

Abstract
The DBF2 gene of the budding yeast Saccharomyces cerevisiae encodes a cell cycle-regulated protein kinase that plays an important role in the telophase/G1 transition. As a component of the multisubunit CCR4 transcriptional complex, DBF2 is also involved in the regulation of gene expression. We have found that MOB1, an essential protein required for a late mitotic event in the cell cycle, genetically and physically interacts with DBF2. DBF2 binds MOB1 in vivo and can bind it in vitro in the absence of other yeast proteins. We found that the expression of MOB1 is also cell cycle regulated, its expression peaking slightly before that of DBF2 at the G2/M boundary. While overexpression of DBF2 suppressed phenotypes associated with mob1 temperature-sensitive alleles, it could not suppress a mob1 deletion. In contrast, overexpression of MOB1 suppressed phenotypes associated with a dbf2-deleted strain and suppressed the lethality associated with a dbf2 dbf20 double deletion. A mob1 temperature-sensitive allele with a dbf2 disruption was also found to be synthetically lethal. These results are consistent with DBF2 acting through MOB1 and aiding in its function. Moreover, the ability of temperature-sensitive mutated versions of the MOB1 protein to interact with DBF2 was severely reduced, confirming that binding of DBF2 to MOB1 is required for a late mitotic event. While MOB1 and DBF2 were found to be capable of physically associating in a complex that did not include CCR4, MOB1 did interact with other components of the CCR4 transcriptional complex. We discuss models concerning the role of DBF2 and MOB1 in controlling the telophase/G1 transition.
In eukaryotic cells, many of the cell cycle stages are regulated by phosphorylation, and a number of protein kinases involved in the cell cycle are known to date. The activities of these kinases are regulated by different mechanisms, including but not limited to formation of complexes with other proteins and cell cycle-dependent control of their expression. The DBF2 protein kinase from the yeast Saccharomyces cerevisiae is required for the proper progression through late mitosis, specifically during telophase (11, 24). dbf2 temperature-sensitive mutant cells arrest at the restrictive temperature with a terminal “dumbbell” phenotype in which they display an elongated spindle and divided chromatin (11). DBF2 protein kinase activity is cell cycle controlled, peaking after the metaphase-to-anaphase transition, which is consistent with its late mitotic role (11, 20). While a dbf2 deletion is not lethal, DBF2 plays an essential role in cells that also lack DBF20, a close homolog of DBF2 (23).
We have recently shown that the DBF2 protein not only regulates cell cycle progression but also controls gene expression as one of the components of the CCR4 transcriptional complex (17). The CCR4 protein is a general transcriptional regulator which affects expression of a number of genes both positively and negatively (16). It is required for full expression of ADH2 and other nonfermentative genes under glucose-derepressed conditions (5). Both dbf2 and ccr4 disruptions affect genes involved in cell wall integrity and under glucose conditions are able to suppress enhanced ADH2 expression caused by an spt10 defect (16, 17, 19). The CCR4 complex contains a number of proteins in addition to DBF2 (6, 7, 16). One of these is CAF1 (POP2) (8, 22), which binds to both CCR4 and DBF2. ccr4 and caf1 defects also cause a partial block in late mitosis at a point similar to that observed for dbf2 defects (17). These results suggest that one of DBF2 functions during late mitosis is to control gene expression through its association with the CCR4 complex.
While previous studies suggest that DBF2 plays an important role in the regulation of the cell cycle and gene expression, the mechanisms by which DBF2 is regulated and the identification of its cellular target proteins remain unclear. We have, therefore, undertaken a search for proteins interacting with DBF2. In this paper, we report that MOB1 binds to DBF2. The MOB1 gene was initially identified in a separate screen for proteins that interact with MPS1 (18). The yeast MPS1 protein is an essential protein kinase which is required for spindle pole body duplication (14, 27) and an M-phase checkpoint function (26). Mutated alleles of MOB1 result in a cell cycle arrest phenotype identical to that observed with dbf2 alleles (18). We have found that MOB1 is periodically expressed during the cell cycle at nearly the same time as DBF2 and that dbf2 and mob1 defects elicit similar cell cycle and other phenotypes. Genetic and biochemical studies suggest that DBF2 acts through and aids MOB1 function in the control of the M-phase transition.
MATERIALS AND METHODS
Yeast strains, growth conditions, enzyme assays, and transformations.
The yeast strains used in this study are listed in Table 1. Yeast strains were generally cultured on minimal medium lacking uracil, histidine, and/or tryptophan and containing either 8% glucose or 2% each raffinose and galactose. Alcohol dehydrogenase II and β-galactosidase enzyme assays were conducted as described elsewhere (2). All yeast transformations were conducted by the lithium acetate method (10).
TABLE 1.
Yeast strains used in this study
| Strain | Genotype |
|---|---|
| EGY188 | MATa ura3 his3 trp1 LexA-LEU2 |
| EGY188-c1 | Isogenic to EGY188 except caf1::URA3 |
| EGY188-1 | Isogenic to 188 except ccr4::URA3 |
| EGY191 | MATα ura3 his3 trp1 LexA-LEU2 |
| 991-1-1b | MATa ura3 his3 trp1 leu2 dbf2::URA3 |
| 1045-2b | MATα ura3 his3 trp1 leu2 dbf20::TRP1 |
| 1300-1a | MATa ura3 his3 trp1 leu2 mob1-77 |
| CG378 | MATa ura3 leu2 trp1 |
| FLY30 | MATa ura3 his3 trp1 leu2 mob1-77 |
| FLY59 | MATa ura3 his3 trp1 leu2 mob1::HIS3; contains plasmid pRS316-MOB1 (URA3) |
| S7-4A | MATa dbf2::URA3 ura3 leu2 ade5 trp1 his7 |
| S7-4A-c-myc | Isogenic to S7-4A except trp1::c-myc-DBF2-TRP1 |
Two-hybrid screen.
A yeast interaction library containing yeast genomic sequences fused to the B42 activation domain (28) was used to transform strain EGY188 containing the LexA-DBF2 fusion and the LexAop-lacZ reporter p34, which has eight LexA binding sites upstream of the GAL1-lacZ reporter (3). Identification of colonies and screening for galactose dependence were done as described elsewhere (8, 28).
Plasmid constructions.
The LexA-MOB1 fusion was constructed by placing a 1.3-kb EcoRI fragment of the MOB1 library clone at the EcoRI site of the LexA202-1 vector (3), resulting in LexA-MOB1(9–314). The B42-DBF2 full-length fusion was constructed by cloning a 2-kb SalI fragment of pRS314-DBF2-c-myc (25) into the pJG4-5 vector at the XhoI site. To construct glutathione S-transferase (GST)-MOB1 and T7-DBF2 fusions, the polylinker sites of the pGEX-KG and pGEM-3zf vectors were modified to change a frame of the EcoRI site by cutting them with EcoRI and SalI and inserting a fragment which was produced by annealing two oligonucleotides with the sequences 5′-AATTATGGAATTCTGAGCGGCCGC-3′ and 5′-TCGAGGGGCCGCTCAGAATTCCAT-3′. The resultant plasmids were then digested with EcoRI, and a 1.3-kb EcoRI fragment of the MOB1 library clone was ligated into both of them.
The construction of B42-MOB1(79–314) and the corresponding mutant alleles was conducted by ligating PCR products cut with EcoRI into the EcoRI site of the pJG4-5 vector. The PCR products were synthesized from pRS314-MOB1, pRS314–mob1-55, -77, and -95 (18) with the oligonucleotides 5′-CGGAATTCATGTCTCCCGTCCTCACTAC-3′ and 5′-GCGAATTCCTACCTATCCCTCAACTCCAT-3′.
The construction of the LexA-DBF20 full-length fusion from pRS305-DBF20 was conducted by PCR with oligonucleotides 5′-CGGAATTCATGTTTTCACGAAGTGAT-3′ and 5′-GTAGGTACCTGGTCTTAATAAAAA-3′. The PCR product was cut with EcoRI and KpnI and ligated into the pSP72 vector cut with EcoRI and KpnI. The resultant plasmid was then cut with EcoRI and SalI, and the DBF20-containing piece was ligated into the LexA202-1 plasmid cut with EcoRI and SalI.
Immunoprecipitation.
Yeast strains EGY188 containing the plasmid pair B42-DBF2(1–561) and LexA-MOB1(9–314), B42-DBF2 and LexA, B42 and LexA-MOB1, or LexA-DBF20 and B42-MOB1(79–314) were grown overnight on minimal medium lacking uracil, tryptophan, and histidine and containing 2% each galactose and raffinose. Cells were pelleted, and the whole-cell protein was extracted in lysis buffer (8 mM K2HPO4, 17 mM KH2PO4, 150 mM KCl, 1 mM sodium pyrophosphate 1 mM NaF, 1% Nonidet P-40, 10% glycerol, 5 mM MgCl2, 1 mM EDTA plus protease inhibitors; pH 7.6). Protein A-agarose (20 mg) was incubated with 0.03 mg of LexA antibody or 0.02 mg of HA1 (Babco) antibody for 30 min and then washed once with 1 ml of the lysis buffer. A 700-mg portion of protein was incubated with the antibody-coupled beads at 4°C for 60 min. The beads were then pelleted by centrifugation in a microcentrifuge and washed twice with 1 ml of the lysis buffer. Sodium dodecyl sulfate (SDS) sample buffer (20 ml) was added to the beads, the beads were boiled for 5 min, and the eluted protein was loaded on an SDS–10% polyacrylamide gel. Western blot analysis with HA1 and LexA antibodies was carried out as described previously (8), and enhanced chemiluminescence analysis (Pierce) was conducted in accordance with the manufacturer’s instructions. Polyclonal antibody to MOB1 was prepared against GST-MOB1(9–314) and affinity purified following binding to GST-MOB1(9–314) bound to glutathione-agarose beads.
GST-MOB1 binding experiments with yeast extracts.
Yeast strain EGY188 containing B42-DBF2 or B42-CAF1 was grown on minimal medium lacking tryptophan and containing 2% each galactose and raffinose. The cells were pelleted, and the whole-cell protein was extracted in a lysis buffer (20 mM HEPES, 1 mM sodium pyrophosphate, 1 mM NaF, 0.1% Tween 80, 5% glycerol, 1 mM EDTA, 5 mM MgCl2 plus proteinase inhibitors; pH 7.6). GST fusion proteins were expressed in Escherichia coli and bound to glutathione-agarose beads (Sigma) in binding buffer (1× phosphate-buffered saline, 1% Triton X-100). The beads were washed three times in 1 ml of binding buffer and once in 1 ml of the lysis buffer containing 150 mM KCl. A 700-mg portion of yeast protein was added to 20 ml of the beads and incubated at 4°C for 60 min. The beads were pelleted by centrifugation, washed twice in 1 ml of the lysis buffer containing 150 mM KCl, and then boiled with 20 ml of the SDS sample buffer, and the eluted protein was loaded on an SDS–10% polyacrylamide gel. Western blotting and enhanced chemiluminescence analysis were carried out as described above.
In vitro binding assay.
GST fusion proteins were expressed and bound to glutathione-agarose beads (Sigma) in binding buffer (1× phosphate-buffered saline, 1% Triton X-100). The beads were washed four times with binding buffer and then incubated for 1 h at 4°C in A300 buffer (20 mM HEPES [pH 7.6], 1 mM EDTA, 1 mM dithiothreitol, 300 mM potassium acetate, 1% Triton X-100) containing 1 mg of E. coli extract per ml and 40 to 200 ng of [35S]methionine-labeled in vitro-translated protein. In vitro translation of T7 fusion proteins was carried out with the TNT coupled transcription-translation system (Promega). Unbound proteins were removed by four washes with A300 buffer, and specifically bound proteins were analyzed by SDS–8% polyacrylamide gel electrophoresis after the beads were boiled in sample buffer.
RESULTS
Isolation of DBF2-interacting proteins.
To identify proteins interacting with DBF2, we used a yeast two-hybrid screen with the LexA-DBF2 fusion protein as the bait. The interaction library contained the E. coli-derived B42 activator fused to yeast genomic DNA fragments under the control of a GAL1 promoter (28). Seven colonies that displayed galactose-dependent activation of both the LexAop-LEU2 and the LexAop-lacZ reporters were isolated from about 106 transformants. Of these, four were found to encode the same protein, which was designated DBI1 (for DBF2-interacting protein 1). A database search revealed that DBI1 was the same protein as MOB1. MOB1 had been isolated in an independent screen for proteins interacting with the protein kinase MPS1, which is required for spindle pole body duplication (18). The MOB1 gene was found to be essential (18). Temperature-sensitive alleles of MOB1 result in a terminal phenotype very similar to that of dbf2-arrested cells: dumbbell-shaped cells that contain duplicated chromatin and an elongated spindle (18). These phenotypes suggest that MOB1 is required for an essential function in late mitosis and that it acts at the same execution point as does DBF2, or one similar to it.
B42-MOB1(9–314) displayed a strong interaction with the LexA-DBF2 protein and failed to interact with LexA alone (Table 2). Smaller B42-MOB1 fusions such as B42-MOB1(79–314) and B42-MOB1(145–314) (18) also interacted with LexA-DBF2 in the two-hybrid system (Table 2). To confirm that the interaction depended on the DBF2 and MOB1 moieties and not on the fortuitous configurations of the LexA-DBF2 and B42-MOB1 fusions, we constructed LexA-MOB1 and B42-DBF2 chimeras and retested their interaction. As shown in Table 2, LexA-MOB1 interacted with B42-DBF2(1–561) but not with B42 alone. LexA-MOB1(9–314) was also capable of activating a LexA-lacZ reporter by itself (yielding 100 U of β-galactosidase per mg under glucose growth conditions) (Table 2; see also Table 5). Since DBF2 is a component of the CCR4 transcriptional complex and both CCR4 and CAF1 can activate transcription when fused to LexA, our results suggested that MOB1 might also be a component of the CCR4 complex. In fact, LexA-MOB1 displayed a two-hybrid interaction with B42-CAF1 (Table 2) as well as two other components of the CCR4 complex, CAF16 and CAF17 (data not shown). The ability of LexA-MOB1 to activate transcription from the LexAop-lacZ reporter, however, was unaffected by a ccr4, caf1, or dbf2 deletion (data not shown).
TABLE 2.
Two-hybrid interaction of MOB1 with DBF2 and CAF1a
| LexA fusion | B42 fusion | β-gal (U/mg)b |
|---|---|---|
| LexA-DBF2(1–561) | B42-MOB1(9–314) | 260 |
| LexA-DBF2(1–561) | B42-MOB1(79–314) | 530 |
| LexA-DBF2(1–561) | B42-MOB1(145–314) | 66 |
| LexA-DBF2(1–561) | B42 | <1 |
| LexA | B42-MOB1(9–314) | <1 |
| LexA-DBF2(1–220) | B42-MOB1(9–314) | 59 |
| LexA-DBF2(1–220) | B42-MOB1(145–314) | 120 |
| LexA-DBF2(1–220) | B42 | <1 |
| LexA-MOB1(9–314) | B42-DBF2(1–561) | 6,700 |
| LexA-MOB1(9–314) | B42-DBF2(205–561) | 350 |
| LexA-MOB1(9–314) | B42-CAF1 | 300 |
| LexA-MOB1(9–314) | B42 | 81 |
Strains were grown on minimal medium lacking uracil, histidine, and tryptophan and supplemented with 2% raffinose and 2% galactose as previously described (8). All assays were done in EGY188/EGY191 diploids containing the p34 reporter (eight LexA operators upstream of the GAL1-lacZ promoter). B42-DBF2(1–561) and B42-DBF2(205–561) were expressed to comparable levels as determined by Western analysis (data not shown).
β-Galactosidase (β-Gal) activities represent the average of determinations for at least three separate transformants. The standard error of the mean was less than 20% in each case.
TABLE 5.
Transactivation effects of LexA-MOB1 variants
| LexA fusiona | β-Gal (U/mg)b |
|---|---|
| LexA-MOB1 | 230 |
| LexA–MOB1-55 (T85P, Q167R, Y183H) | 100 |
| LexA–MOB1-77 (E151K) | 440 |
| LexA–MOB1-95 (L157P, A158I) | 36 |
Assays were conducted as described in Table 2. All LexA-MOB1 fusions were expressed to comparable levels as determined by Western analysis (data not shown). LexA-MOB1 fusions contain residues 79 to 314 of MOB1 and were expressed in EGY188 containing the p34 reporter.
β-Galactosidase (β-Gal) activities represent the average of the determinations for at least three transformants. The standard error of the mean was less than 10% in each case except for LexA–MOB1-95, in which it was 21%.
To determine what portion of the DBF2 protein was responsible for the interaction with MOB1, we tested N- and C-terminal regions of DBF2 fused to either the B42 activator or LexA for their ability to interact with MOB1 in the two-hybrid system (Table 2). When fused to LexA, the N-terminal 220 amino acids of DBF2 were sufficient for interaction with B42-MOB1. Also, B42-DBF2(205–561) displayed a much weaker interaction with LexA-MOB1 than did B42-DBF2(1–561) (Table 2). Since the protein kinase domain of DBF2 extends from residue 164 to 453, these results indicate that this domain does not have to be intact for DBF2 to interact with MOB1. Similarly, a B42-DBF2 fusion containing a mutation in the DBF2 protein kinase domain that blocks DBF2 protein kinase function (17) interacted as well with LexA-MOB1(9–314) as did wild-type B42-DBF2 (data not shown).
MOB1 physically binds to DBF2.
The two-hybrid assay results indicated that MOB1 and DBF2 interact with each other in vivo. We used coimmunoprecipitation and GST binding experiments to analyze their physical association. Whole-cell extract containing the B42-DBF2 (full-length) fusion protein with the HA1 tag and LexA-MOB1(9–314) was incubated with HA1 or LexA antibodies. The immunoprecipitated samples were subsequently analyzed by Western blotting with HA1 and LexA antibodies (Fig. 1). In the LexA immunoprecipitation, the B42-DBF2 protein was specifically coimmunoprecipitated with the LexA-MOB1 fusion (lane 6) but was not coimmunoprecipitated from extracts expressing LexA protein alone (lane 5). In a control experiment, B42 protein did not coimmunoprecipitate with LexA-MOB1 when extracts were treated with the LexA antibody (data not shown). In addition, the LexA-MOB1 protein was specifically coimmunoprecipitated from extracts expressing B42-DBF2 and LexA-MOB1 when the HA1 antibody was used to immunoprecipitate B42-DBF2 (lane 9). In contrast, the HA1 antibody did not immunoprecipitate LexA protein in extracts containing LexA alone and B42-DBF2 (lane 8), nor did this antibody immunoprecipitate LexA-MOB1 when extracts contained only B42 and LexA-MOB1 (lane 7). These experiments indicate that the B42-DBF2 and LexA-MOB1 fusions interact specifically via the DBF2 and MOB1 moieties.
FIG. 1.

Coimmunoprecipitation of LexA-MOB1 with B42-DBF2. Crude extracts (Cr. Ex.) from strain EGY188 containing either B42 and LexA-MOB1 (lanes 1, 4, and 7), B42-DBF2 and LexA (lanes 2, 5, and 8), or B42-DBF2 and LexA-MOB1 (lanes 3, 6, and 9) were incubated with either anti-LexA antibody (lanes 4 to 6) or anti-HA1 antibody (lanes 7 to 9), and the resulting immunoprecipitates (Ip) were subjected to electrophoresis on an SDS–10% polyacrylamide gel. Western analysis was conducted as described previously (8), and the blot was probed with HA1 antibody. Lanes 1 to 3 have crude extracts containing B42 (lane 1) or B42-DBF2 (lanes 2 and 3). The same extracts were immunoprecipitated and analyzed as described above. The blot was probed with LexA antibody. Lanes 1 and 3 contain LexA-MOB1 from crude extracts, and lane 2 has crude extract containing LexA. LexA-MOB1 was capable of being immunoprecipitated with anti-LexA antibody from a strain containing B42 and LexA-MOB1 (data is not shown). Molecular masses are as follows: B42, 10 kDa; B42-DBF2, 72 kDa; LexA, 22 kDa; and LexA-MOB1, 54 kDa.
A GST-MOB1 binding experiment with yeast crude extracts was performed to independently examine the physical association between MOB1 and DBF2. A GST-MOB1 fusion purified from E. coli extracts was used as the bait to isolate MOB1-binding proteins from yeast crude extracts. For this purpose, we prepared crude extracts from strains containing the B42-DBF2 (full-length) fusion, B42-CAF1 (17), or B42 protein alone. Proteins bound specifically to GST or GST-MOB1 were then analyzed by Western blotting with HA1 antibody (Fig. 2). The B42-DBF2(1–561) and B42-CAF1 fusions were found to specifically bind to GST-MOB1 (lanes 8 and 9) but not to GST alone (lanes 5 and 6). The B42 moiety was not involved in binding to GST-MOB1, since B42-DBF2(205–561) (data not shown), B42-SPO20 (data not shown), and B42 protein alone (lanes 4 and 7) were unable to bind to GST-MOB1 or GST. The presence of CCR4 in these bound fractions was analyzed by Western analysis with an antibody raised against CCR4. While the CCR4 protein was present in crude extracts, it was not found to bind GST-MOB1 (data not shown). These experiments indicate that DBF2 and CAF1 can specifically interact with MOB1 and that MOB1-DBF2 and MOB1-CAF1 interactions can occur de novo in vitro. They also suggest that MOB1 can bind DBF2 and CAF1, components of the CCR4 complex, separately from CCR4, in agreement with the results of our two-hybrid analysis described above.
FIG. 2.

GST-MOB1 binds B42-DBF2 and B42-CAF1 from crude extracts. The GST and GST-MOB1 proteins expressed in E. coli were bound to glutathione-agarose beads and then incubated with crude extracts (Cr. Ex.) from the EGY191 strain containing either B42 (lane 1), B42-DBF2 (lane 2), or B42-CAF1 (lane 3). The beads were then boiled with SDS sample buffer, the eluted protein was loaded on an SDS–10% polyacrylamide gel. HA1-containing proteins were detected by Western analysis as previously described (8). Lanes: 1 to 3, crude extracts; 4 to 6, GST incubated with crude extracts from EGY191/B42, EGY191/B42-DBF2, and EGY191/B42-CAF1, respectively; 7 to 9, same as 4 to 6, respectively, except crude extracts were incubated with GST-MOB1. The molecular mass of B42-CAF1 is 54 kDa.
Because the above-described MOB1-DBF2 interactions were analyzed with overproduced and hybrid proteins, we also examined the interaction of DBF2 and MOB1 at their physiological concentrations under the control of their own promoters. Using a c-myc-tagged DBF2 protein, anti-MOB1 antibody was used to immunoprecipitate MOB1 from crude extracts, and the presence of DBF2–c-myc was detected by Western analysis with c-myc antibody. DBF2–c-myc specifically coimmunoprecipitated with MOB1 (Fig. 3, lane 6), whereas MOB1 preimmune serum failed to immunoprecipitate DBF2–c-myc. Conversely, immunoprecipitation of DBF2–c-myc with c-myc antibody brought down MOB1 (Fig. 3, lane 8). As a control, the c-myc antibody did not immunoprecipitate MOB1 from extracts that contained MOB1 (lane 1) but lacked DBF2–c-myc (lane 7). These results confirm that DBF2 and MOB1 bind to each other in vivo under physiological conditions.
FIG. 3.

DBF2 binds to MOB1 at physiological concentrations. Extracts from strains S7-4A (wt) and S7-4A-c-myc were incubated with either preimmune MOB1 serum (pI) (lanes 3 and 4), anti-MOB1 antibody (lanes 5 and 6), or anti-c-myc antibody (lanes 7 and 8), and resulting immunoprecipitates (Ip) were subjected to electrophoresis on an SDS–8% polyacrylamide gel. Western analysis was conducted as described previously (8). The upper portion of the blot was probed with c-myc antibody (Ab), and the lower portion was probed with MOB1 antibody. Lanes 1 and 2, crude extracts (CE) containing DBF2-c-myc (lane 2) and/or MOB1 (lanes 1 and 2). DBF2-c-myc is 62 kDa and MOB1 is 34 kDa in size.
We further examined whether the MOB1-DBF2 interaction was direct. We tested the ability of the GST-MOB1 fusion purified from E. coli to bind to radiolabeled in vitro-translated DBF2. As shown in Fig. 4, GST-MOB1 was able to bind the DBF2 protein. DBF2 did not bind the control GST or GST-Vpu protein. In a control experiment, in vitro-translated luciferase was incubated with the GST-MOB1, GST-Vpu, and GST proteins individually, and no binding was observed in any of these cases. GST-MOB1 can therefore bind DBF2 alone, without the aid of other yeast proteins.
FIG. 4.

Binding of MOB1 to DBF2. (A) Coomassie-stained GST, GST-MOB1, and GST-Vpu. GST fusions were induced as described elsewhere (9), bound to glutathione-agarose beads, eluted from the beads by boiling, and fractionated on an SDS–8% polyacrylamide gel. (B) T7 fusion proteins were translated in vitro with [35S]methionine as described in Materials and Methods. One milliliter of each radioactive protein was separated by SDS-polyacrylamide gel electrophoresis and identified following fluorography. Ten milliliters of each in vitro-translated protein was incubated with 50 mg of a GST fusion and, after washing, eluted by boiling. Molecular masses (in kilodaltons) are indicated on the left.
MOB1 is cell cycle regulated.
Expression of MOB1 RNA across the cell cycle was analyzed to further relate MOB1 function to that of DBF2. RNA extracted from an α-factor-synchronized culture was analyzed by Northern analysis (Fig. 5). MOB1 mRNA expression was found to be cell cycle controlled in a manner similar to that observed for DBF2, occurring coincidentally with expression of CDC5 (data not shown), a gene expressed at the G2/M interphase (1, 13). The peak level for MOB1 mRNA was observed, however, to occur slightly before that of DBF2. Also, a significant level of MOB1 mRNA was found to be present throughout the cell cycle. This result suggests that MOB1 may play roles in addition to that in late mitosis, a conclusion consistent with binding of MOB1 to MPS1 and to the effects of mob1 on ploidy (18).
FIG. 5.
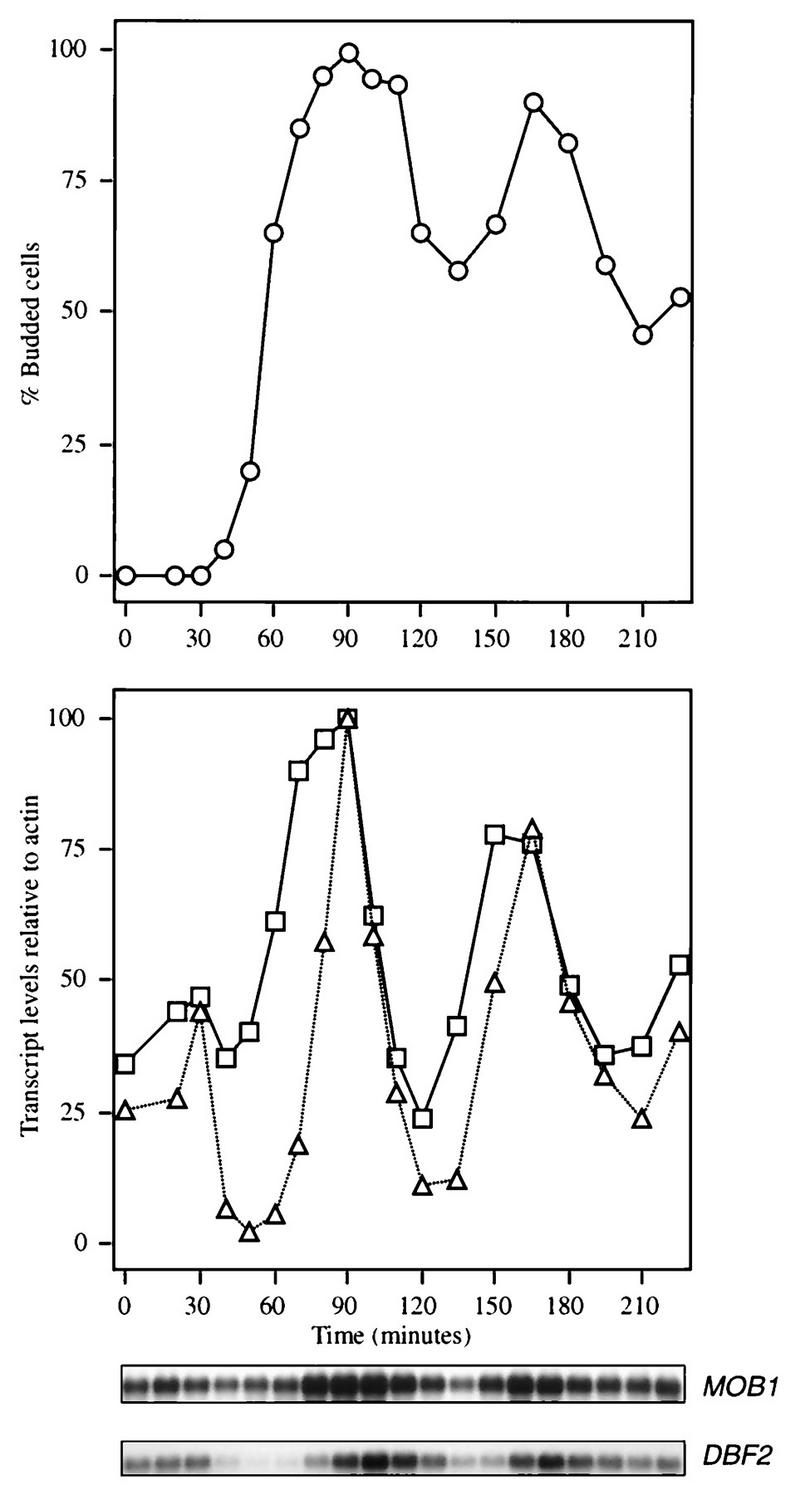
MOB1 is expressed under cell cycle control. A culture of strain CG378 was synchronized by use of α-factor, and samples were taken for RNA hybridization analysis to determine the levels of the MOB1 (□) and DBF2 (▵) transcripts (12). Actin transcript levels were also determined as a control and used to normalize the MOB1 and DBF2 levels in the graph. The percentages of buds in the synchronized population are also shown as an indication of culture synchrony (○).
DBF2 acts through MOB1 in affecting progression through mitosis.
The phenotypic similarity of MOB1 and DBF2 and their ability to bind each other suggest that MOB1 may be either regulated by or a regulator of DBF2. To investigate the genetic interactions between DBF2 and MOB1, we examined the effects of overexpression of DBF2 and MOB1 in strains containing mob1 and dbf2 defects, respectively. While overexpression of DBF2 as a B42-DBF2 fusion under the control of the GAL1 promoter (Table 3) complemented the temperature-sensitive and caffeine-sensitive phenotypes associated with mob1 temperature-sensitive alleles (Table 3), it failed to complement a mob1 deletion (Fig. 6). Overexpression of B42-MOB1(9–314) and B42-MOB1(79–314) did complement the mob1 knockout mutation. In contrast, overexpression of MOB1 was capable of complementing defects associated with a dbf2 deletion (Table 3). Two smaller fragments of MOB1 (145 to 314 and 79 to 314) also complemented a dbf2 defect when overexpressed, although it was found that B42-MOB1(145–314) displayed only a weakened complementation ability (data not shown). Overexpression of B42-CCR4 or B42-CAF1 did not complement any of these phenotypes (data not shown). In addition, the lethality caused by a dbf20 dbf2 double deletion was rescued by coexpression of LexA-MOB1(9–314): only dbf2 dbf20 segregants containing LexA-MOB1 were viable following sporulation and tetrad analysis of a diploid containing dbf2 and dbf20 alleles and LexA-MOB1. Such segregants were also shown to be unable to lose the LexA-MOB1-containing plasmid (data not shown). The LexA moiety was not responsible for complementing the dbf2 dbf20 double deletion, since overexpression of a GAL1-controlled GST-MOB1(79–314) (18) protein also allowed a dbf2 dbf20 strain to maintain viability (data not shown). These results suggest that a large dose of MOB1 bypasses the essential requirement for DBF2 and DBF20 and that DBF2 acts through MOB1 in regulating late mitosis.
TABLE 3.
Suppression analysis of MOB1 and DBF2
| Genotype | Suppression with plasmida:
|
|||||
|---|---|---|---|---|---|---|
| B42-MOB1
|
B42-DBF2
|
B42
|
||||
| 37°C | Caffeine | 37°C | Caffeine | 37°C | Caffeine | |
| mob1-77b | + | + | + | + | − | − |
| dbf2c | + | + | + | + | − | − |
Growth was monitored on yeast extract-peptone agar plates supplemented with 2% each galactose and raffinose and incubated at 37°C and on the same plates additionally supplemented with 8 mM caffeine but incubated at 30°C. B42-MOB1 refers to MOB1(9–314), although B42-MOB1(79–314) complemented as well. Similar results were obtained for a mob1-95 allele or for a dbf2 temperature-sensitive allele. +, suppression evident; −, no suppression evident.
Strain 1300-1a.
Strain 991-1-1b.
FIG. 6.

Ability of B42-DBF2 and B42-MOB1 to suppress the mob1 defect. Strain FLY59 mob1(pRS316-MOB1) was transformed with five different plasmids, expressing B42 alone, B42-DBF2, B42-MOB1(145–314), B42-MOB1(79–314), or B42-MOB1(9–314) as indicated. Transformants were grown overnight in medium lacking uracil, and about 15 × 104 cells of each strain, including the FLY59 strain without any B42 plasmids (−), were plated on medium containing fluoroorotic acid. The picture was taken after 72 h of incubation at 30°C.
To examine these genetic interactions further, a mutation in mob1-77 resulting in temperature sensitivity was combined with those in dbf2, dbf20, ccr4, and caf1 to identify potential synthetic phenotypes. No exacerbation of phenotypes or synthetic lethality was observed for any of these strains carrying mob1 temperature-sensitive alleles and the other mutated alleles except for the mob1-77 dbf2 combination. No meiotic segregants of this latter type were obtained (of 20 tetrads analyzed) unless the diploid also carried the plasmid expressing B42-MOB1 (data not shown). That is, mob1-77 dbf2 B42-MOB1 segregants were found to be viable. Such segregants were also found to be unable to lose the B42-MOB1 plasmid, confirming the lethality of mob1-77 dbf2. These results are consistent with the above-mentioned data showing that DBF2 aids or regulates MOB1 function.
Since our data indicate that MOB1 and DBF2 function together at the same stage of the cell cycle and DBF2 aids MOB1 functioning, we examined by a DBF2 protein kinase assay (17) whether MOB1 was an in vitro substrate of DBF2, as it is for MPS1 (18). When GST-MOB1 isolated from E. coli was incubated with B42-DBF2 that had been immunoprecipitated from yeast extracts, MOB1 protein was not phospholabeled, although H1 histone was capable of being phosphorylated (data not shown). It was also observed that MOB1 addition did not affect the ability of B42-DBF2 to phosphorylate H1 histone or to autophosphorylate, implying that MOB1 does not regulate B42-DBF2 protein kinase activity (data not shown).
Mutations in MOB1 block binding to DBF2 and DBF20 but not to MPS1.
To further analyze the interaction of MOB1 with DBF2, we examined the binding capabilities of different mutated MOB1 proteins. B42-MOB1 derivatives (residues 79 to 314) were constructed for wild-type and three mutated mob1 alleles. Two alleles, mob1-77 and mob1-95, result in a late mitotic block at the restrictive temperature when present either integrated into the genome or on a centromeric plasmid, whereas mob1-55 results in a late mitotic block on a centromeric plasmid but in an increase in ploidy when integrated into the genome (18). As shown in Table 4, the ability of LexA-DBF2 to interact with B42–MOB1-77 and B42–MOB1-95 was reduced 13- and 26-fold, respectively, by the mob1 mutations. In contrast, B42–MOB1-77 and B42–MOB1-95 were unaffected in their ability to interact with LexA-MPS1. B42–MOB1-55, which results in both an increase in ploidy and in a late mitotic block, reduced interaction with LexA-MPS1 by 2.5-fold and that with LexA-DBF2 by nearly 8-fold. No effect of these mutated MOB1 proteins on CAF1 binding was observed (data not shown). These results suggest that the late mitotic block conferred by the mob1-77 and -95 alleles results from defects in DBF2 binding.
TABLE 4.
Two-hybrid interactions of wild-type and mutated forms of MOB1 with DBF2, MPS1, and DBF20
| LexA fusiona | B42 fusionb | β-gal activity (U/mg)c |
|---|---|---|
| LexA-DBF2 | B42-MOB1 | 530 |
| LexA-DBF2 | B42–MOB1-55 (T85P, Q167R, Y183H) | 70 |
| LexA-DBF2 | B42–MOB1-77 (E151K)d | 40 |
| LexA-DBF2 | B42–MOB1-95 (L157P, A158I) | 20 |
| LexA-MPS1 | B42-MOB1 | 100 |
| LexA-MPS1 | B42–MOB1-55 (T85P, Q167R, Y183H) | 40 |
| LexA-MPS1 | B42–MOB1-77 (E151K)d | 120 |
| LexA-MPS1 | B42–MOB1-99 (L157P, A158I) | 120 |
| LexA-DBF20 | B42-MOB1 | 7.7 |
| LexA-DBF20 | B42–MOB1-55 (T85P, Q167R, Y183H) | <1 |
| LexA-DBF20 | B42–MOB1-77 (E151K)d | <1 |
| LexA-DBF20 | B42–MOB1-95 (L157P, A158I) | <1 |
LexA-DBF2 contains full-length DBF2(1–561), LexA-MPS1 contains full-length MPS1(1–764), LexA-DBF20 contains full-length DBF20(1–564), and B42-MOB1 fusions contain residues 79 to 314 of MOB1.
Specific mutations associated with the MOB moieties of the constructs are given in parentheses; e.g., T85P indicates a substitution of a proline for the threonine at position 85.
β-Galactosidase (β-Gal) activities represent the average of three to six separate determinations. The standard error of the mean was less than 15% in each case. Assays were conducted as described in Table 2. All B42-MOB1 fusions were found to be expressed to comparable levels, as determined by Western analysis, and the LexA fusions were comparably expressed as well (data not shown). Assays were conducted in EGY188/EGY191 diploids containing the p34 reporter.
The original mob1-77 temperature-sensitive mutant contained an additional mutation, N65I, but the E151K alteration is sufficient to make MOB1 unable to fully complement a mob1 defect (data not shown).
We have also analyzed whether DBF20, a close homolog of DBF2, can bind wild-type and temperature-sensitive versions of MOB1. As shown in Table 4, the β-galactosidase values for the interactions between LexA-DBF20 full-length and B42 fusions of MOB1 suggest that LexA-DBF20 can interact with B42-MOB1 but not with mutated versions of MOB1. We performed coimmunoprecipitation experiments with strains containing LexA-DBF20 and versions of B42 fusions with MOB1 to confirm a physical interaction between DBF20 and MOB1. We observed that B42-MOB1 coimmunoprecipitated with LexA-DBF20 (Fig. 7, lane 5) whereas the B42–MOB1-77 and B42–MOB1-95 fusions failed to bind LexA-DBF20 (lanes 7 and 6, respectively). The B42–MOB1-55 fusion displayed a decreased ability to bind LexA-DBF20. B42 alone and other B42 fusions did not coimmunoprecipitate with LexA-DBF20, and LexA alone did not bind B42-MOB1 (data not shown). Taken together, these data suggest that the temperature-sensitive mob1 versions are inactive at the restrictive temperature due to defects in binding both DBF2 and DBF20.
FIG. 7.

Coimmunoprecipitation of LexA-DBF20 with B42-MOB1 and B42-mob1 temperature-sensitive fusions. Extracts from diploid strain EGY188/EGY191 containing LexA-DBF20 (full length) and either B42-MOB1(79–314) (lane 1), B42–mob1-95 (lane 2), B42–mob1-77 (lane 3), or B42–mob1-55 (lane 4) were incubated with LexA antibody, and the resulting immunoprecipitates (Ip) were subjected to electrophoresis on an SDS–10% polyacrylamide gel. Western analysis was conducted as described previously (8), and the blot was probed with HA1 antibody. The positions of molecular mass markers (in kilodaltons) are indicated on the left.
We tested the ability of these temperature-sensitive mob1 proteins to activate transcription. As shown in Table 5, the ability of one of the mutants, LexA–MOB1-77, to activate transcription was increased by twofold. In contrast, the transactivation abilities of LexA–MOB1-55 and LexA–MOB1-95 were reduced by two- and sevenfold, respectively. These data indicate that the transactivation activity of temperature-sensitive mob1 proteins does not correlate with their ability to bind DBF2 or DBF20, in agreement with the previous results indicating that a dbf2 allele does not affect LexA-MOB1 transactivation function. Other proteins or interactions appear to be affected by these mutated MOB1 proteins.
The mob1 alleles were analyzed for several CCR4 transcriptionally related phenotypes. Strains containing mob1 alleles were also caffeine sensitive, a phenotype associated with dbf2, ccr4, and caf1 and indicative of a defect in cell wall integrity (data not shown) (17, 21). However, mob1 alleles were not cold sensitive, nor did they affect the ability of ADH2 to derepress (data not shown). We also tested the effect of mob1-77 and mob1-95 alleles on gene expression from several reporter genes, CYC1-lacZ, FKS1-lacZ, and HO-lacZ, that were affected by ccr4 and caf1 alleles (16). However, little or no effect of the mob1 alleles on these reporters was observed (data not shown).
DISCUSSION
MOB1 binds DBF2 and associates with the CCR4 transcriptional complex.
In this paper, we report the identification of a novel protein, MOB1, that interacts with the protein kinase DBF2. We demonstrated that MOB1 is physically associated with DBF2 in vivo and that these two proteins can physically interact in vitro in the absence of other yeast proteins. The two-hybrid interaction between MOB1 and DBF2 was similarly unaffected by deletion of CCR4 or CAF1, two components with which DBF2 interacts. The ability of GST-MOB1 to retain B42-DBF2 from a yeast extract under conditions in which CCR4 is not bound is a further indication of a fairly cohesive association between MOB1 and DBF2 in the absence of other associated proteins. Similarly, we found that MOB1 associated in vivo and coimmunoprecipitated with DBF20, the DBF2 homolog. This result is not surprising considering the extensive sequence homology between DBF2 and DBF20 (23).
MOB1 not only was capable of binding DBF2 but could physically interact with CAF1. Moreover, by the two-hybrid assay, MOB1 was found to interact with four CCR4 complex components: DBF2, CAF1, CAF16, and CAF17. The CCR4 complex consists of at least two recognizable forms, 1.2 × 106 and 1.9 × 106 Da in size (16). The smaller core complex consists of CCR4, CAF1, and the five NOT proteins (16). While we have not been able to demonstrate physical association of CCR4 and MOB1, MOB1 through these described interactions is likely to be a component of the CCR4 transcriptional complex, although perhaps of the larger complex. The fact that the LexA-MOB1 fusion was also able to activate transcription by itself, a phenotype shared by the LexA-CAF1 and LexA-CCR4 fusions (8), supports a role for MOB1 in transcription. We do not rule out the possibility, however, that MOB1 interacts with only a subset of the proteins of the CCR4 complex (such as DBF2 and CAF1), separate from the CCR4 complex, or that MOB1 has roles separate from the CCR4 complex, as in its association with MPS1.
DBF2 acts through MOB1.
The primary phenotype of a mob1 defect is a late mitotic block that is phenotypically very similar to that observed with dbf2 mutants (18). MOB1 mRNA expression, which is cell cycle regulated, was found to peak slightly in advance of the peak of DBF2 mRNA expression, consistent with a role for MOB1 in the late mitotic segment of the cell cycle. The physical association of MOB1 and DBF2 and their near coexpression during the cell cycle strongly suggest that they function together in regulating late mitosis. Our genetic analysis of MOB1 and DBF2 further suggests that MOB1 executes its function with the help of DBF2. Four observations support this: (i) overexpression of DBF2 complemented the temperature and caffeine sensitivities of a mob1 mutant allele but not the deletion of mob1, (ii) overexpression of MOB1 suppressed dbf2 defects, (iii) overexpression of MOB1 rescued the lethality caused by deleting both dbf2 and dbf20, and (iv) a combination of a mob1 temperature-sensitive allele and a dbf2 deletion was lethal. These analyses suggest that DBF2 regulates a crucial step in telophase through its interaction with MOB1. A role for MOB1 in late mitosis coincident with that of DBF2 is further supported by the observation that mob1 temperature-sensitive alleles were synthetically lethal with an lte1 deletion and with cdc5 and cdc15 conditional alleles (18). These other genes also function at telophase.
The functional interaction between MOB1 and DBF2 was further illuminated by our characterization of the mob1 temperature-sensitive alleles. Our finding that the proteins encoded by the mob1-77 and -95 alleles displayed weakened interactions with DBF2 and DBF20 suggests that the principal defect of these mutant alleles is their reduced ability to bind DBF2 and DBF20. Our model for the interaction between MOB1 and DBF2 states that MOB1 requires DBF2, and presumably DBF20, for its function. The observation that overexpression of MOB1 can suppress the late mitotic defect caused by a dbf2 deletion suggests that MOB1, when present in a relatively high concentration in the cell, can overcome a late mitotic block even without its normal regulator, DBF2. When only one copy of wild-type MOB1 is present in the dbf2 strain, the dbf2Δ defect is not lethal, apparently because DBF20 may partially substitute for DBF2 (23) and interact with MOB1. This results in a defect in late mitosis but not in lethality. On the other hand, the mob1 temperature-sensitive allele leads to the same kind of a late mitotic block as observed in a dbf2 strain, since DBF2 would have a weakened ability to bind the mutated version of MOB1. Similarly, overexpression of DBF2 in a strain with a mob1 temperature-sensitive allele overcomes the late mitotic block because under these conditions, mutated MOB1 protein can bind more of the DBF2 regulator and be able to perform its normal function. This model also explains why the mob1 temperature-sensitive allele combined with the dbf2 deletion is lethal: in the absence of DBF2, DBF20 is not able to bind mutated MOB1 as well as DBF2 does, and subsequently the defective MOB1 protein cannot fulfill its function. In agreement with this, it was observed in the two-hybrid system that LexA-DBF20 displays a much weaker interaction with B42-MOB1 than does LexA-DBF2 and that MOB1 mutations abrogate this weakened interaction. A couple of other observations support our model. First, the dbf2 dbf20 double knockout might be lethal since a single copy of wild-type MOB1 would not be able to function properly if neither DBF2 nor DBF20 could bind and/or regulate it. Second, overexpression of MOB1 would be able to rescue the dbf2 dbf20 phenotype for the same reason that it rescues the dbf2 defect (see above). In conclusion, this model strongly supports the idea that DBF2 functions through MOB1 and that this interaction is crucial for the telophase/G1 transition.
We also observed that the transactivation activity of the mutated MOB1 proteins does not correlate with their ability to interact with DBF2. Since MOB1 appears to function within a multiprotein complex, the MOB1 defects may result in altered interactions in addition to those caused by its effects on DBF2. While our results indicate that MOB1 is involved in the regulation of the cell cycle in late mitosis, the level of MOB1 mRNA was found to be significant throughout the entire cell cycle. This result suggests again that MOB1 may also be involved in other processes. The observations that MOB1 interacts with MPS1 and can be phosphorylated by it in vitro suggest that MOB1 has other contacts and sites of action.
ACKNOWLEDGMENTS
We thank H.-Y. Liu for helpful discussions throughout this project and Julie Farrell for technical assistance.
This research was supported by NIH grant GM41215, NSF grant MCB95-13412, Hatch project 291 to C.L.D., the Leukemia Society of America (F.C.L.), and NIH grant GM51312 and Pew Scholars Program in the Biomedical Sciences (P0020SC) to M.W.
Footnotes
Publication 1976 of the New Hampshire Agricultural Experiment Station.
REFERENCES
- 1.Althoefer H, Schleiffer A, Wassmann K, Nordheim A, Ammerer G. Mcm1 is required to coordinate G2-specific transcription in Saccharomyces cerevisiae. Mol Cell Biol. 1995;15:5917–5928. doi: 10.1128/mcb.15.11.5917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cook W J, Chase D, Audino D C, Denis C L. Dissection of the ADR1 protein reveals multiple, functionally redundant activation domains interspersed with inhibitory regions: evidence for a repressor binding to the ADR1c region. Mol Cell Biol. 1994;14:629–640. doi: 10.1128/mcb.14.1.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Costigan C, Kolodrubetz D, Snyder M. NHP6A and NHP6B, which encode HMG1-like proteins, are candidates for downstream components of the yeast SLT2 mitogen-activated protein kinase pathway. Mol Cell Biol. 1994;14:2391–2403. doi: 10.1128/mcb.14.4.2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chiang Y-H, Komarnitsky P, Chase D, Denis C L. ADR1 activation domains contact the histone acetyltransferase GCN5 and the core transcriptional factor TFIIB. J Cell Biol. 1996;271:32359–32365. doi: 10.1074/jbc.271.50.32359. [DOI] [PubMed] [Google Scholar]
- 5.Denis C L. Identification of new genes involved in the regulation of yeast alcohol dehydrogenase II. Genetics. 1984;108:833–834. doi: 10.1093/genetics/108.4.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Denis C L, Draper M P, Liu H-Y, Malvar T, Vallari R C, Cook W J. The yeast CCR4 protein is neither regulated by nor associated with the SPT6 and SPT10 proteins and forms a functionally distinct complex from that of the SNF/SWI transcription factors. Genetics. 1994;138:1–9. doi: 10.1093/genetics/138.4.1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Draper M P, Liu H-Y, Nelsbach A H, Mosley S P, Denis C L. CCR4 is a glucose-regulated transcription factor whose leucine-rich repeat binds several proteins important for placing CCR4 in its proper promoter context. Mol Cell Biol. 1994;14:4522–4531. doi: 10.1128/mcb.14.7.4522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Draper M P, Salvadore C, Denis C L. Identification of a mouse protein whose homolog in Saccharomyces cerevisiae is a component of the CCR4 transcriptional regulatory complex. Mol Cell Biol. 1995;15:3487–3495. doi: 10.1128/mcb.15.7.3487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guan K, Dixon J E. Eukaryotic proteins expressed in Escherichia coli: an improved thrombin cleavage and purification procedure of fusion proteins with glutathione S-transferase. Anal Biochem. 1991;192:262–267. doi: 10.1016/0003-2697(91)90534-z. [DOI] [PubMed] [Google Scholar]
- 10.Ito H, Fukuda Y, Murata K, Kimura A. Transformation of intact cells treated with alkali cations. J Bacteriol. 1983;153:163–168. doi: 10.1128/jb.153.1.163-168.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Johnston L H, Thomas A P. The isolation of new DNA synthesis mutants in the yeast Saccharomyces cerevisiae. Mol Gen Genet. 1982;186:439–444. doi: 10.1007/BF00729466. [DOI] [PubMed] [Google Scholar]
- 12.Johnston L H, Eberly S L, Chapman J W, Araki H, Sugino A. The product of the Saccharomyces cerevisiae cell cycle gene DBF2 has homology with protein kinases and is periodically expressed in the cell cycle. Mol Cell Biol. 1990;10:1358–1366. doi: 10.1128/mcb.10.4.1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kitada K, Johnson A L, Johnston L H, Sugino A. A multicopy suppressor gene of the Saccharomyces cerevisiae G1 cell cycle mutant gene dbf4 encodes a protein kinase and is identified as CDC5. Mol Cell Biol. 1993;13:4445–4457. doi: 10.1128/mcb.13.7.4445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lauze E, Stoelcker B, Luca F C, Weiss E, Schutz A R, Winey M. Yeast spindle pole body duplication gene MPS1 encodes an essential dual specificity protein kinase. EMBO J. 1995;14:1655–1663. doi: 10.1002/j.1460-2075.1995.tb07154.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu H, Krizek J, Bretscher A. Construction of a GAL1-regulated yeast cDNA expression library and its application to the identification of genes whose overexpression causes lethality in yeast. Genetics. 1992;132:665–673. doi: 10.1093/genetics/132.3.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu H-Y, Badarinarayana V, Audino D C, Rappsilber J, Mann M, Denis C L. The NOT proteins are part of the CCR4 transcriptional complex and affect gene expression both positively and negatively. EMBO J. 1998;17:1096–1196. doi: 10.1093/emboj/17.4.1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu H-Y, Toyn J H, Chiang Y-C, Draper M P, Johnston L H, Denis C L. DBF2, a cell cycle-regulated protein kinase, is physically and functionally associated with the CCR4 transcriptional regulatory complex. EMBO J. 1997;16:5289–5298. doi: 10.1093/emboj/16.17.5289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Luca F C, Winey M. MOB1, an essential yeast gene required for completion of mitosis and maintenance of ploidy. Mol Biol Cell. 1998;9:29–46. doi: 10.1091/mbc.9.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Malvar T, Biron R W, Kaback D B, Denis C L. The CCR4 protein from Saccharomyces cerevisiae contains a leucine-rich repeat region which is required for its control of ADH2 gene expression. Genetics. 1992;132:951–962. doi: 10.1093/genetics/132.4.951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Parkes V, Johnston L H. SPO12 and SIT4 suppress mutations in DBF2, which encodes a cell cycle protein kinase that is periodically expressed. Nucleic Acids Res. 1992;20:5617–5623. doi: 10.1093/nar/20.21.5617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Posas F, Casamayor A, Arino J. The PPZ protein phosphatases are involved in the maintenance of osmotic stability of yeast cells. FEBS Lett. 1993;318:282–286. doi: 10.1016/0014-5793(93)80529-4. [DOI] [PubMed] [Google Scholar]
- 22.Sakai A, Chibazakura T, Shimuzu Y, Hishinuma F. Molecular analysis of POP2 gene, a gene required for glucose-derepression of gene expression in Saccharomyces cerevisiae. Nucleic Acids Res. 1992;20:6227–6233. doi: 10.1093/nar/20.23.6227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Toyn J H, Araki A, Sugino A, Johnston L H. The cell-cycle-regulated budding yeast gene DBF2, encoding a putative protein kinase, has a homolog that is not under cell cycle control. Gene. 1991;102:63–70. doi: 10.1016/0378-1119(91)90465-n. [DOI] [PubMed] [Google Scholar]
- 24.Toyn J H, Johnston L H. Spo12 is a limiting factor that interacts with the cell cycle protein kinases Dbf2 and Dbf20, which are involved in mitotic chromatin disjunction. Genetics. 1993;135:936–971. doi: 10.1093/genetics/135.4.963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Toyn J H, Johnston L H. The Dbf2 and Dbf20 protein kinases of budding yeast are activated after metaphase to anaphase cell cycle transition. EMBO J. 1994;13:1103–1113. doi: 10.1002/j.1460-2075.1994.tb06359.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weiss E, Winey M. The Saccharomyces cerevisiae spindle pole body duplication gene MPS1 is part of a mitotic checkpoint. J Cell Biol. 1996;132:111–123. doi: 10.1083/jcb.132.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Winey M, Goetsch L, Baum P, Byers B. MPS1 and MPS2: novel yeast genes defining distinct steps of spindle pole body duplication. J Cell Biol. 1991;114:745–754. doi: 10.1083/jcb.114.4.745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zervos A S, Gyuris J, Brent R. Mwi1, a protein that specifically interacts with Max to bind Myc-Max recognition sites. Cell. 1993;72:232–233. doi: 10.1016/0092-8674(93)90662-a. [DOI] [PubMed] [Google Scholar]