

Abstract
The SH2 domain-containing SHP-1 tyrosine phosphatase has been shown to negatively regulate a broad spectrum of growth factor- and cytokine-driven mitogenic signaling pathways. Included among these is the cascade of intracellular events evoked by stem cell factor binding to c-Kit, a tyrosine kinase receptor which associates with and is dephosphorylated by SHP-1. Using a series of glutathione S-transferase (GST) fusion proteins containing either tyrosine-phosphorylated segments of the c-Kit cytosolic region or the SH2 domains of SHP-1, we have shown that SHP-1 interacts with c-Kit by binding selectively to the phosphorylated c-Kit juxtamembrane region and that the association of c-Kit with the larger of the two SHP-1 isoforms may be mediated through either the N-terminal or C-terminal SHP-1 SH2 domain. The results of binding assays with mutagenized GST-Kit juxtamembrane fusion proteins and competitive inhibition assays with phosphopeptides encompassing each c-Kit juxtamembrane region identified the tyrosine residue at position 569 as the major site for binding of SHP-1 to c-Kit and suggested that tyrosine 567 contributes to, but is not required for, this interaction. By analysis of Ba/F3 cells retrovirally transduced to express c-Kit receptors, phenylalanine substitution of c-Kit tyrosine residue 569 was shown to be associated with disruption of c-Kit–SHP-1 binding and induction of hyperproliferative responses to stem cell factor. Although phenylalanine substitution of c-Kit tyrosine residue 567 in the Ba/F3–c-Kit cells did not alter SHP-1 binding to c-Kit, the capacity of a second c-Kit-binding tyrosine phosphatase, SHP-2, to associate with c-Kit was markedly reduced, and the cells again showed hyperproliferative responses to stem cell factor. These data therefore identify SHP-1 binding to tyrosine 569 on c-Kit as an interaction pivotal to SHP-1 inhibitory effects on c-Kit signaling, but they indicate as well that cytosolic protein tyrosine phosphatases other than SHP-1 may also negatively regulate the coupling of c-Kit engagement to proliferation.
The pivotal role of the SH2 domain-containing SHP-1 (PTP1C, HCP, or SHPTP1) tyrosine phosphatase in the regulation of hemopoietic cell growth and development is now well recognized (1). In contrast to the structurally similar, ubiquitously expressed SHP-2 (Syp or PTP1D) tyrosine phosphatase and its Drosophila csw homolog (10, 37), SHP-1 appears to exert primarily inhibitory effects on the signaling cascades in which it participates (34). SHP-1 has been shown, for example, to suppress the growth-promoting properties of the activated interleukin 3 (IL-3), erythropoietin, and colony-stimulating factor 1 receptors, an effect mediated either directly by receptor dephosphorylation or indirectly by dephosphorylation of receptor-associated protein tyrosine kinases (PTKs) (4, 19, 55, 56). SHP-1 has also been implicated in downregulation of the signaling pathways evoked by engagement of the B- and T-lymphocyte antigen receptors (5, 32, 33), antigen receptor comodulators such as CD22, FcγRIIB, and CD5, and cytosolic signaling molecules such as Vav and Grb2/Sos1 which are involved in Ras activation (6, 8, 20). The capacity of SHP-1 to negatively modulate this broad spectrum of signaling effectors is consistent with the enormous overexpansion of multiple hemopoietic cell populations manifested by motheaten (me) and viable motheaten (mev) mice, animals now known to be homozygous for loss-of-function mutations in the SHP-1 gene (21, 46). The presence of two SH2 domains in SHP-1, as well as the possibility for altering its C-terminal SH2 domain by alternative splicing of a 39-amino-acid segment (46), provides a structural explanation for the diverse range of molecular interactions in which this phosphotyrosine phosphatase (PTP) appears to participate. Thus, while the precise structural basis for and physiologic effects of SHP-1 interactions with the molecules with which it has been shown to associate require further investigation, the current data concerning SHP-1 functions identify this PTP as a critical player in the modulation of hemopoietic cell growth and function.
In addition to the regulation of cell proliferation, SHP-1 has also been implicated in the control of signaling cascades coupling growth factor receptors to hemopoietic cell differentiation. This role for SHP-1 has become particularly well appreciated in the context of data derived from studies of SHP-1 interactions with the transmembrane PTK receptor encoded by the c-Kit proto-oncogene. The latter receptor subserves pivotal functions in promoting the development, survival, and proliferation of hemopoietic stem cells, neural crest-derived cells, and germ cells, a role well illustrated by the depletion of erythroid precursors and mast cells and associated macrocytic anemia, sterility, and hypopigmentation manifested by c-Kit-deficient mice bearing the W mutation (7, 9, 11, 29, 31). The signaling events induced by c-Kit engagement to its cognate ligand, stem cell factor (SCF), have been extensively investigated and shown to involve the initial induction of c-Kit autophosphorylation and consequent association of the activated receptor with phosphatidylinositol 3-kinase, phospholipase Cγ-1, megakaryocyte-associated tyrosine kinase (MATK), and a number of other cytosolic signaling effectors that act in the downstream delivery of the ligand-binding signal (16, 24, 40–42). While these data primarily identify biochemical events which promote c-Kit signaling, the demonstration that SHP-1 not only associates with but also tyrosine dephosphorylates activated c-Kit receptors strongly suggests that c-Kit represents another growth factor receptor subject to negative regulation by SHP-1 (54). This contention is supported by recent data showing that bone marrow progenitor cells from me mice hyperproliferate in response to SCF as well as the finding that defects in c-Kit signaling associated with the Wv mutation are ameliorated in Wv mice carrying the me or mev mutation (25, 35). Together, these data strongly suggest that SHP-1 interaction with c-Kit is relevant to the modulation of c-Kit functions in vivo. Furthermore, in view of the oncogenic potential putatively conferred by alterations in c-Kit activity (12, 15, 18, 22, 47, 48) as well as data revealing lymphoma frequency to be increased in mice heterozygous for the me or mev mutation (1) and thereby implying a tumor suppressor role for SHP-1, the inhibition of c-Kit signaling by SHP-1 may also represent a molecular event relevant to hemopoietic cell transformation.
Although the cumulative data indicate a significant role for SHP-1 in modulating c-Kit signaling capacity, the structural basis for SHP-1 interaction with c-Kit is not well defined. Thus, although this interaction has been ascribed to the binding of the SHP-1 N-terminal and, to a lesser extent, C-terminal SH2 domains to a phosphorylated tyrosine site(s) on c-Kit (54), the precise phosphotyrosine residues within c-Kit which mediate this association are unknown. Accordingly, to further elucidate the molecular basis for SHP-1 effects on c-Kit, we have used a series of glutathione S-transferase (GST) fusion proteins that span the tyrosine-containing segments of the c-Kit cytosolic domain to investigate the site on the c-Kit receptor with which SHP-1 associates. Analysis of SHP-1 interactions with phosphorylated forms of these fusion proteins as well as with phosphorylated peptides encompassing the phosphotyrosine sites within selected regions of c-Kit has allowed the identification of Tyr569 within the c-Kit juxtamembrane region as the binding site for SHP-1. SHP-1 association with this single phosphotyrosine residue was confirmed biochemically, by data from competitive binding studies using wild-type tyrosine-phosphorylated as well as tyrosine→phenylalanine-mutated peptides. By contrast, the structurally similar SHP-2 tyrosine phosphatase, which also associates with activated c-Kit, was found to bind selectively to Tyr567, the tyrosine residue immediately upstream of Tyr569. Importantly, phenylalanine substitution of tyrosine 569 or 567 not only disrupted c-Kit interactions with SHP-1 and SHP-2, respectively, but also was associated with expression of hyperproliferative responses to SCF stimulation. These findings therefore confirm the capacity of SHP-1 to downregulate c-Kit signaling and indicate that the inhibitory effect of SHP-1 on c-Kit is realized through SHP-1 binding with tyrosine 569 in the c-Kit juxtamembrane domain. The data also implicate SHP-2 binding to tyrosine residue 567 in the downregulation of c-Kit signaling and thus suggest that the physiologic outcome of c-Kit receptor engagement can be tempered by the negative influence of each of these PTPs.
MATERIALS AND METHODS
Reagents.
Polyclonal anti-SHP-1 antibodies were generated in rabbits immunized with GST–SHP-1 SH2 domain fusion proteins as previously described (21). The polyclonal anti-c-Kit antibody was purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, Calif.), monoclonal antiphosphotyrosine 4G10 and anti-GST antibodies were obtained from Upstate Biotechnology Inc. (Lake Placid, N.Y.), and polyclonal anti-SHP-2 antibodies (generated against SHP-2 SH2 fusion proteins) were kindly provided by Gen-Sheng Feng (Indiana University Medical Center) (10). Murine recombinant SCF was obtained from Genzyme (Cambridge, Mass.), recombinant human SCF and human and murine IL-3 were purchased from Life Technologies (Burlington, Ontario, Canada), and concanavalin A (ConA) was obtained from Sigma Chemical Co. (St. Louis, Mo.). Protein A-Sepharose, glutathione S-Sepharose, and NHS-Sepharose were purchased from Pharmacia (Baie d’Urfé, Quebec, Canada). All other chemicals used for immunoprecipitation and immunoblotting analyses were obtained from Sigma.
Cells and cell culture.
The Mo7e megakaryocytic and FMA3 mastocytoma cell lines were kindly provided by L. Pegorraro (University di Torino, Turin, Italy) and T. Tsujimura (Osaka University, Osaka, Japan), respectively. The HEL human erythroleukemia and EL4 thymoma cell lines were obtained from the American Type Culture Collection. All cells were maintained in opti-MEM (Life Technologies) medium supplemented with 100 μg of penicillin-streptomycin per ml and 10% fetal calf serum (FCS). The growth factor-dependent Mo7e cells were also grown in opti-MEM supplemented with 100 μg of penicillin streptomycin per ml, 10% FCS, and 50 ng of recombinant human IL-3 per ml. For stimulation of these lines, cells were cultured for 16 h in medium lacking FCS and IL-3 and then treated for 10 min in the presence of 20 μg of ConA per ml (EL4 cells) or for 5 min with 100 ng of SCF per ml (all other cell lines). Ba/F3 cells expressing mutant and wild-type forms of c-Kit were derived as follows. Tyr567→Phe-mutated forms of the c-Kit cDNA were first generated by PCR mutagenesis (14), and these and the wild-type c-Kit cDNA were then cloned into the LXSN retrovirus (26). Retroviral plasmids were calcium phosphate transfected into the retroviral packaging cell line BOSC-23 (36), and supernatants containing greater than 106 infectious units/ml were recovered 48 h after retroviral transfection and used to infect the IL-3-dependent pro-B-cell line Ba/F3. At 72 h following retroviral infection, infected Ba/F3 cell populations were stained with the c-Kit-specific monoclonal antibody ACK2 (28) and sorted for equivalent c-Kit expression by fluorescence-activated cell sorting (FACS) (FACstar; Becton Dickinson). For proliferation assays, 104 Ba/F3 c-Kit-transfected cells were stored overnight in RPMI plus 0.5% FCS, suspended at a density of 2.5 × 105 cells/ml in culture medium, and cultured in 0.2-ml microtiter plates in the presence of various concentrations of SCF (0 to 400 ng/ml). After 48 h of culture in SCF, the cells were pulsed for 6 h with 1 mCi of [3H]thymidine (Dupont, Wilmington, Del.). Cells were then harvested onto microplate filters, and radioactivity was measured by scintillation counting (Top Count; Canberra, Downers Grove, Ill.). Data are presented as the means ± standard deviations (SD) of counts per minute. Alternatively, Ba/F3 cells and Ba/F3 c-Kit-transfected cells were suspended in opti-MEM supplemented with 10% FCS and G418 (for c-Kit transfectants) and plated at 104 cells/well in 96-well microtiter plates. IL-3 at a final concentration of 5 ng/100 μl was added on day 0 and every other day thereafter, and cell proliferation was evaluated every 48 h by a Cell Titer 96 nonradioactive assay (Promega, Madison, Wis.) based on conversion of a tetrazolium salt to formazen. Proliferation was quantitated by enzyme-linked immunosorbent assay at 570 nm with a microtiter cell reader (Thermomax; Molecular Devices).
Generation of GST–c-Kit proteins.
GST–c-Kit fusion proteins were generated by subcloning PCR-amplified murine c-Kit sequences into pGEX-2TK (Pharmacia). The amplified fragments (see Fig. 3A) subcloned into this expression plasmid included the c-Kit juxtamembrane region (residues 545 to 571), the kinase insert (residues 684 to 761), and the C-tail region (residues 917 to 975). The first methionine codon was designated residue 1. Point mutations for tyrosine conversion to phenylalanine, tyrosine deletion, or other amino acid substitutions were generated by overlap extension with PCR and oligonucleotide primers encoding the desired mutation.
FIG. 3.

Association of SHP-1 with the c-Kit juxtamembrane region. (A) Top, schematic diagram showing domain structure of the c-Kit cytosolic region, including the juxtamembrane (JUX), kinase insert (KI), and carboxy-tail (C-TAIL) domains and two kinase domains. Numbers indicate the positions of the amino acid residues bordering each domain. TM, transmembrane. Bottom, pGEX2-T–Kit constructs encoding GST–c-Kit–JUX, –KI, or –C-TAIL fusion proteins were used for transformation into TKX1 cells, and the bacterial cells were treated sequentially with IPTG and IAA. Tyrosine-phosphorylated GST fusion proteins were then purified by affinity chromatography with glutathione-Sepharose beads, fractionated by SDS-PAGE, and visualized by Coomassie blue staining (left) or anti-pTyr immunoblotting analysis (right). (B) Top, cell lysates (1,800 ng) prepared from ConA (20 μg/ml)-treated EL4 cells were either immunoprecipitated with anti-SHP-1 antibody (IP: SHP-1) or incubated for 2 h with 5 μg of tyrosine-phosphorylated GST or GST-JUX, -KI or –C-TAIL fusion proteins immobilized on glutathione-Sepharose beads. Complexes and lysate protein alone were fractionated by SDS-PAGE and subjected to immunoblotting analysis with anti-SHP-1 antibody. Bottom, cell lysates (1,800 μg) prepared from ConA-treated EL4 cells were incubated with various amounts (3, 6, 9, or 12 μl) of glutathione-Sepharose–GST-JUX fusion protein, and the complexes were then subjected to SDS-PAGE and anti-SHP-1 immunoblotting analysis. Mobilities of molecular mass (MW) standards are shown on the right, and the position of SHP-1 is indicated on the left.
To generate GST fusion proteins containing the SHP-1 SH2 domains, the N-terminal (SH2-N, residues 1 to 100), C-terminal (SH2-C, residues 109 to 205), and N- plus C-terminal (SH2-NC, residues 1 to 213) SH2 domains of SHP-1 were PCR amplified from the murine SHP-1 cDNA (the methionine encoded by the first ATG codon is considered residue 1). To generate a GST fusion protein representing the SHP-1 C-terminal SH2 domain splice variant (SH2-C′), cDNA was prepared from 500 ng of RNA isolated from murine splenocytes by using an RNA extraction kit from Qiagen (Chatsworth, Calif.) and subjected to PCR amplification with a primer pair corresponding to SHP-1 nucleotides 348 to 362 and 613 to 628, which encompass the SH2-C′ region, together with a Tth7 DNA polymerase kit (Boehringer Mannheim) and reaction conditions of 1 min at 94°C, 1 min at 65°C, and 1 min at 72°C for 35 cycles. Following BamHI and EcoRI digestion, the reverse transcription-PCR product was subcloned into pGEX-2T, and the insert sequence was confirmed by direct sequence analysis.
Bacterial cells transformed by pGEX2T-SH2 expression plasmids were induced with 2 mM isopropyl-1-thio-β-d-galactopyranoside (IPTG), and the fusion proteins were purified with glutathione-conjugated Sepharose beads (Pharmacia). To derive tyrosine-phosphorylated GST–c-Kit fusion proteins, the various pGEX–c-Kit constructs were transformed into TKX1 (Pharmacia), and the tyrosine-phosphorylated fusion proteins were purified from bacterial cells following sequential incubations with IPTG and indole acetic acid (IAA) (to induce the expression of the tyrosine kinase gene cloned into TKX1 bacteria). Fusion protein tyrosine phosphorylation was confirmed by immunoblotting with 4G10 monoclonal antiphosphotyrosine antibodies (Upstate Biotechnology).
Immunoprecipitation and Western immunoblotting.
Protein lysates were prepared by resuspending 108 resting or SCF (100 ng/ml)-treated HEL, Mo7e, FMA3, EL4, and Ba/F3-Kit cells in 1 ml of lysis buffer (phosphate-buffered saline containing 1% Triton X-100, 1% Tween 20, 1 mM sodium orthovanadate, 1 μg of leupeptin per ml, 1 μg of aprotinin per ml, and 0.001 mM dithiothreitol) (21). Lysate proteins (500 μg) were electrophoresed through sodium dodecyl sulfate (SDS)–8 or 10% polyacrylamide gels and electroblotted onto nitrocellulose, and the blots were then incubated overnight at 4°C in 10 mM Tris (pH 8.0)–150 mM NaCl–0.05% Tween 20 (TBST) containing 5% skim milk. Proteins were detected by incubating blots for 2 h at room temperature with primary antibodies in TBST followed by 125I-protein A (Dupont). Blots were then washed with TBST and exposed to Kodak XAR film at −70°C. For immunoprecipitations, cell lysates were clarified by centrifugation for 20 min at 10,000 × g at 4°C, and 1,800 μg of cell lysate protein was incubated for 2 h at 4°C with selected antibodies and then with 100 μl of protein A-Sepharose (Pharmacia) for 30 min at 4°C. The immune complexes were collected by centrifugation, washed three times with lysis buffer, boiled for 5 min in SDS sample buffer, and then subjected to electrophoresis and immunoblotting as described above.
In vitro binding assays.
To evaluate SHP-1 or c-Kit binding to GST–c-Kit or GST-SHP1 fusion proteins, protein lysates prepared from 1,800 μg of EL4 or SCF-treated HEL cells were incubated at 4°C for 2 h with 5 μg of fusion protein immobilized on glutathione-Sepharose beads. After several washes in lysis buffer, complexes were resuspended in sample buffer, boiled, and analyzed by SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and immunoblotting with either anti-SHP-1 or anti-c-Kit antibodies and 125I-protein A.
Peptide competition.
The following peptides encompassing tyrosine sites within the juxtamembrane region of c-Kit were synthesized in the tyrosine-phosphorylated and unphosphorylated states (Ottawa Regional Cancer Center): peptide 1 (VLTpYKpYLQKPMK) and peptide 2 (VLTpYKYLQKPMK), which correspond to residues 541 to 552; peptide 3 (KPMpYEVQWKVVE), representing residues 549 to 560; and peptide 4 (GNNpYVpYIDPTQK), peptide 5 (GNNpYVYIDPTQK), and peptide 6 (GNNYVpYIDPTQK), which encompass residues 564 to 575 of the c-Kit juxtamembranous region. For competition studies, EL4 cell lysates (1,800 μg) were incubated with 5 μg of GST–c-Kit-juxtamembrane fusion protein in the presence of either 10 μM each peptide, various amounts of peptide 4, or 10 μM peptide 4 preincubated with antiphosphotyrosine antibodies. Protein complexes were washed four times and then subjected to SDS-PAGE followed by immunoblotting with anti-SHP-1 antibody.
Direct binding of SHP-1 to synthetic phosphopeptides.
Synthetic phosphopeptides 3 and 4 were coupled to NHS-Sepharose beads (Pharmacia) as recommended by the manufacturer, and the immobilized peptides were then individually mixed with EL4 cell lysate. Following four washes, the proteins were resolved by SDS-PAGE and blotted with anti-SHP-1 antibodies.
RESULTS
SHP-1 binding to the c-Kit receptor is enhanced following SCF stimulation.
To extend previous data indicating the capacity of SHP-1 to associate with c-Kit in SCF-treated Mo7e cells (54), binding of SHP-1 to c-Kit was investigated in four cell lines known to express high levels of c-Kit receptor and to proliferate in response to SCF stimulation. These included Mo7e cells, HEL cells, Ba/F3 pro-B cells engineered to express c-Kit receptors (Ba/F3-Kit), and FMA3 cells (a cell line which contains a constitutively active form of c-Kit). As shown in Fig. 1A, anti-SHP-1 immunoblotting analysis of anti-c-Kit immunoprecipitates derived from each of the four lines under study revealed coimmunoprecipitation of SHP-1 with c-Kit in both resting and stimulated cells and confirmed previous data suggesting that the association of these molecules is increased in conjunction with SCF stimulation (Fig. 1A). The c-Kit protein was also detected in anti-SHP-1 immunoprecipitates from Mo7e and HEL cells, and again the interaction of these molecules was increased following SCF treatment (Fig. 1B). To determine whether c-Kit engagement in these cells is associated with tyrosine phosphorylation of not only c-Kit but also SHP-1, these proteins were individually immunoprecipitated from resting and SCF-stimulated Mo7e and HEL cells, and their phosphorylation status was assessed by antiphosphotyrosine immunoblotting analysis. As shown in Fig. 1C and D, respectively, SHP-1 and c-Kit are both tyrosine phosphorylated at low levels constitutively, an observation which may reflect a relatively high level of basal tyrosine phosphorylation in the lines under study. However, the phosphorylation status of each protein was also found to be markedly enhanced following Kit engagement by SCF. These findings suggest that SHP-1 associates with the c-Kit receptor and that this interaction is increased by receptor activation and tyrosine phosphorylation.
FIG. 1.

Increases in association and tyrosine phosphorylation of SHP-1 and c-Kit following SCF stimulation. Cell lysates were prepared from unstimulated (−) and SCF (100 ng/ml)-treated Mo7e, FMA3, and HEL cells and from Ba/F3 cells stably expressing the full-length wild-type c-Kit cDNA (Ba/F3-Kit). (A) Protein lysates (1,800 μg) prepared from unstimulated or SCF-stimulated Mo7e, Ba/F3-Kit FMA3, and HEL cells were immunoprecipitated (Ip) with anti-c-Kit antibody, and the immune complexes were subjected to SDS-PAGE, immunoblotting with anti-SHP-1 antibody, and visualization with 125I-protein A. Ba/F3 total cell lysate protein (500 μg) was included as a control for SHP-1 expression. (B) Cell lysate proteins (1,800 μg) prepared from unstimulated or SCF-stimulated Mo7e and HEL cells were immunoprecipitated with anti-SHP-1 antibody, and the immunoprecipitated and cell lysate proteins (500 μg) were resolved by SDS-PAGE and immunoblotted with anti-c-Kit antibody. (C) SHP-1 immunoprecipitates prepared from unstimulated or SCF-treated Mo7e and HEL cells were analyzed by SDS-PAGE and immunoblotting with antiphosphotyrosine antibody. (D) Cell lysate proteins (500 μg) as well as c-Kit immunoprecipitates from unstimulated or SCF-stimulated HEL cells (1,800 μg of lysate protein) were analyzed by SDS-PAGE and antiphosphotyrosine immunoblotting. For each panel, mobilities of molecular mass (MW) standards are shown on the right and positions of SHP-1 and c-Kit are indicated by arrows.
SHP-1–c-Kit interaction involves association of the 71-kDa SHP-1 isoform with the 145-kDa c-Kit isoform.
Previous studies of SHP-1 expression have revealed the existence of two SHP-1 species of ≈67 and 71 kDa, which are differentially expressed among various hemopoietic and epithelial cell types (21, 49). Although both of these SHP-1 species appear to be expressed in the cell lines used in this study, c-Kit immunoprecipitates from these cells appeared to contain primarily the high-molecular-weight species (Fig. 1A). This species, which is generated by alternative splicing of the SHP-1 gene, has been shown to be distinguished from the smaller SHP-1 isoform by the presence of a 39-amino-acid insert in the SHP-1 C-terminal SH2 domain (46). To investigate the potential role for the 71-kDa SHP-1 species in c-Kit binding, the SHP-1 region encompassing the C-terminal SH2 domain of this splice variant (SH2-C′) was amplified by reverse transcription-PCR from murine splenocytes and subcloned into pGEX-2T, and the capacities of bacterial GST–SH2-C′ fusion proteins as well as GST fusion proteins containing the SHP-1 N-terminal, C-terminal, or N- and C-terminal SH2 domains (Fig. 2A) to bind c-Kit derived from SCF-stimulated HEL cells were then assessed. As shown in Fig. 2B and consistent with previous data concerning the binding of SHP-1 to c-Kit (54), the results of this analysis revealed the interaction of c-Kit with the N-terminal, but not the C-terminal, SHP-1 SH2 domain. However, in contrast to the GST–SH2-C proteins, fusion proteins carrying the larger SH2-C′ C-terminal variant precipitated c-Kit to a degree similar to that achieved with either the GST–N-terminal or GST–N- plus C-terminal SHP-1 SH2 domain fusion proteins. These observations are consistent with the immunoblotting data indicating that c-Kit associates with the 71-kDa SHP-1 isoform and suggest that both the N-terminal and C-terminal variant (SH2-C′) SH2 domains play a role in mediating SHP-1 binding to c-Kit.
FIG. 2.

The Kit receptor preferentially associates with the SHP-1 SH2-N and SH2-C′ domains. (A) Schematic diagram showing the structures of the 71- and 67-kDa splice variants of SHP-1 and the SHP-1 SH2 domain sequences present in the GST–SHP-1 fusion proteins used for in vitro binding assays. The shaded region represents a 39-amino-acid segment present in the C-terminal SH2 domain (SH2-C′) of the 71-kDa, but not the 67-kDa, SHP-1 species. (B and C) Cell lysates (1,800 μg) prepared from 108 SCF-stimulated HEL cells were incubated for 2 h at 4°C with 5 μg of purified GST–SHP-1 fusion protein immobilized on glutathione-Sepharose beads. Complexes as well as lysate alone were fractionated by SDS-PAGE and subjected to immunoblotting with anti-c-Kit (B) or anti-GST (C) antibodies. Mobilities of molecular mass (MW) standards are shown on the right.
SHP-1 associates with the juxtamembrane region of the activated c-Kit receptor.
As an initial step toward defining the phosphotyrosine site(s) on c-Kit that interacts with SHP-1, a series of GST fusion proteins carrying various portions of the c-Kit cytosolic region were derived. As illustrated in Fig. 3A, these fusion proteins contained the c-Kit juxtamembrane (GST-JUX), kinase insert (GST-KI), and C-terminal tail (GST-TAIL) regions, encompassing residues 544 to 574, 685 to 762, and 915 to 975, respectively.
To derive tyrosine-phosphorylated versions of each protein, the relevant recombinant plasmids were transformed into a TKXI bacterial strain expressing tyrosine kinase activity, and their expression and phosphorylation status were then assessed following IPTG and IAA induction (Fig. 3A). The phosphorylated fusion proteins were then individually immobilized on gluthathione-Sepharose beads and incubated with lysates prepared from EL4 thymoma cells, and their binding to SHP-1 was examined by anti-SHP-1 immunoblotting analysis. As shown in Fig. 3B, the results of this analysis revealed the precipitation of SHP-1 by the GST-JUX protein. By contrast, SHP-1 was not precipitated by either the GST-KI or GST-TAIL fusion protein (Fig. 3B), even when the experiment was performed with amounts of the latter two fusion proteins three times greater than the amount of GST-JUX protein required to precipitate SHP-1 (data not shown). The precipitation of SHP-1 by GST-JUX was enhanced by the use of larger amounts of fusion protein and, as is consistent with the contention that c-Kit selectively associates with the 71-kDa SHP-1 isoform, involved only the larger of the two SHP-1 species expressed in EL4 cells (Fig. 3B). These data strongly suggest that SHP-1 interactions with c-Kit are mediated by the interactions of this PTP with the c-Kit juxtamembrane region.
Identification of Tyr569 as the binding site for SHP-1 on c-Kit.
As the 30-amino-acid juxtamembrane region of c-Kit contains five tyrosine residues (located at positions 544, 546, 552, 567, and 569), the specific tyrosine(s) involved in SHP-1 binding to c-Kit was next investigated by examining SHP-1 interactions with GST-JUX fusion proteins in which these tyrosine residues had been individually deleted or phenylalanine substituted. Analysis of tyrosine-phosphorylated versions of these fusion proteins for the capacity to precipitate SHP-1 from EL4 cell lysates revealed GST-JUX binding to SHP-1 to be abrogated by deletion of Tyr569 but unaffected by a Tyr→Phe substitution at position 552 or by deletion of Tyr544 or Tyr567 (Fig. 4). By contrast, SHP-1 was not precipitated by unphosphorylated versions of these GST-JUX proteins. To extend these observations, a series of synthetic phosphotyrosyl peptides representing sequences encompassing the tyrosine-containing regions within the c-Kit juxtamembrane region were assessed for their capacities to compete with phosphorylated GST-JUX fusion protein for SHP-1 binding and thereby interfere with GST-JUX-mediated precipitation of SHP-1 from EL4 lysates. As shown in Fig. 5, peptides containing phosphorylated Tyr544 (peptide 2), Tyr544 and Tyr546 (peptide 1), or Tyr552 (peptide 3) did not interfere with the precipitation of SHP-1 from the stimulated cells by the GST-JUX fusion proteins. By contrast, SHP-1 precipitation by phosphorylated GST-JUX protein was abrogated in the context of competition with phosphopeptide 4, a synthetic peptide containing phosphorylated Tyr567 and Tyr569. Inhibition of GST-JUX binding to SHP-1 by phosphopeptide 4 was found to be dose dependent and was reduced by preincubation of peptide 4 with antiphosphotyrosine antibody (Fig. 5B). Direct binding of SHP-1 to immobilized peptides 3 and 4 was also examined, and as shown in Fig. 5C, the results of this analysis revealed that phosphopeptide 4, but not 3, efficiently bound SHP-1. Because phosphopeptide 4 contains two phosphotyrosine residues (Tyr567 and Tyr569) which might serve as SHP-1 binding sites, an analysis of whether one or both of these residues are involved in coupling c-Kit to SHP-1 was performed by further competition analyses with two phosphopeptides (5 and 6) which contained either one or the other of these two residues in a phosphorylated state. As shown in Fig. 5D, of these two latter phosphopeptides, only peptide 6, in which Tyr569 but not Tyr567 was phosphorylated, inhibited GST-JUX binding to SHP-1. These observations provide strong evidence that SHP-1 binding to the phosphorylated tyrosine at position 569 is responsible for the association of SHP-1 with the c-Kit juxtamembrane region.
FIG. 4.

Deletion of c-Kit Y569 abrogates binding of SHP-1 to the c-Kit juxtamembrane region. Tyrosine-phosphorylated or unphosphorylated GST–c-Kit–JUX fusion proteins containing either wild-type (JUX) or mutated versions of the c-Kit juxtamembrane domain were immobilized on glutathione-Sepharose beads and incubated with cell lysates (1,800 μg) prepared from ConA-treated EL4 cells. Complexes and lysate protein (500 μg) were then resolved by SDS-PAGE and immunoblotted with anti-SHP-1 antibody. Sites of the JUX domain mutations are indicated above the lanes and include deletions of tyrosine residues at position 544, 567, or 569 and, in the left panel only, replacement of Tyr552 with phenylalanine (Y552F). For each panel mobilities of molecular mass (MW) standards are shown on the right, and the position of SHP-1 is indicated on the left.
FIG. 5.
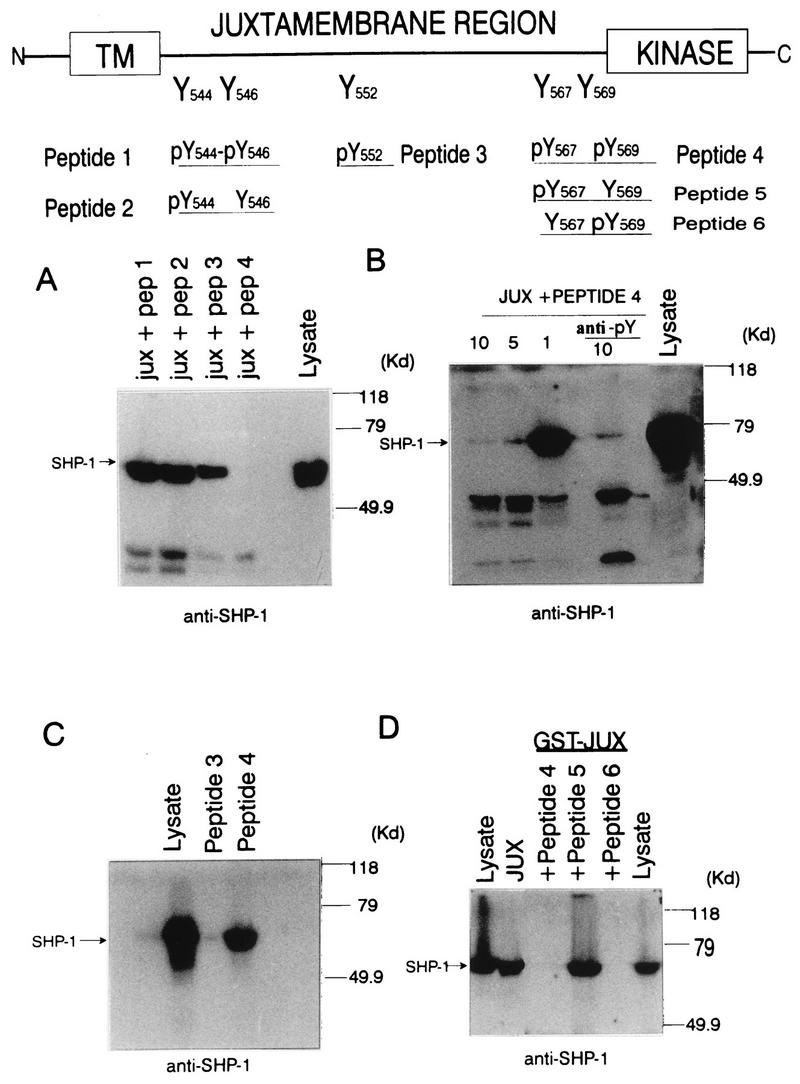
Identification of Tyr569 as the c-Kit binding site for SHP-1. (A) Phosphopeptides (12-mers) spanning the five tyrosine sites contained in the c-Kit juxtamembrane region were synthesized with tyrosines in phosphorylated or unphosphorylated states (upper diagram), and the individual phosphopeptides (10 μM) were then incubated with cell lysates (1,800 μg) from EL4 cells in the presence of 5 μg of glutathione-Sepharose–GST-JUX fusion protein. Complexes were washed four times, and the complexes and lysate protein were then subjected to SDS-PAGE followed by anti-SHP-1 immunoblotting analysis. (B) Lysates prepared from ConA-stimulated EL4 cells were incubated with glutathione-Sepharose–GST-JUX fusion proteins (5 μg) in the presence of various amounts (1, 5, or 10 μM) of phosphopeptide 4 and with 10 μM phosphopeptide 4 preincubated with anti-pTyr antibody (anti-pY). Following washing, complexes and lysate protein (500 μg) were subjected to SDS-PAGE and anti-SHP-1 immunoblotting analysis. (C) c-Kit phosphopeptides 3 and 4 were individually coupled to NHS-Sepharose beads and then incubated with ConA-treated EL4 cell lysates. Following washing, the complexes and lysate protein were resolved by SDS-PAGE and subjected to anti-SHP-1 immunoblotting analysis. (D) Glutathione-Sepharose–GST-JUX fusion protein (5 μg) was incubated with lysates from ConA-treated EL4 cells in the absence or presence of 10 μM phosphopeptide 4, 5, or 6, and the complexes and lysate proteins were subjected to SDS-PAGE and anti-SHP-1 immunoblotting analysis. In each panel, mobilities of molecular mass (MW) standards are shown on the right and the position of SHP-1 is indicated on the left.
Data from studies of another cytosolic PTP, SHP-2, have indicated that this enzyme also interacts via its SH2 domains with activated c-Kit in hemopoietic cells (45). The binding of SHP-2 to c-Kit has been predicted to be mediated through a tyrosine residue located in the c-Kit juxtamembrane region, in this instance Tyr567 (44). Accordingly, to further address the specific role for c-Kit juxtamembrane tyrosine residues in mediating PTP binding to c-Kit, the respective capacities of SHP-1 and SHP-2 to bind Tyr567 and Tyr569 were investigated by using a second set of TXK1-phosphorylated GST-JUX fusion proteins containing Phe substitutions at one or both of these tyrosine sites. As illustrated by the anti-SHP-1 immunoblotting analysis shown in Fig. 6A, the GST-JUX fusion proteins in which Tyr569 alone or Tyr569 together with Tyr567 was replaced with Phe failed to precipitate SHP-1 from EL4 lysates. By contrast, GST-JUX proteins containing only a Tyr567→Phe substitution did precipitate this phosphatase, albeit to a lesser extent than the wild-type JUX protein. SHP-2 was also precipitated by wild-type GST-JUX and not by the Tyr567 Tyr569 double mutant GST-JUX proteins (Fig. 6B). However in contrast to SHP-1, the SHP-2 PTP was also precipitated by the Tyr569→Phe mutant fusion protein and was barely detectable in the precipitates derived by using GST-JUX proteins containing the Tyr567→Phe substitution. While alanine replacement of the valine residue at position 568 had no effect on GST-JUX binding to either SHP-1 or SHP-2, both of these interactions were abrogated by mutation of the hydrophobic residue at codon 570, a residue which occupies the first and third sites carboxy terminal to Tyr567 and Tyr569, respectively. Taken together, these data identify Tyr569 as the pivotal binding site on c-Kit for SHP-1 and identify Tyr567 as the site through which c-Kit interacts with SHP-2. In addition, these data indicate that the interactions of both SHP-1 and SHP-2 with c-Kit depend on the isoleucine residue at position 570.
FIG. 6.

Definition of SHP-1 and SHP-2 binding sites within the c-Kit juxtamembrane region. (A) GST-c–Kit–JUX fusion proteins containing either the wild-type (JUX) or mutated versions of the c-Kit juxtamembrane region were expressed in TKXI cells, and the tyrosine-phosphorylated proteins were then immobilized on gluathione-Sepharose beads and incubated with lysates prepared from 108 ConA-treated EL4 cells. Complexes and lysate proteins were then subjected to SDS-PAGE and anti-SHP-1 immunoblotting analysis. The specific JUX domain mutations are indicated above the lanes and include phenylalanine replacement of tyrosine 567 and 569 individually (Y567F and Y569F, respectively) or together [DM (Y567, Y569F)], alanine replacement of valine 568, and alanine replacement of isoleucine 570. The position of SHP-1 is indicated on the left. (B) The filter shown in panel A was stripped and reblotted with anti-SHP-2 antibody. The position of SHP-2 is indicated on the left.
Expression of Tyr569→Phe- or Tyr567→Phe-mutated c-Kit receptors enhances SCF-induced proliferation of Ba/F3-Kit cells.
To investigate whether the interaction between SHP-1 and the c-Kit Tyr569 or Tyr567 residue is responsible for the previously demonstrated inhibitory influence of SHP-1 on c-Kit signaling, Ba/F3 cells lacking endogenous c-Kit were infected with retroviral vectors carrying cDNAs for either the full-length wild-type (Ba/F3-Kit), Tyr569→Phe-mutated (Ba/F3-Kit Y569F), or Y567→Phe-mutated (Ba/F3-Kit Y567F) c-Kit receptor. FACS (Fig. 7C, bottom panel) and Western analysis (data not shown) revealed comparable levels of c-Kit expression in these transfectants. However, as shown in Fig. 7C (top panel), the cells expressing the Tyr569→Phe mutant c-Kit protein exhibited a markedly enhanced proliferative response to SCF relative to that detected in cells expressing wild-type c-Kit receptors. As is consistent with the identification of Tyr569 on c-Kit as a required residue for c-Kit binding to SHP-1, SHP-1 and c-Kit were not coimmunoprecipitated from the SCF-treated Ba/F3-Kit Y569F cells but were co-immunoprecipitated from stimulated Ba/F3-Kit and Ba/F3-Kit Y567F cells (Fig. 7A). By contrast, the capacity of SHP-2 to associate with activated c-Kit in the Ba/F3–c-Kit transfectants was unaffected in the context of Tyr569 mutation but was markedly reduced by mutation of the tyrosine residue at position 567 (Fig. 7B). Most significantly, as shown in Fig. 7C, expression of c-Kit proteins carrying Phe substitutions at either Tyr567 or Tyr569 was associated with markedly enhanced proliferation of the Ba/F3-Kit cells in response to various amounts of SCF. These increases in proliferation were specifically related to interactions between SCF and the mutant Kit receptors, as proliferative responses to IL-3 in Ba/F3-Kit Y567F and Ba/F3-Kit Y569F cells were comparable to those detected in Ba/F3-Kit cells (Fig. 7D). These observations indicate a critical role for Tyr569 and Tyr567 in mediating the association of c-Kit with SHP-1 and SHP-2, respectively, in vivo and indicate that SHP-2 as well as SHP-1 can negatively modulate c-Kit signaling by interacting with these specific tyrosine residues.
FIG. 7.

Mutations at the SHP-1 or SHP-2 binding sites on c-Kit enhance SCF-driven proliferation of Ba/F3-Kit cells. Cell lysates were prepared from unstimulated (−) or SCF (100 ng/ml)-treated (+) Ba/F3 transfectants infected with a retroviral vector carrying the full-length wild-type (Ba/F3-Kit), Y567→F-mutated (Ba/F3-Kit Y567F), or Y569→F-mutated (Ba/F3-Kit Y569F) c-Kit cDNA. (A and B) Lysate proteins (1,800 μg) from the unstimulated and stimulated cells were immunoprecipitated (Ip) with anti-c-Kit antibody, and the immune complexes and lysate proteins were then subjected to SDS-PAGE and anti-SHP-1 (A) and anti-SHP-2 (B) immunoblotting analysis. (C) Top, Ba/F3-Kit wild-type (Wt), Kit Y567F, and Kit Y569F cells were suspended at 2.5 × 105 cells/ml in culture medium in the presence of various concentrations (0-400 ng/ml) of SCF. Cultures were harvested at 48 h following a 6-h pulse with 1 mCi of [3H]thymidine, and proliferation was measured by beta scintillation counting. Results (means ± SD) represent averages for triplicate cultures and three independent experiments. Bottom, Ba/F3 c-Kit wild-type (WT), Y567F, and Y569F cells (105) were stained with the ACK2 anti-Kit antibody and examined for expression of c-Kit by FACS analysis. (D) Ba/F3, Ba/F3-Kit wild-type (Wt), Kit Y567F, and Kit Y569F cells were suspended at 104 cells/ml in culture medium, and IL-3 (50 ng/ml) was added at day zero and every second day thereafter. Proliferation was evaluated every 48 h by the Cell Titer assay and enzyme-linked immunosorbent assay at 570 nm. Results (means ± SD) represent averages of triplicate cultures. OD, optical density.
DISCUSSION
Recent data derived from biochemical and genetic studies of SHP-1 interactions with the c-Kit PTK receptor have revealed that SHP-1 negatively regulates c-Kit signaling and thereby mitigates the signaling events linking c-Kit engagement to hemopoietic cell proliferation and differentiation (25, 35). We have investigated the structural basis for SHP-1 binding to and consequent inhibitory influence on c-Kit and report here that SHP-1 binds to a specific tyrosine-containing peptide sequence within the juxtamembrane region of c-Kit. Our data indicate that both SHP-1 SH2 domains participate in its interaction with c-Kit and identify the tyrosine residue at position 569 within the phosphorylated c-Kit juxtamembrane region as the major SHP-1 binding site on c-Kit. Mutation of Tyr569 not only abrogates SHP-1 interaction with c-Kit but also results in enhanced c-Kit signaling in response to SCF stimulation. Therefore, binding of SHP-1 to Tyr569 appears to be critical to the capacity of SHP-1 to both associate with c-Kit and negatively modulate the signaling pathways coupling the activated receptor to cellular responses.
The data reported here confirm previous observations indicating that tyrosine-phosphorylated c-Kit interacts with the SHP-1 N-terminal, but not C-terminal, SH2 domain (54). Binding of the alternatively spliced C-terminal SHP-1 SH2 domain (SH2-C′) to phosphorylated c-Kit in vitro was also demonstrated in the current study and appeared to be equivalent to the interaction detected between c-Kit and the SHP-1 N- and C-terminal SH2 domains together (Fig. 2B). Thus, c-Kit association with SHP-1 can be mediated through either the SH2-N, SH2-C′, or tandem SH2-N and -C (or -C′) domains. These data provide the first direct evidence that the ligand binding properties of the SHP-1 C and C′ SH2 domains and, by extension, the two SHP-1 isoforms may be distinct, a conclusion consistent with structural data on SH2 domains indicating that the 39-amino-acid SH2 domain segment distinguishing the SHP-1 isoforms maps within a region forming EFβ strands implicated in defining SH2 domain peptide-binding specificity (2, 50). Along similar lines, the SHP-1 SH2-N and SH2-C domains also diverge in terms of their binding specificities, SHP-1 association with the activated erythropoietin, CD22, and IL-3β receptors being mediated through the N-terminal SHP-1 SH2 domain (23, 55, 56), while its interaction with the natural killer inhibitory and FcγRIIβ receptors is mediated by the C-terminal SH2 domain (3, 6). Such divergence in terms of the peptide-binding specificities of its individual SH2 domains gives SHP-1 the potential to associate with and modulate a broad array of signaling effectors and, accordingly, to assume pivotal roles in regulating many facets of hemopoietic and epithelial cell behavior.
In the current study, the tyrosine residue at position 569 in the c-Kit juxtamembrane region was identified as the critical site for SHP-1 association with activated c-Kit receptors. This tyrosine, in turn, is flanked by Tyr and Val residues at the −2 and −1 positions, respectively, followed by Ile, Asp, and Pro at the +1, +2, and +3 positions, respectively. By contrast, previous studies of the binding motifs for SHP-1 interactions with the CD22 and FcγRIIB receptors on B lymphocytes and KIR, the p58 receptor on natural killer cells, have identified the sequence Val/IleX[pTyr]XXLeu as a consensus motif for association of SHP-1 with each of these receptors. Engagement of the latter receptors and their coincident recruitment of SHP-1 inhibit activation through the B-cell antigen (in the cases of FcγRIIB and CD22) or CD16 (in the case of p58) receptors, and accordingly, this conserved motif has been designated the immunoreceptor tyrosine-based inhibitory motif (6, 30). The SHP-1 N-terminal SH2 domain has also been shown to select the peptide sequence [pTyr]-hydrophobic-X-hydrophobic from a degenerate phosphopeptide library (43). This finding is consistent with the identification here of [pTyr]IsoAspPro as the SHP-1 binding site on c-Kit as well as data identifying [pTyr]ThrIsoLeu as the SHP-1 binding site on the erythropoietin receptor (19). Together these data indicate the capacity for SHP-1 to recognize phosphotyrosines in a multiplicity of amino acid contexts, a property not solely attributable to structural differences between the SHP-1 N- and C-terminal SH2 domains, as receptors such as the erythropoietin and CD22 receptors both bind SHP-1 via its N-terminal SH2 domain despite the differences in their SHP-1 binding site sequences.
In contrast to SHP-1, the SHP-2 PTP was shown to bind c-Kit by interacting with a tyrosine residue (Tyr567) within the sequence [pTyr]ValTyrIle, a motif which matches the peptide sequence ([pTyr]Val/IleX Val/Ile) that the SHP-2 N-terminal SH2 domain preferentially selects from a degenerate peptide library (44). Importantly, phenylalanine replacement of this tyrosine in GST–c-Kit–JUX fusion proteins was associated not only with disruption of SHP-2 binding, but also with some reduction in SHP-1 binding to the phosphorylated fusion protein (Fig. 6). By contrast, Phe replacement of Tyr569 had no effect on the capacity of either GST–c-Kit–JUX fusion proteins (Fig. 6B) or c-Kit receptors expressed in Ba/F3 cells (Fig. 7B) to associate with SHP-2. Thus, unlike Tyr569, which is required for SHP-1 but not involved in SHP-2 interactions with c-Kit, Tyr567 may play both an essential role in SHP-2 binding and a facilitory role in SHP-1 binding to phosphorylated c-Kit. In this context, it is possible that SHP-1 and SHP-2 compete for binding to the latter site on the c-Kit juxtamembrane region.
The involvement of Tyr567 in SHP-2 and, potentially, SHP-1 binding to c-Kit is of particular interest in view of previous data showing that deletion of the comparable tyrosine and juxtaposed valine residues (Tyr568 and Val569) substantially enhances the mitogenic and transforming properties of the feline c-Kit receptor and also represents one of the mutations which distinguishes the wild-type receptor from the oncogenic counterpart, v-Kit (13). Although a similar link between Tyr569 mutation and c-Kit transforming capacity has not been described, the finding that SCF-induced proliferation of Ba/F3-Kit cells is enhanced by either Tyr569→Phe or Tyr567→Phe substitutions of the c-Kit receptors on these cells indicates that SHP-1 and SHP-2 can independently exert negative regulatory effects on c-Kit signaling and, by extension, that mutations of c-Kit which reduce or abrogate its binding to SHP-1 or SHP-2 can engender enhanced mitogenic and potentially oncogenic c-Kit activity. This apparent overlap in the effects of SHP-1 and SHP-2 on c-Kit signaling suggests that in at least some cell lineages c-Kit signaling may be unaffected by loss of function of one of these PTPs and thus provides a molecular explanation for the cell lineage-dependent effects of SHP-1 on c-Kit function observed in Wv/motheaten mice (25).
While our data provide evidence for the capacity of both SHP-1 and SHP-2 to negatively regulate c-Kit signaling, the mechanisms whereby this inhibitory influence is realized remain to be defined. For example, although c-Kit has been identified as an SHP-1 substrate in vitro, it is unclear whether SHP-1 or SHP-2 directly dephosphorylates c-Kit in vivo and/or elicits dephosphorylation of the receptor indirectly by dephosphorylating and inhibiting cytosolic PTKs that act on c-Kit. Downregulation of c-Kit signaling by these PTPs may also reflect the dephosphorylation of signaling effectors involved in downstream transduction of the ligand-binding activation signal, a paradigm recently demonstrated with respect to SHP-1 interactions with the erythropoietin receptor (17). In the latter example, the negative influence of SHP-1 on receptor signaling has been linked to SHP-1-mediated dephosphorylation of the cytosolic JAK2 PTK (17, 19, 52). As JAK2 has also been shown to associate with and modulate the activated c-Kit receptor (51, 53), it is possible that SHP-1 dephosphorylation of JAK2 either impairs c-Kit phosphorylation following ligand engagement or, by analogy with the erythropoietin receptor, interferes with JAK2-mediated activation and recruitment of signaling effectors required to evoke a cellular response. In addition to these possibilities, previous data indicating that the association of Src family tyrosine kinases with the c-Kit related platelet-derived growth factor receptor is mediated through Tyr579 and Tyr581, sites which represent homologs of Tyr567 and Tyr569 on c-Kit (27, 39), also suggest that SHP-1 and/or SHP-2 inhibitory effects on c-Kit signaling may reflect the capacity of these PTPs to compete with and displace Src PTKs. Resolution of these issues should elucidate the molecular mechanisms whereby c-Kit signaling is regulated and translated to particular biological outcomes.
ACKNOWLEDGMENTS
This work was supported in part by grants from the Medical Research Council of Canada and the National Cancer Institute of Canada and by the Health Canada Bureau of Drug Research. Katherine A. Siminovitch is a Senior Scientist and Robert Rottapel is a Research Scholar of the Arthritis Society of Canada, and Louise Larose is a recipient of a Medical Research Council of Canada/Canadian Research Society scholarship.
REFERENCES
- 1.Bignon J S, Siminovitch K A. Identification of PTP1C mutation as the genetic defect in motheaten and viable motheaten: a step toward defining the roles of protein tyrosine phosphatases in the regulation of hemopoietic cell differentiation and function. Clin Immunol Immunopathol. 1994;73:168–179. doi: 10.1006/clin.1994.1185. [DOI] [PubMed] [Google Scholar]
- 2.Birge R B, Hanafusa H. Closing in on SH2 specificity. Science. 1993;262:1522–1524. doi: 10.1126/science.7504323. [DOI] [PubMed] [Google Scholar]
- 3.Burshtyn D N, Scharenberg A M, Wagtmann N, Raja Apopalan S, Berrada K, Alan T, Berrada K, Yi T, Kinet J-P, Long E O. Recruitment of tyrosine phosphatase HCP by the killer cell inhibitory receptor. Immunity. 1996;4:77–85. doi: 10.1016/s1074-7613(00)80300-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen H E, Chang S, Trub T, Neel B G. Regulation of colony-stimulating factor 1 receptor signaling by the SH2 domain-containing tyrosine phosphatase SHPTP1. Mol Cell Biol. 1996;16:3685–3697. doi: 10.1128/mcb.16.7.3685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cyster J G, Goodnow L G. Protein tyrosine phosphatase 1C negatively regulates antigen receptor signaling in B lymphocytes and determines thresholds for negative selection. Immunity. 1995;2:13–24. doi: 10.1016/1074-7613(95)90075-6. [DOI] [PubMed] [Google Scholar]
- 6.D’Ambrosio D, Hippen K L, Minskoff S A, Mellman I, Pani G, Siminovitch K A, Cambier J C. Recruitment and activation of PTP1C in negative regulation of antigen receptor signaling by FcγRIIB1. Science. 1995;268:293–297. doi: 10.1126/science.7716523. [DOI] [PubMed] [Google Scholar]
- 7.der Mujum S K, Brown K, Qui F-H, Besmer P. c-Kit protein, a transmembrane kinase: identification in tissues and characterization. Mol Cell Biol. 1988;8:4896–4903. doi: 10.1128/mcb.8.11.4896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Doody G M, Justement L B, Delibrias C C, Mathews J R, Lin J, Thomas M L, Fearon D T. A role in B cell activation for CD22 and the protein tyrosine phosphatase SHP. Science. 1995;269:242–244. doi: 10.1126/science.7618087. [DOI] [PubMed] [Google Scholar]
- 9.Dutflinger R, Manova K, Berrozpe G, Chu T-Y, DeLeon V, Timokhina I, Chaganti R S K, Zelentz A D, Bachvarova R F, Besmer P. The Wsh and Ph mutations affect the c-Kit expression profile: c-Kit misexpression impairs melanogenesis in Wsh and Ph mutant mice. Proc Natl Acad Sci USA. 1995;92:3754–3758. doi: 10.1073/pnas.92.9.3754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Feng G-S, Hui C C, Pawson T. SH2-containing phosphotyrosine phosphatase as a target of protein-tyrosine kinases. Science. 1993;259:1607–1611. doi: 10.1126/science.8096088. [DOI] [PubMed] [Google Scholar]
- 11.Geissler E N, Ryan M A, Housman E D. The dominant-white spotting (w) locus of the mouse encodes the c-Kit proto-oncogene. Cell. 1988;55:185–192. doi: 10.1016/0092-8674(88)90020-7. [DOI] [PubMed] [Google Scholar]
- 12.Gokkel E, Grossman Z, Ramot B, Yarden Y, Rechavi G, Givol D. Structural organization of the murine c-kit proto-oncogene. Oncogene. 1992;7:1423–1429. [PubMed] [Google Scholar]
- 13.Herbst R, Munemitsu S, Ullrich A. Oncogenic activation of v-kit involves deletion of a putative tyrosine-substrate interaction site. Oncogene. 1995;10:369–379. [PubMed] [Google Scholar]
- 14.Ho S N, Hunt H D, Horton R M, Pullen J K, Pease L R. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 1989;77:51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- 15.Ikeda H, Kanakura Y, Tamaki T, Kuriu A, Kitayama H, Ishikawa J, Kanayama Y, Yonezawa T, Tarui S, Griffin J D. Expression and functional role of the proto-oncogene c-Kit in acute myeloblastic leukemia cells. Blood. 1991;78:2962–2968. [PubMed] [Google Scholar]
- 16.Jhun B H, Rivnay B, Price D, Avraham H. The MATK tyrosine kinase interacts in a specific and SH2-dependent manner with c-Kit. J Biol Chem. 1995;270:9661–9666. doi: 10.1074/jbc.270.16.9661. [DOI] [PubMed] [Google Scholar]
- 17.Jiao H, Berrada K, Yang W, Tabrizi M, Platanias L C, Yi T. Direct association with and dephosphorylation of Jak2 kinase by the SH2-domain-containing protein tyrosine phosphatase SHP-1. Mol Cell Biol. 1996;16:6985–6992. doi: 10.1128/mcb.16.12.6985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kitayama H, Kanakura Y, Furitsu T, Tsujimura T, Oritani K, Ikede H, Sugahara H, Mitsui H, Kanayama Y, Matsuzawa Y. Constitutively activating mutations of c-Kit receptor tyrosine kinase confer factor-independent growth and tumorigenicity of factor dependent hematopoietic cell lines. Blood. 1995;85:790–798. [PubMed] [Google Scholar]
- 19.Klingmuller U, Lorenz U, Cantley L, Neel B, Lodish H. Specific recruitment of SH-PTP1 to the erythropoietin receptor causes inactivation of JAK2 and termination of proliferative signals. Cell. 1995;80:729–738. doi: 10.1016/0092-8674(95)90351-8. [DOI] [PubMed] [Google Scholar]
- 20.Kon-Kozlowski M, Pani G, Pawson T, Siminovitch K A. The tyrosine phosphatase PTP1C associates with Vav, Grb2, and mSos1 in hematopoietic cells. J Biol Chem. 1996;271:3856–3862. doi: 10.1074/jbc.271.7.3856. [DOI] [PubMed] [Google Scholar]
- 21.Kozlowski M, Mlinaric-Rascan I, Feng G-S, Shen R, Pawson T, Siminovitch K A. Expression and catalytic activity of the tyrosine phosphatase PTP1C is severely impaired in motheaten and viable motheaten mice. J Exp Med. 1993;178:2157–2163. doi: 10.1084/jem.178.6.2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kuriu A, Ikeda H, Kanakura Y, Griffin J D, Druker B, Yagura H, Kitayama H, Ishikawa J, Nishihura J, Kanayama Y, Yonezawa T, Tarui S. Proliferation of human leukemia cell line associated with the tyrosine phosphorylation and activation of the proto-oncogene c-Kit product. Blood. 1991;78:2834–2840. [PubMed] [Google Scholar]
- 23.Law C L, Sidorenki S P, Chandran K A, Zhao Z, Shen S H, Fischer E H, Clark E A. CD22 associates with protein tyrosine phosphatase 1C, Syk and phospholipase C-γ1 upon B cell activation. J Exp Med. 1996;183:547–560. doi: 10.1084/jem.183.2.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lev S, Givol D, Yarden Y. A specific combination of substrates is involved in signal transduction by the Kit-encoded receptor. EMBO J. 1991;10:647–654. doi: 10.1002/j.1460-2075.1991.tb07993.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lorenz U, Bergemann A D, Steinberg H N, Flanagan J G, Li X, Galli S J, Neel B G. Genetic analysis reveals cell type-specific regulation of receptor tyrosine kinase c-Kit by the protein tyrosine phosphatase SHP1. J Exp Med. 1996;184:1111–1126. doi: 10.1084/jem.184.3.1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miller A D, Rosman G J. Improved retroviral vectors for gene transfer and expression. BioTechniques. 1989;7:980–982. [PMC free article] [PubMed] [Google Scholar]
- 27.Mori S, Ronnstrand L, Yokote K, Engstrom A, Courtneidge S A, Claesson-Welsh L, Heldin C-H. Identification of two juxtamembrane autophosphorylation sites in the PDGF β-receptor; involvement in the interaction with Src family tyrosine kinases. EMBO J. 1993;12:2257–2264. doi: 10.1002/j.1460-2075.1993.tb05879.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nishikawa S, Kusakabe M, Yoshinaga K, Ogawa M, Hayashi S, Kunisada T, Era T, Sakakura T, Nishikawa S. In utero manipulation of coat color formation by a monoclonal anti-c-kit antibody: two distinct waves of c-kit-dependency during melanocyte development. EMBO J. 1991;10:2111–2118. doi: 10.1002/j.1460-2075.1991.tb07744.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nocka K, Majumder S, Chabot B, Ray P, Gervonne M, Bernstein A, Besmer P. Expression of the c-Kit proto-oncogene in known cellular targets of W mutations in normal and W mutant mice: evidence for an impaired c-Kit kinase in mutant mice. Genes Dev. 1989;3:816–826. doi: 10.1101/gad.3.6.816. [DOI] [PubMed] [Google Scholar]
- 30.Olcese L, Lang P, Vely F, Cambiaggi A, Marguet D, Blery M, Hippen K L, Biassoni R, Moretta A, Moretta L, Cambier J C, Vivier E. Human and mouse killer cell inhibitory receptors recruit PTP1C and PTP1D tyrosine phosphatases. J Immunol. 1996;156:4531–4534. [PubMed] [Google Scholar]
- 31.Onoue H, Maeyama K, Nomura S, Kasugai T, Teri H, Kim H M, Watamabe T, Kitamura Y. Absence of immature mast cells in the skin of Ws/Ws rats with a small deletion at the tyrosine kinase domain of the c-Kit gene. J Pathol. 1993;193:1001–1007. [PMC free article] [PubMed] [Google Scholar]
- 32.Pani G, Kozlowski M, Cambier J C, Mills G B, Siminovitch K A. Identification of the tyrosine phosphatase PTP1C as a B cell antigen receptor-associated protein involved in the regulation of B cell signaling. J Exp Med. 1995;181:2077–2084. doi: 10.1084/jem.181.6.2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pani G, Fischer K D, Mlinaric-Rascan I, Siminovitch K A. Signaling capacity of the T cell antigen receptor is negatively regulated by the PTP1C tyrosine phosphatase. J Exp Med. 1996;184:839–852. doi: 10.1084/jem.184.3.839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pani G, Siminovitch K A. Protein tyrosine phosphatase roles in the regulation of lymphocyte signaling. Clin Immunol Immunopathol. 1997;84:1–16. doi: 10.1006/clin.1996.4326. [DOI] [PubMed] [Google Scholar]
- 35.Paulson R F, Vesely S, Siminovitch K A, Bernstein A. Signaling by the W/kit receptor tyrosine kinase is negatively regulated in vivo by the protein tyrosine phosphatase Shp1. Nat Genet. 1996;13:309–315. doi: 10.1038/ng0796-309. [DOI] [PubMed] [Google Scholar]
- 36.Pear W S, Nolan G P, Scott M L, Baltimore D. Production of high-titer helper-free retroviruses by transient transfection. Proc Natl Acad Sci USA. 1993;90:8392–8396. doi: 10.1073/pnas.90.18.8392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Perkins L A, Larsen I, Perrimon N. Corkscrew encodes a putative protein tyrosine phosphatase that functions to transduce the terminal signal from the receptor tyrosine kinase torso. Cell. 1992;70:225–236. doi: 10.1016/0092-8674(92)90098-w. [DOI] [PubMed] [Google Scholar]
- 38.Piao X, Curtis J E, Minkin S, Minden M D, Bernstein A. Expression of the Kit and KitA receptor isoforms in human acute myelogenous leukemia. Blood. 1994;83:476–481. [PubMed] [Google Scholar]
- 39.Qui F, Ray P, Brown K, Barker P E, Jhanwar S, Ruddle F H, Besmer P. Primary structure of c-Kit: relationship with the CSF-1/PDGF receptor kinase family—oncogenic activation of v-kit involves deletion of extracellular domain and C-terminus. EMBO J. 1988;7:1003–1011. doi: 10.1002/j.1460-2075.1988.tb02907.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Reith A D, Ellis C, Lynman D L, Anderson D M, Williams D E, Bernstein A, Pawson T. Signal transduction by normal isoforms and W mutant variants of the Kit receptor tyrosine kinase. EMBO J. 1991;10:2451–2459. doi: 10.1002/j.1460-2075.1991.tb07784.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rottapel R, Reedijk M, Williams D E, Lynman S D, Anderson D M, Pawson T, Bernstein A. The Steel/W transduction pathway: Kit autophosphorylation and its association with a unique subset of cytoplasmic signaling proteins is induced by steel factor. Mol Cell Biol. 1991;11:3043–3051. doi: 10.1128/mcb.11.6.3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shearman M S, Herbst R, Schlessinger J, Ullrich A. Phosphatidylinosital 3′-kinase associates with p145c-Kit as part of a cell type characteristic multimeric signalling complex. EMBO J. 1993;12:3817–3826. doi: 10.1002/j.1460-2075.1993.tb06060.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Song yang Z, Shoelson S E, Chandhuri M, Gish G, Pawson T, Haser W G, King F, Roberts T, Ratnofsky S, Lechleider R J, Neel B G, Birge R B, Fajardo J E, Chou M M, Hanafusa H, Schaffhausen B, Cantley L C. Specific motifs recognized by the SH2 domains of Csk, 3BP2, Fps/Fes, GRB-2, HCP, SHC, Syk, and Vav. Mol Cell Biol. 1994;14:2777–2785. doi: 10.1128/mcb.14.4.2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Songyang Z, Schoelson S E, McGlade H, Oliver P, Pawson T, Bustelo X R, Barbacid M, Sabe H, Hanafusa H, Yi T, Ren R, Baltimore D, Ratnofsky S, Feldman R A, Cantley L C. SH2 domains recognize specific phosphopeptide sequences. Cell. 1994;72:767–778. doi: 10.1016/0092-8674(93)90404-e. [DOI] [PubMed] [Google Scholar]
- 45.Tauchi T, Feng G S, Marshall M S, Shen R, Mantel C, Pawson T, Broxmeyer H E. The ubiquitously expressed Syp phosphatase interacts with c-Kit and Grb2 in hematopoietic cells. J Biol Chem. 1994;269:25206–25211. [PubMed] [Google Scholar]
- 46.Tsui H W, Siminovitch K A, de Souza L, Tsui F W L. Motheaten and viable motheaten mice have mutations in the hemopoietic cell phosphatase gene. Nat Genet. 1993;4:124–129. doi: 10.1038/ng0693-124. [DOI] [PubMed] [Google Scholar]
- 47.Tsujimura T, Furitsu T, Morimoto M, Isozaki K, Nomura S, Matsuzawa Y, Kitamura Y, Kanakura Y. Ligand-independent activation of c-Kit receptor tyrosine kinase in a murine mastocytoma cell line P-815 generated by a point mutation. Blood. 1994;83:2619–2626. [PubMed] [Google Scholar]
- 48.Tsujimura T, Morimoto M, Hashimoto K, Moriyama Y, Kitayama H, Matsuzawa Y, Kitamura Y, Kanakura Y. Constitutive activation of c-Kit in FMA3 murine mastocytoma cells caused by deletion of seven amino acids at the juxtamembrane domain. Blood. 1996;87:273–283. [PubMed] [Google Scholar]
- 49.Uchida T, Matozaki T, Matsuda K, Suzuki T, Mutozaki S, Nakano O, Wada K, Konda Y, Sakamoto C, Kasugo M. Phorbolester stimulates the activity of protein tyrosine phosphatase containing SH2 domains (PTP1C) in the HL-60 leukemia cells by increasing gene expression. J Biol Chem. 1993;267:23447–23450. [PubMed] [Google Scholar]
- 50.Waksman G, Shoelson S E, Pant N, Cowburn D, Kurigan John. Binding of a high affinity phosphotyrosol peptide to the Src SH2 domain: crystal structures of the complexed and peptide free forms. Cell. 1993;72:779–790. doi: 10.1016/0092-8674(93)90405-f. [DOI] [PubMed] [Google Scholar]
- 51.Weiler S R, Mou S, DeBerry C S, Keller J R, Ruscetti F W, Ferris D K, Longo D L, Linnekin D. JAK2 is associated with the c-Kit proto-oncogene product and is phosphorylated in response to stem cell factor. Blood. 1996;87:3688–3693. [PubMed] [Google Scholar]
- 52.Witthuhn B A, Quelle F W, Silvennoinen O, Yi T, Tang B, Miura O, Ihle J N. JAK2 associates with the erythropoietin receptor and is tyrosine phosphorylated and activated following stimulation with erythropoietin. Cell. 1993;74:227–236. doi: 10.1016/0092-8674(93)90414-l. [DOI] [PubMed] [Google Scholar]
- 53.Wu H, Klingmuller U, Besmer P, Lodish H F. Interaction of the erythropoietin and stem-cell-factor receptors. Science. 1995;337:242–246. doi: 10.1038/377242a0. [DOI] [PubMed] [Google Scholar]
- 54.Yi T, Ihle J N. Association of hematopoietic cell phosphatase with c-Kit after stimulation with c-Kit ligand. Mol Cell Biol. 1993;13:3350–3358. doi: 10.1128/mcb.13.6.3350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yi T, Mui A L F, Krystal G, Ihle J N. Hemopoietic cell phosphatase associates with the interleukin-3 (IL-3) receptor β chain and down-regulates IL-3-induced tyrosine phosphorylation and mitogenesis. Mol Cell Biol. 1993;13:7577–7586. doi: 10.1128/mcb.13.12.7577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yi T, Zhang J, Miura O, Ihle J N. Hematopoietic cell phosphatase associates with erythropoietin (Epo) receptor after Epo-induced receptor tyrosine phosphorylation: identification of potential binding sites. Blood. 1995;85:87–95. [PubMed] [Google Scholar]