

Abstract
C/EBPδ (CCAAT/enhancer binding protein δ) has been implicated as a regulator of acute-phase response (APR) genes in hepatocytes. Its expression increases dramatically in liver during the APR and can be induced in hepatic cell lines by interleukin-6 (IL-6), an acute-phase mediator that activates transcription of many APR genes. Here we have investigated the mechanism by which C/EBPδ expression is regulated by IL-6 in hepatoma cells. C/EBPδ promoter sequences to −125 bp are sufficient for IL-6 inducibility of a reporter gene and include an APR element (APRE) that is essential for IL-6 responsiveness. DNA binding experiments and transactivation assays demonstrate that Stat3, but not Stat1, interacts with this APRE. Two Sp1 sites, one of which is adjacent to the APRE, are required for IL-6 induction and transactivation by Stat3. Thus, Stat3 and Sp1 function cooperatively to activate the C/EBPδ promoter. Replacement of the APRE with Stat binding elements (SBEs) from the ICAM-1 or C/EBPβ promoter, both of which recognize both Stat1 and Stat3, confers responsiveness to gamma interferon, a cytokine that selectively activates Stat1. Sequence comparisons suggest that the distinct Stat binding specificities of the C/EBPδ and C/EBPβ SBEs are determined primarily by a single base pair difference. Our findings indicate that the cytokine specificity of C/EBPδ gene expression is governed by the APRE sequence.
Inflammation is a physiological response to tissue injury, trauma, or infection and consists of a systemic reaction to combat further tissue damage, destroy infective organisms, and activate repair processes. The early stage of inflammation, during which metabolic and catabolic changes occur in many organs, is known as the acute-phase response (APR). The APR is characterized by changes in the levels of several serum acute-phase (AP) proteins, which are synthesized primarily in the liver. Serum concentrations of some AP proteins increase as much as 1,000-fold several hours after onset of the APR (44). Activation of AP genes in hepatocytes is triggered by several inflammatory signals, including interleukin-6 (IL-6), IL-1, tumor necrosis factor alpha, and gamma interferon (IFN-γ) (reviewed in references 29 and 34). Of the numerous cytokines and growth factors that are involved in the upregulation of AP gene expression, IL-6 is considered to be the major mediator. This conclusion is supported by (i) a correlation between increased serum IL-6 levels and changes in AP gene expression during the inflammatory response, (ii) the large number of AP proteins synthesized in response to IL-6, and (iii) the observation that the APR is impaired in mice lacking IL-6 (29).
A number of AP gene promoters have been characterized and shown to contain cis-acting elements that mediate responsiveness to IL-1, IL-6, or both. A small number of AP genes respond only to IL-1 and contain sequences that interact with the NF-κB family of transcription factors. The majority of AP genes are activated by the IL-6-type cytokines and are controlled by IL-6 responsive elements (IL-6REs). The IL-6REs can be classified into two groups. The first are recognized by members of the Stat (signal transducer and activator of transcription) family of transcription factors and conform to the consensus sequence TT(C/A)C(T/G)G(G/T)AA (15, 18, 47). These sequences, also called APR elements (APREs), bind Stat3 (originally designated APRF) in nuclear extracts from IL-6-treated hepatocytes (1, 47, 52). Many APREs also bind Stat1, which is activated by IL-6, IFN-γ, and a variety of other cytokines (14).
The second group of IL-6REs is recognized by the C/EBP subfamily of basis leucine zipper transcriptional activators and conform to the consensus sequence (A/T)(G/A)T(G/T)(A/G)NGNAA (34). Three closely related members of the C/EBP family, C/EBPα, C/EBPβ, and C/EBPδ, are differentially expressed in multiple tissues during the APR. Upon induction of the APR by lipopolysaccharide (LPS) administration in mice, the mRNA levels of C/EBPα decrease in liver, lung, and fat. However, C/EBPβ expression increases in tissues such as the spleen, fat, heart, and kidney, and C/EBPδ mRNA levels are dramatically induced in the liver, kidney, spleen, heart, and brain (2). Several lines of evidence implicate C/EBPδ as the most important member of the C/EBP family in activating transcription of APR genes. For example, in an experimental AP model in rabbits, C/EBPδ is the major induced DNA binding activity in liver extracts that binds to a critical cis-regulatory site in the serum amyloid A gene promoter (37). C/EBPδ mRNA is also rapidly induced by IL-6 in human hepatoma Hep3B cells and is the predominant IL-6-induced protein interacting with C/EBP sites in the promoters for complement C3 (23), hemopexin, haptoglobin, and C-reactive protein (36).
Based on these and other data, it has been proposed that both C/EBPβ and C/EBPδ contribute to AP gene induction in hepatocytes. However, the two C/EBP proteins are activated by different mechanisms. C/EBPβ is constitutively expressed in adult hepatocytes and appears to be activated mainly by posttranslational modification in response to mediators such as IL-6 (35). By contrast, expression of C/EBPδ mRNA and protein is negligible in normal liver tissue but is markedly upregulated by IL-6 during the APR (2, 36, 37). Despite this dramatic induction in the liver, however, little is known about the molecular mechanisms involved. Therefore, we have begun to investigate the molecular control of C/EBPδ gene transcription. Here we describe an analysis of the C/EBPδ promoter and the identification of cis-acting elements and cognate factors that regulate C/EBPδ expression in hepatic cells.
(Some of the research reported in this paper was originally published in the master’s degree thesis of Carrie A. Cantwell, sponsored by the Department of Biomedical Science, Hood College, Frederick, Md.)
MATERIALS AND METHODS
Cloning and sequencing of the C/EBPδ promoter region.
The C/EBPδ promoter region was obtained from a mouse (B6/CBA hybrid) partial Sau3AI genomic library in Lambda FIX II (Stratagene) as previously described (48). A 650-bp SmaI fragment spanning the C/EBPδ coding and promoter regions was subcloned into pBlueScript (pBS) and sequenced by the dideoxy method, using a commercial kit (U.S. Biochemicals). Additional sequences were obtained by using specific primers (primer walking). The DNA used to generate the C/EBPδ promoter-reporter plasmids was cloned from a mouse (129SV strain) partial Sau3AI genomic library in Lambda FIX II (Stratagene). The sequence from −127 to +12 of the 129SV allele was found to be identical to that from the B6/CBA strain.
C/EBPδ promoter-reporter constructs.
Unique restriction sites within the C/EBPδ promoter region were used to generate a set of progressive 5′ deletion mutants. PCR was used to introduce a BamHI site at position +12 relative to the C/EBPδ transcription start site. A region between a unique SpeI site at −322 bp and the +12-bp endpoint was amplified by using the primers p3.22 (+12 primer; 5′-GCCGTCGGATCCTTGGGCTGTCACCTC-3′) and p3.21 (SpeI primer; 5′-GACGGCTCTAGAGAACTGTTCTTGTAT-3′). The PCR product was digested with SpeI and BamHI and used for subsequent cloning steps.
(−729)-Luc.
A unique ScaI site at ∼730 bp and the SpeI site at −322 bp were used to generate an ∼400-bp fragment. This fragment, together with the SpeI-BamHI PCR fragment, was inserted in a three-way ligation into pBS digested with BamHI and EcoRV. A SalI-BamHI fragment was then released from this plasmid and inserted into the luciferase vector pXP2 (31), which had been digested with SalI and BglII.
(−322)-Luc.
The SpeI-BamHI fragment described above was inserted into pBS. This plasmid was digested with SacI and BamHI to release a 334-bp fragment, which was then cloned into pXP2 digested with SacI and BglII.
Additional 5′ deletion mutants [(−127)-Luc, (−81)-Luc, and (−36)-Luc] were generated by PCR, using 5′ primers that defined the deletion endpoint and introduced a SalI site for subsequent cloning steps. The 3′ primer was p3.22 and the 5′ primers were −127 (5′-GACGGCGTCGACGGGCAGAGGGCGGGTCGTTCCCAGCAGC-ACCCCAG-3′), −81 (5′-GACGGCGTCGACTCCGGTCTCCGACCCACTGGGGCCGGGG-3′), and −36 (5′-GACGGCGTCGACCTGGGGCTAGAAAAGGCGGCGGTCCGGC-3′). The PCR products were digested with SalI and BamHI and ligated into pXP2 digested with SalI and BglII. The sequences of all deletions and point mutants were confirmed by dideoxy sequencing.
Point mutations were introduced into putative transcription factor binding sites by using PCR mutagenesis.
APREm-Luc and Sp1(−117)m-Luc.
PCR was performed with 5′ mutagenic primers that introduce SalI at −127 bp and mutate the APRE or −117 Sp1 site. The 3′ primer was p3.22, and the mutagenic primers were APREm (5′-GACGGCGTCGACGGGCAGAGGGCGGGTCGTGTTTCACAGC-ACCCCAG-3′) and Sp1(−117)m (5′-GACGGCGTCGACGGGCAGAGAAGAAGTCGTTCCCAGCAGCACCCCAG-3′). The PCR products were digested with SalI and BamHI and ligated into pXP2 digested with SalI and BglII.
Sp1(−53)m-Luc.
This mutant was generated by two rounds of PCR amplification, using the four-primer mutagenesis procedure (19, 20). Reaction 1 used a 5′ mutagenic primer (21731; 5′-ACTGGGGCCGGAAGAAGGCGTGCG-3′) to mutate the Sp1 site at −53 bp and the 3′ primer p3.22. Reaction 2 used the 5′ primer −127 and a 3′ mutagenic primer (21732; 5′-CGCACGCCTTCTTCCGGCCCCAGT-3′). The PCR products from reactions 1 and 2 were mixed and used as the template in a second round of amplification using the −127 and p3.22 primers. The 139-bp product was digested with SalI and BamHI and cloned into pXP2 digested with SalI and BglII.
Sp1(−117/−53)m-Luc and APREm/Sp1(−53)m-Luc.
The Sp1(−53)m mutant was used as a template for PCR amplification using Sp1(−117)m or APREm as the 5′ primers and p3.22 as the 3′ primer. The ∼139-bp products were digested with SalI and BamHI and cloned into pXP2 digested with SalI and BglII (BamHI at +12).
Artificial promoter constructs.
Double-stranded oligonucleotides containing either the C/EBPδ or α2-macroglobulin (α2-m) APREs were concatenated and inserted upstream of the thymidine kinase (TK) promoter fused to luciferase (TK-Luc, which consists of pXP2 containing the TK promoter truncated to position −81 [31]). The oligonucleotides were designed to contain XhoI-SalI or BamHI-BglII ends to allow directional multimerization and cloning into the luciferase vector. Oligonucleotides used to generate (δAPRE)1-TK-Luc and (δAPRE)4-TK-Luc were C/EBPδ APRE top strand (5′-TCGACTCGTTCCCAGCAGCAC-3′) and C/EBPδ APRE bottom strand (5′-TCGAGTGCTGCTGGGAACGAG-3′). Oligonucleotides used to construct (α2-m)6-TK-Luc were α2-m APRE top strand (5′-GATCCTTCTGGGAATTCCTA-3′) and α2-m APRE bottom strand (5′-GATCTAGGAATTCCCAGAAG-3′).
APRE replacement mutants.
Constructs in which Stat binding elements (SBEs) from C/EBPβ or ICAM-1 were used to replace the C/EBPδ APRE [designated (−127)δ-(C/EBPβ)-Luc and (−127)δ-(ICAM)-Luc, respectively] were generated by PCR mutagenesis. Primer A1829 (5′-GACGGCGTCGACGGGCAGAGGGCGGGTCTTTCCCAGAAG-CACCCCAG-3′), which changes the C/EBPδ APRE to the C/EBPβ Stat site, or primer A1830 (5′-GACGGCGTCGACGGGCAGAGGGCGGGTCTTTCCCGGAAA-CACCCCAG-3′), which converts the C/EBPδ APRE to the ICAM SBE, was used with primer p3.22 to amplify the region between −127 and +12. The 139-bp products were digested with SalI and BamHI and ligated into pXP2 digested with SalI and BglII.
Stat expression vectors.
Expression vectors for Stat1 (5) and Stat3 (38) were kindly provided by David E. Levy (New York University School of Medicine, New York, N.Y.).
Cell culture and transfection.
The human hepatoma cell lines Hep3B and HepG2 were obtained from the American Type Culture Collection. The cells were maintained in minimum essential Eagle’s medium (EMEM; BioWhittaker) supplemented with nonessential amino acids, sodium pyruvate, and 10% fetal bovine serum (HyClone Laboratories, Inc.) in the presence of kanamycin, streptomycin, and penicillin (complete medium) at 5% CO2. Hep3B cells were plated at 2 × 105 cells/6-cm-diameter dish 24 h before transfection. One hour prior to transfection, the cells were fed with 3 ml of fresh medium. Transfections were performed by the Lipofectamine procedure (Gibco-BRL). For each dish, 2 μg of reporter plasmid and 0.25 μg of pRSV β-gal (6) were combined with OptiMEM I (Gibco-BRL) in a total volume of 300 μl. For cotransfection with the Stat expression plasmids; 0.4 μg of the vector DNA was included in the transfection. In a separate tube, 10 μl of Lipofectamine reagent was mixed with 290 μl of OptiMEM I. The two mixtures were combined and incubated at room temperature for 30 min, after which the cells were washed with OptiMEM I. Then 2.4 ml of OptiMEM I was added to the DNA-Lipofectamine, and the mixture was applied to the cells. The cells were incubated for 4.5 h at 37°C, washed with unsupplemented EMEM and fed with 3 ml of complete medium. Cells were incubated an additional 24 h, washed with OptiMEM I, fed with 3 ml of OptiMEM I, and incubated 16 to 18 h before IL-6 was added.
Cytokine treatments.
Recombinant human IL-6 (Peprotech) was resuspended in water and added at a final concentration of 100 ng/ml 4 h before harvest (for transfection assays) or 15 min prior to harvest (for nuclear extracts). Human IFN-γ, a gift from Daniel W. McVicar (National Cancer Institute, Frederick, Md.), was added at 500 U/ml for 4 h.
Luciferase and β-galactosidase assays.
Transfected cells were harvested for luciferase assays as follows. Plates were washed twice with 1× phosphate-buffered saline (PBS) and 150 μl of detergent lysis solution (100 mM potassium phosphate [pH 7.8], 0.2% Triton X-100, 1 mM dithiothreitol [DTT]; Clontech Laboratories, Inc.) was added per plate and incubated at room temperature for 5 to 10 min. Lysates were scraped into microcentrifuge tubes and centrifuged at 16,000 × g for 2 min. To perform luciferase assays, 100 μl of substrate A (Analytical Luminescence Laboratory) was placed in a cuvette, and 50 μl of cell extract was added, followed by 100 μl of substrate B. A tube luminometer (Monolight 2010 instrument; Analytical Luminescence Laboratory) was used to record the light emissions from the expressed luciferase at 10-s intervals. Background reading was determined by measuring cell lysate from mock-transfected cells from two independent dishes. β-Galactosidase activity, which was used as an internal standard for transfection efficiency, was assayed according to the protocol for Luminescent β-galactosidase Genetic Reporter System II (Clontech Laboratories). The tube luminometer was used to record the light emissions from the cleaved galactoside at 5-s intervals. The linear range of the assay was determined for each individual experiment by assaying 0.5, 1.0, 2.0, and 4.0 μl of a cell lysate from pRSV β-gal-transfected cells. Background activity was determined by assaying mock-transfected cell lysates from two independent dishes.
Electrophoretic mobility shift assays (EMSAs) and supershift assays.
Nuclear extracts for Stat binding assays were prepared as follows. HepG2 cells were seeded at 2 × 106 per 150-mm-diameter dish and allowed to grow for 72 h. The cells were then washed with OptiMEM I, fed with 20 ml of OptiMEM I, and incubated for an additional 24 h. Human IL-6 was added at 100 ng/ml for 15 min. Extracts were prepared essentially as described by Sadowski and Gilman (39). The cells were washed twice with ice-cold PBS and once with ice-cold PBS containing 1 mM Na3VO4 and 5 mM NaF. Cells were then washed with hypotonic buffer (20 mM HEPES pH 7.9, 20 mM NaF, 1 mM Na3VO4, 1 mM Na4P2O7, 0.125 μM okadaic acid, 1 mM EDTA, 1 mM EGTA, 1 mM DTT, 0.5 mM phenylmethylsulfonyl fluoride, 1 μg of leupeptin per ml, 1 μg of aprotinin per ml, 1 μg of pepstatin per ml), and 300 μl of hypotonic buffer containing 0.2% Nonidet P-40 was added. Lysates were scraped into microcentrifuge tubes, and the nuclei were pelleted by centrifugation at 16,000 × g for 20 s at 4°C. The supernatant was removed, and the pellet was resuspended in 60 μl of high-salt buffer (420 mM NaCl, 20% glycerol, 20 mM NaF, 1 mM Na3VO4, 1 mM Na4P2O7, 0.125 μM okadaic acid, 1 mM EDTA, 1 mM EGTA, 1 mM DTT, 0.5 mM phenylmethylsulfonyl fluoride, 1 μg of leupeptin per ml, 1 μg of aprotinin per ml, 1 μg of pepstatin per ml) and gently rocked at 4°C for 30 min. Nuclear debris was removed by centrifugation at 16,000 × g and 4°C for 20 min. The extracts were aliquoted and stored at −70°C.
Nuclear extracts to analyze Sp1 binding were prepared essentially as described previously (16). Nuclei were prepared from HepG2 cells as described above and resuspended in nuclear lysis buffer (10 mM HEPES [pH 7.9], 100 mM KCl, 3 mM MgCl2, 0.1 mM EDTA, 1 mM DTT, 10% glycerol) supplemented with the phosphatase and protease inhibitors indicated above; 1/10 volume of 4 M (NH4)2SO4 was then added, and the nuclei were rocked at 4°C for 30 min. Samples were centrifuged at 4°C and 14,000 × g for 15 min, and the supernatants were aliquoted and stored at −70°C. Recombinant human Sp1 was obtained from Promega.
Binding reactions for EMSAs experiments were as follows. Eight micrograms (unless otherwise indicated) of nuclear protein was mixed with 4 μl of 5× binding buffer (65 mM HEPES [pH 7.9], 0.75 mM EDTA, 40% glycerol, 0.1% Nonidet P-40 [39]), 1 μg of poly(dI-dC) (Pharmacia Biotechnologies), 1 mM DTT, 0.06% bromophenol blue, and approximately 50,000 cpm (∼0.2 ng) of 32P-labeled probe in a total volume of 20 μl. The binding reactions shown in Fig. 7B included 10 μg of bovine serum albumin. The reaction mixtures were incubated at room temperature for 20 min, and 10 μl was loaded onto a 4% polyacrylamide gel in 0.25× Tris-borate-EDTA. Samples were separated by electrophoresis at 10 V/cm for approximately 2 h, transferred to 3MM paper (Whatman), dried under vacuum, and exposed to X-ray film. For competition binding assays, unlabeled oligonucleotides were added to the reaction in 10×, 30×, and 100× molar excess and incubated at room temperature for 5 min prior to addition of the probe. For supershift experiments, the antibodies (1 to 2 μg) were added to the reaction mixtures, which were then incubated at 4°C for 1 h prior to addition of probe. The following antibodies were purchased from Santa Cruz Biotechnology, Inc.: Stat1 p84/p91 (E-23), Stat3 (C-20), Sp1 (PEP2), and normal rabbit immunoglobulin G (normal rabbit serum [NRS]). A second Stat1 antibody (Stat1α p91), which was used in the experiment shown in Fig. 10A, was obtained from Chengrong Yu (NCI-Frederick Cancer Research and Development Center, Frederick, Md.).
FIG. 7.

Binding of nuclear proteins to the Sp1(−117) site. (A) Supershift analysis using an antibody (Ab) against Sp1. Nuclear extracts were prepared from control or IL-6-treated HepG2 cells by a method that maximizes the extraction of Sp1 (see Materials and Methods). The extracts were incubated with either NRS or an Sp1-specific antibody and the δAPRE/Sp1 or δAPRE probe. The supershift generated by the Sp1 antibody in lanes 2 and 4 is indicated. (B and C) Binding of recombinant Sp1 to the δAPRE/Sp1 (B) and δAPRE (C) probes. Recombinant human Sp1 (50 ng) was used alone (lanes 5 and 10) or mixed with 8 μg of HepG2 nuclear extract (optimized for Stat protein extraction) from control or IL-6-treated cells. Stat3 antibody was added to the indicated reactions. The lower panel is a longer exposure of the top portion of the gel to emphasize the Stat3 antibody supershift complex.
FIG. 10.


Replacement of the C/EBPδ APRE with SBEs from ICAM-1 or C/EBPβ renders the promoter responsive to IFN-γ. (A) The C/EBPβ promoter contains an SBE that binds Stat1 and Stat3. A probe containing the putative SBE from the C/EBPβ promoter and Stat1- or α-Stat3-specific antibody (Ab) (lanes 3 and 4, respectively) were added to nuclear extracts from control (lane 1) or IL-6 treated (lanes 2 to 4) HepG2 cells and the reactions analyzed by EMSA. Antibody supershift species are indicated. (B) Comparison of SBE sequences from the C/EBPδ, C/EBPβ, and ICAM-1 promoters. Bases that differ from the C/EBPδ APRE sequence are underlined. (C) IL-6 and IFN-γ responsiveness of SBE swap mutants. Constructs in which the C/EBPδ APRE was exchanged with SBEs from the C/EBPβ or ICAM-1 genes were generated. These constructs and (−127)-Luc were cotransfected with pRSV β-gal into Hep3B cells and assayed for basal expression and IL-6 or IFN-γ inducibility. The values represent the average of three independent experiments. Relative basal expression was normalized to the (−127)-Luc level.
The oligonucleotides used as EMSA probes were annealed and gel purified prior to labeling. The sequences of the upper strands of the oligonucleotides used were as follows: α2-m, 5′-GATCCTTCTGGGAATTCCTA; δAPRE, 5′-TCGACTCGTTCCCAGCAGCAC; δAPREm, 5′-TCGACTCGTGTTTCACAGCAC; δAPRE/Sp1, 5′-TCGACCAGAGGGCGGGTCGTTCCCAGCAGCACCC; and C/EBPβ Stat, 5′-GGGCATCTGTTTCCCAGAAGTTG. Double-stranded oligonucleotides containing 5′ overhanging ends (α2-m, BamHI/BglII; δAPRE, δAPREm, and δAPRE/Sp1, XhoI/SalI; C/EBPβ SBE, GGG) were labeled with Klenow polymerase and α[32P]-dCTP.
RNA preparation and Northern blot analysis.
Hep3B and HepG2 cells were grown until approximately 75% confluent. Human IL-6 (100 ng/ml) or IFN-γ (500 U/ml) was added for various time intervals, and cells were harvested for RNA. For cycloheximide inhibition experiments, the drug was resuspended in water and used at a final concentration of 100 μg/ml. For RNA preparation, the cells were washed twice with cold PBS, scraped into PBS, transferred to a microcentrifuge tube, and collected by centrifugation at 10,000 × g for 5 min. RNA preparation, electrophoresis, and blotting was performed essentially as described previously (11, 45).
DNA fragments used as probes for Northern blotting were labeled with [α-32P]dCTP, using the Prime-It II kit (Stratagene). The C/EBPδ probe was a HinfI-XhoI fragment corresponding to the 3′ untranslated region from a human C/EBPδ cDNA clone (T69326; Research Genetics, part of the IMAGE Consortium). The human C/EBPβ (NF-IL6) probe was a PstI-EcoRI fragment from a cDNA clone (kindly provided by David Ron, New York University Medical Center, New York, N.Y.). The c-Fos probe fragment was obtained from a cDNA clone (40). The cyclophilin probe was isolated as an EcoRI-HindIII fragment from plasmid pEMBL-cyclophilin (13).
RESULTS
IL-6-specific induction of C/EBPδ expression in hepatoma cells.
RNA was harvested over a time course from IL-6- or IFN-γ-stimulated Hep3B or HepG2 hepatoma cells, and C/EBPδ expression was analyzed by Northern blotting (Fig. 1A). C/EBPδ transcripts were weakly expressed in untreated cells but were highly induced within 1 h of IL-6 treatment in both cell lines. The elevated level of C/EBPδ expression was maintained for at least 8 h. C/EBPδ was induced much less efficiently and with delayed kinetics by another proinflammatory cytokine, IFN-γ. In contrast, C/EBPβ mRNA expression was only weakly stimulated by IL-6 but was more strongly activated by IFN-γ. Thus, the data show that C/EBPδ, but not C/EBPβ, mRNA levels increase in response to IL-6 in hepatic cells (confirming previous observations [36]) and that C/EBPδ and C/EBPβ respond differentially to IL-6 and IFN-γ.
FIG. 1.

Induction of C/EBPδ and C/EBPβ mRNAs by IL-6 or IFN-γ in hepatoma cell lines. (A) Northern blot analysis of 20 μg of total RNA from Hep3B or HepG2 cells cells treated with IL-6 or IFN-γ for the indicated times. Duplicate blots were hybridized with C/EBPδ or C/EBPβ probes and then with cyclophilin. (B) Northern blotting analysis of 20 μg of total RNAs from control Hep3B cells (lane 1), cells treated with cycloheximide (CHX) for 30, 60, and 120 min (lanes 2 to 4), and cells pretreated with cycloheximide for 30, 60, and 120 min followed by the addition of IL-6 for 2 h (lanes 5 to 7), IL-6 alone for 2 h (lane 8), or cycloheximide and IL-6 concurrently for 2 h (lane 9). The blots were hybridized sequentially with the indicated probes.
To determine if the increase in C/EBPδ mRNA was due to increased stability of C/EBPδ mRNA, we performed a cycloheximide inhibition experiment. Unstable messages, such as that of the c-fos gene, are frequently stabilized by protein synthesis inhibitors (3), presumably because proteins required for their degradation are labile. We therefore compared C/EBPδ and c-Fos mRNA expression in Hep3B cells treated with cycloheximide for various times in the presence or absence of IL-6 (Fig. 1B). C/EBPδ mRNA levels were only slightly increased upon the addition of cycloheximide (lanes 1 to 4). By contrast, IL-6 elicited a much larger induction (lane 8). Pretreatment with cycloheximide did not inhibit induction of C/EBPδ expression by IL-6 but rather potentiated it slightly (lanes 5 to 7). The blot was reprobed for c-Fos, whose mRNA is stabilized by cycloheximide. In contrast to C/EBPδ, c-Fos mRNA levels increased significantly in response to cycloheximide alone (middle panel, lanes 1 to 4). Moreover, IL-6 induction of c-Fos expression was dependent on mRNA stabilization by cycloheximide (lanes 5 to 8). These results allow two conclusions. First, the fact that cycloheximide alone had only a modest effect on C/EBPδ mRNA levels implies that the message is not labile, particularly in comparison to c-Fos mRNA. Second, the observation that cycloheximide failed to inhibit activation of C/EBPδ expression by IL-6 indicates that new protein synthesis is not required for this response. Although we have not assessed the transcription rate of the C/EBPδ gene directly, these data are consistent with the notion that increased expression of C/EBPδ mRNA occurs at the transcriptional level and involves the activation of a latent transcription factor(s).
Mapping an IL-6-responsive region in the C/EBPδ promoter.
We next undertook experiments to identify regulatory sequences that mediate induction of C/EBPδ expression by IL-6. Sequences 5′ to the C/EBPδ coding region were cloned from a mouse genomic DNA library, and approximately 750 bp were sequenced (Fig. 2). A set of 5′ deletion mutants with endpoints at −729, −322, −127, −81, and −36 bp relative to the transcription start site were generated (Fig. 2) and fused to a luciferase reporter gene. These constructs were transfected transiently into Hep3B cells, which were subsequently treated with IL-6 and harvested to assay luciferase activity. Luciferase expression was also determined in the absence of IL-6 treatment to assess the basal activity of the promoters.
FIG. 2.

DNA sequence of the murine C/EBPδ promoter and identification of several putative regulatory elements. The sequence extends from a ScaI site at −729 to position +12 relative to the transcription startsite. Endpoints of 5′ deletion mutants are indicated by brackets at −729, −322, −127, −81, and −36. Sequences corresponding to potential regulatory sites are boxed. The arrow denotes the transcription startsite (25).
Expression from the promoter-reporter constructs is shown in Fig. 3. Basal expression was similar for the −729, −322, and −127 constructs, but deletion to position −81 decreased promoter activity threefold. Removal of sequences to position −36, immediately 5′ to the TATA box, eliminated basal expression altogether. These results confirm that the sequences upstream of the coding region constitute a functional promoter and identify a segment between −127 and −36 that directs basal promoter activity. IL-6 treatment increased expression from the −729, −322, and −127 constructs approximately threefold. A similar level of induction was obtained with a construct containing ∼10 kb of 5′ flanking DNA (data not shown), indicating that distal regulatory sequences do not contribute to IL-6-dependent transcription. A significant decrease in IL-6 inducibility was observed when the promoter was truncated to position −81, and a similar reduction was seen with the −36 deletion. These data indicate that the region between −127 and −81 contains an IL-6RE.
FIG. 3.

Identification of an IL-6-responsive region within the C/EBPδ promoter. Luciferase reporter constructs containing the indicated 5′ promoter deletions were cotransfected with pRSV β-gal into Hep3B cells. The luciferase data were normalized to β-galactosidase values to control for differences in transfection efficiencies. The values represent averages ± standard deviations of three independent experiments. Basal luciferase expression levels from each construct are shown relative to the value for (−127)-Luc. Basal expression from (−36)-Luc was detectable but was rounded to 0.0. Fold induction represents luciferase activity after IL-6 treatment relative to the basal level.
Identification of specific regulatory sites within the C/EBPδ promoter.
Since the analysis of deletion mutants indicated the presence of an IL-6RE between −127 and −81, this region was inspected for known IL-6RE motifs (Fig. 2). The sequence between −100 to −114 closely resembles an APRE, which is recognized by members of the Stat family of proteins. Other potential transcription factor binding sites in the proximal promoter region include a cyclic AMP response element (CRE) at −40 bp and two sequences that match the consensus binding site for the transcriptional activator Sp1. The first Sp1 site is located at −53 bp, just 5′ to the CRE, and the second is located at −117 bp, immediately 5′ to the APRE.
To determine whether any of the aforementioned sites regulate basal or IL-6-induced transcription, we introduced point mutations into these sequences within the context of the −127 deletion (Fig. 4A). The mutant and wild-type (−127)-Luc plasmids were transfected into Hep3B cells and tested for IL-6 inducibility. The CRE mutation did not affect basal expression or IL-6 inducibility (data not shown), and this site was not investigated further. However, mutation of the APRE significantly impaired IL-6 inducibility, as did alteration of the Sp1 site adjacent to the APRE [Sp1(−117)]. The Sp1(−53)m mutation also diminished IL-6 inducibility, although to a lesser degree than the other mutants, while a construct containing mutations in both Sp1 sites displayed no IL-6 response. Inactivation of one or both Sp1 sites also reduced basal promoter activity. A construct containing mutations in both the APRE and the proximal Sp1 element [APREm/Sp1(−53)m] was also poorly activated by IL-6. Thus, induction of C/EBPδ promoter activity by IL-6 requires the APRE and at least one of the two Sp1 sites.
FIG. 4.

Analysis of APRE and Sp1 sites. (A) Diagram of mutations. The mutations were introduced into the APRE at −106 bp and the Sp1 sites at −117 and −53 bp. The wild-type sequences are indicated at the top, and the mutated sequences are shown below. The mutations were incorporated into the (−127)-Luc deletion construct. (B) Transient transfection assays of promoter mutants. The indicated promoter-reporter constructs were cotransfected with pRSV β-gal into Hep3B cells. The luciferase data were normalized to β-galactosidase activity. The values represent the averages of three to six independent experiments. The relative basal expression and fold induction values were determined as described in the legend to Fig. 3.
Identification of factors binding to the putative APRE and Sp1 sites.
We next used the EMSA to identify proteins in IL-6-treated cell extracts that bind to the C/EBPδ APRE and Sp1 sites. HepG2 cells were used for these experiments because Stat protein binding activity was reported to be higher in these cells than in Hep3B cells (50). We first examined whether the formation of Stat protein complexes bound to a probe containing the APRE from rat α2-m (12) could be competed by the C/EBPδ APRE. The α2-m APRE has been shown to bind Stat3 homodimers, Stat1 homodimers, and Stat1-Stat3 heterodimers from IL-6-stimulated HepG2 cells (26). Figure 5A shows that multiple α2-m APRE binding complexes are induced by IL-6 in HepG2 cells. Antibody supershift experiments confirmed that these complexes contain Stat3 (lane 4) and Stat1 (data not shown). Increasing amounts of unlabeled C/EBPδ APRE competitor inhibited formation of the Stat3 supershift complex (Fig. 5B, lanes 3 to 5). However, a mutant C/EBPδ APRE (δAPREm) did not compete for binding (lanes 6 to 8), showing that the interaction between the C/EBPδ APRE and Stat3 is specific.
FIG. 5.

The C/EBPδ APRE competes for binding of Stat3 to the α2-m APRE. (A) EMSA using the rat α2-m APRE, nuclear extracts (6.5 μg) from HepG2 cells, and NRS or Stat3-specific antibody (Ab) as indicated. The HepG2 cells were treated with IL-6 for 15 min. An upper complex (u) and a lower complex (l) appear in the IL-6-treated extracts (lane 3). The Stat3 antibody supershift complex (lane 4) is indicated. (B) Competition for Stat3 binding by the C/EBPδ APRE. Nuclear extracts (10 μg) from IL-6 treated HepG2 cells were incubated with Stat3-specific antibody and 10× (lanes 3 and 6), 30× (lanes 4 and 7), or 100× (lanes 5 and 8) molar excess of unlabeled wild-type (δAPRE) or mutant (δAPREm) binding site, as indicated. The rat α2-m APRE was used as a probe. The film was overexposed to emphasize the supershifted complex.
Using a labeled C/EBPδ APRE oligonucleotide as an EMSA probe, we were unable to observe a distinct DNA binding complex induced by IL-6, probably because the complex is obscured by a background binding activity (Fig. 6, lane 4). However, when the Stat3-specific antibody was included, a supershift complex appeared in reactions with extracts from IL-6-treated cells (lane 6) but not from control cells (lane 3). This Stat3 complex was not observed when a mutant C/EBPδ APRE probe was used (lane 12). In addition, no supershift signal was observed with a Stat1-specific antibody (lane 5), whereas Stat1 antibodies did supershift Stat1 complexes formed with the α2-m APRE (data not shown) and an APRE-like element in the C/EBPβ promoter (see Fig. 10A). These results show that the C/EBPδ APRE selectively binds Stat3, in contrast to the α2-m APRE, which binds both Stat1 and Stat3.
FIG. 6.
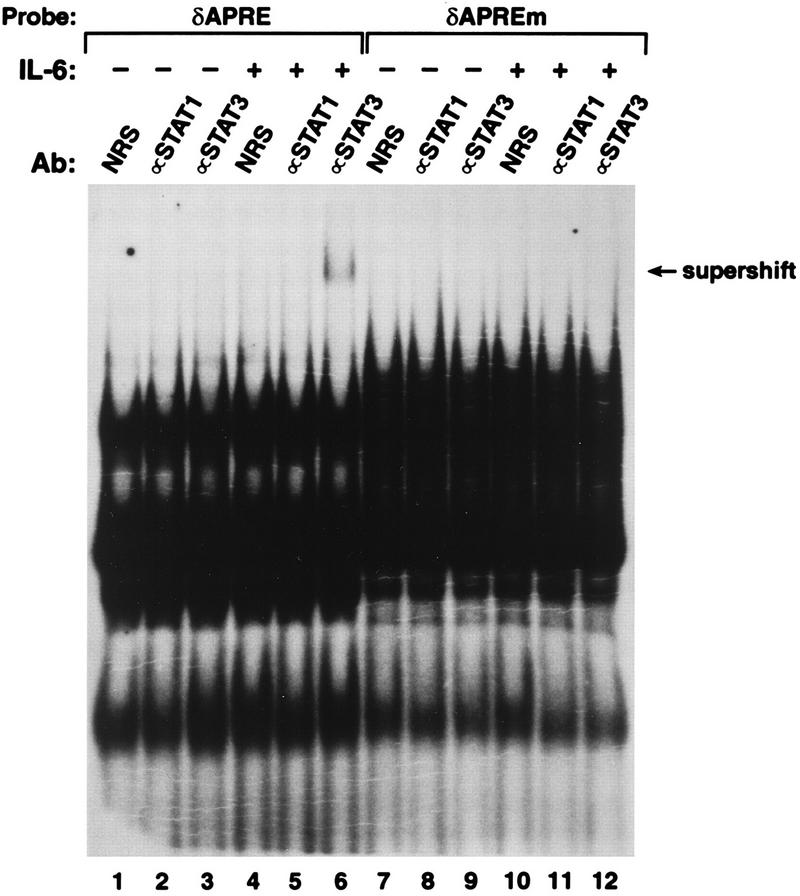
Selective binding of Stat3 to the C/EBPδ APRE. Nuclear extracts from control or IL-6-treated HepG2 cells were analyzed by EMSA using the wild-type or mutant C/EBPδ APRE probes and control antiserum or Stat1- or Stat3-specific antibody (Ab), as indicated. The film was overexposed to emphasize the supershift signal.
Analysis of Sp1 site mutants indicated that both Sp1 sites contribute to the activity of the C/EBPδ promoter and are required for IL-6 inducibility (Fig. 4). To determine whether Sp1 proteins bind to the distal Sp1 site [Sp1(−117)], EMSAs were performed with probes containing either the APRE and Sp1 sites (δAPRE/Sp1) or the APRE alone (δAPRE) and nuclear extracts from untreated or IL-6-stimulated cells (Fig. 7A). Several complexes that were not observed with the δAPRE oligonucleotide were formed with the δAPRE/Sp1 probe. These Sp1-specific bands may reflect the presence of multiple Sp1-like proteins in the nuclear extract (24). A supershifted species was detected when an antibody against human Sp1 was added to the δAPRE/Sp1 binding reaction (lanes 2 and 4). However, no supershift appeared with the δAPRE probe (lanes 6 and 8) or when control serum was used (lanes 1, 3, 5, and 8). Addition of recombinant human Sp1 to the nuclear extracts significantly increased the intensity of the slowest-migrating band (Fig. 7B, lanes 3, 4, 8, and 9), indicating that this complex corresponds to Sp1. In addition, recombinant Sp1 by itself also bound to the δAPRE/Sp1 probe (lanes 5 and 10), whereas there was no evidence of Sp1 binding to the δAPRE probe (Fig. 7C). Collectively, the results of Fig. 7 show that the Sp1(−117) element is a bona fide Sp1 binding site.
The existence of an Sp1 site adjacent to the APRE raised the possibility that cooperative binding interactions may occur between Stat3 and Sp1. Specifically, Sp1 might facilitate binding of Stat3 to the APRE, as was observed for Sp1 and Stat1 on the ICAM-1 promoter (28). However, using Stat3 antibody supershifts to assess binding, the presence of the Sp1 site (δAPRE/Sp1) did not increase Stat3 binding to the δAPRE (Fig. 7B and C, bottom panels). Thus, cooperative DNA binding between Sp1 and Stat3 is apparently not the basis for the synergism between the Sp1 and APRE sites observed in transfection assays.
The C/EBPδ APRE confers IL-6 inducibility to a heterologous promoter.
Having shown that the APRE is a critical regulatory element that specifically binds Stat3, we next examined whether the C/EBPδ APRE could confer IL-6 inducibility to a heterologous promoter. One or four copies of the APRE were inserted upstream of the TK promoter driving the luciferase gene and the resulting constructs were tested for activation by IL-6 (Fig. 8). One copy of the C/EBPδ APRE caused a slight increase in IL-6 responsiveness compared to the parental construct, TK-Luc. Multimerization of the C/EBPδ APRE further enhanced IL-6 induction of the reporter construct (1.9-fold) and also caused a significant increase in basal expression (8.7-fold over TK-Luc). By comparison, a construct bearing six copies of the α2-m APRE [(α2-m)6-TK-Luc] was induced 2.6-fold by IL-6 and exhibited a 2.6-fold increase in basal activity. It is unclear why the multimerized C/EBPδ APRE should function as a strong positive regulatory element in unstimulated cells, although perhaps oligomerization of the APRE sequence created a fortuitous binding site for an unidentified transcriptional regulator. Nonetheless, the data demonstrate that the C/EBPδ APRE functions as an IL-6RE when fused to a heterologous promoter.
FIG. 8.

The C/EBPδ APRE confers IL-6 responsiveness to a heterologous promoter. One or four copies of the C/EBPδ APRE or six copies of the α2-m APRE were inserted upstream of the TK promoter. The constructs were tested for induction by IL-6 after transfection into Hep3B cells as described in the legend to Fig. 3.
Stat3, but not Stat1, can transactivate the C/EBPδ promoter in IL-6-stimulated cells.
The in vitro binding data in Fig. 6 show that the C/EBPδ APRE preferentially interacts with Stat3 and not Stat1. To extend this observation, we tested Stat1 and Stat3 expression vectors (5, 38) for the ability to enhance IL-6 induction of the (−127)-Luc construct in transfected Hep3B cells (Fig. 9A). In the absence of IL-6, neither Stat protein stimulated C/EBPδ promoter activity. However, in IL-6-treated cells, Stat3 increased reporter expression 25-fold while Stat1 had no effect. The Stat1 vector stimulated IFN-γ-induced expression from a promoter containing an SBE from the ICAM-1 promoter (data not shown), demonstrating that a functional Stat1 protein is expressed from this plasmid (see also reference 5). We conclude that Stat3, but not Stat1, can transactivate the C/EBPδ promoter.
FIG. 9.
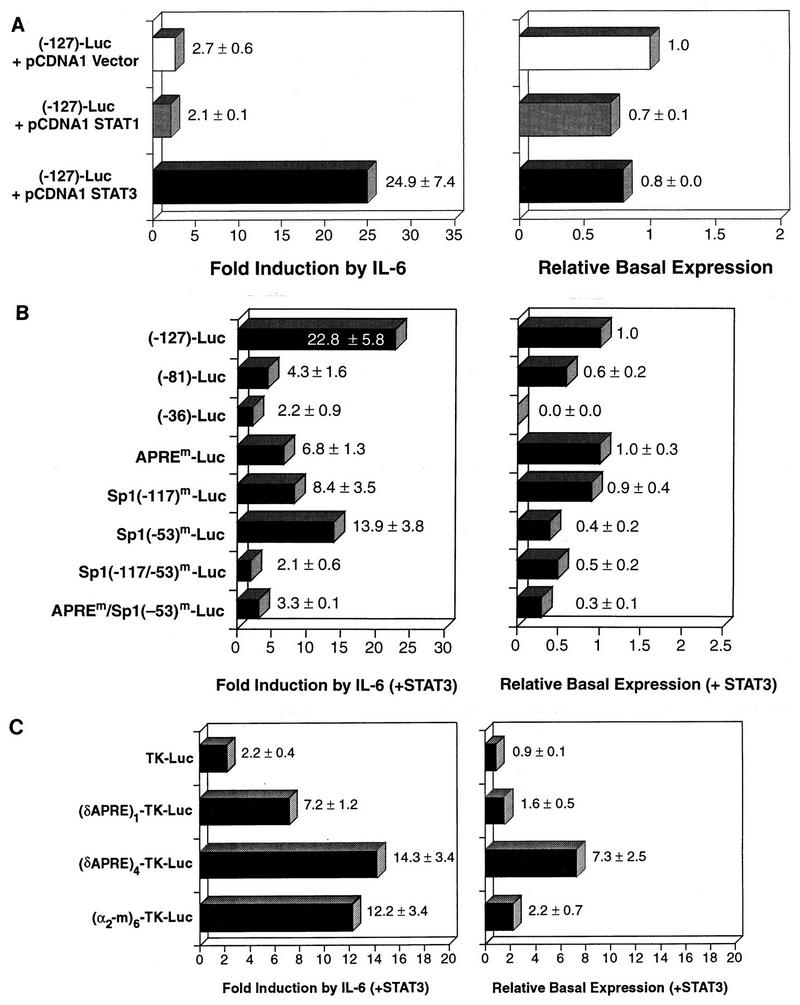
Stat3 mediates IL-6-induced expression from the C/EBPδ promoter. (A) Stat3 but not Stat1 transactivates the C/EBPδ promoter. The (−127)-Luc construct was cotransfected into Hep3B cells with expression vectors for Stat1 or Stat3 or the parental pCDNA1 vector, together with pRSV β-gal as an internal standard, and tested for basal and IL-6-induced luciferase expression. (B) Stat3 transactivation of C/EBPδ promoter mutants. The indicated deletion and point mutants (Fig. 3 and 4) were cotransfected with the Stat3 expression vector into Hep3B cells and tested for basal and IL-6-inducible luciferase expression. (C) Stat3 transactivates a heterologous promoter containing the C/EBPδ APRE. The indicated TK promoter-luciferase reporter constructs (Fig. 8) were cotransfected with the Stat3 expression plasmid into Hep3B cells and tested for basal and IL-6-inducible expression. The cell extracts were assayed for luciferase and β-galactosidase activities as described in Fig. 3. The values represent the averages of three to six independent experiments.
We next tested the ability of Stat3 to transactivate (−81)-Luc, (−36)-Luc, and APREm-Luc, in which the APRE site was either deleted or mutated, and the Sp1 mutants Sp1(−117)m-Luc, Sp1(−53)m-Luc, Sp1(−117/−53)m-Luc, and APREm/Sp1(−53)m-Luc (Fig. 9B). As the region containing the APRE and the adjacent Sp1 site was deleted, the ability of Stat3 to transactivate the promoter was severely decreased. Levels of IL-6-induced expression of (−81)-Luc and (−36)-Luc were reduced 5-fold and 10-fold, respectively, while that of APREm-Luc was reduced more than 3-fold. These results demonstrate that Stat3 acts primarily through the previously identified APRE site. However, the (−81)-Luc and the APREm constructs were still significantly activated by IL-6 in Stat3-transfected cells, compared to no induction without Stat3 (Fig. 3). One possible explanation is that overexpression of Stat3 allows it to interact with a cryptic (low-affinity) site in the promoter or the luciferase vector. Alternatively, Stat3 overexpression could stimulate transcription partly through interactions with proteins bound to the Sp1 sites, independent of its ability to bind DNA.
When the Sp1 sites were mutated individually, transactivation by Stat3 decreased 1.6- to 2.7-fold. However, IL-6 induction was almost completely eliminated in the Sp1(−117/153)m double mutant, demonstrating that at least one Sp1 site is essential for Stat3 transactivation. The APREm/Sp1(−53)m mutant was significantly less responsive to Stat3 transactivation than either of the single mutations, further supporting the notion that the APRE and Sp1 elements function cooperatively to mediate IL-6 induction.
We also examined the ability of Stat3 to transactivate constructs containing multiple copies of the C/EBPδ APRE linked to the TK promoter (Fig. 9C). In the presence of IL-6, Stat3 weakly stimulated the TK-Luc expression (twofold) but enhanced expression of (δAPRE)1-TK-Luc and (δAPRE)4-TK-Luc 7- and 14-fold, respectively. For comparison, (α2-m)6-TK-Luc expression was induced 12-fold. Thus, the C/EBPδ APRE confers Stat3 responsiveness to a heterologous promoter.
Cytokine specificity of the C/EBPδ promoter is governed by the APRE.
The data presented thus far show that IL-6 induction of the C/EBPδ promoter involves Stat3 activation and binding to the APRE. Another proinflammatory cytokine, IFN-γ, was a poor activator of C/EBPδ expression but was a more potent inducer of C/EBPβ mRNA than IL-6 (Fig. 1A). IFN-γ activates Stat1 but not Stat3 (53) and also did not stimulate expression from the (−127)-Luc construct (see below). To determine whether this cytokine specificity was dictated by the C/EBPδ APRE, we replaced the APRE sequence with SBEs from the intercellular adhesion molecule (ICAM-1) or C/EBPβ genes. IFN-γ activates the ICAM-1 gene through an interferon-responsive SBE that binds Stat1 (28). An SBE-like sequence is also present in the promoter region of the C/EBPβ gene, at position −366. As shown in Fig. 10A, the C/EBPβ SBE probe generated three EMSA complexes with nuclear extracts from IL-6-treated HepG2 cells (lane 2). Antibody supershift analysis (lanes 3 and 4) identified these as Stat1 (fast complex) and Stat3 (slow complex) homodimers and Stat1-Stat3 heterodimers (intermediate complex). Thus, the C/EBPβ SBE binds both Stat1 and Stat3 protein complexes in vitro.
The C/EBPδ APRE in (−127)-Luc was replaced by SBEs from the C/EBPβ and ICAM-1 genes (Fig. 10B) to generate the constructs (−127)δ-(C/EBPβ)-Luc and (−127)δ-(ICAM)-Luc, respectively. These reporter genes were tested for inducibility by IL-6 or IFN-γ in Hep3B cells (Fig. 10C). The wild-type (−127) promoter was activated by IL-6 (2.8-fold) but not by IFN-γ (0.82-fold). The (−127)δ-(C/EBPβ) promoter exhibited an increased response to IL-6 (4.4-fold) and was also modestly activated by IFN-γ (2.2-fold). The (−127)δ-(ICAM) promoter was more responsive to IL-6 (4.8-fold) than the wild-type promoter. In addition, expression from this construct was strongly activated by IFN-γ (8.1-fold). These findings demonstrate that the cytokine-specific response of the C/EBPδ promoter is determined by the APRE sequence, in particular its selective interaction with Stat3.
DISCUSSION
C/EBPδ expression is typically lacking in normal cells and tissues but can be induced by a variety of stimuli, including stress and inflammatory signals. In this study, we have focused on the mechanism by which IL-6 activates C/EBPδ gene expression in hepatocytes. We show that IL-6 induces Stat3 to bind to an APRE-like sequence in the C/EBPδ promoter, thereby upregulating hepatic expression of C/EBPδ. The C/EBPδ protein can subsequently bind to C/EBP sites in target AP genes and thus contribute to their transcriptional activation (23, 36, 37). C/EBPδ is therefore a component of a regulatory cascade that controls the synthesis of specific AP proteins in the liver. Promoters of some AP genes, such as C-reactive protein (50), may require both C/EBP and Stat proteins for transcriptional activation. The extent to which other AP genes are combinatorially regulated by these two classes of activators remains to be established.
cis-regulatory sequences mediating induction by IL-6.
IL-6 activation of the C/EBPδ promoter involves sequences located within the first 125 bp upstream of the transcription start site. At least three regulatory sites are required for induced transcription: an APRE centered at position −106 and a pair of Sp1 sites at −117 and −53 bp, respectively. Point mutations in the APRE effectively eliminated IL-6 responsiveness, and four copies of the APRE conferred IL-6 inducibility to a heterologous promoter. Mutation of the Sp1(−117) site also eliminated IL-6 induction, and inactivation of both Sp1 motifs or the APRE and Sp1(−53) sites abolished transactivation by Stat3. Thus, the function of the APRE is strongly dependent on nearby Sp1 elements in the promoter. In accordance with these findings, we found that Stat3 binds to the APRE and Sp1 recognizes the adjacent Sp1 site.
Our data indicate that Sp1 proteins participate in transcriptional synergism with Stat3 but do not promote its binding to the C/EBPδ promoter. It has been reported that Stat3 physically interacts with Sp1, as determined by coimmunoprecipitation assays (28). Thus, transcriptional cooperativity between Sp1 and Stat3 on the C/EBPδ promoter could involve contact between these two proteins, perhaps eliciting conformational changes that expose the activation domains of Stat3 and/or Sp1.
Cytokine specificity results from selective binding of Stat3 to the C/EBPδ APRE.
The C/EBPδ APRE competed for binding of Stat3 to the α2-m APRE, and Stat3 but not Stat1 bound to the C/EBPδ APRE in an IL-6-inducible manner. Stat3 also transactivated the C/EBPδ promoter in response to IL-6, whereas Stat1 did not exhibit this capability. These observations support the notion that Stat3 mediates the induction of C/EBPδ expression in cells exposed to IL-6. Although it is activated by IL-6 in hepatic cells, Stat1 apparently does not functionally interact with the C/EBPδ APRE. This conclusion is supported by our finding that IL-6, but not IFN-γ (which activates Stat1), induces C/EBPδ expression in two hepatoma cell lines. We propose that the preferential binding of Stat3, but not Stat1, to the C/EBPδ promoter limits the spectrum of cytokines and growth factors that can elicit C/EBPδ expression in hepatocytes.
What features of the C/EBPδ APRE determine its selective interaction with Stat3? A comparison of SBE sequences from C/EBPδ, C/EBPβ and several other Stat-regulated genes is shown in Table 1. Stat sites contain a core palindromic TT-AA motif, with a spacer of variable length between the TT and AA dinucleotides. It was previously proposed that the spacer length determines the specificity of Stat sites for the various Stat proteins (43). Our studies show that replacing the C/EBPδ APRE with SBEs from either ICAM-1 or C/EBPβ enables the promoter to respond to IFN-γ, presumably due to the ability of Stat1 to bind to the ICAM-1 (28) and C/EBPβ SBEs. Analysis of the sequences within the core palindromic TT-AA motif of the C/EBPδ and C/EBPβ SBEs reveals a single base change in the AA half-site, from CA in C/EBPδ to AA in C/EBPβ (a second difference in the C/EBPβ sequence occurs 5′ to the core palindrome [Fig. 10B]). The ICAM-1 SBE also features AA in this position. These correlations suggest that the AA dinucleotide is important in determining the ability to bind Stat1. In support of this idea, an SBE in the junB promoter also contains a CA dinucleotide and was found to bind Stat3 but not Stat1, as demonstrated by antibody supershift analysis and lack of a response of the promoter to IFN-γ (12).
TABLE 1.
Sequences and Stat binding properties of the C/EBPδ APRE and several known SBEs
| Gene | SBEa | Binding to:
|
Reference(s) | |
|---|---|---|---|---|
| Stat1 | Stat3 | |||
| Murine C/EBPδ | TCG TT CCC AGCA GCA | − | + | This study |
| Rat C/EBPβ | TGT TT CCC AGAA GTT | + | + | This study |
| Rat α2-m | GAA TT CCC AGAA GGA | + | + | 17, 27 |
| Human hemopexin | TTT TT CCC GGCA GGA | ND | ND | 22 |
| Rat α1-acid glycoprotein | TTT TT CCC AGAA GCC | ND | ND | 49 |
| Human haptoglobin | CTT TT CCA GTAA CAA | ND | ND | 32 |
| Human γ-fibrinogen | TTC TT CCA GTAC ATG | ND | + | 51 |
| Human α1-antichymotrypsin | CTA TT ACA GAAA ATT | ND | ND | 4 |
| Human C-reactive protein | CTC TT CCC GAAG CTC | ND | + | 50 |
| Human LBP | AGA TT CCC AGTG CAG | ND | + | 41 |
| Murine junB | CGC TT CCT GACA GTG | − | + | 12 |
| Human c-fos | CAG TT CCC GTCA ATC | + | + | 8 |
| Human ICAM-1 | GCT TT CCC GGAA ACC | + | + | 7, 28 |
SBEs from a number of cytokine-inducible genes are shown in alignment with the C/EBPδ APRE. The sequences were aligned through the core palindromic TT-AA motif (boldface). In cases where there is more than one possibility for the -AA motif, the alternative bases are boldface and underlined.
Binding (+) or lack of binding (−) of each SBE to Stat1 and Stat3, as determined by use of specific antibodies in supershift assays. ND, not determined.
Table 1 shows that the hemopexin and c-fos genes also contain SBEs that feature the CA dinucleotide in one half of the palindrome. The hemopexin site binds an IL-6-induced complex from HepG2 cells, although the selectivity of this site for Stat3 has not been established. The c-Fos serum-inducible element binds both Stat1 and Stat3, albeit with much lower affinity than a mutant version of the site (m67) in which the C has been converted to A. The ability of the wild-type sequence to bind Stat1 is possibly due to the occurrence of an AA dinucleotide immediately following the C residue. Alternatively, sequence differences at other positions may contribute to the Stat binding specificity of the serum-inducible element. However, our studies of the C/EBPδ and C/EBPβ SBEs suggest that a single base in a critical position within the TT-AA motif dictates specificity for Stat3 alone (CA) or both Stat1 and Stat3 (AA) and, correspondingly, which cytokines are capable of stimulating promoter activity. Thus, the C/EBPδ promoter is designed to respond specifically to IL-6 but not to IFN-γ.
We predict that other cytokines or growth factors that activate Stat3 will also induce C/EBPδ transcription. Indeed, granulocytic differentiation of the 32D c13 cell line in response to granulocyte colony-stimulating factor (G-CSF) is associated with increased C/EBPδ mRNA and protein expression (42). Since activation of the G-CSF receptor stimulates Stat3 phosphorylation and DNA binding activity (46), we propose that Stat3 contributes to the induction of C/EBPδ gene transcription in differentiating granulocytes. Because the C/EBPβ and C/EBPδ Stat elements differ in their protein binding properties, transcription of C/EBPβ may be regulated by distinct, although partially overlapping, signals. The functional importance of the C/EBPβ SBE remains to be demonstrated and is the subject of current investigation in our laboratory.
Multiple transcriptional regulatory elements in the C/EBPδ promoter.
Our analysis has identified an APRE, two Sp1 sites, and a CRE-like motif in the C/EBPδ proximal promoter region. To date we have not observed additional positive regulation by sequences upstream of position −127, including a construct containing 10 kb of 5′ DNA. Therefore, most or all of the relevant cis-regulatory signals that control basal and IL-6-induced transcription lie within 125 bp of the transcription start site. However, regulation by other inductive signals in different cell types may involve upstream sequences or other binding sites not identified in this study.
The CRE motif, which is located 8 bp 5′ of the TATA box, exhibits a five-of-eight-position match to a consensus CRE and includes one perfect half-site (GTCA). This element is similar to a pair of imperfect CREs recently identified in the C/EBPβ promoter that bind CREB and mediate basal promoter activity, as well as induction by the protein kinase A pathway (30). Point mutations inserted into the C/EBPδ CRE did not diminish the ability of IL-6 to induce C/EBPδ promoter activity (data not shown), indicating that this element does not participate in transcriptional regulation by IL-6. The neuropeptides vasoactive intestinal peptide and pituitary adenylate cyclase-activating peptide, in addition to the hormone noradrenaline, induce C/EBPδ expression in cortical astrocytes (10). Since each of these factors causes increased intracellular cyclic AMP levels, it is reasonable to propose that the C/EBPδ CRE motif plays a role in controlling C/EBPδ induction in response to these signals.
The C/EBPδ gene responds to a variety of extracellular signals. For example, C/EBPδ mRNA levels in liver and several other tissues increase dramatically in mice injected with LPS (2). Primary macrophages also induce C/EBPδ expression in response to LPS treatment (21). In addition, C/EBPδ expression is transiently activated during hormonally-induced differentiation of 3T3-L1 preadipocytes (9) and in mammary epithelial cells upon growth arrest elicited by serum withdrawal or contact inhibition (33). In all cases in which it has been examined, induction occurs at the mRNA level, indicating that the C/EBPδ gene promoter is activated by diverse signaling pathways. The C/EBPδ promoter constructs described here should be useful in future studies to elucidate the regulatory mechanisms underlying C/EBPδ induction by these various physiological signals.
ACKNOWLEDGMENTS
We are indebted to David Levy for providing Stat1 and Stat3 expression vectors, Chengrong Yu for providing a Stat1 antibody, Daniel McVicar for a gift of IFN-γ, and David Ron for providing a human C/EBPβ (NF-IL6) probe. We also thank Carla Weinstock and Hilda Marusiodis for expert secretarial assistance.
This research was sponsored by the National Cancer Institute under contract with ABL.
ADDENDUM IN PROOF
We note that Yamada et al. (J. Biochem. 121:731–738, 1997) have also identified the APRE site in the C/EBPδ promoter as an IL-6-inducible element that binds Stat3.
REFERENCES
- 1.Akira S, Nishio Y, Inoue M, Wang X J, Wei S, Matsusaka T, Yoshida K, Sudo T, Naruto M, Kishimoto T. Molecular cloning of APRF, a novel IFN-stimulated gene factor 3 p91-related transcription factor involved in the gp130-mediated signaling pathway. Cell. 1994;77:63–71. doi: 10.1016/0092-8674(94)90235-6. [DOI] [PubMed] [Google Scholar]
- 2.Alam T, An M R, Papaconstantinou J. Differential expression of three C/EBP isoforms in multiple tissues during the acute phase response. J Biol Chem. 1992;267:5021–5024. [PubMed] [Google Scholar]
- 3.Atwater J A, Wisdom R, Verma I M. Regulated mRNA stability. Annu Rev Genet. 1990;24:519–541. doi: 10.1146/annurev.ge.24.120190.002511. [DOI] [PubMed] [Google Scholar]
- 4.Bao J J, Sifers R N, Kidd V J, Ledley F D, Woo S L. Molecular evolution of serpins: homologous structure of the human alpha 1-antichymotrypsin and alpha 1-antitrypsin genes. Biochemistry. 1987;26:7755–7759. doi: 10.1021/bi00398a033. . (Erratum, 27:8508, 1988.) [DOI] [PubMed] [Google Scholar]
- 5.Bluyssen H A, Muzaffar R, Vlieststra R J, van der Made A C, Leung S, Stark G R, Kerr I M, Trapman J, Levy D E. Combinatorial association and abundance of components of interferon-stimulated gene factor 3 dictate the selectivity of interferon responses. Proc Natl Acad Sci USA. 1995;92:5645–5649. doi: 10.1073/pnas.92.12.5645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bonnerot C, Rocancourt D, Briand P, Grimber G, Nicolas J F. A β-galactosidase hybrid protein targeted to nuclei as a marker for developmental studies. Proc Natl Acad Sci USA. 1987;84:6795–6799. doi: 10.1073/pnas.84.19.6795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Caldenhoven E, van Dijk T, Raaijmakers J A, Lammers J W, Koenderman L, De Groot R P. Activation of the STAT3/acute phase response factor transcription factor by interleukin-5. J Biol Chem. 1995;270:25778–25784. doi: 10.1074/jbc.270.43.25778. [DOI] [PubMed] [Google Scholar]
- 8.Campbell G S, Meyer D J, Raz R, Levy D E, Schwartz J, Carter-Su C. Activation of acute phase response factor (APRF)/Stat3 transcription factor by growth hormone. J Biol Chem. 1995;270:3974–3979. doi: 10.1074/jbc.270.8.3974. [DOI] [PubMed] [Google Scholar]
- 9.Cao Z, Umek R M, McKnight S L. Regulated expression of three C/EBP isoforms during adipose conversion of 3T3-L1 cells. Genes Dev. 1991;5:1538–1552. doi: 10.1101/gad.5.9.1538. [DOI] [PubMed] [Google Scholar]
- 10.Cardinaux J R, Magistretti P J. Vasoactive intestinal peptide, pituitary adenylate cyclase-activating peptide, and noradrenaline induce the transcription factors CCAAT/enhancer binding protein (C/EBP)-beta and C/EBP delta in mouse cortical astrocytes: involvement in cAMP-regulated glycogen metabolism. J Neurosci. 1996;16:919–929. doi: 10.1523/JNEUROSCI.16-03-00919.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 12.Coffer P, Lutticken C, van Puijenbroek A, Klop-de Jonge M, Horn F, Kruijer W. Transcriptional regulation of the junB promoter: analysis of STAT-mediated signal transduction. Oncogene. 1995;10:985–994. [PubMed] [Google Scholar]
- 13.Danielson P E, Forss-Petter S, Brow M A, Calvatta L, Milner R J, Sutcliff J G. p1B15: a cDNA clone of the rat mRNA encoding cyclophilin. DNA. 1988;1:261–269. doi: 10.1089/dna.1988.7.261. [DOI] [PubMed] [Google Scholar]
- 14.Darnell J E., Jr The JAK-STAT pathway: summary of initial studies and recent advances. Recent Prog Horm Res. 1996;51:391–403. [PubMed] [Google Scholar]
- 15.Decker T, Kovarik P, Meinke A. GAS elements: a few nucleotides with a major impact on cytokine-induced gene expression. J Interferon Cytokine Res. 1997;17:121–134. doi: 10.1089/jir.1997.17.121. [DOI] [PubMed] [Google Scholar]
- 16.Gorski K, Carneiro M, Schibler U. Tissue-specific in vitro transcription from the mouse albumin promoter. Cell. 1986;47:767–776. doi: 10.1016/0092-8674(86)90519-2. [DOI] [PubMed] [Google Scholar]
- 17.Harroch S, Revel M, Chebath J. Interleukin-6 signaling via four transcription factors binding palindromic enhancers of different genes. J Biol Chem. 1994;269:26191–26195. [PubMed] [Google Scholar]
- 18.Hattori M, Abraham L J, Northemann W, Fey G H. Acute-phase reaction induces a specific complex between hepatic nuclear proteins and the interleukin 6 response element of the rat alpha 2-macroglobulin gene. Proc Natl Acad Sci USA. 1990;87:2364–2368. doi: 10.1073/pnas.87.6.2364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Higuchi R, Krummel B, Saiki R K. A general method of in vitro preparation and specific mutagenesis of DNA fragments: study of protein and DNA interactions. Nucleic Acids Res. 1988;16:7351–7367. doi: 10.1093/nar/16.15.7351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ho S N, Hunt H D, Horton R M, Pullen J K, Pease L R. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 1989;77:51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- 21.Hu, H.-M., M. Baer, S. C. Williams, P. F. Johnson, and R. C. Schwartz. Redundancy of C/EBPα, β, and δ in supporting the lipopolysaccharide-induced transcription of interleukin 6 and monocyte chemoattractant 1. J. Immunol., in press. [PubMed]
- 22.Immenschuh S, Nagae Y, Satoh H, Baumann H, Muller-Eberhard U. The rat and human hemopexin genes contain an identical interleukin-6 response element that is not a target of CAAT enhancer-binding protein isoforms. J Biol Chem. 1994;269:12654–12661. [PubMed] [Google Scholar]
- 23.Juan T S C, Wilson D R, Wilde M D, Darlington G J. Participation of the transcription factor-C/EBP-δ in the acute-phase regulation of the human gene for complement component C3. Proc Natl Acad Sci USA. 1993;90:2584–2588. doi: 10.1073/pnas.90.7.2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kingsley C, Winoto A. Cloning of GT box-binding proteins: a novel Sp1 multigene family regulating T-cell receptor gene expression. Mol Cell Biol. 1992;12:4251–4261. doi: 10.1128/mcb.12.10.4251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kinoshita S, Akira S, Kishimoto T. A member of the C/EBP family, NF-IL6 beta, forms a heterodimer and transcriptionally synergizes with NF-IL6. Proc Natl Acad Sci USA. 1992;89:1473–1476. doi: 10.1073/pnas.89.4.1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kojima H, Nakajima K, Hirano T. IL-6-inducible complexes on an IL-6 response element of the junB promoter contain Stat3 and 36 kDa CRE-like site binding protein(s) Oncogene. 1996;12:547–554. [PubMed] [Google Scholar]
- 27.Liu Z, Fuller G M. Detection of a novel transcription factor for the A alpha fibrinogen gene in response to interleukin-6. J Biol Chem. 1995;270:7580–7586. doi: 10.1074/jbc.270.13.7580. [DOI] [PubMed] [Google Scholar]
- 28.Look D C, Pelletier M R, Tidwell R M, Roswit W T, Holtzman M J. Stat1 depends on transcriptional synergy with Sp1. J Biol Chem. 1995;270:30264–30267. doi: 10.1074/jbc.270.51.30264. [DOI] [PubMed] [Google Scholar]
- 29.Mackiewicz A. Acute phase proteins and transformed cells. Int Rev Cytol. 1997;170:225–300. doi: 10.1016/s0074-7696(08)61623-x. [DOI] [PubMed] [Google Scholar]
- 30.Niehof M, Manns M P, Trautwein C. CREB controls LAP/C/EBPβ transcription. Mol Cell Biol. 1997;17:3600–3613. doi: 10.1128/mcb.17.7.3600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nordeen S K. Luciferase reporter gene vectors for analysis of promoters and enhancers. BioTechniques. 1988;6:454–457. [PubMed] [Google Scholar]
- 32.Oliviero S, Cortese R. The human haptoglobin gene promoter: interleukin-6-responsive elements interact with a DNA-binding protein induced by interleukin-6. EMBO J. 1989;8:1145–1151. doi: 10.1002/j.1460-2075.1989.tb03485.x. . (Erratum, 8:2121.) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.O’Rourke J, Yuan R, DeWille J. CCAAT/enhancer-binding protein-δ (C/EBP-δ) is induced in growth-arrested mouse mammary epithelial cells. J Biol Chem. 1997;272:6291–6296. doi: 10.1074/jbc.272.10.6291. [DOI] [PubMed] [Google Scholar]
- 34.Poli V, Ciliberto G. Transcriptional regulation of acute phase genes by IL-6 and related cytokines. In: Tronche F, Yaniv M, editors. Liver gene expression. R. G. Austin, Tex: Landes Company; 1994. pp. 131–151. [Google Scholar]
- 35.Poli V, Mancini F P, Cortese R. IL-6DBP, a nuclear protein involved in interleukin-6 signal transduction, defines a new family of leucine zipper proteins related to C/EBP. Cell. 1990;63:643–653. doi: 10.1016/0092-8674(90)90459-r. [DOI] [PubMed] [Google Scholar]
- 36.Ramji D P, Vitelli A, Tronche F, Cortese R, Ciliberto G. The 2 C/EBP isoforms, IL-6DBP/NF-IL6 and C/EBPdelta/NF-IL6beta, are induced by IL-6 to promote acute phase gene transcription via different mechanisms. Nucleic Acids Res. 1993;21:289–294. doi: 10.1093/nar/21.2.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ray A, Ray B K. Serum amyloid A gene expression under acute-phase conditions involves participation of inducible C/EBP-beta and C/EBP-delta and their activation by phosphorylation. Mol Cell Biol. 1994;14:4324–4332. doi: 10.1128/mcb.14.6.4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Raz R, Durbin J E, Levy D E. Acute phase response factor and additional members of the interferon-stimulated gene factor 3 family integrate diverse signals from cytokines, interferons, and growth factors. J Biol Chem. 1994;269:24391–24395. [PubMed] [Google Scholar]
- 39.Sadowski H B, Gilman M Z. Cell-free activation of a DNA-binding protein by epidermal growth factor. Nature. 1993;362:79–83. doi: 10.1038/362079a0. [DOI] [PubMed] [Google Scholar]
- 40.Sambucetti L C, Schaber M, Kramer R, Crowl R, Curran T. The fos gene product undergoes extensive post-translational modification in eukaryotic but not in prokaryotic cells. Gene. 1986;43:69–77. doi: 10.1016/0378-1119(86)90009-0. [DOI] [PubMed] [Google Scholar]
- 41.Schumann R R, Kirschning C J, Unbehaun A, Aberle H P, Knope H P, Lamping N, Ulevitch R J, Herrmann F. The lipopolysaccharide-binding protein is a secretory class 1 acute-phase protein whose gene is transcriptionally activated by APRF/STAT/3 and other cytokine-inducible nuclear proteins. Mol Cell Biol. 1996;16:3490–3503. doi: 10.1128/mcb.16.7.3490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Scott L M, Civin C I, Rorth P, Friedman A D. A novel temporal expression pattern of three C/EBP family members in differentiating myelomonocytic cells. Blood. 1992;80:1725–1735. [PubMed] [Google Scholar]
- 43.Seidel H M, Milocco L H, Lamb P, Darnell J E, Jr, Stein R B, Rosen J. Spacing of palindromic half sites as a determinant of selective STAT (signal transducers and activators of transcription) DNA binding and transcriptional activity. Proc Natl Acad Sci USA. 1995;92:3041–3045. doi: 10.1073/pnas.92.7.3041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Steel D M, Whitehead A S. The major acute phase reactants: C-reactive protein, serum amyloid P component and serum amyloid A protein. Immunol Today. 1994;15:81–88. doi: 10.1016/0167-5699(94)90138-4. [DOI] [PubMed] [Google Scholar]
- 45.Sterneck E, Kaplan D R, Johnson P F. Interleukin-6 induces expression of peripherin and cooperates with Trk receptor signaling to promote neuronal differentiation in PC12 cells. J Neurochem. 1996;67:1365–1374. doi: 10.1046/j.1471-4159.1996.67041365.x. [DOI] [PubMed] [Google Scholar]
- 46.Tian S S, Lamb P, Seidel H M, Stein R B, Rosen J. Rapid activation of the STAT3 transcription factor by granulocyte colony-stimulating factor. Blood. 1994;84:1760–1764. [PubMed] [Google Scholar]
- 47.Wegenka U M, Buschmann J, Lutticken C, Heinrich P C, Horn F. Acute-phase response factor, a nuclear factor binding to acute-phase response elements, is rapidly activated by interleukin-6 at the posttranslational level. Mol Cell Biol. 1993;13:276–288. doi: 10.1128/mcb.13.1.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Williams S C, Cantwell C A, Johnson P F. A family of C/EBP-related proteins capable of forming covalently linked leucine zipper dimers in vitro. Genes Dev. 1991;5:1553–1567. doi: 10.1101/gad.5.9.1553. [DOI] [PubMed] [Google Scholar]
- 49.Won K A, Baumann H. The cytokine response element of the rat alpha 1-acid glycoprotein gene is a complex of several interacting regulatory sequences. Mol Cell Biol. 1990;10:3965–3978. doi: 10.1128/mcb.10.8.3965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang D, Sun M, Samols D, Kushner I. STAT3 participates in transcriptional activation of the C-reactive protein gene by interleukin-6. J Biol Chem. 1996;271:9503–9509. doi: 10.1074/jbc.271.16.9503. [DOI] [PubMed] [Google Scholar]
- 51.Zhang Z, Fuentes N L, Fuller G M. Characterization of the IL-6 responsive elements in the gamma fibrinogen gene promoter. J Biol Chem. 1995;270:24287–24291. doi: 10.1074/jbc.270.41.24287. [DOI] [PubMed] [Google Scholar]
- 52.Zhong Z, Wen Z, Darnell J E., Jr Stat3: a STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science. 1994;264:95–98. doi: 10.1126/science.8140422. [DOI] [PubMed] [Google Scholar]
- 53.Zhong Z, Wen Z L, Darnell J E. Stat3 and Stat4: members of the family of signal transducers and activators of transcription. Proc Natl Acad Sci USA. 1994;91:4806–4810. doi: 10.1073/pnas.91.11.4806. [DOI] [PMC free article] [PubMed] [Google Scholar]