J. 1996;15:2174–2183. [PMC free article] [PubMed] [Google Scholar]
- 6.Chen R-H, Abate C, Blenis J. Phosphorylation of the c-Fos transrepression domain by mitogen-activated protein kinase and 90-kDa ribosomal S6 kinase. Proc Natl Acad Sci USA. 1993;90:10952–10956. doi: 10.1073/pnas.90.23.10952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen R-H, Chung J, Blenis J. Regulation of pp90rsk phosphorylation and S6 phosphotransferae activity in Swiss 3T3 cells by growth factor-, phorbol ester-, and cyclic AMP-mediated signal transduction. Mol Cell Biol. 1991;11:1861–1867. doi: 10.1128/mcb.11.4.1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dauvois S, Daneilian P S, White R, Parker M G. Antiestrogen ICI 164,384 reduces cellular estrogen receptor content by increasing its turnover. Proc Nat Acad Sci USA. 1992;89:4037–4041. doi: 10.1073/pnas.89.9.4037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eldar-Finkelman H, Seger R, Vandenheede J R, Krebs E G. Inactivation of glycogen synthase kinase-3 by epidermal growth factor is mediated by mitogen-activated protein kinase/p90 ribosomal protein S6 kinase signaling pathway in NIH 3T3 cells. J Biol Chem. 1995;270:987–990. doi: 10.1074/jbc.270.3.987. [DOI] [PubMed] [Google Scholar]
- 10.Fisher T L, Blenis J. Evidence for two catalytically active kinase domains in pp90rsk. Mol Cell Biol. 1996;16:1212–1219. doi: 10.1128/mcb.16.3.1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Font de Mora J, Porras A, Ahn N, Santos E. Mitogen-activated protein kinase activation is not necessary for, but antagonizes, 3T3-L1 adipocytic differentiation. Mol Cell Biol. 1997;17:6068–6075. doi: 10.1128/mcb.17.10.6068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Joel P B, Traish A M, Lannigan D A. Estradiol and phorbol ester cause phosphorylation of serine 118 in the human estrogen receptor. Mol Endocrinol. 1995;9:1041–1052. doi: 10.1210/mend.9.8.7476978. [DOI] [PubMed] [Google Scholar]
- 13.Joel, P. B., A. M. Traish, and D. A. Lannigan. Submitted for publication.
- 13a.Joel, P. B., and D. A. Lannigan. Unpublished data.
- 14.Kameshita I, Fujisawa H. A sensitive method for detection of calmodulin-dependent protein kinase II activity in sodium dodecyl sulfate-polyacrylamide gel. Anal Biochem. 1989;183:139–143. doi: 10.1016/0003-2697(89)90181-4. [DOI] [PubMed] [Google Scholar]
- 15.Kato S, Endoh H, Masuhiro Y, Kitamoto T, Uchiyama S, Sasaki H, Masushige S, Gotoh Y, Nishida E, Kawashima H, Metzger D, Chambon P. Activation of the estrogen receptor through phosphorylation by mitogen-activated protein kinase. Science. 1995;270:1491–1494. doi: 10.1126/science.270.5241.1491. [DOI] [PubMed] [Google Scholar]
- 16.Khokhlatchev A, Xu S, English J, Wu P, Schaefer E, Cobb M H. Reconstitution of mitogen-activated protein kinase phosphorylation cascades in bacteria. Efficient synthesis of active protein kinases. J Biol Chem. 1997;272:11057–11062. doi: 10.1074/jbc.272.17.11057. [DOI] [PubMed] [Google Scholar]
- 17.Le Goff P, Montano M M, Schodin D J, Katzenellenbogen B S. Phosphorylation of the human estrogen receptor. J Biol Chem. 1994;269:4458–4466. [PubMed] [Google Scholar]
- 18.Mangelsdorf D J, Thummel C, Beato M, Herrlich P, Schütz G, Umesono K, Blumberg B, Kastner P, Mark M, Chambon P, Evans R M. The nuclear receptor superfamily: the second decade. Cell. 1995;83:835–839. doi: 10.1016/0092-8674(95)90199-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moller D E, Xia C-H, Tang W, Zhu A X, Jakubowski M. Human rsk isoforms: cloning and characterization of tissue-specific expression. Am J Physiol. 1994;266:351–359. doi: 10.1152/ajpcell.1994.266.2.C351. [DOI] [PubMed] [Google Scholar]
- 20.Nakajima T, Fukamizu A, Takahashi J, Gage F H, Fisher T, Blenis J, Montminy M R. The signal-dependent coactivator CBP is a nuclear target for pp90RSK. Cell. 1996;86:465–474. doi: 10.1016/s0092-8674(00)80119-1. [DOI] [PubMed] [Google Scholar]
- 21.Nguyen T T, Scimeca J C, Filloux C, Peraldi P, Carpentier J L, Van Obberghen E. Co-regulation of the mitogen-activated protein kinase, extracellular signal-regulated kinase 1, and the 90-kDa ribosomal S6 kinase in PC12 cells. Distinct effects of the neurotrophic factor, nerve growth factor, and the mitogenic factor, epidermal growth factor. J Biol Chem. 1993;268:9803–9810. [PubMed] [Google Scholar]
- 22.Pumiglia K, Chow Y H, Fabian J, Morrison D, Decker S, Jove R. Raf-1 N-terminal sequence necessary for Ras-Raf interaction and signal transduction. Mol Cell Biol. 1995;15:398–406. doi: 10.1128/mcb.15.1.398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reese J C, Katzenellenbogen B S. Differential DNA-binding abilities of estrogen receptor occupied with two classes of antiestrogens: studies using human estrogen receptor overexpressed in mammalian cells. Nucleic Acids Res. 1991;19:6595–6602. doi: 10.1093/nar/19.23.6595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Richards S A, Lounsbury K M, Macara I G. The C terminus of the nuclear RAN/TC4 GTPase stabilizes the GDP-bound state and mediates interactions with RCC1, RAN-GAP, and HTF9A/RANBP1. J Biol Chem. 1995;270:14405–14411. doi: 10.1074/jbc.270.24.14405. [DOI] [PubMed] [Google Scholar]
- 25.Schouten G J, Vertegaal A C O, Whiteside S T, Israel A, Toebes M, Dorsman J C, van der Eb A J, Zantema A. IκBα is a target for the mitogen-activated 90 kDa ribosomal S6 kinase. EMBO J. 1997;16:3133–3144. doi: 10.1093/emboj/16.11.3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Smith C, Onate S A, Tsai M-J, O’Malley B W. CREB binding protein acts synergistically with steroid receptor coactivator-1 to enhance steroid receptor-dependent transcription. Proc Natl Acad Sci USA. 1996;93:8884–8888. doi: 10.1073/pnas.93.17.8884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tora L, Mullick A, Metzger D, Ponglikitmongkol M, Park I, Chambon P. The cloned oestrogen receptor contains a mutation which alters its hormone binding properties. EMBO J. 1989;8:1981–1986. doi: 10.1002/j.1460-2075.1989.tb03604.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Traish A M, Pavao M. Binding of site-directed monoclonal antibodies to an epitope located in the A/B region (amino acids 140-154) of human estrogen receptor-induced conformational changes in an epitope in the DNA-binding domain. Steroids. 1996;61:549–556. doi: 10.1016/s0039-128x(96)00109-2. [DOI] [PubMed] [Google Scholar]
- 29.Tzeng D Z, Klinge C M. Phosphorylation of purified estradiol-liganded estrogen receptor by casein kinase II increases estrogen response element binding but does not alter ligand stability. Biochem Biophys Res Commun. 1996;223:554–560. doi: 10.1006/bbrc.1996.0933. [DOI] [PubMed] [Google Scholar]
- 30.Tzukerman M T, Esty A, Santiso-Mere D, Danielian P, Parker M G, Stein R B, Pike J W, McDonnell D P. Human estrogen receptor transcriptional capacity is determined by both cellular and promoter context and mediated by two functionally distinct intramolecular regions. Mol Endocrinol. 1994;8:21–30. doi: 10.1210/mend.8.1.8152428. [DOI] [PubMed] [Google Scholar]
- 31.Vik T A, Sweet L J, Erikson R L. Coinfection of insect cells with recombinant baculovirus expressing pp60v-src results in the activation of a serine-specific protein kinase pp90rsk. Proc Natl Acad Sci USA. 1990;87:2685–2689. doi: 10.1073/pnas.87.7.2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Weigel N. Steroid hormone receptors and their regulation by phosphorylation. Biochem J. 1996;319:657–667. doi: 10.1042/bj3190657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yue W, Santen R J. Aromatase inhibitors: rationale for use following antiestrogen therapy. Semin Oncol. 1996;23:21–27. [PubMed] [Google Scholar]