

Abstract
NF-κB2 (p100/p52), a member of the NF-κB/Rel family of transcription factors, is involved in the regulation of a variety of genes important for immune function. Previously, we have shown that the NF-κB2 gene is regulated in a positive and a negative manner. Two κB elements within the NF-κB2 promoter mediate tumor necrosis factor alpha-inducible transactivation. In addition, we have shown that there exists a transcriptional repression in the absence of NF-κB. To identify a DNA binding activity responsible for this transcriptional repression, we have partially purified a nuclear complex, named Rep-κB. Here we further analyze this putative repressive binding activity. Detailed examination of Rep-κB–DNA interaction revealed the sequence requirements for binding to be almost identical to those of recombination signal binding protein Jκ (RBP-Jκ), the mammalian homolog of the protein encoded by Drosophila suppressor of hairless [Su(H)]. In addition, in electromobility shift assays, Rep-κB binding activity is recognized by an antibody directed against RBP-Jκ. By performing transient-transfection assays, we show that human RBP-Jκ represses basal as well as RelA (p65)-stimulated NF-κB2 promoter activity. Studies in Drosophila melanogaster have shown that Su(H) is implicated in the Notch signaling pathway regulating cell fate decisions. In transient-transfection assays we show that truncated Notch-1 strongly induces NF-κB2 promoter activity. In summary, our data clearly demonstrate that Rep-κB is closely related or identical to RBP-Jκ. RBP-Jκ is a strong transcriptional repressor of NF-κB2. Moreover, this repression can be overcome by activated Notch-1, suggesting that NF-κB2 is a novel putative Notch target gene.
NF-κB/Rel proteins comprise a family of inducible transcription factors which control the expression of numerous genes involved in the immune, inflammatory, and acute-phase responses (for reviews, see references 3, 5, and 18). There is increasing evidence that members of this family are also involved in the regulation of normal and malignant cellular growth, especially in hematopoietic cells (16). More recently, it was demonstrated that inhibition of NF-κB/Rel induces apoptosis in various cell types, suggesting an antiapoptotic potential of NF-κB/Rel proteins (6, 36, 56, 60, 62).
In higher vertebrates, the NF-κB/Rel family encompasses five different genes: those encoding NF-κB1 (p105/p50), NF-κB2 (p100/p52), RelA (p65), and RelB, and the proto-oncogene encoding c-Rel. To a limited extent, these proteins can form homo- and heterodimers with distinct DNA-binding specificity (39, 43, 49). In most cell types, NF-κB/Rel dimers are sequestered in the cytoplasm by a member of the IκB family of inhibitory proteins. IκB proteins mask the nuclear localization signal of NF-κB/Rel, thereby preventing the nuclear translocation of NF-κB/Rel. Upon stimulation, IκB proteins are phosphorylated on specific serine residues, ubiquitinated, and degraded through proteasome-dependent proteolysis, thereby allowing NF-κB/Rel dimers to translocate into the nucleus and bind to their cognant DNA sequences (for reviews, see references 4, 5, 54, and 57).
This initial activation of NF-κB can occur without de novo protein synthesis. However, it was shown that maintenance of NF-κB activity requires ongoing protein synthesis and continuous stimulation, indicating that NF-κB/Rel is also regulated at a transcriptional or translational level (23).
In a previous study, we demonstrated that NF-κB2 is positively autoregulated via two κB-responsive elements (34), as shown for other family members, including NF-κB1 (53) and IκBα (28). In addition, mutation of the κB elements resulted in a dramatic increase in the basal NF-κB2 promoter activity in various cell lines. Therefore, we postulated that there is a negative regulation of NF-κB2 transcription mediated by the κB elements. A putative repressive DNA binding activity, Rep-κB, which interacts with a κB motif in the NF-κB2 promoter was identified. Rep-κB binding activity was partially purified from different cell sources, indicating that its expression is ubiquitous (34).
Recombination signal binding protein Jκ (RBP-Jκ), also designated KBF2 or CBF1, was originally purified based on its binding to the recombination signal of the Jκ immunoglobulin gene (38). Subsequently it was demonstrated that RBP-Jκ acts as a transcriptional regulator, via binding to specific DNA motifs, rather than as a recombinase in V(D)J rearrangement (55). RBP-Jκ proteins have been highly conserved during evolution not only in vertebrates but also in invertebrates. Genetic analysis revealed that the gene encoding RBP-Jκ is conserved as the Drosophila melanogaster suppressor of hairless [SuH)], a member of the neurogenic gene family including Notch, Delta, Enhancer of split [E(spl)], and Hairless (15, 50). These genes participate in a lateral inhibition mechanism whereby singled-out sensory mother cells prevent their neighbors from adopting the neuronal fate (2). The first evidence that RBP-Jκ acts as a transcription factor came from studies on viral gene expression. A cellular protein with 97% identity to a RBP-Jκ splice variant (RBP-2N), which binds specifically to a 10-bp motif in the promoter region of the adenovirus capsid protein pIX, was purified. Transcriptional repression of the pIX promoter by RBP-2N was shown in in vitro as well as in vivo experiments (11). A transcriptional repression domain within RBP-Jκ which might directly interact with the basal transcription machinery has been identified (24). Alternatively, this domain might function as a binding interface for an as-yet-unidentified corepressor protein (58).
RBP-Jκ-mediated repression is overcome by interaction with the Epstein-Barr virus activator protein EBNA2. EBNA2 by itself is not able to bind DNA but is targeted to its DNA-responsive elements by direct physical interaction with RBP-Jκ, thereby masking the RBP-Jκ repression domain and acting as a strong transcriptional activator (19, 22, 24, 59, 64).
The gene encoding RBP-Jκ/Su(H) is genetically mapped downstream of the Notch receptor in a signaling cascade controlling cell fate decisions during Drosophila development (14). One current model of Notch signaling postulates that after ligand binding the Notch receptor is activated by a proteolytic cleavage step. This activated form of Notch, Notch-IC, exerts its function by targeting DNA-bound RBP-Jκ/Su(H), thereby masking the repression domain of RBP-Jκ and activating transcription (25). In mammals, Notch homologs are implicated in neurogenesis and myogenesis (41), granulocyte differentiation (40), and CD4 versus CD8 as well as αβ versus γδ lineage decisions during T-lymphocyte development (45, 61). Direct physical interaction between activated forms of the murine Notch-1 receptor and RBP-Jκ have been demonstrated (26, 29, 52), making this signal transduction cascade an exceptional short and simple communication strategy between the cell surface and the nucleus. So far, the only known Notch-1 target gene in mammals is the hairy enhancer of split (HES-1) gene, which encodes a basic helix-loop-helix protein that is able to block MyoD-induced myogenesis (32).
Here we demonstrate, on the basis of its DNA binding requirements, its gel mobility shift pattern, and its recognition by a specific antibody, that Rep-κB is closely related or identical to RBP-Jκ. Rep-κB binds specifically to an 8-bp DNA motif in the NF-κB2 promoter with a high degree of similarity to the RBP-Jκ consensus sequence (55). This RBP-Jκ binding site overlaps the 5′-half site of a functional κB element. In transient-transfection assays, we show that RBP-Jκ strongly inhibits basal as well as RelA (p65)-induced NF-κB2 promoter activity. These data show that RBP-Jκ behaves biochemically and functionally identically to the previous proposed Rep-κB as a repressor of NF-κB2 transcription. In addition, we show in transient-transfection assays that truncated Notch-1 overcomes RBP-Jκ-mediated repression and strongly activates NF-κB2 transcription through the RBP-Jκ binding site.
MATERIALS AND METHODS
Plasmids.
A 1.9-kb BamHI-XhoI fragment of the mouse RBP-Jκ gene, kindly provided by Tasuku Honjo (Kyoto, Japan), was isolated and used to screen a cDNA library derived from HeLa cells (Stratagene). Screening was performed as described previously (35). Two independent cDNA clones with 1.8- and 1.5-kb insert sizes were isolated, subcloned into the EcoRI and XhoI sites of Bluescript SK(+) (Stratagene), and sequenced. Both clones represent 3′ fragments of the RBP-Jκ cDNA truncated in exon 2. To generate a full-length cDNA of RBP-2N, the missing 5′ sequence was introduced by PCR, using BamHI and NdeI restriction sites. Using BamHI and XhoI, the full-length cDNA of RBP-2N was cloned into the pcDNA3 vector (Invitrogen), resulting in pCMV-RBP-2N. To generate an RBP-1 expression plasmid, a PCR fragment representing the 5′ end of the RBP-1 gene, designated aPCR1 (1), was digested with ClaI, treated with the Klenow fragment of DNA polymerase, and digested with SphI to generate a fragment which was ligated to SpeI-digested RBP-2N DNA, blunted, and digested with SphI. The full-length RBP-1 gene was subcloned into the NotI and XhoI sites of pcDNA3. In addition, we searched a database of sequencing tags and identified a sequence identical to that of the full-length RBP-3 gene (GenBank accession no. Z36843). The plasmid was kindly provided by T. Gress (Ulm, Germany). The insert was cut out and cloned into the BamHI and NotI sites of Bluescript SK(+) and pcDNA3, resulting in pCMV-RBP-3. The identities of PCR fragments and cloning junctions were confirmed by sequencing.
The reporter plasmid pASwt was constructed by subcloning a 363-bp AvaI-to-SalI fragment of the NF-κB2 promoter (positions −198 to +165) (34) into the MluI and XhoI sites of the luciferase vector pGL2-basic (Promega). pAS-SL49 was generated by site-directed mutagenesis using the oligodeoxynucleotide SL49.
The expression plasmid pRSVRelA and plasmids for in vitro transcription of RelA/p65 and NF-κB1/p50 were described previously (12, 49). The expression plasmid for truncated murine Notch-1, pSV-mNotch-1-IC, corresponds to pSG5mNotch1IC (25) and was kindly provided by Thomas Henkel (Munich, Germany).
Cell lines.
The cell lines HEK-293 (ATCC CRL 1573) and COS-7 (DSM ACC 60) were grown at 37°C under 5% CO2 in Dulbecco’s modified Eagle’s medium (Gibco) supplemented with 10% fetal calf serum (FCS). Jurkat T cells (ATCC TIB 152) were cultivated at 37°C under 5% CO2 in RPMI 1640 medium (Gibco) supplemented with 5% FCS.
Preparation of cell extracts.
Nuclear extracts were prepared as described elsewhere (34). Whole-cell lysates were prepared as follows. Cells were washed three times in phosphate-buffered saline (PBS) and pelleted by centrifugation at 300 × g. The pellet was resuspended in 5 volumes of ice-cold CHAPS lysis buffer, consisting of 10 mM 3-[(cholamidopropyl)-dimethylammonio]-1-propanesulfonate (CHAPS), 50 mM Tris-HCl (pH 7.9), 150 mM NaCl, 2 mM EDTA, 5 mM NaF, 10 μg of aprotinin per ml, 10 μg of leupeptin per ml, 1 mM dithiothreitol (DTT), and 0.5 mM phenylmethylsulfonyl fluoride; this was followed by incubation on ice for 40 min. The lysate was cleared by centrifugation at 80,000 × g for 30 min. Protein concentrations were determined by the Bradford method (Bio-Rad), and extracts were assayed for DNA binding activity by electromobility gel shift assays (EMSAs) and Western blotting.
Translation of recombinant proteins.
In vitro-translated proteins were synthesized in a reticulocyte lysate-coupled transcription-translation system in accordance with the manufacturer’s instructions (Promega). The quality of translation was monitored by simultaneously labeling with [35S]methionine. After separation of the labeled translation products by sodium dodecyl sulfate (SDS) gel electrophoresis, the gels were dried and exposed to X-ray film.
Protein purification and in vitro interaction assay.
Purification of Rep-κB was performed essentially as described previously (34). Nuclear extracts from unstimulated Jurkat-T cells were applied to DNA-cellulose columns equilibrated at 100 mM NaCl. After several washing steps, protein fractions were eluted with increasing NaCl concentrations and analyzed by EMSA. Purification of a bacterially expressed glutathione S-transferase (GST)–Notch-1-IC fusion protein and in vitro interaction assays were performed as described in references 8 and 29.
EMSAs.
Approximately 5 to 10 μg of cell extract was used for EMSAs in a binding buffer consisting of 10 mM Tris-HCl (pH 7.5), 100 mM NaCl, 0.1 mM EDTA, 0.5 mM DTT, and 4% glycerol. For binding reactions, 2 μg of poly(dI-dC) (Pharmacia) and approximately 0.5 ng of 32P-labeled oligonucleotides were added. For competition experiments, a 10- to 60-fold molar excess of unlabeled competitor was used. For antibody perturbation experiments, 0.5 μg of antibody directed against RBP-Jκ (K0043) (21, 46), 10 μg of antibody directed against RelA (p65) (SC109X; Santa Cruz Biotechnology), or 10 μg of antibody directed against the FLAG epitope (M-5; Kodak) was added to the reaction mixture. The reaction products were separated on 5% polyacrylamide gels with 1× Tris-glycine-EDTA at room temperature. The gels were dried and exposed to X-ray film.
Oligonucleotides.
The sequences of the oligodeoxynucleotides SL350 and SL366 are as follows. SL350, 5′-CTAGTAACGTCATGGGAATTCCCCCCTCCGGGGGGCCGAGAAGGGGCTTTCCCGGCCCT-3′, corresponds to positions −119 to −64 of the human NF-κB2 gene and contains two NF-κB binding sites (34). SL366, 5′-GATCGTTACTGTGGGAAAGAAAGTTTGGGAAGTTTCACAC-3′, spans positions −91 to −47 of the murine HES-1 promoter and contains two RBP-Jκ binding sites. SL49, 5′-GCACGGAAACGTGAGGGGAATTCCCCC-3′, was used for site-directed mutagenesis to introduce a mutation into the RBP-Jκ consensus sequence in the reporter construct pASwt to generate pAS-SL49. The sense strand of each oligonucleotide is shown. Sequences of other oligodeoxynucleotides are shown in the respective figures.
DNA transfection and luciferase assay.
A total of 106 HEK-293 cells were transfected in 90-mm-diameter culture dishes with 5 to 10 μg of plasmid DNA expressing RBP-Jκ splice forms and murine Notch-1-IC by calcium phosphate coprecipitation, as described previously (48). Forty-eight hours after transfection, nuclear proteins were prepared and the extracts were assayed for DNA binding activity and protein expression. COS-7 cells were transfected with DEAE-dextran essentially as described previously (49). Cells (2 × 105) in 35-mm-diameter culture dishes were transfected with 2 μg of reporter plasmid DNA together with various amounts of plasmid expressing RelA (p65), RBP-2N, RBP-3, or murine Notch-1-IC. Transfection of Jurkat-T cells was performed as follows. Cells (107) were electroporated in a total volume of 300 μl of RPMI 1640 medium supplemented with 5% FCS. Electroporations were performed with a Gene Pulser II apparatus (Bio-Rad) at 260 V and 975 μF in 40-mm electroporation cuvettes. To quantitate luciferase activity, cells were harvested 48 h after transfection and lysed in 100 μl of a buffer containing 25 mM Tris-HCl (pH 7.8), 2 mM EDTA, 2 mM DTT, 10% glycerol, and 1% Triton X-100 for 10 min at room temperature. Lysates were centrifuged at 7,000 × g for 5 min. Luciferase activity of at least four independent transfections with 20 μl of cleared lysate was determined in an LB 9501 luminometer (Berthold), using a luciferase assay system (Promega). All transfections were normalized for the level of total cellular protein (Bradford assay; Bio-Rad).
Western blotting.
SDS-polyacrylamide gels (7.5%) were transferred to a nitrocellulose filter (BA85; Schleicher & Schuell) for 20 min at 4°C and 150 mA, using a Tris-glycine buffer system. The membrane was blocked for 2 to 3 h in a solution of 5% dry milk powder in PBS-T (0.05% Tween 20 in PBS), incubated with the primary antibody directed against the FLAG epitope (M-5; Kodak) for 1 to 2 h in PBS-T containing 5% dry milk powder, and then washed three times in PBS-T (10 min each). The secondary antibody (1:7,000 dilution of peroxidase-conjugated sheep anti-mouse immunoglobulin G (IgG; Sigma) was incubated with the membrane for 30 min. After the membrane was washed, specific proteins were detected by using an enhanced chemiluminescence system (Amersham).
RESULTS
Purification of Rep-κB DNA binding activity.
Previously, we had shown that two κB elements within the human NF-κB2 promoter are critical for responsiveness of NF-κB2 to tumor necrosis factor alpha (TNF-α) and phorbol myristate acetate. Interestingly, mutation of the κB elements led to a strong induction of basal promoter activity in unstimulated cells. Therefore, we postulated that there is a κB element DNA binding activity which mediates transcriptional repression of the NF-κB2 gene in the absence of NF-κB. A nuclear complex, which we named Rep-κB, likely to be responsible for this transcriptional repression could be identified. To further characterize this putative repressive DNA binding activity, we partially purified protein complexes from nuclear extracts of unstimulated Jurkat-T cells, using DNA-cellulose columns. Proteins were eluted with increasing NaCl concentrations. DNA binding activity in the eluted fractions was monitored by EMSA using the 32P-labeled oligonucleotide SL350, which contains both κB sites (positions −119 to −63) of the NF-κB2 promoter. The specificity of DNA binding was tested by competition with increasing amounts of unlabeled oligonucleotide SL350 (Fig. 1A, lanes 3 and 4). Two major complexes, A and B, turned out to bind specifically to the NF-κB2 probe. On the basis of previous experiments, we identified complex A as Rep-κB activity. Rep-κB was eluted at 280 to 320 mM NaCl (Fig. 1A, fractions 10 to 13). Further characterizations of Rep-κB were performed with fractions 11 and 12.
FIG. 1.




(A) Purification of Rep-κB activity from Jurkat-T cells. Nuclear extracts (NE), flowthrough (Flow), and eluted fractions were tested for binding to the oligonucleotide SL350, which contains both κB elements in the human NF-κB2 promoter and flanking regions (positions −119 to −63). The specificity of binding was analyzed by adding 20- and 60-fold molar excesses of a homologous competitor (lanes 3 and 4). A and B indicate the positions of specific DNA binding complexes. Complex A was identified as Rep-κB activity. (B) Sequence requirements for Rep-κB binding. Competition experiments were performed with eluted fraction 11 (5 μl) and 20- to 60-fold molar excesses of the unlabeled competitor (Comp.) oligonucleotides shown in panel C. As a probe, the 32P-labeled oligonucleotide SL350 was used. −, no competitor added. (C) Sequences and Rep-κB binding abilities of different oligonucleotides derived from the first κB element in the NF-κB2 promoter. Only the positive strand of each oligonucleotide is shown. Lowercase letters represent added linker sequences; dashes indicate identity with the consensus (Consens.) sequence. (D) Rep-κB activity is recognized by an antibody directed against RBP-Jκ. Treatment of eluted fractions 11 and 12 (5 μl) with an RBP-Jκ-specific antibody (α-RBP) specifically abolished band A. Instead, a more slowly migrating complex (band C, lanes 2 and 4) appeared. Another DNA binding activity (complex B) was not affected by the antibody. The probe was a 32P-labeled oligonucleotide, SL332, containing the first κB element (positions −119 to −90) within the NF-κB2 promoter. −, control (antibody not added). (E) Rep-κB binds specifically to the RBP-Jκ site in the murine HES-1 promoter. Nuclear extracts and eluted fractions from Jurkat-T cells were analyzed for specific DNA binding activity, using the 32P labeled oligonucleotide HES-1 as a probe. This oligonucleotide contains the known RBP-Jκ consensus sequence 5′-CTGTGGGAAAGA-3′. As a competitor, a 50-fold molar excess of unlabeled oligonucleotide SL332 (lane 2) or HES-1 (lane 3) was used. Band A represents Rep-κB binding activity. −, control (competitor not added).
Sequence requirements for Rep-κB binding.
To determine the DNA binding properties of Rep-κB in more detail, we carried out competition experiments with eluted fraction 11 and the 32P-labeled oligonucleotide SL350 as a probe (Fig. 1B). For competition, the oligonucleotides shown in Fig. 1C were used at a 20- or 60-fold molar excess. The oligonucleotide SL332 represents the first κB element (positions −119 to −90) in the human NF-κB2 promoter (see also Fig. 3). Since the labeled probe contains both κB elements, the competition experiment with SL332 (Fig. 1B, lanes 2 and 3) suggests that the first κB element is the major target site for Rep-κB binding. Therefore, we introduced various nucleotide changes in the flanking region or within the κB element and used the double-stranded oligonucleotides shown in Fig. 1C as competitors. As shown in Fig. 1B, excesses of oligonucleotides SL336, SL338, SL340 and SL342 did not titrate out Rep-κB binding. However, the competitors SL346 and SL348 were still able to bind Rep-κB activity. Interestingly, we identified a binding consensus element (5′-CATGGGAA-3′ [Fig. 1C]) with obvious similarity to that of RBP-Jκ (5′-CGTGGGAA-3′), the mammalian homolog of Drosophila melanogaster suppressor of hairless (54). Indeed, oligonucleotide SL344, containing the ideal RBP-Jκ consensus sequence (Fig. 1C), did compete for Rep-κB binding (Fig. 1B, lanes 16 and 17). These experiments clearly demonstrate that the 5′-half site of the first κB element in the NF-κB2 promoter, rather than the whole κB element, is necessary for Rep-κB binding. In addition, the sequence requirements for Rep-κB DNA interaction are almost identical to those of RBP-Jκ.
FIG. 3.

Schematic representation of the luciferase (LUC) reporter constructs used for transfection assays. The constructs pASwt and pAS-SL49 contain a DNA fragment (positions −198 to +165) derived from the human NF-κB2 promoter. The sequences of the κB elements (black boxes) and the overlapping putative RBP-Jκ elements (open boxes) are shown. In the construct pAS-SL49, the putative RBP-Jκ consensus element is mutated as indicated. Abbreviations: A, AvaI; E, EcoRI; N, NruI; Pv, PvuI; S, SalI.
Rep-κB is most likely identical to RBP-Jκ.
Additional evidence that Rep-κB is closely related to RBP-Jκ was obtained from gel shift analyses performed in the presence of an RBP-Jκ-specific antibody (Fig. 1D). Eluted fractions 11 (lanes 1 and 2) and 12 (lanes 3 and 4) were incubated with or without the monoclonal rat antibody K0043 (21, 46) prior to gel shift analysis. The presence of the antibody led to the disappearance of complex A in both fractions and to the appearance of a supershifted, novel band. This complex (band C) most likely contains RBP-Jκ. Furthermore, we performed a band shift experiment with crude nuclear extracts from Jurkat-T cells and eluted fractions 9 to 15. As a probe for DNA binding, we used a 32P-labeled oligonucleotide derived from the murine HES-1 promoter. This promoter sequence contains a perfect RBP-Jκ consensus sequence, 5′-CTGTGGGAAAGA-3′ (positions −82 to −71), without an overlapping κB element (8). As shown in Fig. 1E, complex A from Jurkat-T cell nuclear extracts binds specifically to the RBP-Jκ site of the HES-1 promoter (lane 1). Competition for this binding activity was provided by a 50-fold molar excess of unlabeled oligonucleotide SL332 (lane 2) as well as by a 50-fold molar excess of the homologous competitor HES-1 (lane 3). Furthermore, the DNA binding activity associated with the HES-1 site in the eluted fractions corresponds exactly to the NF-κB2-specific Rep-κB activity shown in Fig. 1A, suggesting that Rep-κB is identical to RBP-Jκ.
Finally, we tested whether recombinant RBP-Jκ proteins bind to the κB element in the NF-κB2 promoter. Therefore, cDNAs coding for the human RBP-Jκ splice forms RBP-1, RBP-2N, and RBP-3 were inserted into appropriate vectors (see Materials and Methods) and expressed in a cell-free translation system (Promega). Lysates (4 μl) were assayed for protein-DNA interaction in band shift experiments using the 32P-labeled oligonucleotide SL332 as a probe. Binding specificity was monitored by incubating programmed lysates with an antibody directed against RBP-Jκ. For comparison, we used Rep-κB activity from eluted fraction 12. The results of this experiment are shown in Fig. 2. As previously shown, Rep-κB binding activity (complex A) is recognized by the RBP-Jκ-specific antibody, supershifting a novel band (C) (lanes 1 and 2). Unfortunately, reticulocyte lysate itself contains abundant NF-κB activity which binds to the κB element within the oligonucleotide SL332 (complex B). NF-κB binding activity was confirmed by competition experiments (data not shown) and supershift analysis with a RelA (p65)-specific antiserum (see Fig. 6B, lane 8). However, additional κB binding complexes could be detected in lysates programmed with the human RBP-Jκ proteins RBP-2N (Fig. 2, lane 5), RBP-1 (lane 7), and RBP-3 (lane 9). Incubation with an RBP-Jκ-specific antibody completely abolished complex A (lanes 6, 8, and 10), suggesting that these bands correspond to RBP-Jκ. This complex (A) was absent from a lysate programmed with a control plasmid, Bluescript SK(+) (Fig. 2, lanes 3 and 4).
FIG. 2.
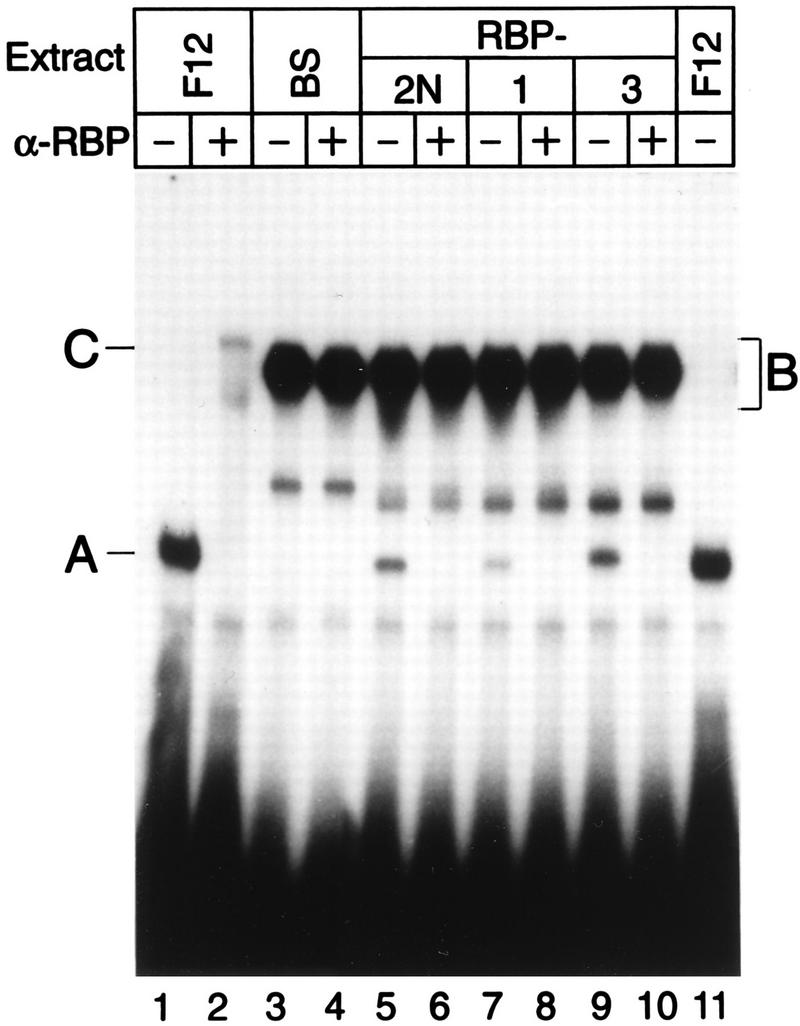
The RBP-Jκ splice variants RBP-2N, RBP-1, and RBP-3 bind specifically to the first κB element in the NF-κB2 promoter. Eluted fraction 12 (5 μl) and RBP-Jκ splice variants synthesized in a cell-free system were incubated with (+) or without (−) an antibody directed against RBP-Jκ (α-RBP) prior to gel electrophoresis. Treatment of lysates with the antibody specifically abolished complex A (lanes 2, 6, 8, and 10). This complex was absent from the lysate in which a control plasmid [Bluescript SK(+) (BS)] was used in the translation assay (lane 4). The added reticulocyte lysate (5 μl) contained NF-κB binding activity (complex B) which covered the supershifted antibody-RBP complex (band C) detected in lane 2. The 32P-labeled oligonucleotide SL332, containing the first κB element of the NF-κB2 promoter, was used as a probe.
FIG. 6.

Cell-free system-synthesized Notch-1-IC interacts with Rep-κB/RBP-Jκ from Jurkat T cell nuclear extracts (Jurkat-NE) at the HES-1 and NF-κB2 promoters. (A) Extracts (2 μg) were incubated without (lane 1) or with increasing amounts of (lanes 2 to 5) reticulocyte lysate programmed with Notch-1-IC (mNotch-1IC). Volumes were equalized by addition of pure reticulocyte lysate. A 32P-labeled HES-1-specific double-stranded oligonucleotide (SL366) was used as a probe. Rep-κB/RBP-Jκ binding activity (complex A) is evidenced by an RBP-Jκ-specific antibody (α-RBP) supershifting a novel band (complex C). Note that this more slowly migrating band (lane 5) does not represent a Notch-1-IC-containing complex. (B) Extracts (2 μg) were incubated without (lane 1) and with increasing amounts of (lanes 4 to 8) reticulocyte lysate programmed with Notch-1-IC. 32P-labeled NF-κB2-specific oligonucleotide SL332 was used as a probe. In addition to the Rep-κB/RBP-Jκ-specific DNA binding activity (complex A) that was recognized by a specific antibody (α-RBP), a second complex (complex B) was detected in nuclear extracts and was more abundant after addition of reticulocyte lysate. Treatment with an antiserum directed against RelA (p65) (α-p65) interfered with this binding activity and supershifted band F (lanes 3 and 8).
These experiments clearly demonstrate that (i) Rep-κB is closely related, if not identical, to RBP-Jκ and that (ii) recombinant RBP-Jκ proteins bind specifically to the first κB element within the NF-κB2 promoter.
The human NF-κB2 promoter is repressed by RBP-2N.
Previously, the human RBP-Jκ splice form RBP-2N was shown to act as a transcriptional repressor of adenovirus pIX gene expression via sequence-specific DNA binding (11). To test the regulation of the human NF-κB2 gene in response to RBP-Jκ, NF-κB2-specific luciferase constructs were transiently cotransfected with plasmids expressing RBP-2N, under the control of the cytomegalovirus (CMV) promoter, into COS-7 or Jurkat-T cells. As shown in Fig. 3, the reporter construct pASwt contains NF-κB2-specific promoter sequences (positions −198 to +165), including both κB elements. The first κB element overlaps the putative RBP-Jκ consensus site, 5′-CATGGGAA-3′. In pAS-SL49, this site has been mutated to 5′-GAGGGGAA-3′ without affecting the functional κB element. Coexpression of increasing amounts of RBP-2N into COS-7 cells resulted in a 70% repression of the basal luciferase activity of pASwt. No significant effect was observed with the reporter construct pAS-SL49, which contains a mutated RBP-Jκ site (Fig. 4A).
FIG. 4.

Repression of basal as well as RelA (p65)-stimulated NF-κB2 promoter activity by RBP-2N depends on a functional RBP-Jκ consensus sequence. (A) Portions (2 μg) of reporter constructs pASwt (black bars) and pAS-SL49 (open bars) were cotransfected into COS-7 cells with increasing amounts of an RBP-2N expression plasmid. (B and C) Portions (2 μg) of reporter constructs pASwt (black bars) and pAS-SL49 (open bars) were cotransfected with 500 ng of pRSVRelA and increasing amounts of CMV-RBP-2N into COS-7 cells (B) or Jurkat-T cells (C). Luciferase activity was determined from 100 μg of total cell extract, and the basal promoter activity of each construct was set to one. Mean values and standard deviations for at least four independent experiments are shown.
Next, we asked whether RBP-2N would also repress prestimulated NF-κB2 transcription. Therefore, we stimulated NF-κB2 promoter activity by transfecting a RelA (p65)-expressing plasmid together with increasing amounts of RBP-2N. The luciferase activity of pASwt was stimulated 10-fold following cotransfection with 500 ng of pRSVRelA. This activation could be abrogated to basal levels by gradually increasing the amount of RBP-2N. Using the reporter plasmid pAS-SL49, a ninefold increase in luciferase activity was detected after cotransfection with pRSVRelA. However, the repressive effect of RBP-2N was completely lost (Fig. 4B). Similar results were obtained with Jurkat-T cells (Fig. 4C).
Taken together, these results demonstrate that RBP-2N is able to suppress basal as well as RelA (p65)-stimulated NF-κB2 promoter activity. This transcriptional repression depends on a functional RBP-Jκ binding site within the NF-κB2 promoter.
Rep-κB/RBP-Jκ and NF-κB/Rel (p50/p65) compete for binding to the first κB element within the NF-κB2 promoter.
To investigate the binding of NF-κB/Rel complexes and Rep-κB/RBP-Jκ to their overlapping sites in more detail, we first performed a semiquantitative binding analysis for Rep-κB/RBP-Jκ and NF-κB/Rel (p50/p65) by the method of Scatchard (12). Fraction 12, containing partially purified Rep-κB protein (Fig. 1A, lane 14), was used as the source of Rep-κB/RBP-Jκ DNA binding activity, and cell-free system-cotranslated p50/p65 heterodimers were used to determine NF-κB/Rel-specific affinity for the first κB site. Figure 5A shows a graph of the binding of the NF-κB/Rel (p50/p65) heterodimers to the labeled oligonucleotide SL332, with saturation for values exceeding 1 nM free probe. For Rep-κB/RBP-Jκ, no saturation could be obtained with a probe concentration of 2 nM. Although we could not precisely determine the binding affinity of Rep-κB/RBP-Jκ for the NF-κB2-specific oligonucleotide, this experiment shows that the affinity of NF-κB/Rel (p50/p65) for the κB element is at least sixfold higher than that of Rep-κB/RBP-Jκ (Fig. 5A). To investigate whether NF-κB/Rel proteins and Rep-κB/RBP-Jκ could bind to their overlapping sites simultaneously, we carried out band shift experiments; the results are shown in Fig. 5B. Fraction 12 was incubated with increasing amounts of cell-free system-synthesized p50/p65 heterodimers and constant amounts of 32P-labeled oligodeoxynucleotide SL332, resulting in the subsequent appearance of the NF-κB-specific complex B (Fig. 5B, lanes 3 to 5). The specificity of this complex was tested by treatment with antiserum directed against RelA (p65), which induced a supershifted novel band, band F (Fig. 5B, lane 6). Interestingly, augmenting NF-κB/Rel (p50/p65)-specific DNA binding activity (complex B) was associated with a gradual loss of Rep-κB/RBP-Jκ-specific DNA binding (complex A), suggesting that NF-κB/Rel and Rep-κB/RBP-Jκ compete for binding to overlapping sites within the NF-κB2 promoter.
FIG. 5.
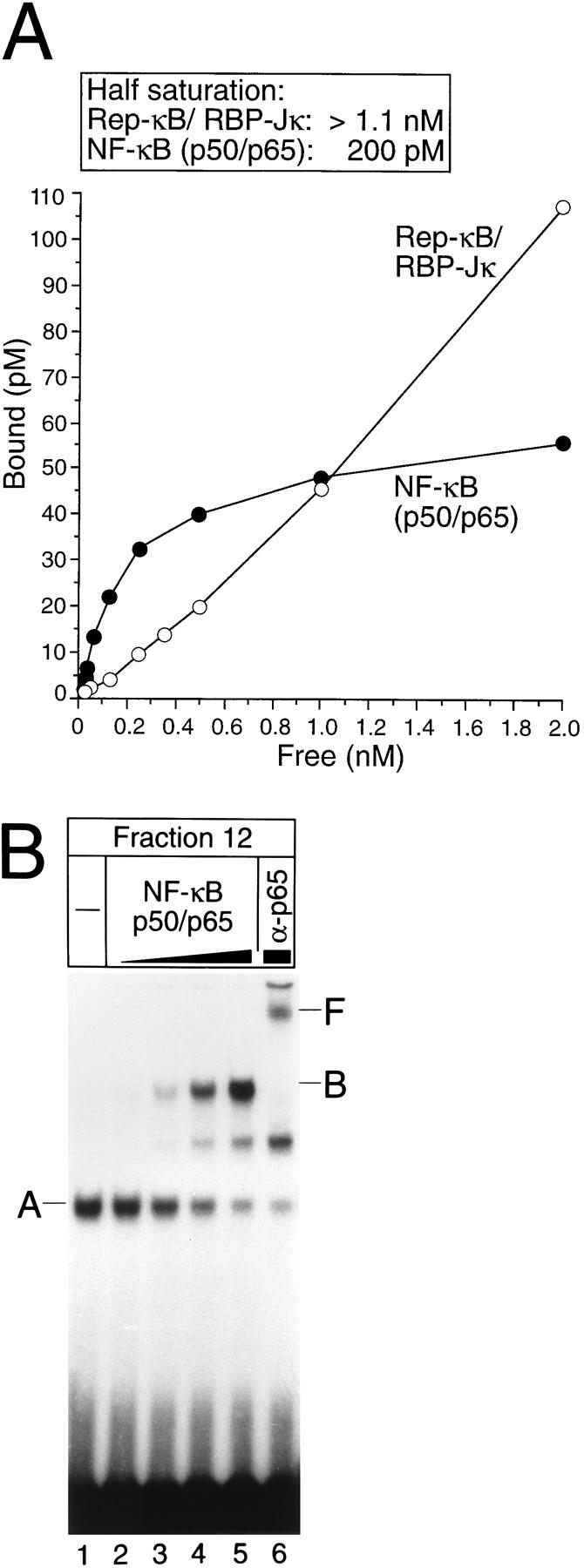
(A) Affinity of Rep-κB/RBP-Jκ and NF-κB/Rel (p50/p65) for the first κB element and the overlapping RBP-Jκ site within the NF-κB2 promoter, evaluated by Scatchard analysis. Protein-DNA binding studies were performed with a range of concentrations of labeled oligonucleotide SL332 in a 20-μl reaction volume. Bound and free probe species were quantitated with a PhosphorImager (Molecular Dynamics). Reactions were performed in triplicate. (B) Competitive binding of NF-κB/Rel (complex B) and Rep-κB/RBP-Jκ (complex A). The band shift experiment was performed with fraction 12 of partially purified nuclear extracts from Jurkat-T cells and increasing amounts of cell-free cotranslated NF-κB/Rel (p50/p65) heterodimers. Band F represents a RelA (p65)-containing complex which was supershifted after treatment with antiserum (α-p65). Radiolabeled oligonucleotide SL332 was used as a probe. −, control (no NF-κB/Rel added).
Activated mammalian Notch-1 interacts with Rep-κB/RBP-Jκ.
Truncated Notch-1 was shown to activate transcription mediated by physical interaction with RBP-Jκ, thereby masking the repressive effect of RBP-Jκ (25). In mammals, this was demonstrated with the murine HES-1 promoter (29). To investigate whether activated Notch-1 is also involved in the transcriptional regulation of NF-κB2, we first performed band shift experiments (Fig. 6). Nuclear extracts from Jurkat-T cells were incubated with and without increasing amounts of cell-free system-synthesized truncated Notch-1 protein (Notch-1-IC). As a probe, we used a 32P-labeled oligonucleotide derived from the murine HES-1 promoter (positions −91 to −47), which contains two RBP-Jκ consensus sequences (−80 to −74 [5′-GTGGGAA-3′] and −66 to −60 [5′-TTGGGAA-3′]) (Fig. 6A), or the NF-κB2-specific oligonucleotide SL332 (Fig. 6B). Specific protein-DNA complexes were identified by using antibodies directed against RBP-Jκ and RelA (p65). Complex A turned out to contain RBP-Jκ (Fig. 6A, lane 5). This complex binds to the NF-κB2 probe (Fig. 6B) as well as to the HES-1 probe (Fig. 6A). In contrast, an additional, more slowly migrating complex (Fig. 6B, band B) could be identified with the NF-κB2-specific oligonucleotide but not the HES-1 probe, which contains no κB site. A RelA (p65)-specific antiserum supershifted this additional complex (Fig. 6B, band F, lanes 3 and 8), suggesting that this complex contains RelA. The strong NF-κB binding activity is in part due to intrinsic NF-κB proteins in the reticulocyte lysate added to the binding reactions shown in lanes 4 to 8 (compare with Fig. 2, lanes 3 to 10). Increasing amounts of cell-free system-synthesized truncated Notch-1 protein led to the disappearance of the RBP-Jκ-specific complex A, which bound to the HES-1 probe (Fig. 6A; compare lanes 2 to 4 with lane 1) and to the NF-κB2 probe (Fig. 6B; compare lanes 4 to 7 with lane 1), suggesting that activated Notch-1 interacts with Rep-κB/RBP-Jκ in both cases.
Since we could not detect an additional, higher-order complex which contains Notch-1 together with RBP-Jκ bound to DNA, we used a bacterially expressed GST–Notch-1-IC fusion protein (Fig. 7A). Fraction 12, containing partly purified Rep-κB/RBP-Jκ DNA binding activity, was incubated with increasing amounts of purified GST–Notch-1-IC prior to the gel shift experiment. Radiolabeled oligonucleotide SL332 was used as a probe. As expected, increasing amounts of GST–Notch-1-IC led to a clear reduction of Rep-κB/RBP-Jκ DNA binding activity (complex A). In addition, a higher-order complex (band D) could be detected (Fig. 7A, lanes 5 to 9). This complex was absent when GST–Notch-1-IC was incubated with fraction 25, which does not contain Rep-κB binding activity (lanes 1 to 3). GST protein incubated with fraction 12 (lanes 12 and 13) or fraction 25 (lanes 10 and 11) served as controls.
FIG. 7.
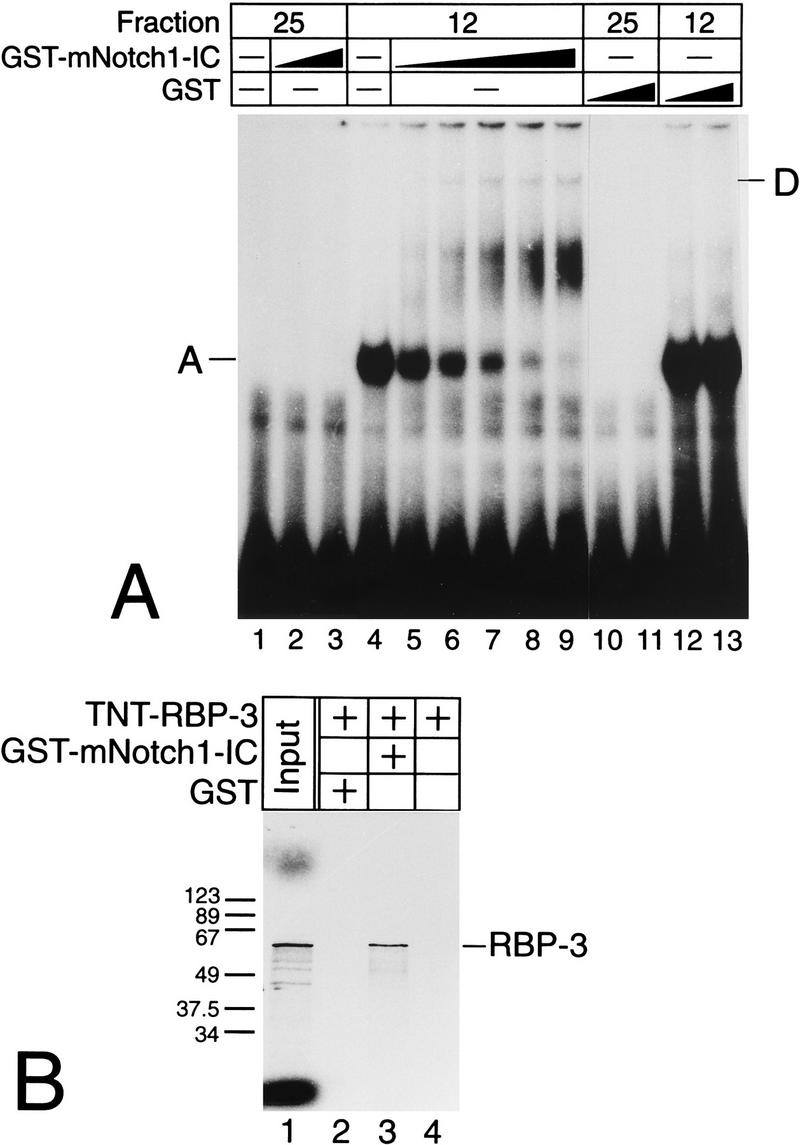
(A) GST–Notch-1-IC forms a higher-order complex with Rep-κB/RBP-Jκ bound to the RBP-Jκ site within the NF-κB2 promoter. Fractions 12 and 25 were incubated with the GST proteins as indicated. Only the addition of GST–Notch-1-IC to a Rep-κB/RBP-Jκ-containing fraction resulted in a decrease in RBP-Jκ-specific DNA binding activity (complex A), and a novel, more slowly migrating complex (band D) appeared. (B) GST–Notch-1-IC interacts with recombinant RBP-Jκ in vitro. GST proteins were immobilized with Sepharose beads and incubated with cell-free system-synthesized RBP-3 (TNT-RBP-3). After extensive washing steps, the reaction mixtures were boiled and proteins were separated by SDS-polyacrylamide gel electrophoresis. The positions of molecular size markers (in kilodaltons) are shown on the left.
Furthermore, the interaction of GST–Notch-1-IC with recombinant RBP-Jκ protein was verified in vitro. GST–Notch-1-IC was immobilized with glutathione-Sepharose (Pharmacia) and incubated with cell-free system-synthesized, 35S-labeled RBP-3. After extensive washing, the reaction mixture was boiled and separated by SDS-polyacrylamide gel electrophoresis (Fig. 7B). Radiolabeled RBP-3 protein could be detected only when GST–Notch-1-IC was added to the reaction (Fig. 7B, lane 3). RBP-Jκ did not bind to GST protein (Fig. 7B, lane 2) or to Sepharose beads (Fig. 7B, lane 4).
To investigate this interaction in a cellular context, we transiently transfected HEK-293 cells with plasmids expressing the RBP-Jκ splice form RBP-3 (pCMV-RBP-3) and a truncated Notch-1 protein with an N-terminal FLAG epitope (pSV-mNotch-1-IC). Protein extracts were prepared 48 h after transfection. Expression of the activated Notch-1 protein was verified by Western blot analysis with an antibody directed against the FLAG epitope (Fig. 8B). In addition, extracts (5 μg) were assayed for protein-DNA interactions in band shift experiments using the 32P-labeled oligonucleotides SL366 (Fig. 8A) and SL332 (Fig. 8C) as probes. Using the HES-1-specific oligonucleotide SL366, an RBP-Jκ-specific complex (band A) could already be identified in extracts of untransfected HEK-293 cells (Fig. 8A, lane 1). Transfection of 5 μg of pCMV-RBP-3 led to an increased RBP-Jκ-specific DNA binding activity (Fig. 8A, lane 3). The endogenous RBP-Jκ-specific DNA binding activity was almost abolished in extracts from HEK-293 cells transfected with 5 μg of pSV-mNotch-1-IC. In these extracts, a novel, more slowly migrating complex (band D) appeared (Fig. 8A, lane 5). Incubation of cell extracts with an antibody directed against the FLAG epitope supershifted complex D, resulting in complex E, which most likely contains Notch-1-IC interacting with RBP-Jκ (Fig. 8A, lane 6). The anti-FLAG antibody did not interfere with RBP-Jκ binding to DNA (Fig. 8A, lanes 2 and 4). The higher-order complex D and the supershifted complex E could also be identified in nuclear extracts from HEK-293 cells cotransfected with Notch-1-IC and an RBP-3 expression plasmid (Fig. 8A, lanes 7 and 8).
FIG. 8.
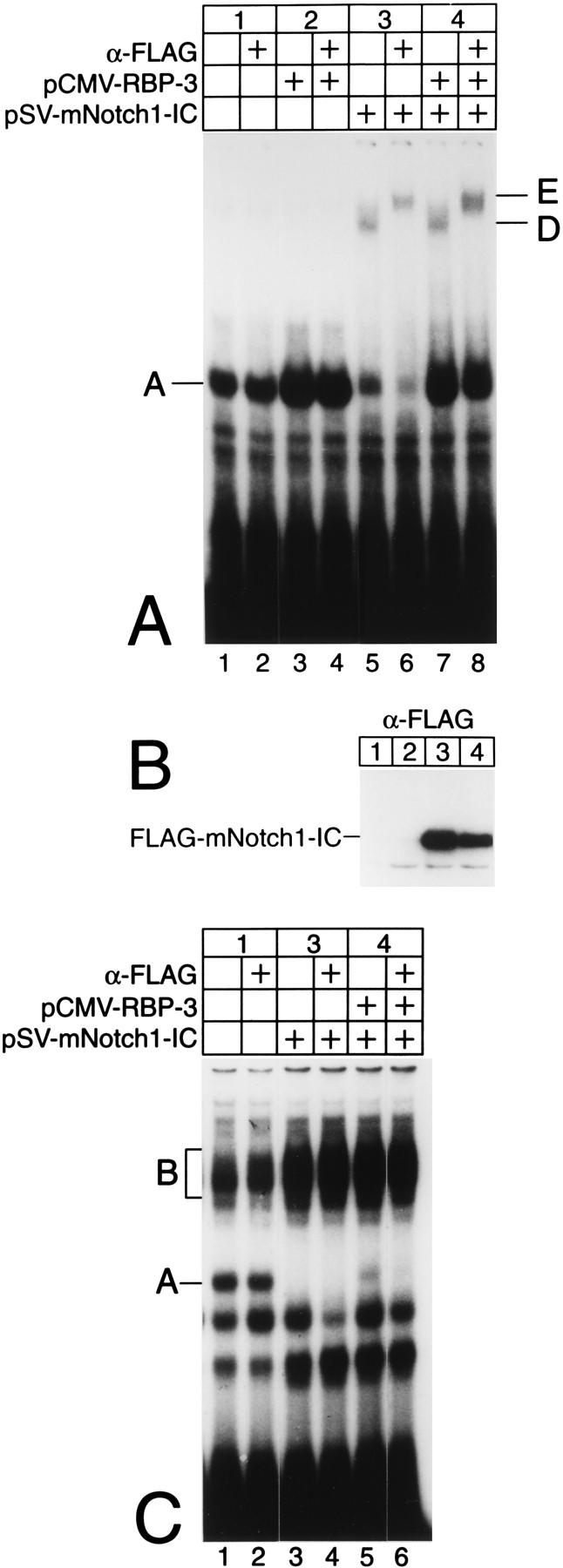
Truncated mammalian Notch-1 interacts with Rep-κB/RBP-Jκ DNA binding activity in transfected HEK-293 cells. (A) In cell extracts from HEK-293 cells transfected with a Notch-1-IC expression plasmid, Rep-κB/RBP-Jκ-specific DNA binding activity bound to the HES-1-specific oligonucleotide (complex A) was decreased and a more slowly migrating complex appeared (complex D). Treatment with an anti-FLAG antibody (α-FLAG) recognized complex D and supershifted a novel complex, labeled E. (B) Cell extracts were prepared 48 h after transfection of 5 μg of pCMV-RBP-3 and/or 5 μg pSV-mNotch-1-IC and assayed for Notch-1-IC expression by Western blotting. (C) In cell lysates transfected with a Notch-1-IC expression plasmid, Rep-κB/RBP-Jκ-specific DNA binding activity bound to the NF-κB2-specific oligonucleotide (complex A) was abolished, but no higher-order complex, could be identified; it was probably masked by additional NF-κB-specific DNA binding activity (complex B).
To investigate the interaction of RBP-Jκ with Notch-1-IC in the context of the NF-κB2 promoter, similar experiments were performed. The oligonucleotide SL332 was used as a probe. The results are shown in Fig. 8C. Again, the RBP-Jκ-specific complex A could be detected in lysates of untransfected HEK-293 cells (Fig. 8C, lane 1). The specificity of complex A was determined by competition experiments and supershift experiments with an RBP-Jκ-specific antibody (data not shown). In lysates transfected with pSV-mNotch-1-IC, this complex was completely abolished, suggesting the occurrence of an interaction of truncated Notch-1 with Rep-κB/RBP-Jκ at the NF-κB2 promoter (Fig. 8C, lane 3). A similar decrease in RBP-Jκ binding activity was observed in extracts from HEK-293 cells transfected with a plasmid expressing the truncated form of Notch-1 together with CMV-RBP-3 (Fig. 8C, lane 5). However, we did not detect an additional, higher-order DNA binding complex (Fig. 8C, lanes 3 and 5). After lysates were incubated with the anti-FLAG antibody, a supershifted complex could not be identified (lanes 4 and 6).
Taken together, the results shown in Fig. 6 to 8 suggest the occurrence of a physical interaction between Rep-κB/RBP-Jκ and truncated Notch-1, thereby changing RBP-Jκ DNA binding complexes, both at the murine HES-1 promoter and at the human NF-κB2 promoter.
Activated mammalian Notch-1 stimulates NF-κB2 promoter activity.
Since the interaction of Rep-κB/RBP-Jκ with truncated Notch-1 results in alterations of the RBP-Jκ-specific DNA binding, we were interested in the functional consequences. Therefore, we carried out transient-transfection experiments with NF-κB2-specific reporter constructs (Fig. 3) and expression plasmids for Notch-1-IC, RBP-2N, and RelA (p65). Luciferase activity of pASwt was stimulated 21-fold after transfecting 2 μg of pSV-mNotch-1-IC into COS-7 cells. This strong stimulation mediated by truncated Notch-1 was clearly diminished when pAS-SL49, which has a mutated RBP-Jκ binding site, was used as a reporter (Fig. 9A). Therefore, transactivation of NF-κB2 by truncated mammalian Notch-1 depends on the functional RBP-Jκ site within the NF-κB2 promoter.
FIG. 9.

(A) Stimulation of the NF-κB2 promoter by activated Notch-1. Portions (2 μg) of reporter construct pASwt (black bars) and pAS-SL49 (open bars) were cotransfected into COS-7 cells with the indicated amounts of plasmid DNA expressing truncated Notch-1. (B) The RelA (p65)-mediated promoter activity is not further increased by the addition of Notch-1-IC, but Notch-1-IC-stimulated NF-κB2 promoter activity is abrogated by RBP-2N. Portions (2 μg) of reporter construct pASwt were cotransfected into COS-7 cells with the indicated amounts of plasmid DNA expressing RelA (p65), Notch-1-IC, and RBP-2N. Luciferase activity in 100 μg of total cellular extract was determined. The basal promoter activity of each construct was set to one. Mean values and standard deviations for at least four independent experiments are shown.
Cotransfection of pASwt with 1 μg of pRSVRelA stimulated the NF-κB2 promoter activity 16-fold, and this stimulation was only marginally increased by the addition of pSV-mNotch-1-IC. These data indicate that activated Notch-1 and RelA do not have an additive effect on the NF-κB2 promoter (Fig. 9B). However, the Notch-1-IC-mediated stimulation of the NF-κB2 promoter was greatly reduced with increasing amounts of RBP-2N, suggesting that activation versus repression is mediated by the ratio of Notch-1-IC/RBP-2N complexes to RBP-2N at the binding site (14-fold versus 1.5-fold) (Fig. 9B).
Rep-κB/RBP-Jκ binds to a subset of NF-κB-responsive elements.
In a previous study, we investigated whether Rep-κB binding was specific for the NF-κB2 κB elements or could also bind to other known κB elements. At that time, we did not know that Rep-κB was identical to RBP-Jκ. Furthermore, we believed that the observed repression was mediated by κB elements in the NF-κB2 promoter. The oligonucleotides used in the former competition experiments were not designed in view of an RBP-Jκ site overlapping the κB element. For these reasons, new oligonucleotides representing κB elements and flanking sequences from various genes were designed (Fig. 10B). Partially purified Rep-κB/RBP-Jκ binding activity (fraction 12 [Fig. 1A]) was used in competition experiments with the oligonucleotide SL332 as a probe. The various competitors were added in 10- and 60-fold molar excesses, and the HES-1-specific oligonucleotide SL366 served as a control. The results are shown in Fig. 10A and summarized in Fig. 10B. The tested sequences could be divided into three subclasses with regard to Rep-κB/RBP-Jκ DNA binding affinity: sequences with high, intermediate, and low levels of binding activity. Oligonucleotides with high affinity for Rep-κB/RBP-Jκ DNA were derived from the promoters of the following genes, as listed in Fig. 10B: HES-1, used as a control (lanes 6 and 7); mRBP-Jκ (lanes 10 and 11); Bcl-3 (lanes 8 and 9); interleukin-6 (IL-6) (lanes 30 and 31); IκBα (lanes 28 and 29); β2-microglobulin (lanes 20 and 21); beta interferon (IFN-β) (lanes 24 and 25); and NF-κB2 (first κB site) (lanes 2 and 3). Moderate-level binding of Rep-κB/RBP-Jκ was observed with oligonucleotides derived from the promoters of the following genes: angiotensinogen (one κB site) (lanes 18 and 19), invariant chain (lanes 32 and 33), major histocompatibility complex (MHC) class I (lanes 34 and 35), serum amyloid A1 (SAA-1) (lanes 14 and 15), and TNF-α (lanes 38 and 39). Weak binding or no affinity for Rep-κB/RBP-Jκ was detected with oligonucleotides derived from the following promoters: A20 (lanes 16 and 17), immunoglobulin heavy chain (lanes 22 and 23), Igκ enhancer (lanes 26 and 27), NF-κB1 (lanes 36 and 37), and NF-κB2 (second κB site) (lanes 12 and 13).
FIG. 10.
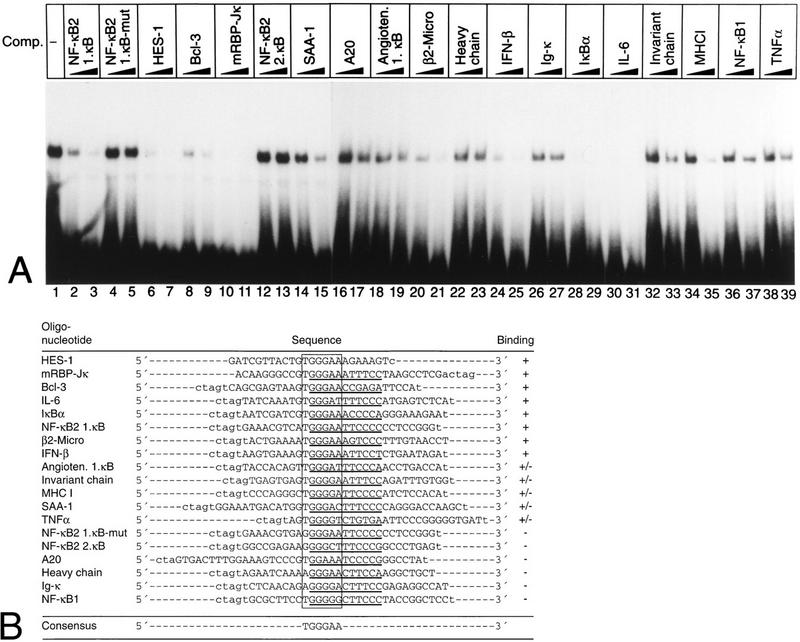
Rep-κB/RBP-Jκ binds to a subset of κB elements. (A) Competition experiments were performed with fraction 12 of partially purified nuclear extract from Jurkat-T cells; a radiolabeled oligonucleotide (SL332) encompassing the first κB element of the human NF-κB2 promoter (positions −119 to −90) was used as a probe. As competitors, the unlabeled double-stranded oligonucleotides (shown in panel B) were added at 20- and 60-fold molar excesses. (B) Oligonucleotides used for competition analysis. The sequences were derived from the promoters-enhancers of the genes indicated. In each case, the putative κB element (underlined) and the overlapping RBP-Jκ site are marked. Their GenBank accession numbers are as follows: A20, M96756; angiotensinogen (Angioten.), M31673; Bcl-3, M31731; β2-microglobulin (β2-Micro), M12485; immunoglobulin heavy chain, U17387; HES-1, D16464; IFN-β, V00534; Igκ, X00268; IκBα, U08468; IL-6, M22111; invariant chain, M35872; MHC-I, M11847; NF-κB1, X63942; RBP-Jκ, X58337; SAA-1, M64088; and TNF-α, X59352. Symbols: +, strong binding; +/−, intermediate-level binding; −, weak or no binding.
Summarizing these data, we can reveal a Rep-κB/RBP-Jκ consensus sequence, 5′-TGGGAA-3′, which overlaps with a κB binding site. The concept of overlapping RBP-Jκ binding sites with κB elements could allow the cell to tightly control κB-mediated gene expression in an unstimulated status through transcriptional repression by Rep-κB/RBP-Jκ. However, the functional consequences of RBP-Jκ binding to the promoters of these various genes have to be elucidated.
DISCUSSION
In a previous report, we demonstrated a negative transcriptional regulation of NF-κB2 mediated by κB elements in its promoter region. A ubiquitously expressed DNA binding activity which binds specifically to this region was identified. Because of its putative repressive effect, this binding activity was named Rep-κB. Rep-κB activity did not bind solely to the NF-κB2 κB element; it also bound to κB sites present in the IFN-β and β2-microglobulin enhancers. In contrast, no binding to the canonical κB site was observed. Therefore, we postulated the existence of a novel mode of κB-dependent regulation mediated by Rep-κB (34).
Here we have provided biochemical and functional evidence that Rep-κB binding activity is identical or closely related to RBP-Jκ. This assumption is based on the following findings. (i) By using partially purified Rep-κB binding activity in competition experiments, we identified an 8-bp Rep-κB binding motif (CATGGGAA) with strong sequence homology to the RBP-Jκ consensus site (CGTGGGAA). This Rep-κB/RBP-Jκ binding site partially overlaps a functional κB element (GGGAATTCCC). (ii) Partially purified Rep-κB activity binds with high affinity to a known RBP-Jκ-responsive site in the murine HES-1 promoter. (iii) Different in vitro-translated RBP-Jκ splice variants bind to the Rep-κB binding site in the NF-κB2 promoter. (iv) In supershift assays, Rep-κB activity is recognized by a specific antibody raised against RBP-Jκ. (v) In transient-transfection assays, RBP-2N represses NF-κB2 promoter activity.
Several observations suggesting a role for RBP-Jκ in the regulation of NF-κB-inducible genes have been published. (i) KBF2 was purified with a β2-microglobulin κB affinity column and found to be identical to RBP-Jκ (27). (ii) A DNA binding activity with affinity for the MHC class I probe was purified from thymic nuclear extracts and identified as RBP-Jκ (51). Binding of RBP-Jκ to the MHC class I oligonucleotide was not evident in splenic extracts, suggesting the occurrence of tissue-specific binding. (iii) Recently, RBP-Jκ was shown to be involved in transcriptional repression of the IL-6 gene. An RBP-Jκ binding site overlapping with a functional κB element was identified as being responsible for this repression (30, 44).
In this report, we have shown that human RBP-Jκ (RBP-2N) strongly represses basal as well as RelA (p65)-induced NF-κB2 promoter activity, suggesting that RBP-Jκ acts as a repressor of NF-κB2 transcription. Previously, we did not detect any repression of this promoter in B lymphocytes, in which NF-κB/Rel proteins are constitutively activated, although large amounts of Rep-κB/RBP-Jκ were detected in the nuclei of these cells (34). Therefore, we suggest the following model. In the absence of NF-κB (e.g., in unstimulated T lymphocytes or nonlymphoid cells), Rep-κB/RBP-Jκ binds to the RBP-Jκ site and inhibits basal NF-κB2 transcription. In the presence of NF-κB (e.g., in B lymphocytes or stimulated T or nonlymphoid cells), NF-κB/Rel proteins replace Rep-κB/RBP-Jκ and activate transcription. We have shown that RBP-Jκ and NF-κB/Rel proteins compete for occupation of their recognition sites and thus may dictate activation versus repression. While this work was in progress, Plaisance et al. found a similar RBP-Jκ-dependent regulation when studying the IL-6 promoter (44). As for NF-κB2, the constitutive occupancy of the IL-6 κB site by RBP-Jκ is thought to be responsible for the low basal levels of IL-6 promoter activity. In this report, it was shown that RBP-Jκ is more abundant than NF-κB in the nuclei of cells tested, but NF-κB has a higher affinity for the κB element within the IL-6 promoter. These results are consistent with our observations for the κB element within the NF-κB2 promoter.
Other mechanisms controlling κB-mediated repression have been reported. The expression of the rat serum amyloid A1 gene is tightly regulated by YY1, which binds to a site overlapping an NF-κB and C/EBP binding site (37). An NF-κB site-mediated suppression of the IL-8 gene by IFN-β has been described (42). Dorsal, responsible for dorsoventral patterning in D. melanogaster, acts as both an activator and a repressor of transcription. This dual effect seems to be controlled by an HMG1-like protein (33). In addition, differential binding of specific NF-κB/Rel subunits has been shown to mediate a negative regulation of the MHC complex class II invariant-chain promoter in a cell-specific fashion (9). These different examples underline the importance of tight control of κB-regulated genes. Loss of this control might become harmful to the cell and result in inflammation or autoimmune disease.
The RBP-Jκ gene is genetically mapped downstream of the Notch receptor (14). In mammals, all four known Notch family members (for a review, see reference 2) can physically interact with RBP-Jκ (25, 29, 31, 52). Truncated Notch-1 (Notch-1-IC), which consists of only the intracellular domains, localizes to the nucleus and functions as an activated receptor (25, 29, 52). In mammals, HES, a basic helix-loop-helix protein involved in the regulation of myogenesis, is activated through Notch (29, 47). In the present study, we have shown that Notch-1-IC stimulates NF-κB2 transcription severalfold. This activation is dependent on the presence of a functional RBP-Jκ site and is almost abolished when a mutated NF-κB promoter construct is used. Activation versus repression of the NF-κB2 promoter seems to be determined by the ratio of RBP-Jκ to Notch-1-IC, since the addition of increasing amounts of RBP-Jκ can antagonize the stimulatory effect of truncated Notch.
Several possibilities for Notch-mediated activation of transcription have been considered. It has been suggested that Notch-1 does not destabilize RBP-Jκ DNA binding but rather impairs RBP-Jκ-mediated repression by associating with and masking the repression domain of RBP-Jκ (25, 52). On the other hand, Notch itself has activation domains which are thought to interact with the basal transcription apparatus. Most investigators favor a combination of both models (52).
By EMSAs, we were able to show that RBP-Jκ and truncated Notch-1 form a higher-order DNA binding complex on the HES-1 promoter. This observation is consistent with previous findings by Jarriault et al. (29). The appearance of this higher-order complex is paralleled by a decrease in the RBP-Jκ complex. A similar dose-dependent decrease in RBP-Jκ binding activity was observed on the NF-κB2 promoter when Notch-1-IC was added. An additional higher-order complex could be identified only with recombinant protein. Different models of regulation can be hypothesized to explain this observation. (i) On the NF-κB2 promoter, interaction of Notch-1-IC with RBP-Jκ leads to the loss of RBP-Jκ DNA binding activity. Transcriptional activation by Notch-1-IC would then solely be due to a derepression mechanism. (ii) Notch-1-IC and RBP-Jκ form a higher-order DNA binding complex on the NF-κB2 promoter. However, this complex is not detectable under our experimental conditions due to the presence of a strong κB binding activity in reticulocyte lysates as well as in HEK-293 extracts, an activity which runs at the same position as that expected for an RBP-Jκ–Notch-1-IC complex. This model would suggest that derepression and transactivation contribute to Notch-1-IC-mediated transcription of NF-κB2. Since we observed a strong transcriptional activation in cotransfection experiments with Notch-1-IC and detected a higher-order complex with recombinant protein, we favor the latter model.
The structural similarity between the cytoplasmic portion of Notch-1 and members of the IκB family suggested that Notch has functional properties of an IκB-like regulator (20). Overexpression of truncated Notch-1 was proposed to have a dose-dependent effect on κB-mediated transactivation of a multimerized reporter gene. Whereas low doses attenuated the inhibitory effect of an excess of p50 homodimer, higher doses of truncated Notch prevented κB-dependent transactivation in a manner similar to Bcl-3. In vitro, binding of p50-containing NF-κB complexes was shown to be inhibited by Notch-1-IC (20). However, on the NF-κB2 promoter, Notch-1-IC seems to act only as a transcriptional activator and not as an IκB-like protein, since inhibition of promoter activity was not observed at any concentration of Notch-1-IC transfected.
By freezing cells in an immature state, Notch has been shown to function in cell fate decisions. The human homolog of Drosophila Notch was isolated out of a translocation with the T-cell receptor β locus from a T-lymphoblastic leukemia (13). This chromosomal rearrangement predicts the formation of a truncated Notch-1 with most of the extracellular domains deleted. The resulting gain-of-function protein is thought to block precursor cells from responding to the proper developmental signals. In agreement with the Drosophila expression pattern, Notch activity is associated with proliferating cell populations. By using the Moloney murine leukemia virus, a frequent viral integration site was located between the ankyrin repeats and the transmembrane domain of Notch-1. This event synergizes with Myc expressed in transgenic mice to form CD4+ CD8+ thymomas (17). In addition, overexpression of Notch-1 was also detected in human cervical and colon carcinoma cells but was absent from the respective differentiated epithelial cells (63). Because activated Notch and NF-κB2 are implicated in malignant transformation, it is tempting to speculate that upregulation of NF-κB2 by truncated Notch could at least in part contribute to this phenotype. Indeed, overexpression of NF-κB2 (p100/p52) and a lack of processing have been found in breast cancer cells but not in untransformed human mammary epithelial cells. Overexpressed NF-κB2 (p100/p52) seemed to sequester heterodimeric p50/p65 in the cytoplasm (10). A similar overexpression of NF-κB2 (p100/p52) was detected in colon cancer cell lines (7). Furthermore, rearrangements of the nfkb2 gene have been found at a low frequency in B-cell lymphomas, multiple myelomas, and, more commonly, cutaneous T-cell lymphomas. Consistently, these alterations led to deletions of the C-terminal ankyrin repeats, resulting in a constitutively active form of NF-κB2 (16).
Taken together, our data strongly suggest an important role for RBP-Jκ in repression of NF-κB2 transcription. The finding of RBP-Jκ binding motifs overlapping with κB elements in promoters of various NF-κB-inducible genes indicates a novel role for RBP-Jκ in silencing NF-κB-inducible transcription. Interestingly, the human NF-κB2 promoter can be activated by truncated Notch-1. This does not prove that NF-κB2 has a definitive role in the Notch signaling pathway; however, the hypothesis that NF-κB2 can act as a nuclear Notch target in the appropriate context is attractive. Which of the other NF-κB-regulated genes mentioned above may be transactivated by members of the Notch family remains to be elucidated. This pathway might introduce another level of complexity in the transcriptional regulation of NF-κB-responsive genes.
ACKNOWLEDGMENTS
F.O. and S.L. contributed equally to this work.
We thank U. Wegenka for critically reading the manuscript. We also thank T. Henkel for providing the plasmid pSG5mNotch1IC, B. Ehring for the gift of pGST-TKmNotch1IC, and T. Honjo for providing the RBP-Jκ-specific antibody K0043 and the expression vector for murine RBP-2. For excellent technical assistance we thank E. Rüber and S. Schirmer. We also thank E. Ebert for providing reagents.
This study was supported by the Deutsche Forschungsgemeinschaft Sonderforschungsbereich 322 to S.L. and R.M.S. and by the Bundes- ministerium für Bildung und Forschung to R.M.S.
REFERENCES
- 1.Amakawa R, Jing W, Ozawa K, Matsunami N, Hamaguchi Y, Matsuda F, Kawaichi M, Honjo T. Human Jκ recombination signal binding protein gene (IGKJRB): comparison with its mouse homologue. Genomics. 1993;17:306–315. doi: 10.1006/geno.1993.1326. [DOI] [PubMed] [Google Scholar]
- 2.Artavanis-Tsakonas S, Matsuno K, Fortini M E. Notch signaling. Science. 1995;268:225–232. doi: 10.1126/science.7716513. [DOI] [PubMed] [Google Scholar]
- 3.Baeuerle P A, Baltimore D. NF-κB: ten years after. Cell. 1996;87:13–20. doi: 10.1016/s0092-8674(00)81318-5. [DOI] [PubMed] [Google Scholar]
- 4.Baeuerle P A, Henkel T. Function and activation of NF-κB in the immune system. Annu Rev Immunol. 1994;12:141–179. doi: 10.1146/annurev.iy.12.040194.001041. [DOI] [PubMed] [Google Scholar]
- 5.Baldwin A S., Jr The NF-κB and IκB proteins: new discoveries and insights. Annu Rev Immunol. 1996;14:649–683. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- 6.Beg A A, Baltimore D. An essential role for NF-κB in preventing TNF-α-induced cell death. Science. 1996;274:782–784. doi: 10.1126/science.274.5288.782. [DOI] [PubMed] [Google Scholar]
- 7.Bours V, Dejardin E, Goujon-Letawe F, Merville M P, Castronovo V. The NF-κB transcription factor and cancer: high expression of NF-κB- and IκB-related proteins in tumor cell lines. Biochem Pharmacol. 1994;47:145–149. doi: 10.1016/0006-2952(94)90448-0. [DOI] [PubMed] [Google Scholar]
- 8.Brou C, Logeat F, Lecourtois M, Vandekerckhove J, Kourilsky P, Schweisguth F, Israel A. Inhibition of the DNA-binding activity of Drosophila suppressor of hairless and of its human homolog, KBF2/RBP-Jκ, by direct protein-protein interaction with Drosophila hairless. Genes Dev. 1994;8:2491–2503. doi: 10.1101/gad.8.20.2491. [DOI] [PubMed] [Google Scholar]
- 9.Brown A M, Linhoff M W, Stein B, Wright K L, Baldwin A S, Jr, Basta P V, Ting J P-Y. Function of NF-κB/Rel binding sites in the major histocompatibility complex class II invariant chain promoter is dependent on cell-specific binding of different NF-κB/Rel subunits. Mol Cell Biol. 1994;14:2926–2935. doi: 10.1128/mcb.14.5.2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dejardin E, Bonizzi G, Bellahcene A, Castronovo V, Merville M P, Bours V. Highly-expressed p100/p52 (NFKB2) sequesters other NF-κB-related proteins in the cytoplasm of human breast cancer cells. Oncogene. 1995;11:1835–1841. [PubMed] [Google Scholar]
- 11.Dou S, Zeng X, Cortes P, Erdjument-Bromage H, Tempst P, Honjo T, Vales L D. The recombination signal sequence-binding protein RBP-2N functions as a transcriptional repressor. Mol Cell Biol. 1994;14:3310–3319. doi: 10.1128/mcb.14.5.3310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Duckett C S, Perkins N D, Kowalik T F, Schmid R M, Huang E-S, Baldwin A S, Jr, Nabel G J. Dimerization of NF-KB2 with RelA(p65) regulates DNA binding, transcriptional activation, and inhibition by an IκB-α (MAD-3) Mol Cell Biol. 1993;13:1315–1322. doi: 10.1128/mcb.13.3.1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ellisen L W, Bird J, West D C, Soreng A L, Reynolds T C, Smith S D, Sklar J. TAN-1, the human homolog of the Drosophila notch gene, is broken by chromosomal translocations in T lymphoblastic neoplasms. Cell. 1991;66:649–661. doi: 10.1016/0092-8674(91)90111-b. [DOI] [PubMed] [Google Scholar]
- 14.Fortini M E, Artavanis-Tsakonas S. The suppressor of hairless protein participates in Notch receptor signaling. Cell. 1994;79:273–282. doi: 10.1016/0092-8674(94)90196-1. [DOI] [PubMed] [Google Scholar]
- 15.Furukawa T, Maruyama S, Kawaichi M, Honjo T. The Drosophila homolog of the immunoglobulin recombination signal-binding protein regulates peripheral nervous system development. Cell. 1992;69:1191–1197. doi: 10.1016/0092-8674(92)90640-x. [DOI] [PubMed] [Google Scholar]
- 16.Gilmore T D, Koedood M, Piffat K A, White D W. Rel/NF-κB/IκB proteins and cancer. Oncogene. 1996;13:1367–1378. [PubMed] [Google Scholar]
- 17.Girard L, Hanna Z, Beaulieu N, Hoemann C D, Simard C, Kozak C A. Frequent provirus insertional mutagenesis of Notch1 in thymomas of MMTVD/myc transgenic mice suggests a collaboration of c-myc and Notch1 for oncogenesis. Gene Dev. 1996;10:1930–1944. doi: 10.1101/gad.10.15.1930. [DOI] [PubMed] [Google Scholar]
- 18.Grilli M, Chiu J J, Lenardo M J. NF-κB and Rel: participants in a multiform transcriptional regulatory system. Int Rev Cytol. 1993;143:1–62. doi: 10.1016/s0074-7696(08)61873-2. [DOI] [PubMed] [Google Scholar]
- 19.Grossman S R, Johannsen E, Tong X, Yalamanchili R, Kieff E. The Epstein-Barr virus nuclear antigen 2 transactivator is directed to response elements by the Jκ recombination signal binding protein. Proc Natl Acad Sci USA. 1994;91:7568–7572. doi: 10.1073/pnas.91.16.7568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guan E, Wang J, Laborda J, Norcross M, Baeuerle P A, Hoffman T. T cell leukemia-associated human Notch/translocation-associated Notch homologue has IκB-like activity and physically interacts with nuclear factor-κB proteins in T cells. J Exp Med. 1996;183:2025–2032. doi: 10.1084/jem.183.5.2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hamaguchi Y, Yamamoto Y, Iwanari H, Maruyama S, Furukawa T, Matsunami N, Honjo T. Biochemical and immunological characterization of the DNA binding protein (RBP-Jκ) to mouse Jκ recombination signal sequence. J Biochem. 1992;112:314–320. doi: 10.1093/oxfordjournals.jbchem.a123898. [DOI] [PubMed] [Google Scholar]
- 22.Henkel T, Ling P D, Hayward S D, Peterson M G. Mediation of Epstein-Barr virus EBNA2 transactivation by recombination signal-binding protein Jκ. Science. 1994;265:92–95. doi: 10.1126/science.8016657. [DOI] [PubMed] [Google Scholar]
- 23.Hohmann H-P, Remy R, Scheidereit C, van Loon A P G M. Maintenance of NF-κB activity is dependent on protein synthesis and the continuous presence of external stimuli. Mol Cell Biol. 1991;11:259–266. doi: 10.1128/mcb.11.1.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hsieh J J, Hayward S D. Masking of the CBF1/RBPJκ transcriptional repression domain by Epstein-Barr virus EBNA2. Science. 1995;268:560–563. doi: 10.1126/science.7725102. [DOI] [PubMed] [Google Scholar]
- 25.Hsieh J J-D, Henkel T, Salmon P, Robey E, Peterson M G, Hayward S D. Truncated mammalian Notch1 activates CBF1/RBPJκ-repressed genes by a mechanism resembling that of Epstein-Barr virus EBNA2. Mol Cell Biol. 1996;16:952–959. doi: 10.1128/mcb.16.3.952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hsieh J J-D, Nofziger D E, Weinmaster G, Hayward S D. Epstein-Barr virus immortalization: Notch2 interacts with CBF1 and blocks differentiation. J Virol. 1997;71:1938–1945. doi: 10.1128/jvi.71.3.1938-1945.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Israel A, Yano O, Logeat F, Kieran M, Kourilsky P. Two purified factors bind to the same sequence in the enhancer of mouse MHC class I genes: one of them is a positive regulator induced upon differentiation of teratocarcinoma cells. Nucleic Acids Res. 1989;17:5245–5257. doi: 10.1093/nar/17.13.5245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ito C Y, Kazantsev A G, Baldwin A S. Three NF-κB sites in the IκB-α promoter are required for induction of gene expression by TNFα. Nucleic Acids Res. 1994;22:3787–3792. doi: 10.1093/nar/22.18.3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jarriault S, Brou C, Logeat F, Schroeter E H, Kopan R, Israel A. Signalling downstream of activated mammalian Notch. Nature (London) 1995;377:355–358. doi: 10.1038/377355a0. [DOI] [PubMed] [Google Scholar]
- 30.Kannabiran C, Zeng X, Vales L D. The mammalian transcriptional repressor RBP (CBF1) regulates interleukin-6 gene expression. Mol Cell Biol. 1997;17:1–9. doi: 10.1128/mcb.17.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kato H, Sakai T, Tamura K, Minoguchi S, Shirayoshi Y, Hamada Y, Tsujimoto Y, Honjo T. Functional conservation of mouse Notch receptor family members. FEBS Lett. 1996;395:221–224. doi: 10.1016/0014-5793(96)01046-0. [DOI] [PubMed] [Google Scholar]
- 32.Kopan R, Nye J S, Weintraub H. The intracellular domain of mouse Notch: a constitutively activated repressor of myogenesis directed at the basic helix-loop-helix region of MyoD. Development. 1994;120:2385–2396. doi: 10.1242/dev.120.9.2385. [DOI] [PubMed] [Google Scholar]
- 33.Lehming N, Thanos D, Brickman J M, Ma J, Maniatis T, Ptashne M. An HMG-like protein that can switch a transcriptional activator to a repressor. Nature (London) 1994;371:175–179. doi: 10.1038/371175a0. [DOI] [PubMed] [Google Scholar]
- 34.Liptay S, Schmid R M, Nabel E G, Nabel G J. Transcriptional regulation of NF-κB2: evidence for κB-mediated positive and negative autoregulation. Mol Cell Biol. 1994;14:7695–7703. doi: 10.1128/mcb.14.12.7695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liptay S, Schmid R M, Perkins N D, Meltzer P, Altherr M R, McPherson J D, Wasmuth J, Nabel G J. Related subunits of NF-κB map to two distinct loci associated with translocations in leukemia, NFKB1 and NFKB2. Genomics. 1992;13:287–292. doi: 10.1016/0888-7543(92)90244-m. [DOI] [PubMed] [Google Scholar]
- 36.Liu Z G, Hsu H, Goeddel D V, Karin M. Dissection of TNF receptor 1 effector functions: JNK activation is not linked to apoptosis while NF-κB activation prevents cell death. Cell. 1996;87:565–576. doi: 10.1016/s0092-8674(00)81375-6. [DOI] [PubMed] [Google Scholar]
- 37.Lu S-Y, Rodriguez M, Liao W S-L. YY1 represses rat serum amyloid A1 gene transcription and is antagonized by NF-κB during acute-phase response. Mol Cell Biol. 1994;14:6253–6263. doi: 10.1128/mcb.14.9.6253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Matsunami N, Hamaguchi Y, Yamamoto Y, Kuze K, Kangawa K, Matsuo H, Kawaichi M, Honjo T. A protein binding to the Jκ recombination sequence of immunoglobulin genes contains a sequence related to the integrase motif. Nature (London) 1989;342:934–937. doi: 10.1038/342934a0. [DOI] [PubMed] [Google Scholar]
- 39.Mercurio F, DiDonato J A, Rosette C, Karin M. p105 and p98 precursor proteins play an active role in NF-κB-mediated signal transduction. Genes Dev. 1993;7:705–718. doi: 10.1101/gad.7.4.705. [DOI] [PubMed] [Google Scholar]
- 40.Milner L A, Bigas A, Kopan R, Brashem-Stein C, Bernstein I D, Martin D I. Inhibition of granulocytic differentiation by mNotch1. Proc Natl Acad Sci USA. 1996;93:13014–13019. doi: 10.1073/pnas.93.23.13014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nye J S, Kopan R, Axel R. An activated Notch suppresses neurogenesis and myogenesis but not gliogenesis in mammalian cells. Development. 1994;120:2421–2430. doi: 10.1242/dev.120.9.2421. [DOI] [PubMed] [Google Scholar]
- 42.Oliveira I C, Mukaida N, Matsushima K, Vilček J. Transcriptional inhibition of the interleukin-8 gene by interferon is mediated by the NF-κB site. Mol Cell Biol. 1994;14:5300–5308. doi: 10.1128/mcb.14.8.5300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Perkins N D, Schmid R M, Duckett C S, Leung K, Rice N R, Nabel G J. Distinct combinations of NF-κB subunits determine the specificity of transcriptional activation. Proc Natl Acad Sci USA. 1992;89:1529–1533. doi: 10.1073/pnas.89.5.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Plaisance S, Vanden Berghe W, Boone E, Fiers W, Haegeman G. Recombination signal sequence binding protein Jκ is constitutively bound to the NF-κB site of the interleukin-6 promoter and acts as a negative regulatory factor. Mol Cell Biol. 1997;17:3733–3743. doi: 10.1128/mcb.17.7.3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Robey E, Chang D, Itano A, Cado D, Alexander H, Lans D, Weinmaster G, Salmon P. An activated form of Notch influences the choice between CD4 and CD8 T cell lineages. Cell. 1996;87:483–492. doi: 10.1016/s0092-8674(00)81368-9. [DOI] [PubMed] [Google Scholar]
- 46.Sakai T, Furukawa T, Iwanari H, Oka C, Nakano T, Kawaichi M, Honjo T. Loss of immunostaining of the RBP-Jκ transcription factor upon F9 cell differentiation induced by retinoic acid. J Biochem. 1995;118:621–628. doi: 10.1093/oxfordjournals.jbchem.a124955. [DOI] [PubMed] [Google Scholar]
- 47.Sasai Y, Kageyama R, Tagawa Y, Shigemoto R, Nakanishi S. Two mammalian helix-loop-helix factors structurally related to Drosophila hairy and Enhancer of split. Genes Dev. 1992;6:2620–2634. doi: 10.1101/gad.6.12b.2620. [DOI] [PubMed] [Google Scholar]
- 48.Schmid R M, Liptay S, Betts J C, Nabel G J. Structural and functional analysis of NF-κB. Determinants of DNA binding specificity and protein interaction. J Biol Chem. 1994;269:32162–32167. [PubMed] [Google Scholar]
- 49.Schmid R M, Perkins N D, Duckett C S, Andrews P C, Nabel G J. Cloning of an NF-κB subunit which stimulates HIV transcription in synergy with p65. Nature (London) 1991;352:733–736. doi: 10.1038/352733a0. [DOI] [PubMed] [Google Scholar]
- 50.Schweisguth F, Posakony J W. Suppressor of Hairless, the Drosophila homolog of the mouse recombination signal-binding protein gene, controls sensory organ cell fates. Cell. 1992;69:1199–1212. doi: 10.1016/0092-8674(92)90641-o. [DOI] [PubMed] [Google Scholar]
- 51.Shirakata Y, Shuman J D, Coligan J E. Purification of a novel MHC class I element binding activity from thymus nuclear extracts reveals that thymic RBP-Jκ/CBF1 binds to NF-κB-like elements. J Immunol. 1996;156:4672–4679. [PubMed] [Google Scholar]
- 52.Tamura K, Taniguchi Y, Minoguchi S, Sakai T, Tun T, Furukawa T, Honjo T. Physical interaction between a novel domain of the receptor Notch and the transcription factor RBP-Jκ/Su(H) Curr Biol. 1995;5:1416–1423. doi: 10.1016/s0960-9822(95)00279-x. [DOI] [PubMed] [Google Scholar]
- 53.Ten R M, Paya C V, Israel N, Le Bail O, Mattei M G, Virelizier J L, Kourilsky P, Israel A. The characterization of the promoter of the gene encoding the p50 subunit of NF-κB indicates that it participates in its own regulation. EMBO J. 1992;11:195–203. doi: 10.1002/j.1460-2075.1992.tb05042.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Thanos D, Maniatis T. NF-κB: a lesson in family values. Cell. 1995;80:529–532. doi: 10.1016/0092-8674(95)90506-5. [DOI] [PubMed] [Google Scholar]
- 55.Tun T, Hamaguchi Y, Matsunami N, Furukawa T, Honjo T, Kawaichi M. Recognition sequence of a highly conserved DNA binding protein, RBP-Jκ. Nucleic Acids Res. 1994;22:965–971. doi: 10.1093/nar/22.6.965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Van Antwerp D J, Martin S J, Kafri T, Green D R, Verma I M. Suppression of TNF-α-induced apoptosis by NF-κB. Science. 1996;274:787–789. doi: 10.1126/science.274.5288.787. [DOI] [PubMed] [Google Scholar]
- 57.Verma I M, Stevenson J K, Schwarz E M, Van Antwerp D, Miyamoto S. Rel/NF-κB/IκB family: intimate tales of association and dissociation. Genes Dev. 1995;9:2723–2735. doi: 10.1101/gad.9.22.2723. [DOI] [PubMed] [Google Scholar]
- 58.Waltzer L, Bourillot P Y, Sergeant A, Manet E. RBP-Jκ repression activity is mediated by a co-repressor and antagonized by the Epstein-Barr virus transcription factor EBNA2. Nucleic Acids Res. 1995;23:4939–4945. doi: 10.1093/nar/23.24.4939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Waltzer L, Logeat F, Brou C, Israel A, Sergeant A, Manet E. The human Jκ recombination signal sequence binding protein (RBP-Jκ) targets the Epstein-Barr virus EBNA2 protein to its DNA responsive elements. EMBO J. 1994;13:5633–5638. doi: 10.1002/j.1460-2075.1994.tb06901.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang C Y, Mayo M W, Baldwin A S., Jr TNF- and cancer therapy-induced apoptosis: potentiation by inhibition of NF-κB. Science. 1996;274:784–787. doi: 10.1126/science.274.5288.784. [DOI] [PubMed] [Google Scholar]
- 61.Washburn T, Schweighoffer E, Gridley T, Chang D, Fowlkes B J, Cado D, Robey E. Notch activity influences the αβ versus γδ T cell lineage decision. Cell. 1997;88:833–843. doi: 10.1016/s0092-8674(00)81929-7. [DOI] [PubMed] [Google Scholar]
- 62.Wu M, Lee H, Bellas R E, Schauer S L, Asura M, Katz D, Fitzgerald M J, Rothstein T L, Sherr D H, Sonenshein G E. Inhibition of NF-κB/Rel induces apoptosis of murine B cells. EMBO J. 1996;15:4682–4690. [PMC free article] [PubMed] [Google Scholar]
- 63.Zagouras P, Stifani S, Blaumueller C M, Carcangiu M L, Artavanis-Tsakonas S. Alterations in Notch signaling in neoplastic lesions of the human cervix. Proc Natl Acad Sci USA. 1995;92:6414–6418. doi: 10.1073/pnas.92.14.6414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zimber-Strobl U, Strobl L J, Meitinger C, Hinrichs R, Sakai T, Furukawa T, Honjo T, Bornkamm G W. Epstein-Barr virus nuclear antigen 2 exerts its transactivating function through interaction with recombination signal binding protein RBP-Jκ, the homologue of Drosophila Suppressor of Hairless. EMBO J. 1994;13:4973–4982. doi: 10.1002/j.1460-2075.1994.tb06824.x. [DOI] [PMC free article] [PubMed] [Google Scholar]