

Abstract
The proliferation of lymphocytes in response to cytokine stimulation is essential for a variety of immune responses. Recent studies with signal transducer and activator of transcription 6 (Stat6)-deficient mice have demonstrated that this protein is required for the normal proliferation of lymphocytes in response to interleukin-4 (IL-4). In this report, we show that the impaired IL-4-induced proliferative response of Stat6-deficient lymphocytes is not due to an inability to activate alternate signaling pathways, such as those involving insulin receptor substrates, or to a failure to upregulate IL-4 receptor levels. Cell cycle analysis showed that the percentage of Stat6-deficient lymphocytes that transit from the G1 to the S phase of the cell cycle following IL-4 stimulation is lower than that of control lymphocytes. Although the regulation of many genes involved in the control of cytokine-induced proliferation is normal in Stat6-deficient lymphocytes, protein levels of the cdk inhibitor p27Kip1 were found to be markedly dysregulated. p27Kip1 is expressed at significantly higher levels in Stat6-deficient lymphocytes than in control cells following IL-4 stimulation. The higher level of p27Kip1 expression seen in IL-4-stimulated Stat6-deficient lymphocytes correlates with decreased cdk2-associated kinase activity and is the result of the increased accumulation of protein rather than altered mRNA expression. Similarly, higher levels of p27Kip1 protein expression are also seen following IL-12 stimulation of Stat4-deficient lymphocytes than are seen following stimulation of control cells. These data suggest that Stat proteins may control the cytokine-induced proliferative response of activated T cells by regulating the expression of cell cycle inhibitors so that cyclin-cdk complexes may function to promote transition from the G1 to the S phase of the cell cycle.
The proliferation of lymphocytes is dependent on the receipt of appropriate signals to complete transitions through the cell cycle. Antigen activation of a T lymphocyte, mediated by cross-linking of T-cell receptor complexes, enables the cell to transit from the G0 to the G1 phase of the cell cycle. Activated T lymphocytes then require cytokine stimulation to continue through the cell cycle and progress from the G1 to the S phase (33). In the absence of this second signal, lymphocytes do not proliferate and will undergo apoptosis.
The proliferative response of lymphocytes to interleukin-4 (IL-4) provides a useful model system for studying the mechanisms regulating cytokine-induced proliferation. IL-4-mediated responses result from the interaction of a ligand with a cell surface receptor composed of at least two membrane proteins; one chain specific for interactions with IL-4 (IL-4Rα) and a second common chain (γc) also used by the receptors for IL-2, IL-7, IL-9, and IL-15 (16, 27, 40, 48, 56). Recent studies have shown that a subunit of the high-affinity IL-13 receptor (IL-13R) may also be involved in one form of the IL-4R (18). Engagement of the IL-4R leads to activation of at least two distinct signaling pathways. IL-4 stimulation activates Janus kinases Jak1 and Jak3 (20, 36). The subsequent phosphorylation of the IL-4Rα chain at specific tyrosine residues by Jak kinases results in the activation of Stat6 (19, 28, 50). IL-4R engagement has also been shown to induce the phosphorylation of insulin receptor substrate (IRS) molecules such as IRS-1 and 4PS or IRS-2 (23, 54). Activated IRS-2 associates with phosphatidylinositol 3-kinase and may be responsible for mediating some IL-4-induced responses. A portion of the Ras pathway is also activated in response to IL-4, although studies have shown that this signaling pathway does not contribute to the proliferative response induced by IL-4 in lymphocytes (45).
Analysis of the cytoplasmic portion of the IL-4Rα chain led to the suggestion that separate regions are responsible for activation of the Stat and IRS signaling pathways. The tyrosine at amino acid 497 (Y497, on the basis of a numbering system where 1 is the amino terminus of the mature IL-4R protein) of the human IL-4R is part of the IRS interaction site and has been shown to be crucial for IL-4-induced proliferation. Transfected IL-4Rs, truncated or mutated so that they do not contain Y497, do not support IL-4-induced proliferation (11, 23). Several other tyrosine-phosphorylated sites have been identified as Stat6 docking sites, and any one of them individually can mediate the activation of Stat6 (19, 49, 60). Transfected receptors containing Y497 but lacking the three Stat6 docking sites were able to convey a proliferative signal, leading to the initial conclusion that Stat6 is dispensable for IL-4-mediated proliferative responses (46). However, in later studies, Stat6 phosphorylation could be detected in these transfectants, and presumably occurs through the interaction of Stat6 with the IRS binding site (Y497) which has homology with Stat6 docking sites (49). If the IRS docking site is indeed capable of mediating Stat6 activation, it would not be possible to determine the relative roles of these two signaling pathways in the proliferative response by this approach.
To determine the role of Stat6 in mediating IL-4-induced responses, we and others have recently generated Stat6-deficient mice (21, 53, 55). These mice lack a normal proliferative response to IL-4 but not to other cytokines or polyclonal activators, strongly supporting a role for Stat6 in IL-4-induced proliferation. In this study, we investigate the molecular mechanism by which Stat molecules control cytokine-induced proliferation. We show that Stat proteins regulate p27Kip1 expression and that modulation of p27Kip1 protein levels correlates with the ability of cytokine-stimulated cells to progress from the G1 to the S phase of the cell cycle.
MATERIALS AND METHODS
T-cell culture.
Total splenocytes (2 × 106/ml) were activated with 2.5 μg of concanavalin A (ConA) (Sigma Chemical Co., St. Louis, Mo.) per ml or 1 μg of plate-bound anti-CD3 (Pharmingen, San Diego, Calif.) per ml in RPMI 1640 supplemented as described previously (21). Cells were cultured for 72 h, washed, and purified over Lympholyte-M (Cedarlane Laboratories). Washes and subsequent incubations of cells activated with ConA were in medium containing 0.05 M α-methyl mannoside (Sigma). Cells were then replated at 2 × 106/ml (cell cycle analysis) or 5 × 106/ml (immunoprecipitations and RNA isolation) and incubated for the time periods indicated in the presence of 1,000 U of IL-4 (Genzyme, Cambridge, Mass.), 200 U of IL-2 (Boehringer-Mannheim), or 200 U of IL-12 (M. Gately, Hoffman-LaRoche) per ml or in the absence of interleukin.
Cell extracts.
Cells were lysed in a solution containing 0.5% Nonidet P-40 (NP-40), 50 mM Tris (pH 8.0), 0.1 mM EDTA, 150 mM NaCl, 1 mM Na3VO4, 5 mM NaF, 5 mM β-glycerol phosphate, 1 mM dithiothreitol, 1 mM phenylmethylsulfonyl fluoride, 2 μg of aprotinin per ml, 2 μg of pepstatin A per ml, 2 μg of leupeptin per ml, 1 mM benzamidine, 1 mM iodoacetamide, and 10% (vol/vol) glycerol by mixing vigorously with a pipette. Lysates were incubated on ice for 15 min and cleared of insoluble material by centrifugation at 13,000 × g for 10 min at 4°C.
Immunoprecipitations.
Three milligrams of extract was precleared with protein G-coated beads for 30 min at 4°C. One milligram of extract was used for each immunoprecipitation. Antibodies (anti-IRS-1 [Upstate Biotechnology Inc.], anti-IRS-2 [gift from M. White], and anti-Stat6 [as described in reference 51]) were added to extracts and incubated at 4°C for 1 h in IP buffer (0.5% NP-40, 50 mM HEPES [pH 7.6], 1 mM EDTA, 200 mM NaCl, 1 mM Na3VO4, 1 mM NaF, 1 mM β-glycerol phosphate, 1 mM phenylmethylsulfonyl fluoride, 1 μg of aprotinin per ml, 1 μg of leupeptin per ml, 10% (vol/vol) glycerol). Protein G was added, and the solution was rocked at 4°C for 1 h. Precipitates were washed two times in IP buffer and four times in IP buffer plus 0.5% deoxycholate. Precipitates were resuspended in polyacrylamide gel loading buffer.
Immunoblot analysis.
Proteins were separated on a polyacrylamide gel (8% for Stat6 and IRS immunoprecipitations; 10 to 15% for all other proteins examined) and transferred to nitrocellulose. Immunoblots for phosphotyrosine analysis were blocked for 2 h in TBST (50 mM Tris [pH 7.5], 100 mM NaCl, 0.05% NP-40, 0.03% Tween 20) plus 3% bovine serum albumin (BSA). Blots were then incubated with a mouse monoclonal antibody specific for phosphotyrosine (UBI) diluted 1/1,000 in TBS plus 1.5% BSA overnight at 4°C. Filters were washed four times in TBS and incubated with a horseradish peroxidase (HRP)-labelled antimouse secondary reagent (Amersham) diluted 1/3,000 in TBS plus 1.5% BSA for 2 h at room temperature. Filters were then washed five times in TBS, and detection was carried out with the Amersham enhanced chemiluminescence (ECL) detection kit.
Immunoblots for protein analysis were washed once in 2× TBST and blocked for 1 h in 1× TBST plus 4% dry milk. All subsequent incubations were performed with 2× TBST. Rabbit polyclonal antibodies (specific for cyclin E, cdk2, bcl-2, p21CIP1/WAF1, p27Kip1, and p57Kip2) were purchased from Santa Cruz Biotechnology (Santa Cruz, Calif.). Mouse monoclonal antibodies were used to detect p27Kip1 and bcl-x (Transduction Laboratories, Lexington, Ky.). Filters were incubated for 1 h at room temperature, washed twice, and incubated for an additional hour with HRP-labelled anti-rabbit antibody (Santa Cruz Biotechnology) or HRP-labelled antimouse antibody (Transduction Laboratories). Filters were washed once in incubation buffer and once in 2× TBST, and detection was carried out as described above. Filters were stripped and reblotted according to ECL detection kit directions.
Propidium iodide analysis.
At the indicated time points, cells were washed in phosphate-buffered saline and fixed in 80% ethanol. Cells were treated with 50 μg of RNase A per ml at 37°C for 30 min. Cells were then stained with 700 μM propidium iodide for 30 min at room temperature. Analysis was performed on a FACScan flow cytometer (Becton Dickenson). Percentages of cells undergoing apoptosis were determined with markers in the Lysis analysis package. Percentages of cells in specific stages of the cell cycle were determined by using the CellFit analysis program on the staining profile of live cells.
Northern analysis.
Cells were activated with ConA or anti-CD3 as indicated in the figure legends, and after further incubation in the presence or absence of IL-4 for the times indicated, cells were washed with phosphate-buffered saline and total RNA was isolated with Trizol (Gibco/BRL). RNA was separated on a 1.2% formaldehyde-agarose gel and transferred to GeneScreen (New England Nuclear) membranes. Membranes were hybridized with a genomic probe for c-myc (a gift from L. Jackson-Grusby), a glyceraldehyde-3-phosphate dehydrogenase cDNA, or a p27Kip1 cDNA (a gift from M. Fero and J. Roberts). A T-cell receptor α constant-region probe was used as a control probe where indicated since its levels vary less during T-cell activation than do those of many housekeeping genes.
Kinase assay.
Whole-cell extracts (60 μg) were immunoprecipitated with anti-cdk2 by incubation at 4°C overnight. Complexes were then incubated with protein G-conjugated agarose (Boehringer-Mannheim) for 2 h at 4°C, precipitated, and washed four times in kinase buffer (50 mM Tris-HCl [pH 7.4], 10 mM MgCl2, 1 mM dithiothreitol, 100 μg of BSA per ml). Precipitates were then resuspended in 30 μl of kinase buffer and supplemented with 30 μM ATP–1 μCi of [γ-32P]ATP–1 μg of histone H1. Reaction mixtures were incubated at 37°C for 1 h followed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis analysis. Proteins were transferred to a nitrocellulose membrane before autoradiography.
RESULTS
The IRS pathway is activated in the absence of Stat6.
Previous results from our laboratory and others demonstrated that Stat6 is required for a normal proliferative response to IL-4 (21, 53, 55). Although unlikely, it remained possible that in the absence of Stat6, other signaling pathways were not properly functioning. To assess this possibility, we examined the phosphorylation states of other known signaling components activated by IL-4. Control and Stat6-deficient spleen cells were activated with ConA and kept in culture for 3 days. Cells were then incubated for 15 min in the presence of 1,000 U of IL-4 per ml or in the absence of IL-4, and the phosphorylation states of Stat6, IRS-1, and IRS-2 were analyzed by phosphotyrosine immunoblotting of specific immunoprecipitates (Fig. 1). The low level of Stat6 phosphorylation seen in cells from unstimulated control cultures can be attributed to the small amounts of IL-4 produced by the ConA-activated cells. As shown in Fig. 1, stimulation of control lymphocytes with IL-4 leads to a large induction of Stat6 phosphorylation. Stat6 is, of course, not present in immunoprecipitates from Stat6-deficient cells. Both IRS-1 and IRS-2 are also phosphorylated in the absence of cytokine stimulation following ConA activation, a finding which similarly can be attributed to endogenous production of either IL-4 or IL-2. Importantly, IL-4 stimulation of both control and Stat6-deficient cells leads to equivalent increases in the levels of IRS phosphorylation. Thus, the IRS signaling pathway is activated by IL-4 in Stat6-deficient lymphocytes. The integrity of IRS signaling in resting spleen cells from Stat6-deficient mice has also recently been demonstrated (61). Since IRS phosphorylation is dependent on Jak activation and IL-4R phosphorylation, we can conclude that molecular events occurring prior to Stat activation are unaffected by the absence of Stat6.
FIG. 1.

Phosphotyrosine immunoblot analysis of signaling molecules activated by IL-4. ConA-activated control (+/+) and Stat6-deficient (−/−) lymphocytes were incubated in the presence (+) or absence (−) of IL-4 for 15 min. The indicated proteins were immunoprecipitated from total cell lysates and immunoblotted with a phosphotyrosine-specific antibody to determine phosphorylation state. IP, immunoprecipitate.
Impaired proliferation of Stat6-deficient lymphocytes is not due to a failure to upregulate IL-4R levels.
Previously, we demonstrated that IL-4-induced IL-4Rα chain upregulation does not occur in the absence of Stat6 and suggested that this lack of increased expression might be responsible for the impaired proliferation of Stat6-deficient lymphocytes in response to IL-4 (21). To test this possibility, control and Stat6-deficient lymphocytes were activated with ConA for 48 h and then examined for IL-4Rα expression levels by flow cytometry. Figure 2A shows that ConA induces an increase in the expression of IL-4Rα as previously described (12, 67) and that ConA stimulation results in equivalent levels of IL-4Rα expressed on activated lymphocytes from control and Stat6-deficient mice. These cells, which displayed high levels of IL-4Rα, were then assayed for their proliferative response to IL-4. As shown in Fig. 2B, the proliferation of activated, Stat6-deficient lymphocytes with high levels of IL-4Rα is still decreased compared to that of similarly treated control lymphocytes. Thus, the impaired proliferative response of Stat6-deficient lymphocytes to IL-4 stimulation is not due to the inability to upregulate IL-4Rα expression and thus increase cytokine responsiveness.
FIG. 2.
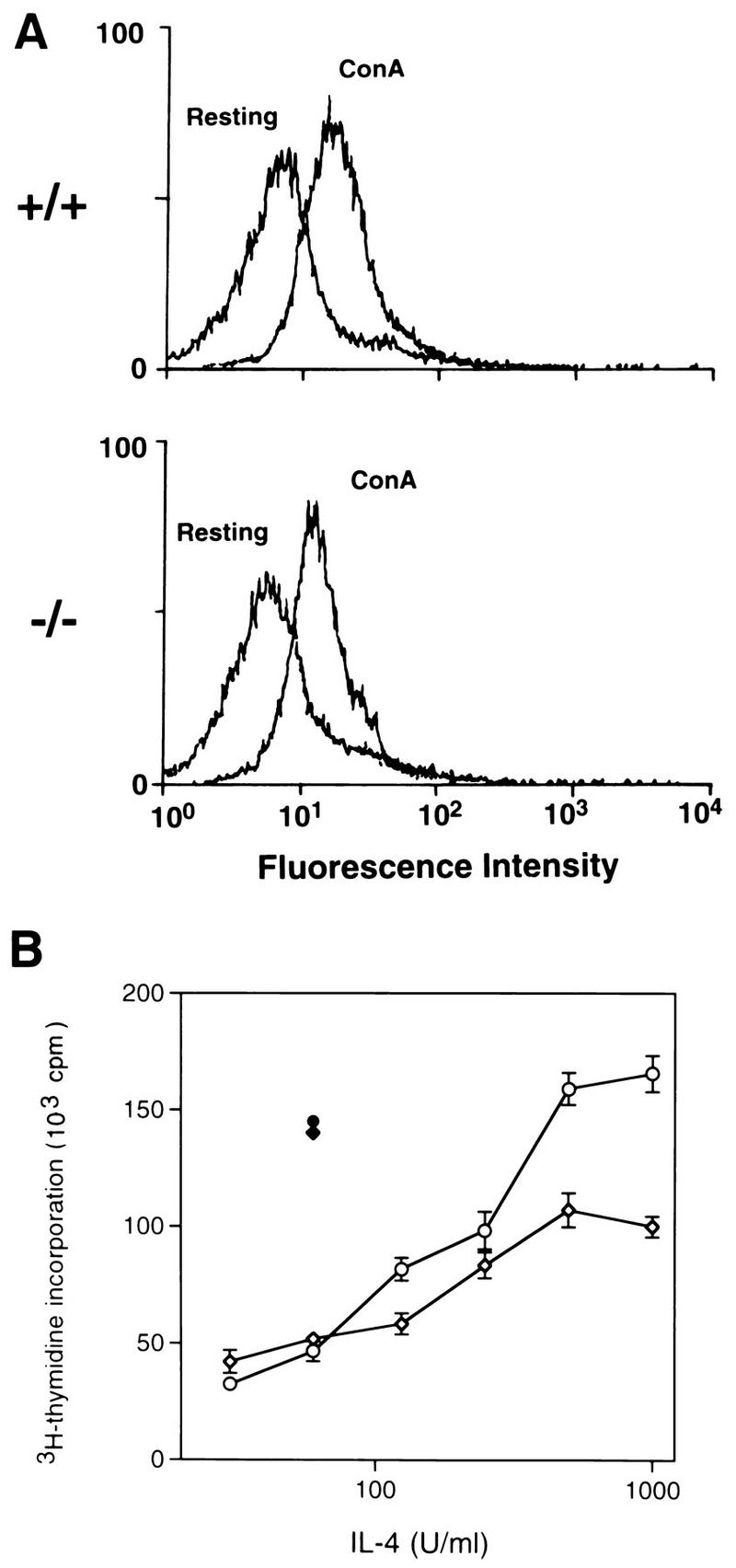
IL-4Rα expression and IL-4-induced proliferation of ConA-activated lymphocytes. (A) Flow cytometric analysis of IL-4Rα expression on control (+/+) and Stat6-deficient (−/−) lymphocytes which were either resting or activated with ConA for 48 h. (B) Control (open circles) and Stat6-deficient (open diamonds) lymphocytes activated with ConA for 48 h were assayed for proliferation in response to increasing concentrations of IL-4. The proliferation of control (solid circle) and Stat6-deficient (solid diamond) cells induced by IL-2 is also shown. Cells were pulsed with [3H]thymidine for the last 18 h of a 48-h culture period.
Stat6-deficient lymphocytes are impaired in their ability to transit from the G1 to the S phase of the cell cycle.
To further characterize the proliferative defect of Stat6-deficient lymphocytes, we examined cell cycle progression by flow cytometric analysis of propidium iodide-stained cells. Control and Stat6-deficient spleen cells were activated with ConA for 72 h, washed, and then incubated for an additional 24, 30, 36, or 48 h in the presence or absence of IL-4. As shown in Fig. 3A, less than 15% of activated cells were in the S or G2/M phase of the cell cycle at any time point analyzed in the absence of cytokine stimulation. In contrast, nearly 50% of control cells, but only 23% of Stat6-deficient cells, were in the S or G2/M phase 30 h after IL-4 stimulation. Given that the maximum percentage of activated control cells were dividing at 30 h after cytokine stimulation, we chose this time point for further cell cycle analysis.
FIG. 3.

Cell cycle analysis of cytokine-stimulated lymphocytes. (A) Time course of entry into the S+G2M stages of the cell cycle. The percentage of dividing cells was determined at each of the indicated time points by propidium iodide staining of control (squares) and Stat6-deficient (circles) lymphocytes which had been activated with ConA for 72 h and then cultured in the presence of 1,000 U of IL-4 per ml (solid symbols) or in the absence of IL-4 (open symbols). The standard error for all points was less than 5%. (B) Cell cycle analysis of lymphocytes following 30 h of cytokine stimulation. Control (+/+) or Stat6-deficient (−/−) lymphocytes were activated with ConA for 72 h and then cultured for an additional 30 h in the presence of 200 U of IL-2 or 1,000 U of IL-4 per ml or in the absence of IL (unstimulated). Numbers to the left of the major G0/G1 peaks represent the numbers of apoptotic events expressed as percentages of all events analyzed by flow cytometry. Numbers to the right of the major G0/G1 peaks represent the numbers of cells in the S+G2M phases of the cell cycle expressed as percentages of all live cells, as calculated by the CellFit analysis program.
Two distinct possibilities could explain the proliferative defect seen in Stat6-deficient lymphocytes. Stat6-deficient lymphocytes, being unable to respond to IL-4, may be growth factor starved and undergoing apoptosis, or, alternatively, cells may be surviving but simply not proliferating. To address these two possibilities, control and Stat6-deficient spleen cells were activated with ConA, washed, and then incubated for an additional 30 h either with IL-2 or IL-4 or without further stimulation. Cells were then stained with propidium iodide and analyzed by flow cytometry. In the absence of exogenous cytokine stimulation (Fig. 3B, left panels), a large percentage of the cells (35 to 45%) were in an apoptotic state, as evidenced by the presence of an amount of DNA less than 2N. By contrast, both control and Stat6-deficient lymphocytes stimulated with IL-2 showed minimal levels (8 to 12%) of apoptosis (Fig. 3B, middle panels). Similarly, there was minimal cell death (7 to 11%) in either control or Stat6-deficient lymphocytes stimulated with IL-4. These results suggest that IL-4 is capable of inhibiting the apoptosis of activated T cells in the absence of Stat6 and are consistent with recent reports suggesting that the IRS signaling pathway is responsible for the antiapoptotic signal delivered by IL-4 in lymphocytes (64) and that IL-4 can protect Stat6-deficient resting T cells from apoptosis (58).
Flow cytometric profiles of propidium iodide-stained lymphocytes were analyzed with the computer program CellFit. Of the live cells cultured in the absence of any cytokines for 30 h, less than 10% were in the S or G2/M phase of the cell cycle (Fig. 3B, left panels). Control and Stat6-deficient lymphocytes stimulated with IL-2 for 30 h progressed through the cell cycle, as both populations had greater than 30% of the cells accumulated in the S or G2/M phase. While control cells showed a similar response to IL-4, there was a 50% decrease in the number of Stat6-deficient lymphocytes in the S or G2/M phase of the cell cycle following IL-4 stimulation (Fig. 3B, right panels). These results demonstrate that the impaired proliferative response of Stat6-deficient lymphocytes to IL-4 correlates with an inability to progress through a control point in the G1-to-S-phase transition.
Stat6 regulates proliferation by altering levels of a cdk inhibitor.
Many of the proteins involved in the control of cellular proliferation have now been characterized. Since some of the genes encoding these proteins are known to be cytokine and cell cycle regulated, we investigated whether a dysregulation of any of these genes or their protein products could explain the proliferative defect seen in Stat6-deficient lymphocytes. c-myc plays a major role in cell cycle progression and is known to be induced within 1 h of IL-4 stimulation (25). Northern analysis of total RNA from ConA-activated control and Stat6-deficient lymphocytes incubated for 1 h either in the presence or absence of IL-4 demonstrated that c-myc induction occurs normally in the absence of Stat6 (Fig. 4).
FIG. 4.

Analysis of c-myc mRNA expression. Control (+/+) and Stat6-deficient (−/−) lymphocytes were activated with ConA for 72 h and then incubated for one additional hour in the presence (+) or absence (−) of IL-4. Ten micrograms of total RNA was loaded in each lane. Following hybridization with a c-myc probe, the blot was stripped and rehybridized with a glyceraldehyde-3-phosphate dehydrogenase (GAPDH) probe as a control for RNA loading.
Cyclin dependent kinase 2 (cdk2) associates with cyclin E during the transition from the G1 to the S phase of the cell cycle, and with cyclin A during the early S phase. Expression of these cyclins leads to an increase in cdk2-associated kinase activity, which is critical for cell cycle transit (52). To determine whether kinase activity was affected in the absence of Stat6, cdk2-associated kinase activities in extracts from anti-CD3-activated control and Stat6-deficient cells which were stimulated for 30 h either in the presence or absence of cytokines were examined. cdk2 was immunoprecipitated with polyclonal antisera and tested in a kinase reaction with histone H1 as a substrate. As shown in Fig. 5A, extracts from unstimulated control cells had small amounts of kinase activity, while stimulation with either IL-2 or IL-4 led to a marked increase in kinase activity. While IL-2 stimulation caused a significant increase in kinase activity in Stat6-deficient lymphocytes, there was only a modest increase in activity following IL-4 stimulation. Immunoblot analysis of these same cell extracts demonstrated that protein levels of cdk2 and cyclin E did not change significantly with cytokine stimulation and were similar for both normal and Stat6-deficient cells (Fig. 5B).
FIG. 5.
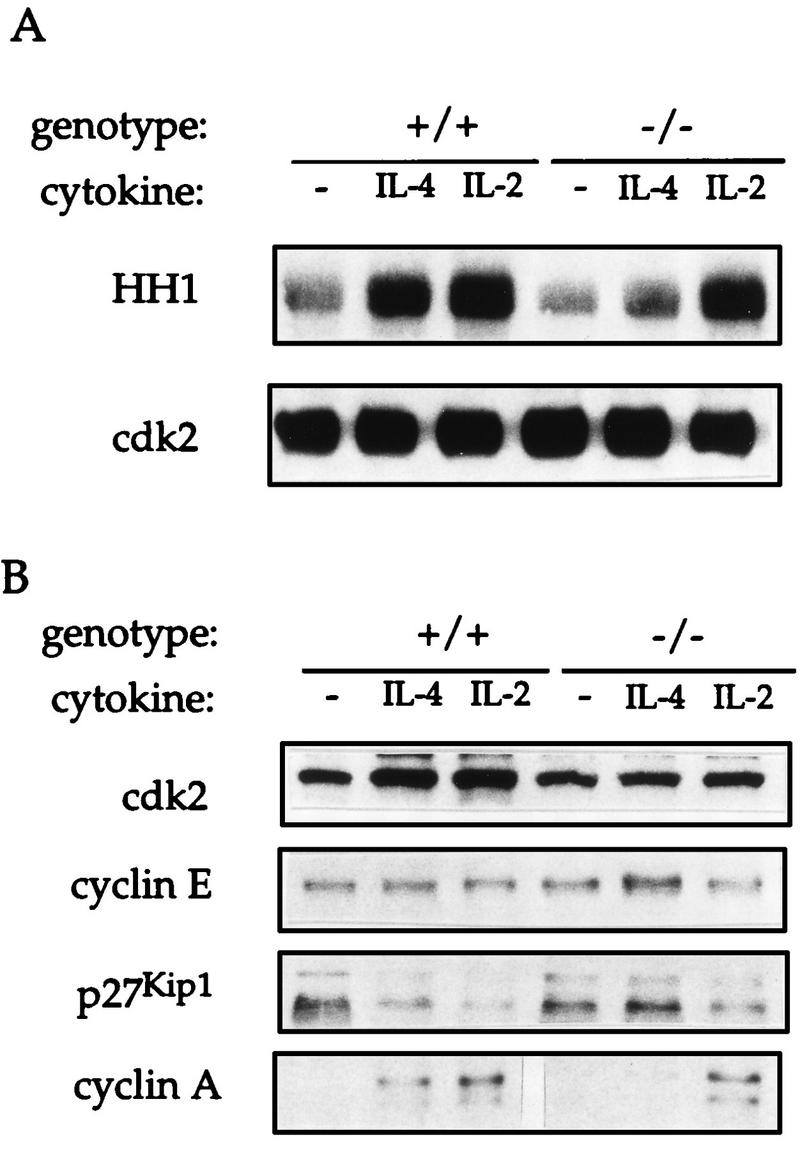
Analysis of cyclin, cdk, and cdk inhibitor expression. (A) Control (+/+) and Stat6-deficient (−/−) lymphocytes were activated with anti-CD3 for 72 h and incubated for an additional 30 h in the presence or absence (−) of the indicated cytokine. Total cell extracts were immunoprecipitated with anti-cdk2 and precipitates were used in a histone H1 (HH1) kinase reaction. Reaction mixtures were electrophoresed on a polyacrylamide gel and transferred to nitrocellulose before exposure to X-ray film. Radioactive histone H1 is shown in the top panel. Nitrocellulose was probed with anti-cdk2 to verify equivalent amounts of cdk2 immunoprecipitated in each sample. (B) Total cell extracts (30 μg) prepared from the cells stimulated as described for panel A were electrophoresed on a polyacrylamide gel and immunoblotted with the indicated antibodies.
The activity of the cdk2 complex is regulated by a family of three cdk inhibitors, p21CIP1/WAF1, p27Kip1, and p57Kip2 (52). Recently, Stat1 has been shown to regulate the expression of p21CIP1/WAF1, which mediates the antiproliferative effect of gamma interferon (6). However, lymphoid cells express very low levels of either p21CIP1/WAF1 or p57Kip2 (32, 43), and, consistent with previous analyses, small amounts of these two proteins were seen in spleen cell extracts and their levels were not altered with exposure to IL-4 (data not shown). Previously, IL-2 has been shown to decrease the expression of p27Kip1 in both a mouse cell line and human peripheral blood lymphocytes (14, 41). As seen in Fig. 5B, immunoblot analysis showed that p27Kip1 was readily detected in cell extracts from both control and Stat6-deficient lymphocytes. Treatment of control cells with IL-4 led to a marked decrease of p27Kip1 expression, consistent with previous studies using IL-2. Strikingly, this response was absent in Stat6-deficient lymphocytes stimulated with IL-4.
The impaired ability of IL-4 to induce cdk2 activity (Fig. 5A) and to downregulate p27Kip1 expression (Fig. 5B) in Stat6-deficient lymphocytes had demonstrable affects on the expression of proteins which regulate downstream stages of the cell cycle. It has previously been suggested that cyclin E-cdk2 complexes may be directly involved in the transcription of cyclin A and that p27Kip1 interferes with upregulation of cyclin A gene expression (65). Immunoblot analysis of cyclin A protein levels showed that, while cyclin A was absent in unstimulated cells, control cells stimulated with either IL-2 or IL-4 had a significant increase in cyclin A protein levels. In the absence of Stat6, however, IL-4 stimulation did not lead to an increase in the level of the cyclin A protein.
p27Kip1 is regulated by other Stat-activating cytokines.
We and others have described a proliferative defect in Stat4-deficient lymphocytes stimulated with IL-12 similar to that seen in Stat6-deficient lymphocytes stimulated with IL-4 (22, 57). To address whether p27Kip1 protein levels may be regulated by other Stat molecules, control cells were stimulated with a variety of cytokines and cell extracts were subjected to immunoblot analysis. As shown in Fig. 6, stimulation of control cells with IL-2, IL-4, or IL-12, which activates Stat5, Stat6, and Stat4, respectively, leads to a significant decrease in p27Kip1 expression. This decrease in p27Kip1 expression, however, is absent in Stat4-deficient lymphocytes stimulated with IL-12. Regulation of p27Kip1 is not completely impaired in Stat4-deficient cells since treatment of Stat4-deficient lymphocytes with IL-2 or IL-4 leads to a decrease in p27Kip1 protein levels.
FIG. 6.

p27Kip1 expression in control and Stat4-deficient lymphocytes. Control (+/+) and Stat4-deficient (−/−) lymphocytes were activated with anti-CD3 for 72 h and incubated for an additional 30 h in the presence or absence (−) of the indicated cytokine. Cell extracts were analyzed by immunoblotting as described in the legend for Fig. 5.
Stat6 regulates protein but not mRNA levels of p27Kip1.
p27Kip1 expression has been shown to be regulated at the transcriptional, translational, and posttranslational levels (1, 17, 26, 29, 31, 35, 42). In murine T cells, anti-CD3 stimulation leads to transcriptional downregulation of p27Kip1 mRNA (29) (Fig. 7A), a response which is normal in Stat6-deficient T cells (data not shown). This elimination of the p27Kip1 message is maintained for at least 72 h of culture in the presence of anti-CD3 (Fig. 7A) and may explain the relatively minor impairment seen when cells from p27Kip1-deficient mice were stimulated in vitro with anti-CD3 (13, 24, 39). To determine whether p27Kip1 mRNA is also downregulated in cytokine-stimulated activated T cells, we performed Northern analysis of total RNA from control and Stat6-deficient T cells activated for 72 h with anti-CD3 followed by incubation in the presence or absence of IL-4 for 20 or 30 h as indicated. Figure 7B demonstrates that p27Kip1 mRNA levels accumulate to a resting level of expression by 20 h after removal from the anti-CD3 stimulus and are not further increased by 30 h. Furthermore, there is no effect of IL-4 on the p27Kip1 message seen in either control or Stat6-deficient lymphocytes. Thus, the dysregulation of p27Kip1 protein levels seen in IL-4-stimulated Stat6-deficient lymphocytes (Fig. 5B) does not correlate with altered levels of mRNA expression.
FIG. 7.

Northern analysis of p27Kip1 mRNA expression. (A) Total RNA was isolated from control purified CD4+ T cells that were unstimulated (0) or that were activated with anti-CD3 for 24 or 72 h. Northern analysis was performed by hybridizing filters with a p27Kip1 cDNA probe; a T cell receptor α (TCRα) probe was used as a control. (B) Total RNA was isolated from control (+/+) and Stat6-deficient (−/−) lymphocytes that were activated with anti-CD3 for 72 h followed by incubation for 20 or 30 h in the presence (+) or absence (−) of IL-4. Northern analysis was performed as described for panel A.
To further examine the mechanism by which p27Kip1 protein levels are regulated, we performed a time course analysis, focusing on time points at which cells were known to be proliferating, as shown in Fig. 3. In the absence of cytokine stimulation, p27Kip1 protein levels reaccumulate to resting levels between 24 and 30 h after removal from anti-CD3 stimulation (Fig. 8), and this correlates with the reexpression of p27Kip1 mRNA (Fig. 7). Stimulation of control cells with IL-4 prolongs the recovery of protein levels seen at the resting phase past the 48-h time point (Fig. 8), correlating with the ability of the cells to proliferate (Fig. 3). In the absence of Stat6 however, IL-4-stimulated p27Kip1 protein levels reaccumulate much faster (Fig. 8), again correlating with an inhibition of cytokine-driven proliferation (Fig. 3).
FIG. 8.

Time course analysis of p27 protein levels. Control (+/+) and Stat6-deficient (−/−) lymphocytes were activated with anti-CD3 for 72 h and incubated for the additional times indicated in the presence (+) or absence (−) of IL-4. Cell extracts were analyzed by immunoblotting as described in the legend for Fig. 5.
DISCUSSION
In this report we have demonstrated that the impaired proliferation of Stat6-deficient lymphocytes in response to IL-4 stimulation correlates with a dysregulation in the expression of p27Kip1, the major cyclin-dependent kinase inhibitor in lymphocytes. Nourse et al. (41) demonstrated that in IL-2–rapamycin-stimulated extracts in which p27Kip1 was not downregulated, removal of p27Kip1 from the extract returned cdk activity to normal IL-2-stimulated levels. This suggests that during cytokine stimulation, p27Kip1 is an important factor controlling cdk function and thus cell cycle progression. This conclusion is also supported by the lymphoproliferative dysregulation seen in p27Kip1 knockout mice in vivo (13, 24, 39) and in vitro (13, 30). In contrast, no lymphoproliferative disorders in p21WAF1/CIP1-deficient mice (4, 9) or in p57Kip2-deficient mice (62, 66) were described, consistent with our data suggesting that these cdk inhibitors play a minor role in regulating the initial lymphocyte proliferative response. p21WAF1/CIP1 has been shown to be upregulated at later time points following lymphocyte stimulation (14, 41), making it possible that p21WAF1/CIP1 may play a role in later stages of the cell cycle or in more differentiated lymphocytes.
The importance of Stat proteins in proliferative responses has been highlighted in work characterizing Stat6- and Stat4-deficient lymphocytes (21, 22, 53, 55, 57). The demonstration that Stat6-deficient lymphocytes lacked a normal proliferative response to IL-4 contrasted with the results of previous studies suggesting that Stat6 was dispensable for mitogenic responses (46). However, more recent studies showing the activation of Stat6 in the absence of characterized Stat6 docking sites (49) and the correlation of Stat6 activation with IL-4-induced proliferation in clones which do not activate the IRS pathway (44) support a role for Stat6 in the proliferative process. In this report we demonstrate that the regulation of the proliferative response by Stat molecules is not restricted to Stat6 in that IL-12 similarly affects p27Kip1 expression through a Stat4-dependent mechanism. Our data are similar to those supporting the previous finding that IL-2 led to downregulation of p27Kip1 (14, 41) and suggest that it is Stat5 in the IL-2 response that mediates this process. As with Stat6, there have been conflicting results regarding the role of Stat5 in cytokine-stimulated proliferation induced by both truncated receptor transfectants and dominant-negative Stat5 proteins (7, 15, 38, 47, 59). The analysis of Stat5-deficient lymphocytes should help to resolve this issue. The results in this report also suggest that some of the differences seen in studies using transfected cell lines may be due to variation in endogenous p27Kip1 levels. The experiments presented here support a paradigm in which p27Kip1 serves as a central check point for regulating lymphocyte proliferation induced by cytokine-stimulated Stat signaling.
Whether Stat proteins regulate p27Kip1 protein levels by a transcriptional, translational, or posttranslational mechanism is still unclear. The transcriptional downregulation associated with anti-CD3 stimulation of lymphocytes (29) (Fig. 7A) does not seem to play a role in the cytokine responses we have discussed in this report since message levels were not altered by cytokine treatment (Fig. 7B). Translational control of p27Kip1 protein levels has been demonstrated by decreased rates of protein synthesis when fibroblast or epithelial cells are stimulated with either serum or platelet-derived growth factor and correlates with the decreased association of the p27Kip1 message with ribosomal complexes (1, 17, 35). Alternatively, p27Kip1 protein levels are posttranslationally regulated by ubiquitin-mediated degradation following entry of a cell into the cell cycle (42). Since no genes that regulate these processes are known to be Stat regulated, it is difficult to immediately determine which pathway is affected in Stat-deficient lymphocytes.
While we have demonstrated that regulation of proliferation is Stat dependent, the primary cell cycle machineries responsible for apoptosis inhibition and cycle progression are activated independent of Stat molecules. The mechanism of protection from apoptosis is still unclear. In contrast to IL-2 stimulation of activated cells, which leads to an increase in the expression of bcl-2 and maintenance of bcl-x expression (3, 10, 37), stimulation of activated T cells with IL-4 did not alter the protein levels of bcl-2 or bcl-x (data not shown). We have shown that IL-4-induced regulation of c-myc and the regulation of cdk-cyclin complexes up to the G1-S restriction point are normal in the absence of Stat6. This further supports the idea that the IRS signaling pathway or other IL-4-stimulated signaling pathways contribute to the mitogenic response. Indeed, both in our previous study (21) and in this study (Fig. 2B and 3), low levels of IL-4-induced proliferation in Stat6-deficient cells that must be a result of these other pathways were found. Since Stat regulation of the proliferative response seems to be restricted to the action of a single cdk inhibitor, the above finding may explain why there is a greater proliferative defect when Stat6-deficient cells are stimulated with IL-4 alone (21) than when IL-4 and costimulators are used (53, 55). First, costimuli may provide additional mitogenic signals that can overcome the maintenance of high-level p27Kip1 expression or regulate the levels of p27Kip1 themselves, thus minimizing the requirement for Stat6 in IL-4-induced proliferation (2). Additionally, these stimuli could lead to the secretion of other cytokines that could act on the cell to decrease p27Kip1 expression.
Most importantly, the results in this report indicate a mechanism through which Stat proteins may contribute to cellular transformation. Constitutively activated Stat proteins have been found in cells transformed by human T-cell leukemia virus type 1 (HTLV-1), src, and abl, and by bcr-abl fusions (5, 8, 34, 63). Cells transformed by HTLV-1 became lymphokine independent as constitutively activated Stat molecules appeared within the cells. Our results suggest that the breakdown of cellular division control must be at two levels: first, by activation of the pathways leading to cyclin-cdk activation, and second, by decreasing expression of cdk inhibitors. In this model, Stat molecules would play the auxiliary role of downregulating the cdk inhibitor so that cell cycle progression activated by the main transforming molecule can proceed unimpeded. In previous reports we have suggested that targeting Stats in vivo may be a useful method of modulating T-helper-cell responses. This study further suggests that intervention of Stat function may be effective in the treatment of tumorigenesis.
ACKNOWLEDGMENTS
We thank L. Jackson-Grusby, M. Fero, J. Roberts, J. Pierce, M. White, and M. Gately for providing probes, antisera, and cytokines. We also thank M. Carroll for critical review of this paper.
M.H.K. is a Special Fellow and M.J.G. is a Scholar of the Leukemia Society of America. This work was supported by the Mathers Foundation and NIH grant AI40171.
REFERENCES
- 1.Agrawal D, Hauser P, McPherson F, Dong F, Garcia A, Pledger W J. Repression of p27Kip1 synthesis by platelet-derived growth factor in BALB/c 3T3 cells. Mol Cell Biol. 1996;16:4327–4336. doi: 10.1128/mcb.16.8.4327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Blanchard D A, Affredou M T, Vazquez A. Modulation of p27Kip1 cyclin-dependent kinase inhibitor expression during IL-4 mediated human B cell activation. J Immunol. 1997;158:3054–3061. [PubMed] [Google Scholar]
- 3.Broome H E, Dargan C M, Krajewski S, Reed J C. Expression of bcl-2, bcl-x and bax after T cell activation and IL-2 withdrawal. J Immunol. 1995;155:2311–2317. [PubMed] [Google Scholar]
- 4.Brugarolos J, Chandrasekaran C, Gordon J I, Beach D, Lacks T, Hannon G J. Radiation-induced cell cycle arrest compromised by p21 deficiency. Nature. 1995;377:552–557. doi: 10.1038/377552a0. [DOI] [PubMed] [Google Scholar]
- 5.Carlesso N, Frank D A, Griffen J D. Tyrosyl phosphorylation and DNA binding activity of signal transducers and activators of transcription (STAT) proteins in hematopoietic cell lines transformed by Bcr/Abl. J Exp Med. 1996;183:811–820. doi: 10.1084/jem.183.3.811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chin Y E, Kitagawa M, Su W-C S, You Z-H, Iwamoto Y, Fu X-Y. Cell growth arrest and induction of cyclin-dependent kinase inhibitor p21Waf1/Cip1 mediated by Stat1. Science. 1996;272:719–722. doi: 10.1126/science.272.5262.719. [DOI] [PubMed] [Google Scholar]
- 7.Damen J E, Wakao H, Miyajima A, Krosl J, Humphries R K, Cutler R L, Krystal G. Tyrosine 343 in the erythropoietin receptor positively regulates erythropoietin-induced cell proliferation and Stat5 activation. EMBO J. 1995;14:5557–5568. doi: 10.1002/j.1460-2075.1995.tb00243.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Danial N N, Pernis A, Rothman P B. Jak-Stat signaling induced by the v-abl oncogene. Science. 1995;269:1875–1877. doi: 10.1126/science.7569929. [DOI] [PubMed] [Google Scholar]
- 9.Deng C, Zhang P, Harper J W, Elledge S J, Leder P. Mice lacking p21Cip1/Waf1 undergo normal development, but are defective in G1 checkpoint control. Cell. 1995;82:675–684. doi: 10.1016/0092-8674(95)90039-x. [DOI] [PubMed] [Google Scholar]
- 10.Deng G, Podack E R. Suppression of apoptosis in a cytotoxic T-cell line by interleukin 2 mediated gene transcription and deregulated expression of the protooncogene bcl-2. Proc Natl Acad Sci USA. 1993;90:2189–2193. doi: 10.1073/pnas.90.6.2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Deutsch H H J, Koettnitz K, Chung J, Kalthoff F S. Distinct sequence motifs within the cytoplasmic domain of the human IL-4 receptor differentially regulate apoptosis inhibition and cell growth control. J Immunol. 1995;154:3696–3703. [PubMed] [Google Scholar]
- 12.Dokter W H, Borger P, Hendricks D, van der Horst I, Halie M R, Vellenga E. Interleukin-4 (IL-4) receptor expression on human T cells is affected by different intracellular signaling pathways and by IL-4 at transcriptional and posttranscriptional levels. Blood. 1992;80:2721–2728. [PubMed] [Google Scholar]
- 13.Fero M L, Rivkin M, Tasch M, Porter P, Carow C E, Firpo E, Polyak K, Tsai L-H, Broudy V, Perlmutter R M, Kaushansky K, Roberts J M. A syndrome of multiorgan hyperplasia with features of gigantism, tumorigenesis, and female sterility in p27Kip1-deficient mice. Cell. 1996;85:733–744. doi: 10.1016/s0092-8674(00)81239-8. [DOI] [PubMed] [Google Scholar]
- 14.Firpo E J, Koff A, Solomon M J, Roberts J M. Inactivation of a Cdk2 inhibitor during interleukin 2-induced proliferation of human T lymphocytes. Mol Cell Biol. 1994;14:4889–4901. doi: 10.1128/mcb.14.7.4889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fujii H, Nakagawa Y, Schindler U, Kawahara A, Mori H, Gouilleux F, Groner B, Ihle J N, Minami Y, Miyazaki T. Activation of Stat5 by interleukin 2 requires a carboxyl-terminal region of the interleukin 2 receptor beta chain but is not essential for proliferative signal transmission. Proc Natl Acad Sci USA. 1995;92:5482–5486. doi: 10.1073/pnas.92.12.5482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Giri J G, Ahdieh M, Eisenman J, Shanebeck K, Grabstein K, Kumaki S, Namen A, Park L S, Cosman D, Anderson D. Utilization of the beta and gamma chains of the IL-2 receptor by the novel cytokine IL-15. EMBO J. 1994;13:2822–2830. doi: 10.1002/j.1460-2075.1994.tb06576.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hengst L, Reed S I. Translational control of p27Kip1 accumulation during the cell cycle. Science. 1996;271:1861–1864. doi: 10.1126/science.271.5257.1861. [DOI] [PubMed] [Google Scholar]
- 18.Hilton D J, Zhang J-G, Metcalf D, Alexander W S, Nicola N A, Wilson T A. Cloning and characterization of a binding subunit of the interleukin 13 receptor that is also a component of the interleukin 4 receptor. Proc Natl Acad Sci USA. 1996;93:497–501. doi: 10.1073/pnas.93.1.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hou J, Schindler U, Henzel W J, Ho T C, Brasseur M, McKnight S L. An interleukin-4-induced transcription factor: IL-4 Stat. Science. 1994;265:1701–1706. doi: 10.1126/science.8085155. [DOI] [PubMed] [Google Scholar]
- 20.Johnston J A, Kawamura M, Kirken R A, Chen Y-Q, Blake T B, Shibuya K, Ortaldo J R, McVicar D W, O’Shea J J. Phosphorylation and activation of the Jak-3 Janus kinase in response to interleukin-2. Nature. 1994;370:151–153. doi: 10.1038/370151a0. [DOI] [PubMed] [Google Scholar]
- 21.Kaplan M H, Schindler U, Smiley S T, Grusby M J. Stat6 is required for mediating responses to IL-4 and for the development of Th2 cells. Immunity. 1996;4:313–319. doi: 10.1016/s1074-7613(00)80439-2. [DOI] [PubMed] [Google Scholar]
- 22.Kaplan M H, Sun Y-L, Hoey T, Grusby M J. Impaired IL-12 responses and enhanced development of Th2 cells in Stat4-deficient mice. Nature. 1996;382:174–177. doi: 10.1038/382174a0. [DOI] [PubMed] [Google Scholar]
- 23.Keegan A D, Nelms K, White M, Wang L-M, Pierce J H, Paul W E. An IL-4 receptor region containing an insulin receptor motif is important for IL-4 mediated IRS-1 phosphorylation and cell growth. Cell. 1994;76:811–820. doi: 10.1016/0092-8674(94)90356-5. [DOI] [PubMed] [Google Scholar]
- 24.Kiyokawa H, Kineman R D, Manova-Todorova K O, Soares V C, Hoffman E S, Ono M, Khanam D, Hayday A C, Frohman L A, Koff A. Enhanced growth of mice lacking the cyclin-dependent kinase inhibitor function of p27Kip1. Cell. 1996;85:721–732. doi: 10.1016/s0092-8674(00)81238-6. [DOI] [PubMed] [Google Scholar]
- 25.Klemsz M J, Justement L B, Palmer E, Cambier J C. Induction of c-fos and c-myc expression during B cell activation by IL-4 and immunoglobulin binding ligands. J Immunol. 1989;143:1032–1039. [PubMed] [Google Scholar]
- 26.Kobayashi K, Phuchareon J, Inada K, Tomita Y, Koizumi T, Hatano M, Miyatake S, Tokuhisa T. Overexpression of c-fos inhibits down-regulation of a cyclin-dependent kinase-2 inhibitor p27Kip1 in splenic B cells activated by surface Ig cross-linking. J Immunol. 1997;158:2050–2056. [PubMed] [Google Scholar]
- 27.Kondo M, Takeshita T, Higuchi M, Nakamura M, Sudo T, Nishikawa S-I, Sugamura K. Functional participation of the IL-2 receptor gamma chain in IL-7 receptor complexes. Science. 1994;263:1453–1454. doi: 10.1126/science.8128231. [DOI] [PubMed] [Google Scholar]
- 28.Kotanides H, Reich N C. Requirement of tyrosine phosphorylation for rapid activation of a DNA binding factor by IL-4. Science. 1993;262:1265–1267. doi: 10.1126/science.7694370. [DOI] [PubMed] [Google Scholar]
- 29.Kwon T K, Buchholz M A, Ponsalle P, Chrest F J, Nordin A A. The regulation of p27Kip1 expression following the polyclonal activation of murine Go T cells. J Immunol. 1997;158:5642–5648. [PubMed] [Google Scholar]
- 30.Luo Y, Marx S O, Kiyokawa H, Koff A, Massague J, Marks A R. Rapamycin resistance tied to defective regulation of p27Kip1. Mol Cell Biol. 1996;16:6744–6751. doi: 10.1128/mcb.16.12.6744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Matsumura I, Ishikawa J, Nakajima K, Oritani K, Tomiyama Y, Miyagawa J-I, Kato T, Miyazaki H, Matsuzawa Y, Kanakura Y. Thrombopoietin-induced differentiation of human megakaryoblastic leukemia cell line, CMK, involves transcriptional activation of p21WAF1/Cip1 by STAT5. Mol Cell Biol. 1997;17:2933–2943. doi: 10.1128/mcb.17.5.2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Matsuoka S, Edwards M C, Bai C, Parker S, Zhang P, Baldini A, Harper J W, Elledge S J. p57Kip2, a structurally distinct member of the p21Cip1 Cdk inhibitor family, is a candidate tumor suppressor gene. Genes Dev. 1995;9:650–662. doi: 10.1101/gad.9.6.650. [DOI] [PubMed] [Google Scholar]
- 33.Meur S C, zum Buschenfelde K H M. T cell receptor triggering induces responsiveness to interleukin 1 and interleukin 2 but does not lead to proliferation. J Immunol. 1986;136:4106–4112. [PubMed] [Google Scholar]
- 34.Migone T-S, Lin J-X, Cereseto A, Mulloy J C, O’Shea J J, Franchini G, Leonard W J. Constitutively activated Jak-Stat pathway in T cells transformed with HTLV-I. Science. 1995;269:79–81. doi: 10.1126/science.7604283. [DOI] [PubMed] [Google Scholar]
- 35.Millard S S, Yan J S, Nguyen H, Pagano M, Kiyokawa H, Koff A. Enhanced ribosomal association of p27Kip1 mRNA is a mechanism contributing to accumulation during growth arrest. J Biol Chem. 1997;272:7093–7098. doi: 10.1074/jbc.272.11.7093. [DOI] [PubMed] [Google Scholar]
- 36.Miyazaki T, Kawahara A, Fujii H, Nakagawa Y, Minami Y, Liu Z-J, Oishi I, Silvennoinen O, Witthuhn B A, Ihle J N, Taniguchi T. Functional activation of Jak1 and Jak3 by selective association with IL-2 receptor subunits. Science. 1994;266:1045–1047. doi: 10.1126/science.7973659. [DOI] [PubMed] [Google Scholar]
- 37.Miyazaki T, Liu Z-J, Kawahara A, Minami Y, Yamada K, Tsujimoto Y, Barsoumian E L, Perlmutter R M, Taniguchi T. Three distinct IL-2 signaling pathways mediated by bcl-2, c-myc and lck cooperate in hematopoietic cell proliferation. Cell. 1995;81:223–231. doi: 10.1016/0092-8674(95)90332-1. [DOI] [PubMed] [Google Scholar]
- 38.Mui A L-F, Wakao H, Kinoshita T, Kitamura T, Miyajima A. Suppression of interleukin-3-induced gene expression by a c-terminal truncated Stat5: role of Stat5 in proliferation. EMBO J. 1996;15:2425–2433. [PMC free article] [PubMed] [Google Scholar]
- 39.Nakayama K, Ishida N, Shirane M, Inomata A, Inoue T, Shishido N, Horii I, Loh D Y, Nakayama K-I. Mice lacking p27Kip1 display increased body size, multiple organ hyperplasia, retinal dysplasia and pituitary tumors. Cell. 1996;85:707–720. doi: 10.1016/s0092-8674(00)81237-4. [DOI] [PubMed] [Google Scholar]
- 40.Noguchi M, Nakamura Y, Russell S M, Ziegler S F, Tsang M, Cao X, Leonard W J. Interleukin-2 receptor gamma chain: a functional component of the interleukin-7 receptor. Science. 1993;262:1877–1880. doi: 10.1126/science.8266077. [DOI] [PubMed] [Google Scholar]
- 41.Nourse J, Firpo E, Flanagan W M, Coats S, Polyak K, Lee M-H, Massague J, Crabtree G R, Roberts J M. Interleukin-2-mediated elimination of the p27Kip1 cyclin-dependent kinase inhibitor prevented by rapamycin. Nature. 1994;372:570–573. doi: 10.1038/372570a0. [DOI] [PubMed] [Google Scholar]
- 42.Pagano M, Tam S W, Theodoras A M, Beer-Romero P, del Sal G, Chau V, Yew P R, Draetta G F, Rolfe M. Role of the ubiquitin-proteosome pathway in regulating the abundance of the cyclin-dependent kinase inhibitor p27. Science. 1995;269:682–685. doi: 10.1126/science.7624798. [DOI] [PubMed] [Google Scholar]
- 43.Parker S B, Eichele G, Zhang P, Rawls A, Sands A T, Bradley A, Olsen E N, Harper J W, Elledge S J. p53-independent expression of p21Cip1 in muscle and other terminally differentiating cells. Science. 1995;267:1024–1027. doi: 10.1126/science.7863329. [DOI] [PubMed] [Google Scholar]
- 44.Pernis A, Witthuhn B, Keegan A D, Nelms K, Garfein E, Ihle J N, Paul W E, Pierce J H, Rothman P. Interleukin 4 signals through two related pathways. Proc Natl Acad Sci USA. 1995;92:7971–7975. doi: 10.1073/pnas.92.17.7971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pruett W, Yuan Y, Rose E, Batzer A G, Harada N, Skolnik E Y. Association between GRB2/Sos and insulin receptor substrate 1 is not sufficient for activation of extracellular signal-regulated kinases by interleukin-4: implications for Ras activation by insulin. Mol Cell Biol. 1995;15:1778–1785. doi: 10.1128/mcb.15.3.1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Quelle F W, Shimoda K, Thierfelder W, Fischer C, Kim A, Ruben S M, Cleveland J L, Pierce J H, Keegan A D, Nelms K, Paul W E, Ihle J N. Cloning of murine Stat6 and human Stat6, Stat proteins that are tyrosine phosphorylated in responses to IL-4 and IL-3 but are not required for mitogenesis. Mol Cell Biol. 1995;15:3336–3343. doi: 10.1128/mcb.15.6.3336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Quelle F W, Wang D, Nosaka T, Thierfelder W E, Stravopodis D, Weinstein Y, Ihle J N. Erythropoietin induces activation of Stat5 through association with specific tyrosines on the receptor that are not required for a mitogenic response. Mol Cell Biol. 1996;16:1622–1631. doi: 10.1128/mcb.16.4.1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Russell S M, Johnston J A, Noguchi M, Kawamura M, Bacon C M, Friedmann M, Berg M, McVicar D W, Witthuhn B A, Silvennoinen O, Goldman A S, Schmalsteig F C, Ihle J N, O’Shea J J, Leonard W J. Interaction of IL-2Rβ and γc chains with Jak1 and Jak 3: implications for XSCID and XCID. Science. 1994;266:1042–1045. doi: 10.1126/science.7973658. [DOI] [PubMed] [Google Scholar]
- 49.Ryan J J, McReynolds L J, Keegan A, Wang L-H, Garfein E, Rothman P, Nelms K, Paul W E. Growth and gene expression are predominantly controlled by distinct regions of the human IL-4 receptor. Immunity. 1996;4:123–132. doi: 10.1016/s1074-7613(00)80677-9. [DOI] [PubMed] [Google Scholar]
- 50.Schindler C, Kashleva H, Pernis A, Pine R, Rothman P. STF-IL-4: a novel IL-4 induced signal transducing factor. EMBO J. 1994;13:1350–1356. doi: 10.1002/j.1460-2075.1994.tb06388.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schindler U, Wu P, Rothe M, Brasseur M, McKnight S L. Components of a Stat recognition code: evidence for two layers of molecular selectivity. Immunity. 1995;2:689–697. doi: 10.1016/1074-7613(95)90013-6. [DOI] [PubMed] [Google Scholar]
- 52.Sherr C J, Roberts J M. Inhibitors of mammalian G1 cyclin-dependent kinases. Genes Dev. 1995;9:1149–1163. doi: 10.1101/gad.9.10.1149. [DOI] [PubMed] [Google Scholar]
- 53.Shimoda K, van Deursen J, Sangster M Y, Sarawar S R, Carson R T, Tripp R A, Chu C, Quelle F W, Nosaka T, Vignali D A A, Doherty P C, Grosveld G, Paul W E, Ihle J N. Lack of IL-4 induced Th2 response and IgE class switching in mice with disrupted Stat6 gene. Nature. 1996;380:630–633. doi: 10.1038/380630a0. [DOI] [PubMed] [Google Scholar]
- 54.Sun X L, Wang L-M, Zhang Y, Yenush L, Myers M G J, Glasheen E, Lane W S, Pierce J H, White M F. Role of IRS-2 in insulin and cytokine signalling. Nature. 1995;377:173–177. doi: 10.1038/377173a0. [DOI] [PubMed] [Google Scholar]
- 55.Takeda K, Tanaka T, Shi W, Matsumoto M, Minami M, Kashiwamura S-I, Nakanishi K, Yoshido N, Kishimoto T, Akira S. Essential role of Stat6 in IL-4 signalling. Nature. 1996;380:627–630. doi: 10.1038/380627a0. [DOI] [PubMed] [Google Scholar]
- 56.Takeshita T, Asao H, Ohtani K, Ishii N, Kumaki S, Tanaka N, Munakata H, Nakamura M, Sugamura K. Cloning of the gamma chain of the human IL-2 receptor. Science. 1992;257:379–382. doi: 10.1126/science.1631559. [DOI] [PubMed] [Google Scholar]
- 57.Thierfelder W E, van Deursen J M, Yamamoto K, Tripp R A, Sarawar S R, Carson R T, Sangster M Y, Vignali D A A, Doherty P C, Grosveld G C, Ihle J N. Requirement for Stat4 in interleukin-12 mediated responses of natural killer and T cells. Nature. 1996;382:171–174. doi: 10.1038/382171a0. [DOI] [PubMed] [Google Scholar]
- 58.Vella A, Teague T K, Ihle J, Kappler J, Marrack P. Interleukin 4 (IL-4) or IL-7 prevents the death of resting T cells: Stat6 is probably not required for the effect of IL-4. J Exp Med. 1997;186:325–330. doi: 10.1084/jem.186.2.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang D, Stravopodis D, Teglund S, Kitazawa J, Ihle J N. Naturally occurring dominant negative variants of Stat5. Mol Cell Biol. 1996;16:6141–6148. doi: 10.1128/mcb.16.11.6141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang H Y, Paul W E, Keegan A D. IL-4 function can be transferred to the IL-2 receptor by tyrosine containing sequences found in the IL-4 receptor alpha chain. Immunity. 1996;4:113–121. doi: 10.1016/s1074-7613(00)80676-7. [DOI] [PubMed] [Google Scholar]
- 61.Wang H Y, Zamorano J, Yoerkie J L, Paul W E, Keegan A D. The IL-4 induced tyrosine phosphorylation of the insulin receptor substrate is dependent on Jak1 expression in human fibrosarcoma cells. J Immunol. 1997;158:1037–1040. [PubMed] [Google Scholar]
- 62.Yan Y, Frisen J, Lee M-H, Massague J, Barbacid M. Ablation of CDK inhibitor p57Kip2 results in increased apoptosis and delayed differentiation during mouse development. Genes Dev. 1997;11:973–983. doi: 10.1101/gad.11.8.973. [DOI] [PubMed] [Google Scholar]
- 63.Yu C-L, Meyer D J, Campbell G S, Larner A C, Carter-Su C, Schwartz J, Jove R. Enhanced DNA-binding activity of a Stat3-related protein in cells transformed by the Src oncoprotein. Science. 1995;269:81–83. doi: 10.1126/science.7541555. [DOI] [PubMed] [Google Scholar]
- 64.Zamorano J, Wang H Y, Wang L-M, Pierce J H, Keegan A D. IL-4 protects cells from apoptosis via the insulin receptor substrate pathway and a second independent signaling pathway. J Immunol. 1996;157:4926–4934. [PubMed] [Google Scholar]
- 65.Zerfass-Thome K, Schulze A, Zwerschke W, Vogt B, Helin K, Bartek J, Henglein B, Jansen-Durr P. p27Kip1 blocks cyclin E-dependent transactivation of cyclin A gene expression. Mol Cell Biol. 1997;17:407–415. doi: 10.1128/mcb.17.1.407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhang P, Liegeois N J, Wong C, Finegold M, Hou H, Thompson J C, Silverman A, Harper J W, DePinho R A, Elledge S J. Altered cell differentiation and proliferation in mice lacking p57Kip2 indicates a role in Beckwith-Wiedemann syndrome. Nature. 1997;387:151–158. doi: 10.1038/387151a0. [DOI] [PubMed] [Google Scholar]
- 67.Zuber C E, Galizzi J P, Harada N, Durand I, Banchereau J. Interleukin-4 receptors on human blood mononuclear cells. Cell Immunol. 1990;129:329–340. doi: 10.1016/0008-8749(90)90209-a. [DOI] [PubMed] [Google Scholar]