

Abstract
The V(D)J recombinase recognizes a pair of immunoglobulin or T-cell receptor gene segments flanked by recombination signal sequences and introduces double-strand breaks, generating two signal ends and two coding ends. Broken coding ends were initially identified as covalently closed hairpin DNA molecules. Before recombination, however, the hairpins must be opened and the ends must be modified by nuclease digestion and N-region addition. We have now analyzed nonhairpin coding ends associated with various immunoglobulin gene segments in cells undergoing V(D)J recombination. We found that these broken DNA ends have different nonrandom 5′-strand deletions which were characteristic for each locus examined. These deletions correlate well with the sequence characteristics of coding joints involving these gene segments. In addition, unlike broken signal ends, these nonhairpin coding-end V(D)J recombination reaction intermediates have 3′ overhanging ends. We discuss the implications of these results for models of how sequence modifications occur during coding-joint formation.
Immunoglobulin (Ig) and T-cell receptor genes are assembled in developing lymphocytes by a series of site-specific DNA recombination reactions known as V(D)J recombination (reviewed in reference 31). Gene segments which undergo V(D)J recombination are flanked by recombination signal sequences (RSSs). RSSs consist of a highly conserved heptamer, a spacer of conserved length (12 or 23 nucleotides [nt]) but not of conserved sequence, and a less well conserved nonamer (Fig. 1). The recombination reaction, which can occur only between gene segments whose RSSs have dissimilar spacer lengths, generates two products, a signal joint and a coding joint (Fig. 1) (reviewed in reference 16). Signal joints are precise, head-to-head fusions of two RSSs without loss or addition of nucleotides. Coding joints are more complex, frequently involving deletions and additions of DNA sequence. These sequence alterations contribute significantly to the diversity of the immune repertoire. Added sequences are of two types, N regions and P nucleotides. N regions are short, nontemplated additions to coding joints made by the lymphoid cell-specific enzyme terminal deoxynucleotidyltransferase (TdT). P nucleotides are short palindromic repeats of DNA sequences at the ends of the rearranging segments (14).
FIG. 1.
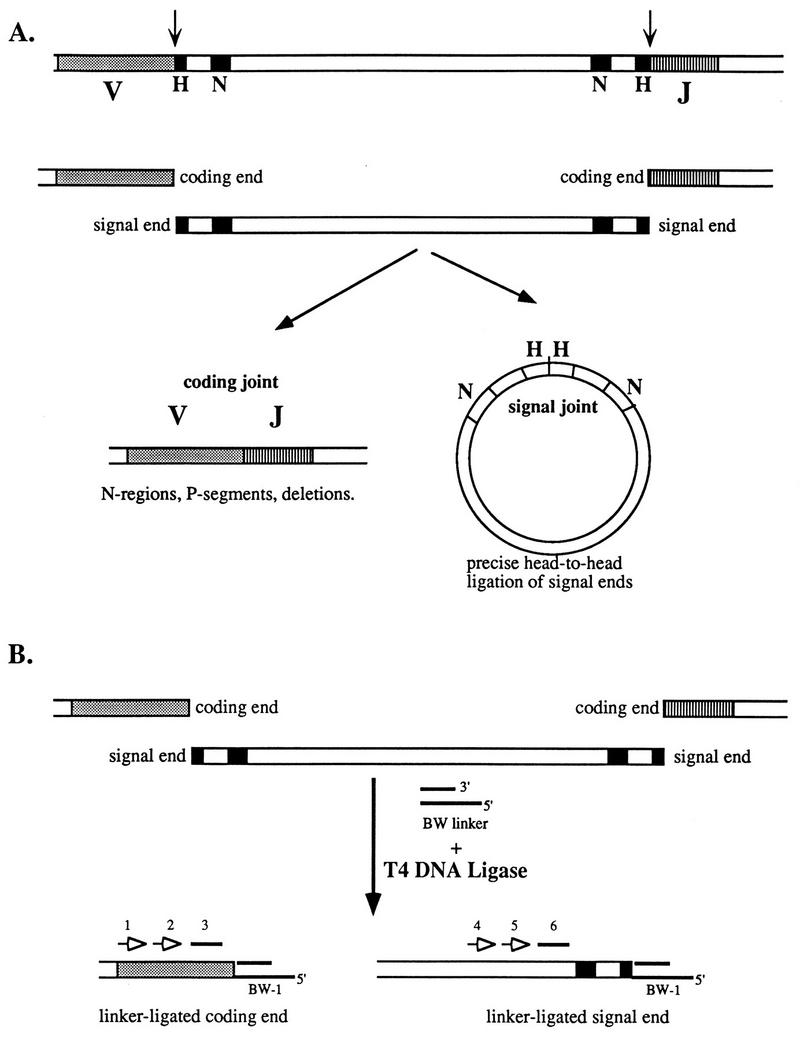
V(D)J recombination reaction pathway and LM-PCR assay for reaction intermediates. (A) Diagram of the reactants (top), broken DNA intermediates (middle), and products (bottom) of V(D)J recombination. V and J gene segments, with their associated RSSs (heptamer [H] and nonamer [N]) are recognized and cleaved by the recombinase at the RSS–coding-segment junction (arrow), generating coding-end and signal-end fragments. These ends are joined to form a coding joint and a signal joint. (B) LM-PCR assay for broken-ended recombination reaction intermediates. The BW linker is ligated to available ends in total genomic DNA by using T4 DNA ligase. The sites of linker ligation are revealed by a set of nested PCR assays with a linker primer (BW-1) and locus-specific primers (open arrows labeled 1, 2, 4, and 5). Blots of PCR products were probed with internal oligonucleotides (solid lines labeled 3 and 6).
Two V(D)J recombination reaction intermediates have been characterized (Fig. 1A) (reviewed in reference 1). Broken signal ends are blunt and 5′ phosphorylated (26, 28). Direct ligation of these ends would account for the structure of signal joints. The metabolism of coding ends is more complex. The broken coding segments that have been characterized in scid thymocytes have covalently closed hairpin ends (24). It has been proposed that hairpin opening by a nuclease followed by fill-in synthesis might account for the generation of P nucleotides (14, 24). TdT would modify coding ends after hairpin opening.
Despite the seemingly more complex nature of coding-joint formation, coding ends have exceptionally short half-lives in wild-type lymphoid precursors (22). Nonhairpin coding ends have been detected at the Jκ1 gene segment in a transformed pro-B-cell line, at the Jκ1 and Jκ2 gene segments in bone marrow B cells, and at the Jα50 gene segment in thymus DNA (4, 18, 22). In contrast, while Dδ2 signal ends are readily detectable by Southern blot hybridization in thymocytes, coding ends are undetectable (25). One study suggested that coding ends were at least 1,000-fold less abundant than signal ends in wild-type thymus DNA (36). However, these Dδ2 coding ends can be detected in thymocytes from scid mice, which have a recessive mutation in an enzyme known to be involved in DNA repair, DNA-dependent protein kinase (10, 13). V(D)J recombination in scid mutant mice is characterized by inefficient coding-joint formation but relatively normal signal joint formation (16). The coding ends detected in scid thymocytes were shown to be covalently closed hairpins (24).
Recent data from a new in vitro system that recapitulates the early steps of V(D)J recombination has shown that two proteins, RAG1 and RAG2, are sufficient for recognition and cleavage of RSSs in plasmid DNA or oligonucleotides. This reaction generates blunt signal ends and hairpin coding ends (19, 32). It remains undetermined, however, how hairpin coding ends are opened, processed, and ultimately ligated to form a coding joint. In the present study, we have used a ligation-mediated PCR (LM-PCR) assay to detect and characterize the structure of coding ends in DNA purified from a pro-B-cell line, bone marrow B cells, and thymus.
MATERIALS AND METHODS
Cell lines and tissues.
103 bcl2/4 cells (2) were obtained from N. Rosenberg (Tufts University) and grown in RPMI 1640 supplemented with 10% fetal calf serum, 50 μM β-mercaptoethanol, 1 mg of G418 (LifeTechnologies) per ml, and antibiotics at 33°C in a 5% CO2 incubator. For induction of rearrangement, the cells were shifted to 39°C for 18 to 24 h. Thymocytes were obtained by mechanical disruption of dissected thymuses from 10-day-old BALB/c mice. CD19+ bone marrow cells were purified as described previously (30).
Purification of DNA. (i) Phenol-chloroform method.
Cells were washed and resuspended in phosphate-buffered saline (PBS) at 107 cells per ml. The cell suspension was mixed with an equal volume of 2× PKB (100 mM Tris [pH 7.7], 50 mM EDTA, 1% sodium dodecyl sulfate), and proteinase K (Boehringer) was added to a final concentration of 400 μg/ml. The cell lysate was incubated at 56°C for 12 to 18 h and then extracted successively with phenol, phenol-chloroform (1:1), and chloroform-isoamyl alcohol (24:1). The DNA was precipitated with 0.8 volume of isopropanol and then resuspended in 300 μl of TE (10 mM Tris [pH 8.0], 0.2 mM EDTA) per 107 cells. RNase was added to a final concentration of 20 μg/ml, and the sample was incubated at 22°C for 30 min. An equal volume of 2× PKB and proteinase K (250 μg/ml) was added, and the preparation was incubated at 56°C for 4 to 6 h. Phenol and chloroform extractions were performed as above, and the DNA was alcohol precipitated, washed, and finally resuspended in TE at approximately 0.5 mg/ml.
(ii) Protein precipitation method.
DNA was purified with a Puregene kit from Gentra. In brief, cells were washed and resuspended, as above, in PBS. An equal volume of lysis solution containing RNase and detergent (Puregene) was added, and the mixture was incubated at 37°C for 60 min. After it had been cooled to room temperature, precipitation solution was added, and precipitated detergent and proteins were cleared by centrifugation. DNA was recovered from the supernatant by isopropanol precipitation and resuspended in TE.
(iii) Agarose plug method.
Cells were washed and resuspended in PBS at 1 × 106 to 3 × 106 cells per 40 μl. Up to 0.5 ml of suspended cells was warmed to 37°C briefly before being mixed with an equal volume of molten 1% agarose (SeaKem LE; FMC Corp.) in PBS, cooled to 50°C. The agarose-cell mixture was immediately dispensed into plug molds (Bio-Rad) and allowed to cool. The plugs were extruded into plug lysis buffer (100 mM Tris [pH 8.0], 25 mM EDTA, 1% Sarkosyl) and incubated at 56°C for 12 to 18 h after the addition of proteinase K to 400 μg/ml. The plugs were washed once in a large volume of TE at 56°C for 30 min, then in TE plus 0.5 mM phenylmethylsulfonyl fluoride for 30 min, and then twice in TE at 4°C over 24 h. DNA embedded in plugs was used directly for LM-PCR. Some plug DNA samples were subjected to T4 DNA polymerase treatment by incubating 40 μl of plug in an 80-μl reaction mixture with manufacturer’s buffer (Life Technologies), 5 U of T4 DNA polymerase, and 100 μM deoxynucleoside triphosphates at 37°C for 1 h. Other plugs were treated with various amounts of mung bean nuclease (BRL) under conditions previously reported by Zhu and Roth (36). Treated plugs were washed extensively in TE and then processed as described below.
LM-PCR assay for coding ends.
DNA (1 to 3 μg), either in solution or in agarose plugs, was subjected to linker ligation for 18 h at 16°C in a 50- to 100-μl reaction mixture containing ligation buffer (Boehringer), 40 pmol of linker, and 2 U of T4 DNA ligase (Boehringer). The ligation reaction mixture was then mixed with an equal volume of PCRL (10 mM Tris [pH 8.8], 50 mM KCl, 0.25% Tween 20, 0.25% Nonidet P-40) and heated to 95°C for 15 min. Agarose plug DNA reactions mixtures were cooled to 56°C and then used for PCR, whereas other DNA preparations were maintained on ice before PCR.
For PCR, 5 μl of linker-ligated DNA was added to 25 μl (final volume) of reaction mixture with appropriate primers (see below) and Taq DNA polymerase (Life Technologies) and cycled 12 times at 94°C for 1 min and 66°C for 2 min. A 1-μl sample of the first PCR product was used as the template for a second 27-cycle PCR under identical conditions with a nested locus-specific primer (see below). One-fifth of the ultimate product was analyzed by electrophoresis on a 1% agarose–1% NuSieve (FMC Corp.) gel and blotted under alkaline conditions to a nylon membrane (ZetaBind; Cuno). The blots were hybridized with 32P-labeled locus-specific internal oligonucleotides and analyzed with a PhosphorImager and ImageQuaNT software (Molecular Dynamics).
DNA sequence analysis of coding-end fragments.
Amplified coding-end fragments were purified on 1% agarose gels. The purified DNA was digested with restriction endonucleases corresponding to sites encoded by the PCR primers. The digested fragments were cloned into pBSK (Stratagene) and sequenced by the dideoxy method with reagents from United States Biochemicals (Sequenase II).
Assessment of coding-joint length heterogeneity.
D-to-JH and V-to-Jκ coding joints were PCR amplified from thymocyte or induced 103 bcl2/4 cell DNA, respectively, with the primers DH and JHB4 and the primers VκS and Jκrev as described previously (27, 29). PCR products were gel purified and then labeled with T4 polynucleotide kinase (Boehringer) and [γ-32P]ATP. The labeled products were digested with EcoRI to remove the label from one end of the fragment and then analyzed by electrophoresis on a denaturing polyacrylamide gel. The gels were dried, and the labeled DNA was visualized with a PhosphorImager.
Oligonucleotide primers, probes, and linkers.
For Jκ1, JH1, and JH2 coding-end assays, the first two primers listed were used successively for nested PCR with the linker primer BW-1. The third primer served as a radiolabeled blot hybridization probe. For the Vκ and DH coding-end assays, the two primers were used successively for nested PCR with linker primer BW-1 and the second primer was used for blot hybridization. VκH-N was used with VκS to display the length heterogeneity of the Vκ repertoire (see Fig. 5). The primers are as follows: for Jκ1 coding-end assay, (outside) JκS (5′ CCAAGCTTT CCAGCTTGGTCCCCCCTCCGAA 3′), (inside) Jκ1-2 (5′ GTGTCCCTTCACTCAACCCCCATAC 3′), and (probe) Jκrev (5′ GAGTAAGATTTTATACATCATTTTTAGACA 3′); for JH1 coding-end assay, (outside) JHB3 (5′ ACACACATTTCCCCCCCAACAAA 3′), (inside) JHB1 (5′ GATCTGAGAATATCTTTTCCCGT 3′), and (probe) JHB2 (5′ GAATGGAATGTGCAGAAAGAAAAAAGCC 3′); for JH2 coding-end assay, (outside) JH-A (5′ TGCCTCAGACTTCAAGCTTCAGTTCTGG 3′), (inside) JHB3 (5′ ACACACATTTCCCCCCCAACAAA 3′), and (probe) JHB4 (5′ GTAAAATCTATCTAAGCTGAATAGAAGA 3′); for Vκ coding-end assay, (outside) VκB (5′ GACATTCAGCTGACCCAGTCTCCA 3′), (inside) VκS (5′ CCG AAT TCG STT CAG TGG CAG TGG RTC WGG RAC 3′), and VκHN (5′ GGCCCGGGTTTWTGTTMWGRBYTGTAKCACAGTG 3′); and for DH coding-end assay, (outside) DHsp (5′ GGCCCCTGACACTGTGCACTGCTACCTC 3′) and (inside) DH (5′ GGAATTCGMTTTTTGTSAAGGGATCTACTACTGTG 3′).
FIG. 5.

Length heterogeneity of amplified coding ends and joints. DH, JH1, JH2, Vκ, and Jκ1 coding ends (ce) (A, B, C, E, and F) and DJH and VJκ joints (D and G) were amplified from T4 polymerase-treated thymus or induced 103 bcl2/4 cell DNA. The amplified fragments were gel purified and end labeled with [γ-32P]ATP by using T4 DNA kinase. Labeled fragments were electrophoresed on 6% denaturing polyacrylamide gels alongside DNA sequencing ladders used as size markers. The diagrams adjacent to each coding-fragment gel image indicate the sequence position based on these comigrating sequence markers. Position zero in the diagrams corresponds to the full-length coding end (i.e., the junction between the RSS and the coding segment), positive numbers indicate coding-end deletions, and negative numbers indicate longer-than-full-length coding-end lengths. The diagrams adjoining DJH and VJκ joints indicate successive nucleotide lengths. In panel E, the lane labeled Vκ mkr contains a radiolabeled amplification product of genomic DNA demonstrating the length heterogeneity of intact Vκ genes (see the text).
The linker oligonucleotides listed below were annealed as previously described to generate linkers for ligation to genomic DNA (28): BW-1, 5′ GCGGTGACCCGGGAGATCTGAATTC 3′; BW-2, 5′ GAATTCAGATC 3′; BW-2N, 5′ GATCTGAATTC(N)2–5; N-BW-2, 5′ (N)2–5GAATTCAGATC, BW-1R, 5′ GAATTCAGATCTCCCGGGAGACCGC 3′.
With the exception of BW-1R, which was 5′ phosphorylated, all the linker oligonucleotides had 5′-OH groups. The BW linker was made by annealing BW-1 and BW-2, the 5′ overhanging linker was made by annealing BW-1 and N-BW-2, and the 3′ overhanging linker was made by annealing BW-1R and BW-2N.
RESULTS
Coding ends can be detected by LM-PCR in wild-type lymphoid progenitors.
In a previous report, we described an LM-PCR technique which sensitively detects broken signal-end DNA in cells undergoing V(D)J recombination (28). Purified total genomic DNA from a tissue active in recombination is ligated to a blunt, unphosphorylated oligonucleotide linker. The linker-ligated DNA is then used in a nested PCR assay to map double-strand DNA breaks relative to Ig gene segments (Fig. 1B). Using this assay, we demonstrated that broken signal ends were blunt and 5′ phosphorylated. With appropriate primers, however, we were unable to detect the corresponding broken coding ends in the same DNA samples (data not shown). Others reported similar difficulties (36). One explanation for this might be their existence in a hairpin structure, incapable of linker ligation. Before joint formation, however, hairpins must be opened by a nuclease and modified by enzymes including TdT. We hypothesized that nonhairpin coding ends might be present in our samples at very low levels but might be undetectable due to inefficiency of the assay.
To increase the efficiency of linker ligation, we purified DNA by embedding cells in agarose and digesting and extracting cellular components in situ, leading to fewer adventitious DNA breaks to compete with potential coding ends for linker ligation and amplification. We prepared DNA from 103 bcl2/4, a pro-B-cell line transformed with a temperature-sensitive Abelson leukemia virus (2). Under restrictive conditions (39°C), these cells activate recombination of their Ig κ loci. As shown in Fig. 2A, regardless of the method used to prepare the DNA, we found that the abundance of broken signal ends detected by LM-PCR was markedly increased by the shift to restrictive conditions. We had great difficulty in identifying amplified products with mobilities corresponding to coding ends in DNA prepared by phenol extraction or by salt precipitation methods (Fig. 2B, lanes 1 to 4).
FIG. 2.

Broken coding-end DNA can be detected in induced 103 bcl2/4 cell DNA. (A and B) 103 bcl2/4 cells were induced to activate V(D)J rearrangement by a temperature shift. DNA was prepared from induced 39°C (lanes +) and uninduced 33°C (lanes −) samples by the indicated techniques as described in the text. The samples were analyzed by LM-PCR for broken signal ends (se) (A) and broken coding ends (ce) (B) at the Jκ1 locus. Phosphorimages of oligonucleotide-probed Southern blots of reaction products are shown with the positions of various signal and coding ends indicated by arrows. (C) Ethidium bromide-stained agarose gel analysis of control amplifications of a nonrearranging locus (CD14) from the same DNA samples used in panels A and B. Controls included buffer in place of template (BL; lane 8) and 63-12 cell DNA (a RAG-2-deficient cell line; lane 9). Lanes 6 and 7 contain two independently purified DNA samples. SDS-Pk, sodium dodecyl sulfate plus proteinase K.
These assays generated a series of amplified fragments which were highly variable in length and not specifically induced in the 39°C DNA sample. When we analyzed DNA prepared by the agarose plug method, however, we detected amplified products in induced samples corresponding in length to Jκ1 coding ends (Fig. 2B, lanes 6 and 7). Similar amplified products were observed with DNA purified from CD19+ bone marrow B cells (4) (see below). We failed to detect these ends in similarly prepared DNA samples from RAG2-deficient pro-B cells (lane 9) and several nonlymphoid tissues and cell lines (data not shown). Using this method, we were also able to detect broken coding ends associated with various JH, DH, and Vκ gene segments (see below).
JH and Jκ coding ends contain overhanging ends and 5′ deletions of nonrandom length.
The blunt-ended BW linker used in our LM-PCR assays ligates only to blunt 5′-phosphorylated DNA ends. To determine if a fraction of coding ends were not detected by this assay because they were not blunt, we pretreated agarose plug DNA prepared from 103 bcl2/4 cells and from newborn-mouse thymocytes with T4 DNA polymerase before linker ligation. 103 bcl2/4 cells, as noted above, are inducible for Jκ rearrangement, and thymocytes undergo frequent DH-to-JH gene rearrangement (5). As shown in Fig. 3 and in our previous work (28), this pretreatment did not affect our ability to detect Jκ1 signal ends in 103 bcl2/4 cell DNA or JH2 signal ends in thymocyte DNA. However, DNA polymerase treatment dramatically increased the coding-end signal in the LM-PCR assay. We conclude from this observation that coding ends contain either 5′ or 3′ overhangs. Coding-end hairpins would not be revealed by T4 DNA polymerase treatment.
FIG. 3.
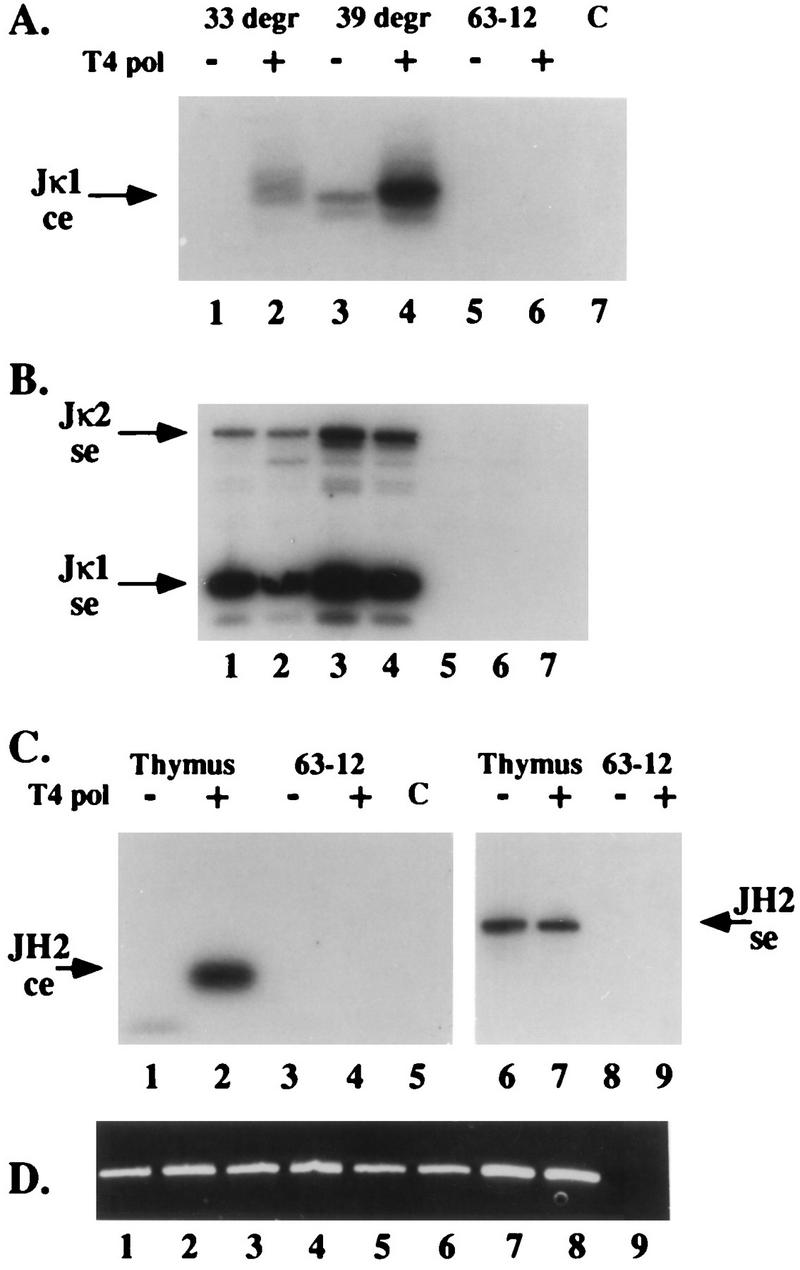
T4 DNA polymerase treatment enhances LM-PCR detection of broken coding ends. (A to C) DNA samples prepared by the agarose plug method from uninduced (33°C [33 degr]) and induced (39°C [39 degr]) 103 bcl2/4 cells (A and B) and from newborn thymus (C) were analyzed by LM-PCR for broken Jκ1 coding (A), Jκ1 and Jκ2 signal (B), and JH2 coding and signal (C) ends without (lanes −) or with (lanes +) T4 DNA polymerase (T4 pol) pretreatment. Controls included identically prepared and treated 63-12 (RAG-2-deficient) cell DNA and buffer (lane C). DNA samples from panel C were amplified with primers specific for a nonrearranging genomic locus, demonstrating the presence of DNA in all samples. (D) Ethidium bromide-stained agarose gel of these control amplifications. Lanes 1 to 4 correspond to samples 1 to 4 in panel C, lanes 5 to 8 correspond to samples 6 to 9 in panel C, and lane 9 is a buffer-only control amplification. Lanes 1 to 6 of panels A and B were shown to contain equivalent amounts of DNA by a similar method (data not shown). ce, coding ends; se, signal ends.
We cloned and determined the DNA sequence of amplified coding-end fragments associated with the Jκ1, JH1, and JH2 gene segments. The results of this sequencing analysis are shown in Fig. 4. In each of the J coding ends, the end of the amplified fragment mapped to several nucleotides 3′ of the RSS–coding-segment junction. This is in contrast to the structure of signal ends, which we and others showed end precisely at the RSS–coding-segment junction. In addition, the positions of DNA breaks in coding segments were nonrandom. For example, Jκ1 coding fragments terminated 4 nt into the coding segment in 9 of 10 sequenced clones (termed +4). Similarly, JH1 and JH2 coding segments showed predominant breakage sites which were different for each locus (Fig. 4). These DNA sequencing analyses have been repeated on cloned PCR products from several independent experiments with essentially identical results (data not shown).
FIG. 4.

DNA sequence analysis of cloned broken coding- and signal-end LM-PCR fragments. Broken coding-end fragments were excised from agarose gels, reamplified, purified, and cloned into the vector pBSK. Randomly selected coding-end clones were sequenced. The arrows above the germ line sequences indicate sites of linker ligation determined by sequencing. The numbers above the arrows indicate the frequencies of individual sequences from sets of consecutive sequences. The solid vertical bar indicates the RSS–coding-segment junction, with arrows to the right indicating coding ends and those to the left indicating signal ends. The RSS heptamer and nonamer sequences are boxed. Signal-end sequences were reported previously and are shown for comparison only (28).
To survey a larger number of molecules for coding-end length heterogeneity, we analyzed radiolabeled amplification products by denaturing polyacrylamide gel electrophoresis (Fig. 5). DNA-sequencing reaction mixtures were electrophoresed in adjoining lanes and used to precisely determine the coding-end fragment lengths. In general, this approach corroborated the sequencing analysis, showing a series of predominant breakage sites for each type of coding end. By using this method, the predominant Jκ1 coding end was mapped to 4 nt into the coding segment (+4, 71% of the signal by PhosphorImager analysis). Predominant coding ends for JH1 mapped to 0, +2, and +4 (17, 40, and 42%), and those for JH2 mapped to +2, +7 and +9 (57, 8, and 34%) with respect to the RSS–coding-segment junction.
Vκ and DH coding ends are predominantly 5′ truncated.
The analysis of Vκ coding ends is more difficult because of possible heterogeneity in the lengths of Vκ gene segments in the genome. To assess this heterogeneity, we amplified genomic DNA with a pair of nested degenerate Vκ framework region primers (VκB and VκS) and the VκHN primer. VκHN is a degenerate primer with homology to the conserved heptamer and spacer 3′ of rearranging Vκ gene segments. Amplified products were labeled and analyzed by electrophoresis on a denaturing polyacrylamide gel. As shown in Fig. 5E, this analysis revealed only very modest heterogeneity (3 nt) among the set of amplified Vκ germ line sequences. LM-PCR of T4 DNA polymerase-treated 103 bcl2/4 cell DNA with the same framework primers and the BW-1 linker primer revealed two predominant Vκ coding-end fragment lengths corresponding to cleavages at the tip of the hairpin intermediate (83%) and at a position 7 nt into the Vκ gene segment (16%) (Fig. 5E). Cloning followed by DNA sequence analysis of these ends showed a similar series of coding-end lengths (data not shown). In addition, this sequencing analysis revealed that this assay detected coding ends involving multiple distinct Vκ gene segments. Interestingly, 3 of 18 sequenced Vκ coding ends showed breaks at the fourth or fifth position within the signal heptamer.
Using the identical approach, we amplified and analyzed, by both denaturing polyacrylamide gel electrophoresis and DNA sequencing, coding ends 3′ to the DSP2 family of DH gene segments. Since D-to-JH rearrangement is invariably deletional (29), the 3′ DH coding ends must come from gene segments undergoing D-to-JH rearrangement. This analysis revealed a predominant broken coding end at position +2 (89% of signal [Fig. 5A]) and lower intensities at positions −5 (9%) and −9 (2%) relative to the coding-segment–RSS junction. Sequence analysis of 12 cloned DH coding ends revealed 10 sequences ending at position +2. As with Vκ gene segments, we found two coding-end breaks at a position 5 nt into the heptamer.
Broken coding-end DNA contains 3′ overhangs.
As noted above, the blunt BW linker will ligate only to blunt DNA ends. The observations that the LM-PCR signal increased after T4 DNA polymerase treatment of genomic DNA (Fig. 3) and that the 5′ ends of broken coding-segment DNA sequences were invariably shorter than the full-length coding segments (Fig. 4 and 5) led us to hypothesize that broken coding segments in vivo have 3′-overhanging ends. Since signal ends are apparently generated by cleavage precisely at the coding-segment–RSS junction, 3′-overhanging coding ends might be generated by asymmetrical nucleolytic processing of a full-length coding end (presumably a hairpin DNA molecule).
To determine whether non-blunt coding ends had 3′ or 5′ overhangs, we performed LM-PCR on agarose plugs containing purified 103 bcl2/4 cell or thymocyte DNA that had been pretreated with either T4 DNA polymerase or mung bean nuclease. T4 polymerase has both 3′-to-5′ exonuclease and 5′-to-3′ polymerase activities, whereas mung bean nuclease digests any single-stranded DNA. Treatment of DNA with either enzyme led to enhanced detection of broken JH2 (Fig. 6A) and Jκ1 (Fig. 6B) coding ends. If broken coding ends have 5′ overhangs, the LM-PCR products of T4 polymerase-treated DNA will be longer than those of mung bean nuclease-treated DNA. If these ends have 3′ overhangs, the two enzyme treatments should yield LM-PCR products of identical length. Amplified coding ends were gel purified, reamplifed with another nested radiolabeled DNA primer, and analyzed on a denaturing polyacrylamide gel with single-nucleotide resolution. As shown in Fig. 6C, T4 polymerase and mung bean nuclease treatments resulted in identical fragment lengths, leading us to conclude that these broken ends had 3′ overhangs.
FIG. 6.
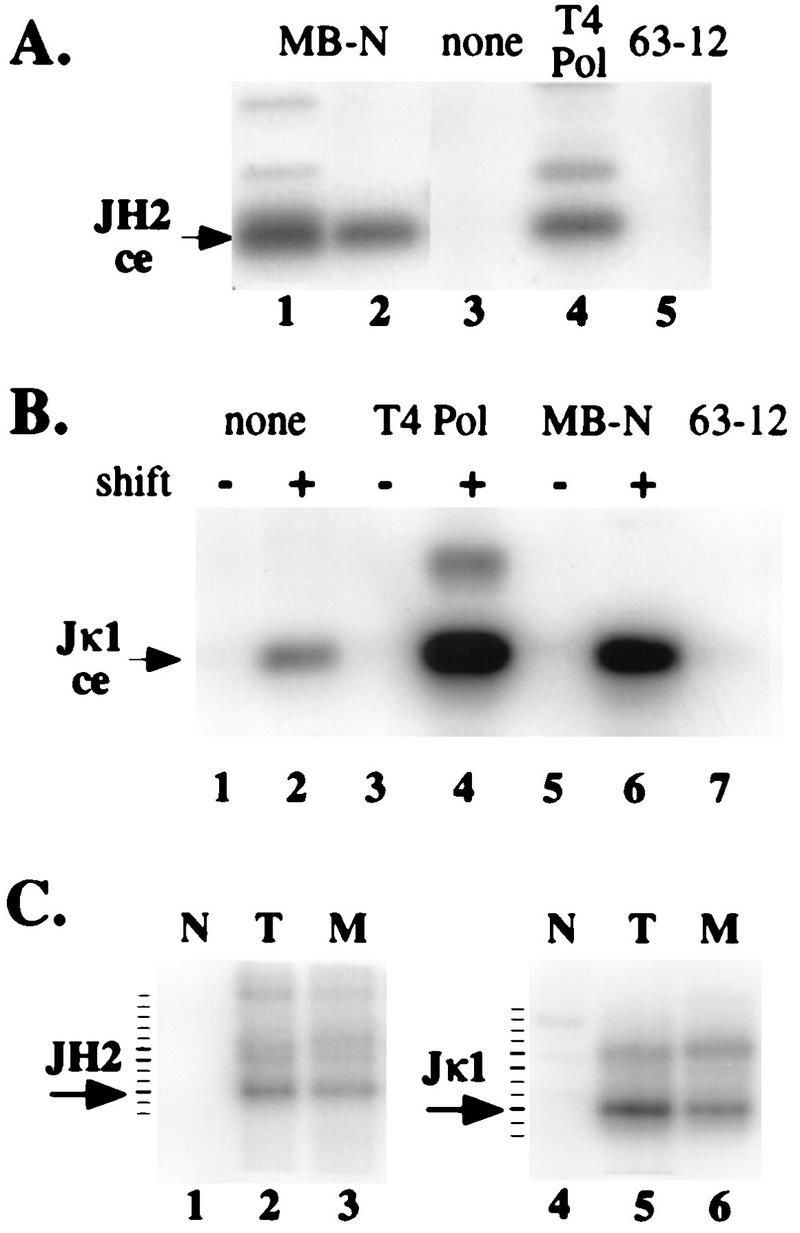
Jκ1 and JH2 coding ends have 3′ overhangs. (A and B) DNA purified in agarose plugs from newborn-mouse thymocytes (A) or 103 bcl2/4 cells cultured at 33 or 39°C (B) (shift − or +) was ligated to the BW linker without pretreatment (lanes none) or after treatment with either T4 DNA polymerase (lanes T4 Pol) or mung bean nuclease (lanes MB-N). Ligated plugs were analyzed by PCR for JH2 (A) or Jκ1 (B) coding-end (ce) breaks (arrows). The lanes labeled 63-12 were control LM-PCR assays with 63-12 cell DNA. Lanes 1 and 2 in panel A represent independent thymocyte DNA samples. (C) Amplified products from lanes 1, 3, and 4 in panel A and lanes 2, 4, and 6 in panel B were gel purified, reamplified for five cycles with a 32P-labeled specific oligonucleotide, and analyzed by denaturing polyacrylamide gel electrophoresis. In each case, the arrow indicates the predominant broken-ended molecule (+9 for JH2 and +4 for Jκ1) and the tick marks indicate 1-nt intervals (determined by the electrophoresis of a DNA sequencing reaction mixture on the same gel). Lanes: N, no pretreatment; T, T4 DNA polymerase pretreatment; M, mung bean nuclease pretreatment.
To confirm this result and to determine the length heterogeneity of the 3′-overhanging ends, we designed a set of modified LM-PCR linkers which can ligate directly to 5′- or 3′-overhanging ends (Fig. 7A). These duplex linkers have fully degenerate 5′- or 3′-overhanging ends 2 to 5 nt in length. Ligation occurs between the long strand of the linker and a 5′ phosphate (5′ overhang) or 3′ hydroxyl (3′ overhang) in genomic DNA. The structures of both these linker mixes are such that overhanging ends longer than 2 to 5 nt should also serve as substrates for linker ligation. We tested the ability of these linkers to ligate to blunt, 5′-overhanging, or 3′-overhanging targets by mixing 0.1 ng of appropriately cut plasmid DNA into 3 μg of purified genomic DNA and performing LM-PCR. As shown in Fig. 7B, target DNA with 5′-overhanging ends was efficiently detected with the 5′-overhanging linkers but not with the 3′-overhanging linker (lanes 13 to 16). The reverse was true of target DNA with 3′-overhanging ends (lanes 19 to 22). Surprisingly, the 5′-overhanging linker could efficiently detect blunt-ended target DNA (lanes 4 and 5). We believe that this is due to transient “breathing” of the ends of blunt DNA fragments, resulting in strand displacement by the linker and ligation.
FIG. 7.
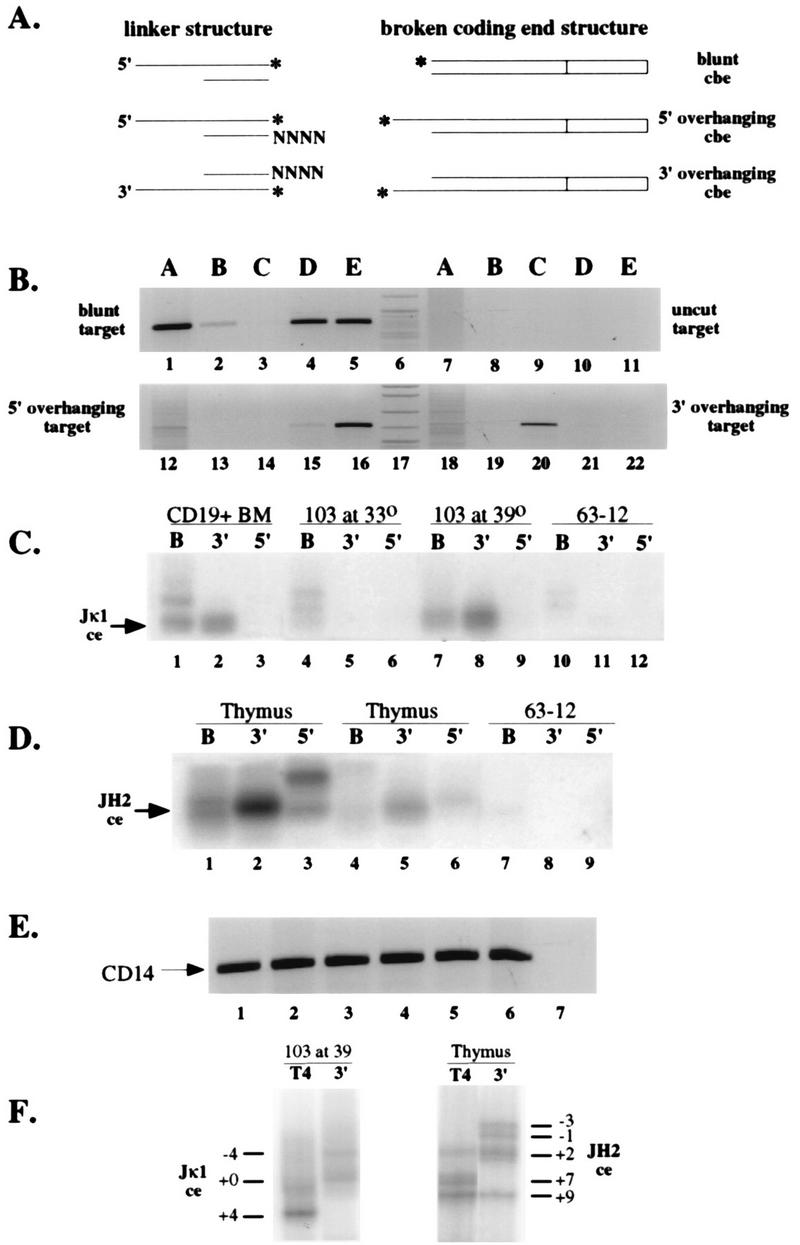
Analysis of nonblunt coding ends by LM-PCR with 5′- and 3′-overhanging linkers. (A) Structure of linkers designed to ligate to 3′- and 5′-overhanging DNA breaks. The asterisks indicate the nucleotides directly ligated in each case. N indicates an equimolar mixture of each deoxynucleoside triphosphate. cbe, coding broken end. (B) LM-PCR assays testing the ability of blunt (lanes A), 3′-overhanging (lanes B and C), or 5′-overhanging (lanes D and E) linkers to detect blunt-ended (lanes 1 to 5), uncut (lanes 6 to 11), 5′-overhanging (lanes 12 to 17), or 3′-overhanging (lanes 18 to 22) plasmid targets mixed with genomic DNA. An ethidium-stained agarose gel of PCR products is shown. Linkers B and D have 2-nt degenerate overhangs, and linkers C and E have 4-nt degenerate overhangs. (C) Jκ1 coding breaks detected with blunt (lanes B) or 3′- or 5′-overhanging linkers (lanes 3′ and 5′). The overhanging linkers were equimolar mixtures of linkers with 2-, 3-, and 4-nt overhangs. CD19+ bone marrow B-cell DNA, uninduced or induced 103 bcl2/4 cell DNA, and 63-12 cell DNA in agarose plugs were ligated with the indicated linkers and subjected to PCR with primers specific for the Jκ1 coding-end break. A phosphorimage of an oligonucleotide-probed Southern blot is shown, with the arrow indicating the position of migration of the Jκ1 coding-end break (ce). (D) JH2 coding-end breaks detected with linkers as indicated for panel C. DNA in agarose plugs purified from two separate thymocyte preparations (lanes 1 to 3 and lanes 4 to 6) or 63-12 cells (lanes 7 to 9) was ligated to the indicated linkers and subjected to PCR with primers specific for the JH2 coding-end break. A phosphorimage of an oligonucleotide-probed Southern blot is shown, with the arrow indicating the position of migration of the JH2 coding-end break (ce). (E) Control PCR assays on DNA samples used in each assay in panels C and D. An ethidium-stained gel analysis is shown, with the arrow indicating the position of the specific CD14 gene amplification product. (F) Denaturing polyacrylamide gel analysis of LM-PCR products generated from T4 DNA polymerase-polished linker-ligated (lanes T4) or 3′-overhanging linker-ligated (lanes 3′) amplified 103 bcl2/4 cell or thymus DNA, demonstrating the range of sizes of the PCR products.
We assayed genomic DNA purified from CD19+ bone marrow B cells, uninduced and induced 103 bcl2/4 pro-B cells, and thymocytes for either Jκ1 or JH2 broken coding ends by using blunt, 3′-overhanging, or 5′-overhanging degenerate linkers (Fig. 7C and D). In each case, the 3′-overhanging linker gave the strongest LM-PCR signal. The 5′-overhanging linker ligation amplification product in Fig. 7D, lane 3, was not reproducible. We compared the sizes of LM-PCR products from T4 DNA-polymerase treated DNA with those obtained by using the 3′-overhanging linker on denaturing polyacrylamide gels (Fig. 7F). The greater length of most of the 3′-overhanging linker ligation products confirms the existence of 3′-overhanging ends in genomic DNA. We were unable to recover and reamplify PCR products from the 5′-overhanging linker ligation shown in Fig. 7D, lane 3.
LM-PCR products of reactions in which the 3′-overhanging linker was used to analyze Jκ1 and JH2 3′-overhanging coding ends were gel purified, cloned, and subjected to DNA sequence analysis (Table 1). We found that the 3′ ends of these DNA breaks contained DNA sequences closer to the coding-segment–RSS border and were more heterogeneous than the sequences of 5′ ends shown in Fig. 4. Notably, two sequences in each set revealed the presence of full-length palindromic 3′ ends, consistent with their being the primary products of hairpin opening.
TABLE 1.
Sequences of 3′ ends of coding-end breaks
| Gene segment | Sequencea | No. of sequences |
|---|---|---|
| Jκ1 | gtggACGTTCGGTGGAGGCACCAA 3′ | |
| CACCTGCAAGCCACCTCCGTGGTT 5′ | ||
| 3′ end | TGCAAGCCACCTCCGTGGTT 5′ | 3 |
| CCTGCAAGCCACCTCCGTGGTT 5′ | 3 | |
| CACCTGCAAGCCACCTCCGTGGTT 5′ | 4 | |
| TGCACCTGCAAGCCACCTCCGTGGTT 5′ | 1 | |
| GGTGCACCTGCAAGCCACCTCCGTGGTT 5′ | 1 | |
| JH2 | actactttgaCTACTGGGGCCAA 3′ | |
| TGATGAAACTGATGACCCCGGTT 5′ | ||
| 3′ end | AACTGATGACCCCGGTT 5′ | 2 |
| TGAAACTGATGACCCCGGTT 5′ | 2 | |
| ATGAAACTGATGACCCCGGTT 5′ | 3 | |
| TGATGAAACTGATGACCCCGGTT 5′ | 2 | |
| CATGATGAAACTGATGACCCCGGTT 5′ | 1 | |
| ATCATGATGAAACTGATGACCCCGGTT 5′ | 1 |
Discrete broken coding-end fragments join to form coding joints of either homogeneous or heterogeneous length.
A goal of these studies is to determine how the structure of broken coding ends might contribute to the formation of corresponding coding joints. Having found that coding ends showed characteristic lengths for each locus, we proceeded to examine the length heterogeneity of DJH and VJκ joints. Rearranged alleles were amplified with previously described primers from murine thymocyte DNA and induced 103 bcl2/4 cell DNA (27, 29). These sources were chosen to avoid the influence of cellular selection on the repertoire of coding joints. Unlike the case with B-cell progenitors, the expression of protein from certain DJH alleles does not result in selection in T cells (27a). Similarly, VJκ expression in the 103 bcl2/4 pro-B-cell line does not result in cell selection, since 103 bcl2/4 cells contain nonproductive V(D)J rearrangements on both heavy-chain loci (data not shown). Expressed κ light chains lack heavy chain for Ig assembly; therefore both in frame and out of frame rearrangements should remain unselected.
Amplified DJH and VJκ fragments were labeled and analyzed by polyacrylamide gel electrophoresis (Fig. 5D and G). We found that despite the similarly limited nature of DH, JH, Vκ, and Jκ coding-end heterogeneity, DJH and VJκ joints displayed strikingly different length heterogeneity. The DJH joint length varied over a range of at least 22 nt, whereas VJκ joints were of a single predominant length. Inspection of the Kabat database of Ig sequences shows a similar limitation of VJκ gene length (11). The difference between heavy- and light-chain heterogeneity cannot be explained solely by N-region addition, since we obtained essentially similar results analyzing TdT knockout thymocyte DNA for DJH length heterogeneity (reference 8 and data not shown). Some of this heterogeneity, however, is attributable to the difference in length of Dfl16 (22 nt) and Dsp (17 nt) DH gene segments, since this PCR assay detects both sets of DH gene segments.
DISCUSSION
In vitro studies have led to a definition of the earliest steps of V(D)J recombination. RAG1 and RAG2 are sufficient to recognize a RSS on an oligonucleotide or plasmid template (19). These proteins then introduce a nick, generating a free 3′ hydroxyl on the coding segment at the junction between the RSS and the coding-segment sequence. The hydroxyl group then carries out a nucleophilic attack on a phosphodiester bond on the strand opposite the nick (33). This concerted step in the reaction yields a hairpin coding end and a blunt signal end. In contrast to our detailed knowledge of the mechanism of these early steps in V(D)J recombination, there is little understanding of the subsequent metabolism of these broken ends to generate signal joints and coding joints. This study presents data regarding the structure of nonhairpin coding ends, the presumed direct precursor of the coding joint.
Structure of nonhairpin coding ends.
We modified a previously described LM-PCR assay, enabling us to detect a variety of nonhairpin coding-end DNA fragments in association with loci undergoing V(D)J recombination. Template dilution experiments led us to estimate that nonhairpin coding ends were as much as 100-fold less abundant than signal ends in the same DNA sample (data not shown). We infer the involvement of these fragments in V(D)J recombination from their induction in 103 bcl2/4 cells in parallel with recombinase activity and joint formation, their presence in thymocytes and bone marrow B cells, and their absence from RAG-deficient lymphocytes and nonlymphoid cells. A previous study presented evidence that similarly defined ends were in fact intermediates in V(D)J recombination (22). These ends might represent hairpin ends which have been nucleolytically opened and possibly further processed. Alternatively, these ends might represent a V(D)J recombination reaction product or intermediate which was not generated from a hairpin coding-end precursor. We think that it is unlikely that these ends represent the action of a nonspecific nuclease on coding-end hairpins, since we were unable to detect these ends in scid thymocytes, a source of cells rich in hairpin coding ends (references 24 and 36 and data not shown).
Sequence analysis of nonhairpin coding ends led to several surprising observations. First, the free 5′ end of the broken coding-segment DNA is almost invariably shorter than full length. This may have implications for the mechanism of hairpin processing. The discovery of hairpin coding-end DNA led immediately to models of hairpin opening that would account for the existence of P nucleotides—a nuclease would open a hairpin, leaving either a 5′ or 3′ extension, which could be filled in by a polymerase before or after joining to generate the observed palindromic duplex (17, 24). If opened coding ends had 5′ extensions, we would detect these ends after T4 DNA polymerase treatment and LM-PCR as fragments longer than full length with palindromic sequence at their termini. Our analyses failed to detect such fragments.
Second, the lengths of 5′ coding ends of Vκ, Jκ, DH, and JH were nonrandom. Sequence analysis and denaturing gel electrophoresis revealed predominant sites of 5′ coding-end breakage. No obvious sequence motif defines the site of breakage, however. We presume that these nonrandom coding-end deletions influence the precise structure of the resultant coding joints. Several reports in the literature demonstrate a role for coding-segment sequence in influencing the precise structure of coding joints (6, 20, 21). Examination of the Kabat database of VJκ joints revealed the frequent deletion of the first 4 nt of Jκ1 from most VJκ1 joints (11). This corresponds exactly to the 4 nt deleted from our sequenced Jκ1 coding ends. Another group has recently reported a similar predominance of 4-nt-deleted Jκ coding ends (22). These observations support our contention that these broken coding ends are true recombination intermediates. The extent of 5′-strand deletion does not define the limit of coding-end sequences which might ultimately be found in a coding joint, however. The 3′-overhanging coding-end sequence, after joining by the recombinase, could serve as a template for resynthesis of some or all of the deleted 5′-strand nucleotides.
Third, after T4 DNA polymerase polishing, the only longer-than-full-length coding ends we observed were actually aberrant cleavages within the RSS heptamer rather than filled-in palindromes. Two recent studies also detected several coding ends of this type (18, 22). This is consistent with previous data demonstrating that the positioning of cleavage by the recombinase is not strictly confined to the junction between the heptamer and the coding segment (19, 32).
Finally, coding ends show 3′ overhangs that, at least for Jκ1 and JH2, correspond to nucleotides between the recessed 5′ end and the coding-segment–RSS junction (Fig. 6 and 7; Table 1). In several instances, we observed palindromic extensions at the ends of full-length 3′ strands. This observation supports our contention that these ends represent the products of hairpin opening. It is possible that our 3′-overhanging linker LM-PCR assay underestimates the frequency of palindromic 3′-overhanging ends because the palindromic nature of these ends might interfere with their efficient ligation to the linker. One previous report failed to detect 3′ overhangs on nonhairpin Jκ1 coding ends but did detect an identical predominant 4-nt deletion from their 5′ ends (22). This discrepancy might be due to adventitious exonuclease digestion of the overhanging ends during DNA purification. A second study reported 3′-overhanging coding ends associated with the Jα50 gene segment in thymus DNA but did not evaluate the structure of the overhanging DNA (18).
The ends we detected by LM-PCR may have been processed by multiple nucleolytic events. For example, regardless of where the hairpin precursor is opened, a 5′-to-3′ exonuclease might generate a series of 3′-overhanging DNA molecules. The 3′ overhangs, generated by exonuclease extension of a double-strand break, have been defined as an intermediate in homologous recombination (9). Although there is little evidence of similarity between V(D)J recombination and homologous recombination, certain enzymatic activities might be used by both processes. In this regard, it is worth noting several reports which show V(D)J joining events directed by short regions of homology (3, 6, 7). Similarly, a 3′-overhanging strand might be trimmed by exonuclease activity prior to joining. This would be consistent with the data presented in Table 1.
Processing of hairpin ends.
It was reported recently that the position of hairpin opening by certain nucleases is a property of the last 4 nt of its DNA sequence (12). If the broken coding-segment ends described in this report were generated from a hairpin precursor, the distribution of the positions of the ends might be a conserved property of the coding-segment DNA sequence. In contrast to model templates, however, we found that hairpin opening in vivo invariably occurs on the strand of the coding segment containing a 5′ end. The initial nick introduced by the recombinase leaves a free 3′ hydroxyl group at the end of the coding segment (19). Therefore, this initial step in the reaction generates an asymmetric recombinase-DNA complex. We suggest that this asymmetric distribution of recombinase components might target the nucleolytic event to the strand opposite this initial nick.
Either 5′- or 3′-overhanging ends can ultimately generate palindromic repeats observed in coding joints. Rather than fill-in synthesis, ligation of a 5′ end in target DNA to a palindromic 3′-overhanging strand can result in P-nucleotide insertion. A major difference between these two modes of P-nucleotide addition is their timing—one occurs by fill-in synthesis before end joining, and the other occurs by fill-in synthesis after end joining. The fact that we were unable to detect any blunt-ended full-length or longer coding segments (in samples not pretreated with T4 DNA polymerase) is consistent with a 3′-end ligation model for recombination.
Another factor favoring the involvement of 3′-overhanging ends in V(D)J recombination is the substrate preference of TdT. TdT adds nontemplated nucleotides (N regions) more efficiently to 3′-overhanging ends than to either blunt or 5′-overhanging ends (1a). The intermediates we report would be ideal templates for N-region addition.
Formation of coding joints.
Coding joints exhibit various degrees of length and sequence heterogeneity. D-to-JH and V-to-DJH rearrangements, for example, vary in length over a range of more than 20 nt, only a small portion of which is due to TdT-mediated N-region addition (Fig. 5) (references 11 and 20 and data not shown). V-to-Jκ rearrangements, in contrast, show exceptionally little length heterogeneity (Fig. 5) (34). Furthermore, the Jκ1 sequences within these joints are deleted in a nonrandom fashion, with approximately 70% of molecules ending at either +3, +4, or +5 relative to the 5′ end of the coding segment (34). Similar nonrandom deletions were described in the JH1, JH3, and JH4 segments of V-D-J joints (20). We propose that the nonrandom distribution of 5′ ends we observed in coding ends contributes to this biased array of joint sequences. The extremely limited length heterogeneity of V-to-Jκ1 joints as compared with the D-to-JH joints might be due to any or all of the following possibilities: (i) the absence of TdT, (ii) the absence of a processing nuclease, or (iii) a conserved structure which promotes more rapid joining, leaving little time for nucleolytic processing.
Assays for coding-end processing.
Several groups have recently reported success in obtaining coding-joint formation in vitro (15, 23, 35). The heterogeneity of coding joints, however, creates a problem in identifying with certainty the enzymatic activities which generate joints. Ends and joints generated in vitro with various reconstituted systems might not involve the proteins and mechanisms used in vivo. For example, if broken ends are generated by RAG1 and RAG2 in vitro, any combination of nuclease and ligase present in a crude nuclear extract might be expected to open, polish, and ligate coding ends in vitro. The present study, as well as previous studies of the structures of coding joints, allowed us to define expected intermediates and products of authentic V(D)J recombination for comparison with those generated in vitro. Our characterization of these intermediates will also focus efforts on purifying enzymatic activities which generate similar molecules upon reaction with synthetic hairpins in vitro.
ACKNOWLEDGMENTS
I thank Naomi Rosenberg (Tufts University) for the 103 bcl2/4 cell line, Fred Alt (Harvard Medical School/HHMI) for the 63-12 cell line, and Stacey Dillon (Johns Hopkins University) for CD19+ bone marrow DNA. The manuscript was improved by the insightful criticisms of various members of the Schlissel lab, Drew Pardoll, and several anonymous reviewers.
This work was funded by a Culpeper Foundation Scholarship in Medical Sciences, a Cancer Research Institute Investigator Award, a Leukemia Society Scholarship and a grant from the NIH (AI40227).
REFERENCES
- 1.Bogue M, Roth D B. Mechanism of V(D)J recombination. Curr Opin Immunol. 1996;8:175–180. doi: 10.1016/s0952-7915(96)80055-0. [DOI] [PubMed] [Google Scholar]
- 1a.Chang L M, Bolum F J. Molecular biology of terminal transferase. Crit Rev Biochem. 1986;21:27–52. doi: 10.3109/10409238609113608. [DOI] [PubMed] [Google Scholar]
- 2.Chen Y Y, Wang L C, Huang M S, Rosenberg N. An active v-abl protein tyrosine kinase blocks immunoglobulin light-chain gene rearrangement. Genes Dev. 1994;8:688–697. doi: 10.1101/gad.8.6.688. [DOI] [PubMed] [Google Scholar]
- 3.Chukwuocha R U, Nadel B, Feeney A J. Analysis of homology-directed recombination in VDJ junctions from cytoplasmic Ig-pre-B cells of newborn mice. J Immunol. 1995;154:1246–1255. [PubMed] [Google Scholar]
- 4.Constantinescu A, Schlissel M S. Changes in locus-specific V(D)J recombinase activity induced by immunoglobulin gene products during B cell development. J Exp Med. 1997;185:609–620. doi: 10.1084/jem.185.4.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cory S, Adams J M, Kemp D J. Somatic rearrangements forming active immunoglobulin mu genes in B and T lymphoid cell lines. Proc Natl Acad Sci USA. 1980;77:4943–4947. doi: 10.1073/pnas.77.8.4943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ezekiel U R, Sun T, Bozek G, Storb U. The composition of coding joints formed in V(D)J recombination is strongly affected by the nucleotide sequence of the coding ends and their relationship to the recombination signal sequences. Mol Cell Biol. 1997;17:4191–4197. doi: 10.1128/mcb.17.7.4191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gerstein R M, Lieber M R. Extent to which homology can constrain coding exon junctional diversity in V(D)J recombination. Nature. 1993;363:625–627. doi: 10.1038/363625a0. [DOI] [PubMed] [Google Scholar]
- 8.Gilfillan S, Dierich A, Lemeur M, Benoist C, Mathis D. Mice lacking TdT: mature animals with an immature lymphocyte repertoire. Science. 1993;261:1175–1178. doi: 10.1126/science.8356452. [DOI] [PubMed] [Google Scholar]
- 9.Haber J E. In vivo biochemistry: physical monitoring of recombination induced by site-specific endonucleases. Bioessays. 1995;17:609–620. doi: 10.1002/bies.950170707. [DOI] [PubMed] [Google Scholar]
- 10.Hartley K O, Gell D, Smith G C, Zhang H, Divecha N, Connelly M A, Admon A, Lees-Miller S P, Anderson C W, Jackson S P. DNA-dependent protein kinase catalytic subunit: a relative of phosphatidylinositol 3-kinase and the ataxia telangiectasia gene product. Cell. 1995;82:849–856. doi: 10.1016/0092-8674(95)90482-4. [DOI] [PubMed] [Google Scholar]
- 11.Kabat E, Wu T, Reid-Miller M, Perry H, Gottesman K. Sequences of proteins of immunological interest. U.S. Washington, D.C: Department of Health and Human Services; 1987. [Google Scholar]
- 12.Kabotyanski E B, Zhu C, Kallick D A, Roth D B. Hairpin opening by single-strand-specific nucleases. Nucleic Acids Res. 1995;23:3872–3881. doi: 10.1093/nar/23.19.3872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kirchgessner C U, et al. DNA-dependent kinase (p350) as a candidate gene for the murine SCID defect. Science. 1995;267:1178–1183. doi: 10.1126/science.7855601. [DOI] [PubMed] [Google Scholar]
- 14.Lafaille J J, DeCloux A, Bonneville M, Takagaki Y, Tonegawa S. Junctional sequences of T cell receptor gamma delta genes: implications for gamma delta T cell lineages and for a novel intermediate of V-(D)-J joining. Cell. 1989;59:859–870. doi: 10.1016/0092-8674(89)90609-0. [DOI] [PubMed] [Google Scholar]
- 15.Leu T M J, Eastman Q M, Schatz D G. Coding joint formation in a cell free V(D)J recombination system. Immunity. 1997;7:303–313. doi: 10.1016/s1074-7613(00)80532-4. [DOI] [PubMed] [Google Scholar]
- 16.Lewis S M. The mechanism of V(D)J joining: lessons from molecular, immunological, and comparative analyses. Adv Immunol. 1994;56:27–150. doi: 10.1016/s0065-2776(08)60450-2. [DOI] [PubMed] [Google Scholar]
- 17.Lieber M R. The mechanism of V(D)J recombination: a balance of diversity, specificity, and stability. Cell. 1991;70:873–876. doi: 10.1016/0092-8674(92)90237-7. [DOI] [PubMed] [Google Scholar]
- 18.Livak F, Schatz D G. Identification of V(D)J recombination coding end intermediates in normal thymocytes. J Mol Biol. 1997;267:1–9. doi: 10.1006/jmbi.1996.0834. [DOI] [PubMed] [Google Scholar]
- 19.McBlane J F, Vangent D C, Ramsden D A, Romeo C, Cuomo C A, Gellert M, Oettinger M A. Cleavage at a V(D)J recombination signal requires only Rag1 and Rag2 proteins and occurs in two steps. Cell. 1995;83:387–395. doi: 10.1016/0092-8674(95)90116-7. [DOI] [PubMed] [Google Scholar]
- 20.Nadel B, Feeney A J. Influence of coding-end sequence on coding-end processing in V(D)J recombination. J Immunol. 1995;155:4322–4329. [PubMed] [Google Scholar]
- 21.Nadel B, Feeney A J. Nucleotide deletion and P addition in V(D)J recombination: a determinant role of the coding-end sequence. Mol Cell Biol. 1997;17:3768–3778. doi: 10.1128/mcb.17.7.3768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ramsden D A, Gellert M. Formation and resolution of double-strand break intermediates in V(D)J rearrangement. Genes Dev. 1995;9:2409–2420. doi: 10.1101/gad.9.19.2409. [DOI] [PubMed] [Google Scholar]
- 23.Ramsden D A, Paull T T, Gellert M. Cell-free V(D)J recombination. Nature. 1997;388:488–492. doi: 10.1038/41351. [DOI] [PubMed] [Google Scholar]
- 24.Roth D B, Menetski J P, Nakajima P B, Bosma M J, Gellert M. V(D)J recombination: broken DNA molecules with covalently sealed (hairpin) coding ends in scid mouse thymocytes. Cell. 1992;70:1–9. doi: 10.1016/0092-8674(92)90248-b. [DOI] [PubMed] [Google Scholar]
- 25.Roth D B, Nakajima P, Menetski J P, Bosma M J, Gellert M. V(D)J recombination in mouse thymocytes: double-strand breaks near T cell receptor δ rearrangement signals. Cell. 1992;69:41–53. doi: 10.1016/0092-8674(92)90117-u. [DOI] [PubMed] [Google Scholar]
- 26.Roth D B, Zhu C, Gellert M. Characterization of broken DNA molecules associated with V(D)J recombination. Proc Natl Acad Sci USA. 1993;90:10788–10792. doi: 10.1073/pnas.90.22.10788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schlissel M, Baltimore D. Activation of immunoglobulin kappa gene rearrangement correlates with induction of germline kappa gene transcription. Cell. 1989;58:1001–1007. doi: 10.1016/0092-8674(89)90951-3. [DOI] [PubMed] [Google Scholar]
- 27a.Schlissel, M. S. Unpublished results.
- 28.Schlissel M S, Constantinescu A, Morrow T, Baxter M, Peng A. Double-strand signal sequence breaks in V(D)J recombination are blunt, 5′ phosphorylated, RAG-dependent and cell cycle regulated. Genes Dev. 1993;7:2520–2532. doi: 10.1101/gad.7.12b.2520. [DOI] [PubMed] [Google Scholar]
- 29.Schlissel M S, Corcoran L M, Baltimore D. Virally-transformed pre-B cells show ordered activation but not inactivation of immunoglobulin gene rearrangement and transcription. J Exp Med. 1991;173:711–720. doi: 10.1084/jem.173.3.711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shaffer A L, Peng A, Schlissel M S. In vivo occupancy of the κ light chain enhancers in primary pro- and pre-B cells: a model for κ locus activation. Immunity. 1997;6:131–143. doi: 10.1016/s1074-7613(00)80420-3. [DOI] [PubMed] [Google Scholar]
- 31.Tonegawa S. Somatic generation of antibody diversity. Nature. 1983;302:575–581. doi: 10.1038/302575a0. [DOI] [PubMed] [Google Scholar]
- 32.van Gent D C, McBlane J F, Ramsden D A, Sadofsky M J, Hesse J E, Gellert M. Initiation of V(D)J recombination in a cell-free system. Cell. 1995;81:925–934. doi: 10.1016/0092-8674(95)90012-8. [DOI] [PubMed] [Google Scholar]
- 33.van Gent D C, Mizuuchi K, Gellert M. Similarities between initiation of V(D)J recombination and retroviral integration. Science. 1996;271:1592–1594. doi: 10.1126/science.271.5255.1592. [DOI] [PubMed] [Google Scholar]
- 34.Victor K D, Vu K, Feeney A J. Limited junctional diversity in kappa light chains. Junctional sequences from CD43+B220+ early B cell progenitors resemble those from peripheral B cells. J Immunol. 1994;152:3467–3475. [PubMed] [Google Scholar]
- 35.Weis-Garcia F, Besmer E, Sawchuk D J, Yu W, Hu Y, Cassard S, Nussenzweig M C, Cortes P. V(D)J recombination: in vitro coding joint formation. Mol Cell Biol. 1997;17:6379–6385. doi: 10.1128/mcb.17.11.6379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhu C, Roth D B. Characterization of coding ends in thymocytes of scid mice: implications for the mechanism of V(D)J recombination. Immunity. 1995;2:101–112. doi: 10.1016/1074-7613(95)90082-9. [DOI] [PubMed] [Google Scholar]