

Abstract
Phosphatidylinositol (PI) 3-kinase has been suggested to mediate cell survival. Consistent with this possibility, apoptosis of conditionally (simian virus 40 Tts) immortalized rat hippocampal H19-7 neuronal cells was increased in response to wortmannin, an inhibitor of PI 3-kinase. Downstream effectors of PI 3-kinase include Rac1, protein kinase C, and the serine-threonine kinase Akt (protein kinase B). Here, we show that activation of Akt is one mechanism by which PI 3-kinase can mediate survival of H19-7 cells during serum deprivation or differentiation. While ectopic expression of wild-type Akt (c-Akt) does not significantly enhance survival in H19-7 cells, expression of activated forms of Akt (v-Akt or myristoylated Akt) results in enhanced survival which can be comparable to that conferred by Bcl-2. Conversely, expression of a dominant-negative mutant of Akt accelerates cell death upon serum deprivation or differentiation. Finally, the results indicate that Akt can transduce a survival signal for differentiating neuronal cells through a mechanism that is independent of induction of Bcl-2 or Bcl-xL or inhibition of Jun kinase activity.
Programmed cell death is a characteristic of the normal developmental process as well as a response of cells to stress or other environmental insults (55, 59). While the mechanisms leading to programmed cell death are not yet understood, several factors have been implicated in apoptosis. For example, expression of exogenous bcl-2 or bcl-xL genes can substitute for neurotrophic survival factors by delaying or preventing apoptosis in primary neuronal cells and established neuronal cell lines, but the mechanism by which this occurs is not known (7, 34, 35, 37, 50, 62, 73). Moreover, p53, originally described as a tumor suppressor protein has been implicated in apoptosis following DNA damage as well as growth factor or nutrient deprivation in cells lacking Rb or expressing an activated oncogene such as c-myc (8, 15, 49, 67). The ICE (interleukin-1β-converting enzyme)-like family of cysteine proteases (also termed caspases [9]) has also been implicated in apoptosis in a wide range of cell types and species (25, 51, 68).
Neuronal cells undergo differentiation-induced apoptosis during development as a mechanism for eliminating nonessential cells (59, 66). Diverse factors have been implicated in neuronal survival, such as neurotrophic factors, glial cell-derived factors, and cell-cell contacts. However, the specific mechanisms by which these factors operate remain largely unknown (71). Treatment of PC12 cells, a pheochromocytoma cell line derived from a vascular tumor of adrenal medulla chromaffin tissue (36), with nerve growth factor (NGF) or fibroblast-derived growth factor (FGF) induces the cells to differentiate into cells with neuronal characteristics that survive in culture for an extended time. PC12 cells transfected with the platelet-derived growth factor (PDGF) receptor also differentiate in response to PDGF. These factors activate phosphatidylinositol (PI) 3-kinase, a lipid and protein kinase that triggers key signaling cascades in growth and development (22, 41). Studies based upon transfections of mutant PDGF receptors or addition of wortmannin, an inhibitor of PI 3-kinase (65), provided evidence that PI 3-kinase activation by NGF or PDGF may be responsible for the survival phenotype exhibited by PC12 cells in response to these factors (72).
Akt kinase, the cellular homolog of the viral oncoprotein v-Akt, is related to protein kinase C (PKC) within the catalytic domain. However, c-Akt differs from the PKC family members by the presence of a pleckstrin homology (PH) domain at its N terminus that is involved in the regulation of the activity of the enzyme by growth factors and intracellular signaling molecules (17). v-Akt results from the fusion of c-Akt and a retroviral Gag protein with the inclusion of an additional 21 amino acids derived from the translation of 63 nucleotides of the c-akt 5′ untranslated region placed in phase between Gag and Akt (10, 11). The myristoylation sites in the Gag sequence target Akt to the plasma membrane and result in high basal kinase activity (2, 43). It has recently been shown that phosphorylations at Thr308 and Ser473 are required for full activation of Akt kinase activity. These phosphorylations are mediated by upstream kinases that are regulated by phospholipid products of PI 3-kinase (6, 63). In addition, interactions of phospholipid with the PH domain of Akt may also be required for full activation (13, 16, 31, 32).
To investigate the potential role of Akt in the survival of differentiating neuronal cells, we utilized a cell line (H19-7) derived from E17 rat hippocampal cells that have been conditionally immortalized by expression of a temperature-sensitive simian virus 40 (SV40) large T antigen (Tts) (30). Thus, the H19-7 cells offer the advantage of temporary immortalization, enabling the cells to be propagated and transfected while they are immortalized and then differentiated to a neuronal phenotype in the absence of an immortalization signal. As is the case for PC12 cells and isolated hippocampal neuronal precursors (60), addition of FGF but not epidermal growth factor induces differentiation of H19-7 cells. The differentiated Tts-immortalized hippocampal cells do not divide in response to serum, express neuronal markers such as neurofilaments and brain type II sodium channels, and display action potentials (27, 28, 30). However, unlike PC12 cells, differentiated H19-7 cells undergo apoptosis within 2 to 6 days following induction of differentiation (26). They can be partially rescued from apoptosis by expression of Bcl-2 or Bcl-xL. The death of H19-7 cells during in vitro differentiation appears to reflect the in vivo process (33, 57, 69), illustrating the utility of these cells as a model system for neuronal cell death within the developing central nervous system.
In the present study, we demonstrate that treatment of differentiating H19-7 neuronal cells with wortmannin, a PI 3-kinase inhibitor, inhibits c-Akt activation and induces apoptosis. Expression of activated Akt rescues cells from death induced by wortmannin, serum deprivation, or neuronal differentiation, suggesting a mechanism whereby neurotrophic factors promote neuronal survival through the successive activation of PI 3-kinase and Akt. Ectopic expression of an Akt mutant which has a dominant-negative phenotype enhances the rate of cell death. Inhibition of apoptosis via the PI 3-kinase–Akt pathway appears to be independent of induction of Bcl-2 and Bcl-xL or suppression of Jun kinase activity.
MATERIALS AND METHODS
Cells.
The parental neuronal cell line H19-7 was derived from embryonic day 17 (E17) rat hippocampal cells by immortalization with SV40 Tts (30). Cell lines H19-7 and Bcl2-R10 have been described previously (26, 30). The immortalized cell lines were induced to differentiate by switching the cells from Dulbecco’s modified essential medium (DMEM) containing 10% fetal bovine serum (FBS) at 33°C, the permissive temperature for the Tts, to serum-free DMEM with N2 supplements (12) and 10 ng of basic FGF (bFGF) per ml at 39°C, the nonpermissive temperature for the Tts. PC12 cells were maintained in DMEM plus 10% FBS plus 5% horse serum. v-akt and c-akt constructs (32) were introduced into H19-7 or Bcl2-R10 cells along with pBabepuro (52) by standard CaPO4 transfection techniques (61). Colonies were isolated following selection in 1 μg of puromycin per ml.
Vectors.
The c-Akt and v-Akt vectors and the hemagglutinin (HA) antigen-tagged vectors used have been described previously (32, 43). The kinase-dead Akt (Akt kin−) is mutated at the ATP binding site (K179M) (10). For the retroviral vectors, the puromycin resistance gene was removed from pBabepuro (52) and replaced with enhanced green fluorescent protein (EGFP) (Clontech). Subsequently, c-Akt was cut from the pLXSN vector (2) at the 5′ EcoRI and 3′ BamHI sites, cloned into pSP73, cut out with BglII and BamHI, and finally cloned into the BamHI site of pBabe 5′ to EGFP. Myristoylated Akt (myrAkt) was removed from the retroviral SRα vector (3) as a BglII-BamHI fragment and cloned into the BamHI site of pBabeEGFP.
Cell viability determinations.
Viability was determined as previously described (26). Cells were plated at 105 per 35-mm-diameter well in DMEM plus 10% FBS. The following day, they were rinsed with phosphate-buffered saline (PBS) and shifted to differentiation or other test conditions. Cell counts for each cell line on day 1 following the shift are defined as 1.0 to normalize for any differences such as plating efficiency or proportion of cells undergoing division at the time of plating.
Immunoblotting.
Immunoblotting was performed as previously described (26) with 15 μg of protein from whole-cell lysates. On the immunoblots, the Akt forms were detected with polyclonal antiserum raised against the 15 C-terminal amino acids of Akt (32). Akt phosphorylated at Ser473 was detected with phospho-specific Akt antibody from New England BioLabs (Beverly, Mass.). On the same blots, total Akt was detected by using a phosphorylation-independent antibody raised against the same peptide as the phospho-specific Akt antibody. Bcl-2 was detected with polyclonal antiserum from Santa Cruz (N-19). Polyclonal antiserum to Bcl-x was obtained from C. Thompson (University of Chicago). On some immunoblots, the optical density was quantified by using an Ambis system.
Detection and quantification of apoptotic nuclei.
For nuclear staining, cells were fixed with 2% formalin in methanol (−20°C) for 10 min, rinsed three times with PBS, and stained with 1 μg of Hoechst 33258 (Molecular Probes) per ml in PBS for 10 min. For quantification, normal, condensed, and fragmented nuclei in 10 randomly chosen fields (20 on very sparse coverslips) were counted at ×40 magnification.
Kinase inhibitors.
Wortmannin (Sigma) was dissolved in dimethyl sulfoxide (DMSO) (2 mM) and stored at 4°C. PD098059 was dissolved in DMSO (10 mM) and stored at −80°C. For viability studies, immortalized cells were plated at 105 per 35-mm-diameter tissue culture well and differentiated for 2 days. Wortmannin or PD098059 in DMSO or DMSO alone was added, and the cells were cultured under differentiation conditions for an additional 24 h. The number of cells in each of triplicate wells was then determined for each concentration of inhibitor.
Microinjections.
H19-7 cells were plated on 15-mm poly-lysine-coated glass coverslips 2 days before injection. Cells were injected by using an Eppendorf (Madison, Wis.) Micromanipulator 5171 and Transjector 5246. Control vector DNA or Akt construct DNA was mixed with a green fluorescent protein (GFP) vector DNA (Green Lantern; Life Technologies, Gaithersburg, Md.), and the mixtures were injected into equal numbers of cells for each experimental construct. The final DNA concentration was 1 μg/μl in 50 mM HEPES (pH 7.4)–40 mM NaCl. Following injection, the coverslips were placed at 33°C in growth medium overnight. The next day, the number of green (GFP-expressing) cells was determined for each injection, and the coverslips were transferred to 39°C in N2 medium or in N2 medium plus 10 ng of bFGF per ml. Twenty-four hours later, the number of surviving green cells was determined; floating and other obviously dead cells were excluded from the counts. Although Hoechst 33258 staining of nuclei was used to detect apoptotic cells (see Fig. 1), this method was not quantitative because of the very rapid detachment and loss of the apoptotic H19-7 cells which are exacerbated by the fixation procedure for Hoechst staining. For each experiment, all the cells analyzed in a culture condition (i.e., N2 or differentiation) were resident on a single coverslip.
FIG. 1.


Apoptosis resulting from differentiation of H19-7 cells is increased by wortmannin. Cells were processed and counted as described in Materials and Methods. (A) Undifferentiated H19-7 cells (left panel) and H19-7 cells differentiated by 10 ng of FGF per ml in N2 medium at 39°C for 3 days (right panel). The cells were fixed and the nuclei were stained with Hoechst 33258. Examples of condensed or fragmented nuclei indicative of apoptosis are indicated (arrowheads). Scale bar = 100 μm. (B) Plot of the survival of H19-7 cells that were differentiated by FGF for 2 days and then treated with the indicated concentrations of wortmannin or PD098059. Cell survival was determined 24 h after treatment. Survival of untreated cells was defined as 100%. Each point is the mean ± standard deviation of triplicate samples. The results are representative of two (PD098059) or three (wortmannin) independent experiments.
Transfections.
For transient transfections of HA–v-Akt, 10 μg of vector DNA mixed with 40 μl of TransIT-LT1 (PanVera Corp., Madison, Wis.) was added to a 10-cm-diameter plate of cells according to the recommendations of the manufacturer. After 1 day, the cells were divided into the number of cultures required. After 24 h, the medium was changed to DMEM with N2 supplements at either 33°C, to maximize protein expression, or 39°C, as indicated. On the third day after transfection, the cells were treated with 1 μM okadaic acid and harvested for Akt assays as described below.
Transductions.
All retroviruses used expressed EGFP. Retroviral supernatants were added to 30% confluent cultures in the presence of 8 μg of hexadimethrine bromide (Sigma) per ml and incubated for 16 h at 33°C. The supernatants were replaced with fresh growth medium, and the cells were transferred to the required number of cultures. The cells were transferred to DMEM plus N2 supplements at 39°C with or without 10 ng of bFGF per ml to induce differentiation. After 4 days, the proportion of green cells in each control or treated culture was determined by counting the green cells in an equal number of randomly chosen microscopic fields for each culture. To verify ectopic Akt expression in these cells, a portion of each population was expanded, and cells expressing GFP were isolated by fluorescence-activated cell sorting. Protein extracts from those populations were analyzed by immunoblotting with anti-Akt antibody (32).
Immunocomplex in vitro kinase assays.
Akt kinase assays were carried out essentially as previously described (32). Protein concentrations in cell extracts were determined by the Bradford assay, and equal amounts of protein were used for each assay in an experiment. The antibodies, anti-C-terminal Akt (32) or anti-HA (12CA5; BAbCo, Richmond, Calif.), were precoupled to protein A-agarose beads for 1 h at 4°C with rotation, and immunoprecipitation was done at 4°C for 3 h. The Akt substrates used were a branched peptide of a modified PKC ɛ pseudosubstrate containing a phosphorylation site (14) or histone H2B (Boehringer Mannheim). In some cases, the resulting autoradiograph bands were quantified by optical density with an Ambis system.
For PI 3-kinase assays, PC12 or H19-7 cells at 70 to 80% confluency in 150-mm-diameter culture dishes were serum starved for 16 h and treated with NGF (100 ng/ml) or bFGF (10 ng/ml) for the times indicated in the figures. The cells were lysed with 1% Nonidet P-40 in 20 mM Tris (pH 7.5)–150 mM NaCl–5 mM EDTA at 4°C. Lysates were precleared with rabbit immunoglobulin G-agarose at 4°C for 15 min. PI 3-kinase activity was immunoprecipitated with antibodies to phosphotyrosine (05-321) from Upstate Biotechnology Inc., Lake Placid, N.Y., and protein A-agarose. The immunoprecipitates were washed three times with lysis buffer, once with Ca2+-free PBS, once with 100 mM Tris (pH 7.5)–0.5 M LiCl, once with H2O, and once with 10 mM Tris (pH 7.5)–100 mM NaCl–0.1 mM EDTA. All washes were done at 4°C, and all buffers and washes contained 10 μg of leupeptin per ml, 10 μg of aprotinin per ml, 200 μM phenylmethylsulfonyl fluoride, and 1 mM Na3VO4 added fresh. The 50-μl reaction mixture contained 10 mM Tris (pH 7.5), 100 mM NaCl, 20 mM MgCl2, 0.2 mM EGTA, 20 μg of PI, 10 μM ATP, 10 μCi of [γ-32P]ATP, and inhibitors as described above. The reactions were allowed to proceed for 20 min at room temperature. The reactions were terminated, and the lipids were extracted by addition of 100 μl of CHCl3-methanol (MeOH)-HCl (100:200:2) and mixing, followed by addition of 100 μl of CHCl3 and then 100 μl of H2O. The mixture was vortexed and centrifuged. The organic phase was collected and dried, then redissolved in 25 μl of CHCl3-MeOH (1:1), and spotted on thin-layer chromatography plates. The plate was developed with CHCl3-MeOH-H2O-NH4OH (43:38:7:5), dried, and exposed to X-Omat film (Kodak).
Jun kinase assays.
Cells were differentiated as described above. Day 0 samples were collected at 6 h following the shift to differentiation conditions. Assays of Jun kinase activities were carried out according to the solid-phase assay protocol of Hibi et al. (40). The glutathione S-transferase (GST)–c-Jun construct pGEX-3XJ1-93 was provided by E. Wattenberg (University of Minnesota). GST–c-Jun was purified from bacterial lysates by using a Pharmacia Biotech Bulk Purification Module according to the instructions provided by the manufacturer.
RESULTS
Wortmannin accelerates cell death in H19-7 cells.
To test whether PI 3-kinase might play a role in the survival of differentiating neuronal cells, we determined the effect of the PI 3-kinase inhibitor wortmannin on H19-7 cells induced to undergo differentiation. Following incubation for 2 days with 10 ng of FGF per ml at 39°C to induce differentiation, H19-7 cells were treated with 0, 50, or 200 nM wortmannin. As shown previously, differentiating H19-7 cells undergo apoptosis as manifested by terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling (TUNEL), condensed nuclei, and a decrease in cell survival (26) (Fig. 1A). After 1-day exposure to wortmannin, the survival of the differentiated H19-7 cells was further decreased by 25% (50 nM wortmannin) or 50% (200 nM wortmannin) relative to that of the untreated H19-7 cells (Fig. 1B). Wortmannin is not acting as a nonspecific toxic agent, since even 10 μM wortmannin had no effect on the survival of differentiated H19-7 cells expressing Bcl-2 (Bcl2-R10 [26]) (see Fig. 3F).
FIG. 3.

v-Akt protects against differentiation-induced cell death. v-Akt and c-Akt constructs were introduced into H19-7 or Bcl2-R10 cells as described in Materials and Methods. (A and B) Immunoblots of cell extracts from cell lines stably transfected with c-Akt or v-Akt; samples were immunoblotted with anti-Akt antibody for enhanced c-Akt or v-Akt expression, respectively. (C) Activity of Akt in wild-type H19-7 cells and H19-7 cells overexpressing c-Akt (HCAP-5). H19-7 and HCAP-5 cells were serum starved at 33°C in DMEM for 24 h and then pretreated with 1 μM okadaic acid for 15 min. Cultures were untreated (DMEM) or treated with 20% FBS for 5 min (Serum). Akt was assayed as described in Materials and Methods, and the final activity is expressed relative to Akt activity in untreated cells. These data are from two experiments. The error bars represent the range and in some cases are too small to be visible. (D) Activity of v-Akt transiently expressed in H19-7 cells. Epitope-tagged v-Akt or the vector control was transfected into H19-7 cells as described in Materials and Methods. The transfected populations were serum starved for 24 h in defined medium (N2) at 33 or 39°C. Cells transfected with HA–v-Akt or the control vector were treated for 25 min with 1 μM okadaic acid and then harvested for Akt assays as described in Materials and Methods. These data are from two independent experiments. The error bars represent the range and in some cases are too small to be visible. (E) Viability of cell lines expressing v-Akt and Bcl-2. The parental cell line (H19-7) and cell lines expressing v-Akt (HAP-5), Bcl-2 (Bcl2-R10), Bcl-2 and v-Akt (BAP-9), or Bcl-xL (XL-12) were differentiated and their viabilities were determined as described in Materials and Methods. Each time point represents the mean ± standard deviation of triplicate samples. In some cases, the standard deviation is too small to be seen beyond the margins of the symbols. These data are representative of at least two independent experiments. (F) Plot of survival of HAP-5 or Bcl2-R10 cells that were differentiated and treated with the indicated concentrations of wortmannin as described in Materials and Methods. Each point represents the mean ± standard deviation of triplicate samples. (G) Immunoblot comparing the levels of v-Akt in H19-7, HAP-5, and HAP-54 cells. Cells were grown and assayed as described in Materials and Methods. (H) Viability of cell lines expressing v-Akt and c-Akt. HCAP-5, a cell line expressing ectopic c-Akt, and HAP-54, a second line expressing v-Akt, were differentiated, and their viabilities were determined as described above. H19-7 and HAP-5 viability curves are included here for comparison. Each time point represents the mean ± standard deviation of triplicate samples. In some cases, the standard deviation is too small to be seen beyond the margins of the symbols.
It has previously been suggested that mitogen-activated protein (MAP) kinase mediates survival of some neuronal cells (70), and wortmannin has been shown to indirectly suppress MAP kinase activity in some cells (38). However, treatment of cells with 30 μM PD098059, an inhibitor of MAP kinase kinase (24, 58), decreases survival by only 17% (Fig. 1B). At this concentration, PD098059 inhibits the activation of MAP kinase completely in H19-7 cells (48). These results indicate that the effects of wortmannin on H19-7 cells cannot be attributed to suppression of MAP kinase activity and are consistent with a role for PI 3-kinase in the survival of neuronal cells upon differentiation by FGF.
Transient activation of Akt in differentiating H19-7 cells.
To determine whether the PI 3-kinase signaling pathway might mediate neuronal survival through activation of Akt, we first examined whether FGF activates PI 3-kinase and Akt in H19-7 cells. Analysis of PI 3-kinase activity showed threefold (mean ± standard deviation, 3.0 ± 1.0; n = 3) stimulation by 10 ng of FGF per ml (Fig. 2A). The level of endogenous Akt activity in H19-7 cells was determined before and after neuronal differentiation. H19-7 cells at 39°C were treated with 10 ng of FGF per ml in N2 medium and then assayed for Akt activity. Treatment of cells for 15 min with FGF resulted in activation of the Akt serine-threonine kinase when a PKC pseudosubstrate peptide or histone H2B (29) was used as a substrate (Fig. 2B, upper panel). The activation of Akt by FGF appeared to be transient. When lysates from H19-7 cells that had been differentiated for 3 days (39°, N2 medium, FGF) were assayed, no significant increase in Akt kinase activity over that observed in unstimulated cells was detected (Fig. 2B, upper panel). Pretreatment of cells with 200 nM wortmannin prior to growth factor addition suppressed FGF-induced activation of c-Akt, a finding consistent with earlier observations that Akt is activated via a PI 3-kinase-dependent pathway (Fig. 2B, lower panel).
FIG. 2.



PI 3-kinase and Akt activities in H19-7 cells. (A) Stimulation of PI 3-kinase activity by differentiation factors in H19-7 and PC12 cells. The cells were either untreated (U) or treated for 1 min with 10 ng of FGF per ml or 100 ng of NGF per ml. PI 3-kinase was immunoprecipitated from cell lysates with antiphosphotyrosine antibody and assayed for PI 3-K activity as described in Materials and Methods. The position of the PI 3-kinase product, PI 3-phosphate, is indicated (PIP). (B) (Upper panel) Stimulation of Akt activity by FGF in H19-7 cells. H19-7 cells were cultured in N2 medium at 39°C for 24 h and then untreated (U), treated for 15 min (15′ FGF), or differentiated for 3 days (3d. FGF) with 10 ng of FGF per ml. Endogenous Akt was immunoprecipitated from cell lysates and assayed by phosphorylation of a pseudosubstrate peptide as described in Materials and Methods. (Lower panel) Inhibition of FGF activation of Akt by wortmannin. Cells were cultured in DMEM at 39°C for 24 h and then untreated (U) or pretreated with 200 nM wortmannin (wort) for 10 min. In some samples, 10 ng of FGF per ml was added to untreated (FGF) and wortmannin-pretreated (wort FGF) cells, which were then incubated for 15 min. Akt activity was assayed as described above. These results are representative of two independent experiments. (C) Transient phosphorylation of Akt during H19-7 differentiation. Whole-cell extracts from cultures treated without FGF (N2) or with FGF for 15 min (15′) or 3 days (3d) were probed with an antibody specific for phosphorylated Akt (inset). The phosphorylated Akt bands were quantified by optical densitometry and normalized to the amounts of Akt protein detected on the same blot with the phosphorylation state-independent antibody (see Materials and Methods). These results are representative of three independent experiments.
The transient activation of Akt during differentiation was also demonstrated by monitoring Akt phosphorylation. It has recently been determined that growth factor-activated Akt is phosphorylated at a minimum of two sites, Thr308 and Ser473, and that a high level of Akt activation is dependent on phosphorylation at these two sites (5). An antibody that specifically recognizes Akt phosphorylated at Ser473 was used in order to determine the activation state of endogenous Akt. Lysates from cultures of cells that were either undifferentiated, treated for 15 min with FGF, or differentiated for 3 days with FGF were probed for phosphorylated Akt. The bands were quantified and normalized to total Akt protein. As shown in Fig. 2C, phosphorylated Akt is induced fourfold (mean ± standard deviation, 3.8 ± 0.6) within 15 min of FGF treatment. After 3 days of differentiation, the amount of phosphorylated Akt was decreased to 40% (39% ± 8%) of that detectable after 15 min of FGF treatment, reducing it to a level slightly above background. Thus, reduced Akt activity during differentiation results, at least in part, from dephosphorylation of the enzyme.
Activated Akt inhibits apoptosis of H19-7 cells.
To determine whether activation of Akt is able to promote survival, H19-7 cells were stably transfected with vectors expressing c-Akt, the wild-type enzyme, or v-Akt, a constitutively active enzyme (13, 32). We have shown previously (26) that the expression of ectopic Bcl-2 enhances survival of differentiating H19-7 cells. Since the action of Akt could be additive or synergistic to that of Bcl-2, v-Akt was also stably introduced into H19-7 cells expressing human Bcl-2 (Bcl2-R10 cells [26]). Akt expression was monitored by immunoblotting with anti-Akt antibody (Fig. 3A and B). Expression of ectopic c-Akt in the stably transfected H19-7 cell lines (e.g., HCAP-2, -5, and -6) was greater than that of the endogenous c-Akt (Fig. 3A). The levels of v-Akt in the highest-expressing clone (HAP-5) and in the Bcl-2-expressing H19-7 cells (e.g., BAP-2 and BAP-9) were low relative to that of the endogenous c-Akt (Fig. 3B), raising the possibility that a high level of expression of oncogenic Akt may not be well tolerated by these cells. c-Akt activity was assayed at 33°C following treatment with serum in the presence of okadaic acid to maximize Akt protein expression and activity (47) (Fig. 3C). The results showed higher Akt activity in HCAP-5 than in the parent H19-7 cell line, indicating that the ectopic c-Akt is functional. Although the low levels of v-Akt expression in these lines prevented detectable immunoprecipitation and assay of v-Akt activity, transient expression of HA-tagged v-Akt in H19-7 cells confirmed that the expressed protein is an active kinase in H19-7 cells (Fig. 3D), as observed previously for other cell lines (32, 43).
Analysis of differentiation-associated apoptosis indicated that H19-7 cells stably expressing v-Akt survive longer than the parental cell line. Whereas >90% of the H19-7 cells normally die after 5 days of FGF treatment in serum-free N2 medium at 39°C, 50% of the v-Akt-expressing HAP-5 cells were still viable at this time (Fig. 3E). This protection against differentiation-associated cell death due to v-Akt expression was comparable to that obtained by expression of Bcl-2 (Fig. 3E) or the Bcl-2-related protein Bcl-xL in H19-7 cells (26). Coexpression of v-Akt and Bcl-2 in H19-7 cells provided protection against differentiation-associated cell death similar to that of either v-Akt or Bcl-2 alone, although a small increase in survival was observed at early time points in two Bcl-2-expressing cell lines (Fig. 3E, BAP-9). Cells expressing v-Akt were also less sensitive to treatment with 200 nM wortmannin (Fig. 3F). This effect is presumably due to wortmannin-insensitive Akt activity, as observed in other v-Akt-expressing cells (43).
The extent of cell survival appeared to reflect the relative expression of v-Akt. Another clonal line (HAP-54) that expresses less v-Akt than HAP-5 exhibited a survival phenotype intermediate between those of HAP-5 and H19-7 (Fig. 3G and H). Expression of additional c-Akt in H19-7 cells conferred a slight increase in survival, but it was significantly less than that obtained with v-Akt (Fig. 3H). The failure of overexpressed c-Akt to significantly enhance survival can be explained by the inactivation of c-Akt that occurs during differentiation (Fig. 2B). Thus, the antiapoptotic action of v-Akt is not due simply to increased expression of Akt protein, since transfected c-Akt was expressed at significantly higher levels. Furthermore, expression of ectopic v-Akt or c-Akt did not affect the rate or extent of morphological differentiation of the cells or promote transformation, indicating that Akt is not acting by blocking the differentiation process (29). These results indicate that v-Akt can protect neuronal cells against differentiation-induced apoptosis, a process which is p53 independent (26, 69).
In order to further demonstrate that activated Akt enhances viability in H19-7 cells undergoing differentiation, we tested a second constitutively active Akt, myrAkt (3), which has previously been shown to enhance viability in other apoptotic systems (3, 43). Since the low v-Akt expression levels in the stable cell lines suggested that activated Akt is lethal at high levels, we used retroviral transduction to introduce myrAkt or c-Akt along with EGFP into H19-7 cells. Expression of the Akt constructs was verified by immunoblotting (Fig. 4A). Following differentiation with FGF, the green cells expressing myrAkt-GFP exhibited better survival than the green cells transduced with viral vectors expressing either c-Akt–GFP or GFP alone (Fig. 4B). As observed previously for the stably transfected cell lines, green cells transduced with virus expressing c-Akt did not exhibit enhanced survival upon differentiation.
FIG. 4.

Retroviral transduction of c-Akt and myrAkt into H19-7 cells. (A) Immunoblot demonstrating expression of transduced Akt vectors. Retroviruses expressing either EGFP alone, EGFP and c-Akt, or EGFP and myrAkt were introduced into H19-7 cells, and protein extracts from those populations were analyzed by immunoblotting with anti-Akt antibody as described in Materials and Methods. (B) Survival of virally transduced cells upon differentiation. Aliquots of the transduced populations were transferred to 39°C in DMEM plus N2 supplements with or without bFGF to induce differentiation. After 4 days, the proportion of green cells in each culture was determined. To factor out differences in viral titers, the proportion of green cells in the differentiating (FGF) cultures was normalized to the proportion of green cells in the untreated (control) cultures for each vector. The numbers above each column are the total number of cells counted in each culture.
Activated Akt inhibits cell death resulting from serum deprivation.
Undifferentiated H19-7 cells undergo cell death upon cultivation in serum-free medium at 39°C (26) but at a much lower rate than during differentiation. A shift of the cells from 33 to 39°C results in the inactivation of the SV40 large T antigen and the release of p53 that had been bound to the large T antigen (26). Cell death incurred upon switching of proliferating, undifferentiated H19-7 cells to 39°C in serum-free medium is rescued by v-Akt to the same extent as by Bcl-2 or Bcl-xL (Fig. 5). These results suggest that v-Akt can also rescue undifferentiated H19-7 cells from cell death resulting from serum deprivation in the presence of p53 and other factors that bind to large T antigen.
FIG. 5.

v-Akt protects against cell death from serum deprivation. Cell lines expressing v-Akt (HAP-5 and BAP-9), their parent lines (H19-7 and Bcl2-R10, respectively), and the Bcl-xL-overexpressing line XL-12 were shifted to 39°C in N2 medium, and their viability was determined.
A dominant-negative Akt mutant accelerates H19-7 cell death.
Expression of a mutant of Akt that has a dominant-negative phenotype would be a useful and direct demonstration of the involvement of Akt in mediating H19-7 neuronal survival. We therefore tested the effect on H19-7 viability of a kinase-dead mutant (Akt kin−) (32, 45). DNA plasmids expressing wild-type Akt or Akt kin− as well as a control vector were each microinjected into cells along with a plasmid expressing GFP. The cells were incubated overnight under growth conditions to allow recovery from microinjection, and the number of green cells was determined. The cells were processed to monitor the effect of the wild-type Akt or Akt kin− on the survival of undifferentiated H19-7 cells in N2 medium at 39°C or of cells undergoing differentiation in response to FGF in N2 medium at 39°C. The number of surviving green cells after 24 h under each of these two conditions was then determined directly by counting. The results indicate that wild-type Akt has minimal effect on survival of undifferentiated or differentiating cells, consistent with the results obtained with cells overexpressing ectopic c-Akt either stably or following transduction. In contrast, the mutant Akt kin− reduced survival of cells by 40 to 55% (Fig. 6). Note that the cells are normally undergoing apoptosis during this time. Thus, loss of Akt function accelerates cell death in both undifferentiated and differentiating cells, indicating that Akt is a mediator of survival in these cells.
FIG. 6.

Effects of microinjection of wild-type or mutant Akt DNA on the survival of H19-7 cells. A control vector and vectors expressing wild-type Akt or kinase-dead Akt (Akt kin−) were each microinjected into H19-7 cells along with GFP DNA as described in Materials and Methods. After incubation at 33°C in growth media overnight, the green (GFP-expressing) cells were counted, and the cells were shifted to defined N2 medium (A) or to FGF differentiation conditions (B) at 39°C. Twenty-four hours later, the remaining green cells were counted. The data for N2-treated cells are derived from two (wild-type Akt) or three (Akt kin− and control vector [Cont.]) independent experiments, and those for FGF-differentiated cells are derived from four (wild-type Akt) or 5 (Akt kin− and control vector) independent experiments. The total number of cells initially expressing GFP is indicated above each column. Each point is the mean ± 1 standard deviation Poisson error. The slight decrease in survival of the wild-type Akt relative to that of the control in panel B is within one ς error and therefore not significant.
v-Akt does not induce bcl-2 or bcl-xL and does not inhibit Jun kinase activity.
One potential mechanism by which v-Akt may enhance neuronal survival is to increase expression of endogenous Bcl-2 or Bcl-xL. To test this possibility, we analyzed extracts from cells expressing v-Akt by Western blotting with antibodies against Bcl-2 or Bcl-xL. The results show no significant increase in either Bcl-2 or Bcl-xL protein in response to activated Akt relative to that in control H19-7 cells (Fig. 7).
FIG. 7.
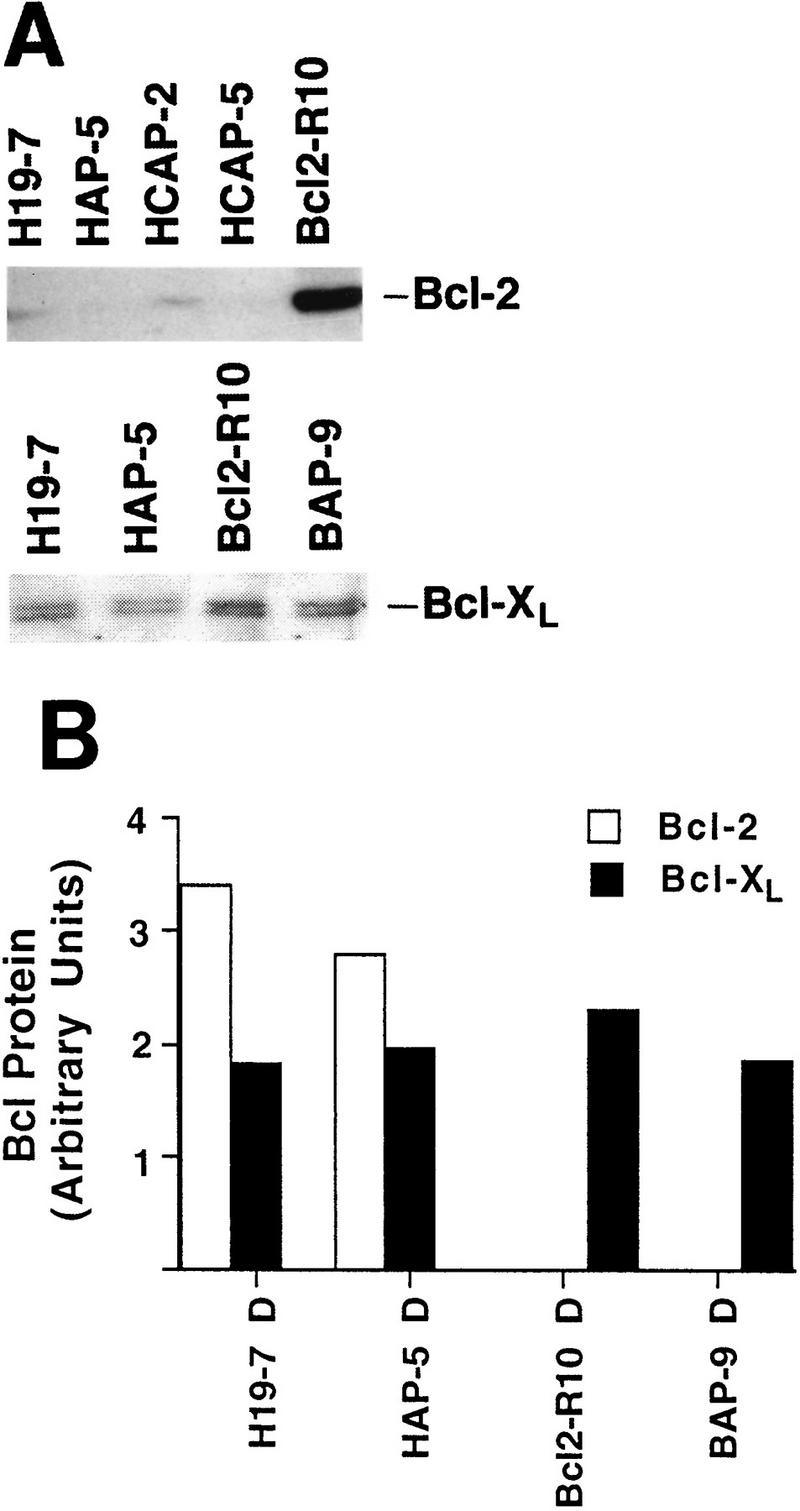
v-Akt expression does not enhance Bcl-2 or Bcl-xL protein levels in H19-7 cells. (A) Whole-cell extracts from cells differentiated for 3 days were fractionated on sodium dodecyl sulfate–10% polyacrylamide gels and immunoblotted for Bcl-2 or Bcl-xL. (B) Plot of relative Bcl-2 or Bcl-xL expression after normalization to tubulin levels. Immunoblots for Bcl-2 and Bcl-xL were reprobed with a monoclonal antibody to tubulin. Following determination of protein levels by optical density using an Ambis scanner, the amount of Bcl protein per lane was normalized to the amount of tubulin per lane. Since the Bcl-2 and Bcl-xL scans were done independently, the plot does not reflect the relative amounts of these proteins in a single cell line. D, differentiated.
Recent studies based upon transient expression of activators and inhibitors of the Jun kinase stress pathway have suggested that Jun kinase can mediate cell death in PC12 cells (70). Analysis of Jun kinase activation in H19-7 cells showed that Jun kinase activity gradually increased for the first several days following the induction of differentiation by FGF (Fig. 8), a time course consistent with a role for Jun kinase in apoptosis. No transient increases in Jun kinase activity were detected in the first few hours following differentiation (1). To test whether v-Akt inhibits apoptosis by suppressing Jun kinase activation, we analyzed Jun kinase activity in v-Akt-expressing cells (HAP-5 and BAP-5 cells) and their parent lines (H19-7 and Bcl2-R10) before and after FGF-induced differentiation. The results indicate that v-Akt does not inhibit Jun kinase activity (Fig. 8). Therefore, the inhibition of apoptosis by v-Akt is also Jun kinase independent.
FIG. 8.
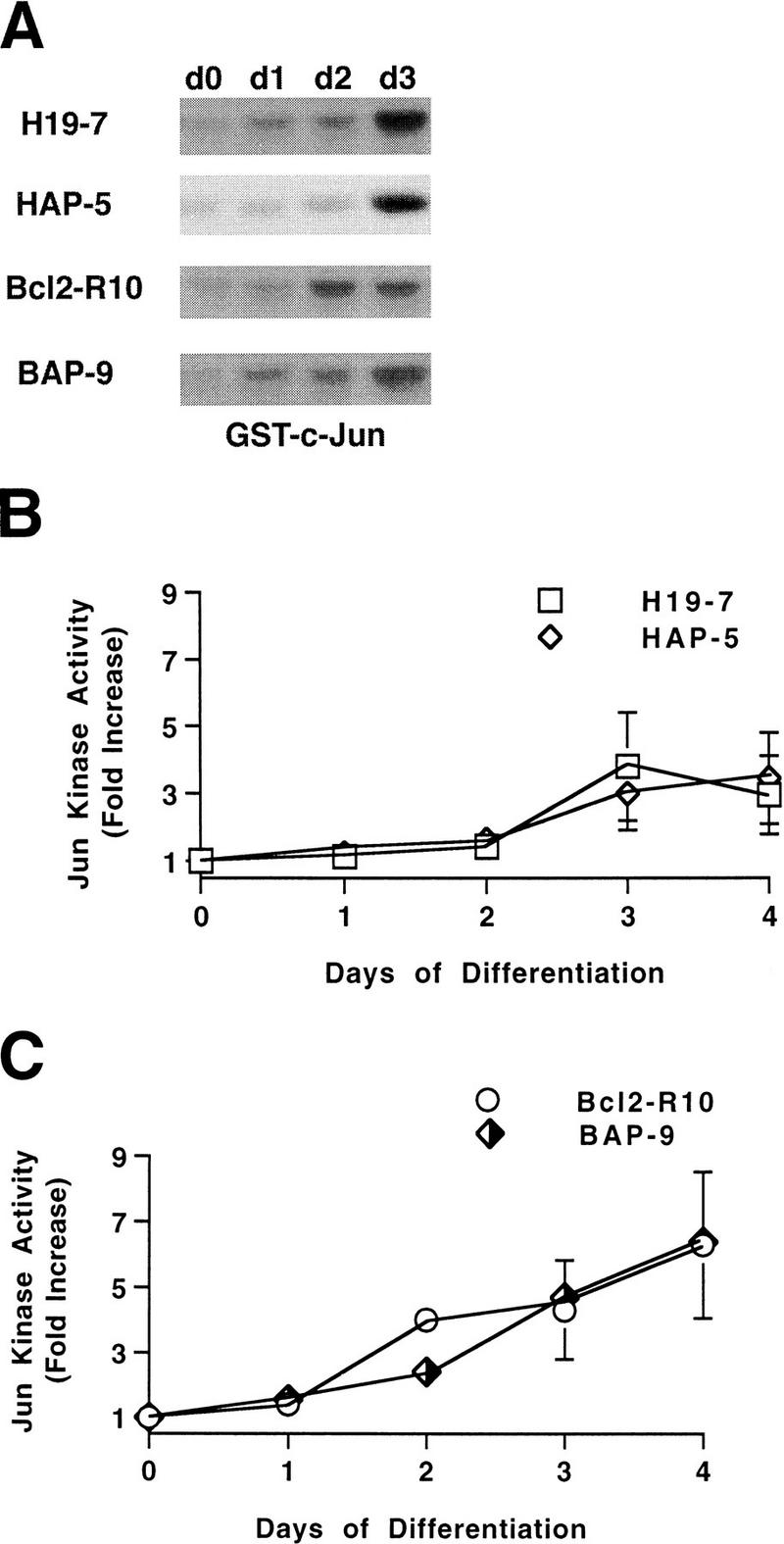
v-Akt does not suppress Jun kinase activity in differentiating H19-7 cells. (A) c-Jun kinase was assayed over 3 days of differentiation (d0 to d3) in cell lines expressing v-Akt (HAP-5 and BAP-9) and their parent lines. (B and C) Plot of time course of Jun kinase activity in H19-7 and HAP-5 cells or Bcl2-R10 and BAP-9 cells, respectively. Jun phosphorylation was analyzed by optical density using an Ambis scanner, and all values were normalized to Jun kinase activity on day 0. The data are from two to five independent experiments.
DISCUSSION
The present studies demonstrate that activated Akt is able to promote survival in differentiating neuronal cells. The activities of both Akt and its upstream activator, PI 3-kinase, are induced by the differentiating factor FGF. Treatment with wortmannin suppresses activation of these enzymes and reduces cell viability. Furthermore, the acceleration of cell death induced by the dominant-negative mutant Akt kin− indicates that endogenous Akt has a role in the survival of H19-7 rat hippocampal cells deprived of serum or undergoing neuronal differentiation. Finally, expression of activated Akt renders the cells resistant to wortmannin treatment and delays cell death during serum deprivation or differentiation.
The mechanism by which Akt promotes cell survival is not yet understood. Jun kinase has been implicated as a mediator of apoptosis in some neuronal cells (70). Consistent with this possibility, Jun kinase activity increases in H19-7 cells during differentiation and subsequent apoptosis. However, our results indicate that Akt does not act as an inhibitor of Jun kinase activity. An alternative possibility is that Akt acts to enhance the activity of MAP kinase, which has been proposed to promote survival in some neuronal cells (70). However, this mechanism is unlikely in H19-7 cells, since inhibition of the MAP kinase pathway has no major effect on survival, in contrast to inhibition of the PI 3-kinase pathway. Previous studies have suggested that Akt activates the p70S6 kinase via a rapamycin-sensitive pathway. Akt therefore may function upstream or in a pathway that parallels the rapamycin-inhibited step (13).
The extent of v-Akt-enhanced survival is similar to that obtained with peptide inhibitors of the ICE family of proteases or with Bcl-2 or Bcl-xL (26). Furthermore, Bcl-2 and v-Akt when expressed together do not significantly enhance the level of protection over that obtained with either factor alone, consistent with a common mechanism of action. Although v-Akt does not appear to be acting by increasing the levels of Bcl-2 or Bcl-xL protein, it is possible that Akt does modulate the function of one or more members of the Bcl-2 family by either changing their phosphorylation state or selectively altering their expression. Recent data indicate that BAD, a proapoptotic Bcl-2-related protein, is a substrate of Akt (18, 19), and phosphorylation of BAD appears to block BAD-induced apoptosis in cerebellar granule neurons in culture (19). Whether a similar pathway is responsible for the action of Akt on H19-7 cell survival remains to be determined.
The ability of activated Akt to promote survival is not limited to differentiating neuronal cells. Recent studies using a similar approach with fibroblasts, epithelial cells, and pro-B cells have shown that activated Akt inhibits apoptosis due to growth factor deprivation, matrix detachment, or c-myc activation (3, 42–44, 46). Constitutive activation of Akt and subsequent cell survival do not require fusion of Akt to a viral Gag protein, since myrAkt has also been shown to promote cell survival in our system and others (3, 43). It was recently reported that transient transfection of dominant-negative Akt promoted death of cerebellar granule neurons in insulin-containing medium, and wild-type Akt promoted limited survival of the neurons under conditions of serum and KCl deprivation (23). In our studies and those cited above, little or no enhancement of survival was observed even with stable overexpression of wild-type Akt. These differences may reflect cell type differences in the induction and maintenance of Akt activation.
Cellular Akt activity is a function of an equilibrium between the rates of enzyme activation and deactivation. Survival could be enhanced either by inducing the activation or inhibiting the inactivation of Akt. The observations that the activation of c-Akt by FGF is transient and that the presence of okadaic acid increases the activity are consistent with the limited effectiveness of Akt as a survival factor in the H19-7 cells. If Akt activity were sustained, as in the case of v-Akt, then FGF would be a more efficient mediator of survival. It is possible that mechanisms modulating Akt activity differ in developing versus mature neurons; thus, multiple neurotrophic factors might be required to act in concert during development to maintain Akt activation and prevent or delay programmed cell death.
Akt is activated via lipid products of PI 3-kinase and at least one other protein kinase (6, 63), its activation is inhibited by wortmannin, and activated Akt can rescue wortmannin-induced death of H19-7 cells; these results together suggest that neuronal survival can be mediated by a signal transduction pathway whereby a receptor activates PI 3-kinase, which in turn activates Akt. However, it is unlikely that activation of Akt is the only mechanism by which PI 3-kinase might promote cell survival. PI 3-kinase has been shown to activate the small GTP-binding protein Rac (39), as well as a number of other kinases, including the atypical PKC subfamily λ/tPKC and ζPKC (4, 54), other nonclassical PKCs (ɛPKC and ηPKC) (53, 64), and PKC-related kinase 1 (56). Interestingly, Akt shows homology with the PKC family within the catalytic domain, and recent evidence supports a role for the atypical PKCs in survival of NIH 3T3 cells (21). It is possible that Akt and these PKCs promote survival by phosphorylating targets that either directly or indirectly regulate mediators of cell death, such as members of the ICE protease (caspase) family. Furthermore, these results do not preclude the possibility that mechanisms protecting neurons or other cells against programmed cell death other than those initiated by PI 3-kinase activation are also utilized in vivo.
Mechanisms of neuronal apoptosis can differ depending on the maturation state of the cell. For example, immature cerebellar granule neurons are rescued from apoptosis by various growth factors and cytokines when maintained in vitro in the absence of depolarizing potassium concentrations, but the same cells allowed to differentiate in culture are refractory to the same factors (20). The studies presented here demonstrate that activated Akt can rescue cells from apoptosis even during differentiation and that down regulation of Akt activity may facilitate the apoptotic process.
ACKNOWLEDGMENTS
We thank Alan Saltiel (Parke-Davis) for generously providing PD098059, Rodney DeKoter and Harinder Singh for pBabeEGFP, Suzana Gomes for technical assistance, Larry Hill for assistance with the manuscript, and Mitchell Villereal for critical reading of the manuscript.
This work was supported by the National Institutes of Health (grants NS 33858 to M.R.R., CA 57436 to P.N.T., and CA 71874 to N.H.), a gift from the Cornelius Crane Fund for Eczema Research to M.R.R., and the American Cancer Society (grant CB-133 to N.H.).
REFERENCES
- 1.Abe, M. K., and M. R. Rosner. 1996. Unpublished data.
- 2.Ahmed N N, Franke T F, Bellacosa A, Datta K, Gonzalez-Portal M E, Taguchi T, Testa J R, Tsichlis P N. The proteins encoded by c-akt and v-akt differ in post-translational modification, subcellular localization and oncogenic potential. Oncogene. 1993;8:1957–1963. [PubMed] [Google Scholar]
- 3.Ahmed N N, Grimes H L, Bellacosa A, Chan T O, Tsichlis P N. Transduction of interleukin-2 antiapoptotic and proliferative signals via Akt protein kinase. Proc Natl Acad Sci USA. 1997;94:3627–3632. doi: 10.1073/pnas.94.8.3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Akimoto K, Mizuno K, Osada S, Hirai S, Tanuma S, Suzuki K, Ohno S. A new member of the third class in the protein kinase C family, PKCλ, expressed dominantly in an undifferentiated mouse embryonal carcinoma cell line and also in many tissues and cells. J Biol Chem. 1994;269:12677–12683. [PubMed] [Google Scholar]
- 5.Alessi D R, Andjelkovic M, Caudwell B, Cron P, Morrice N, Cohen P, Hemmings B A. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 1996;15:6541–6551. [PMC free article] [PubMed] [Google Scholar]
- 6.Alessi D R, James S R, Downes C P, Holmes A B, Gaffney P R, Reese C B, Cohen P. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase B alpha. Curr Biol. 1997;7:261–269. doi: 10.1016/s0960-9822(06)00122-9. [DOI] [PubMed] [Google Scholar]
- 7.Allsopp T E, Wyatt S, Paterson H F, Davies A M. The proto-oncogene bcl-2 can selectively rescue neurotrophic factor-dependent neurons from apoptosis. Cell. 1993;73:295–307. doi: 10.1016/0092-8674(93)90230-n. [DOI] [PubMed] [Google Scholar]
- 8.Almasan A, Yin Y, Kelly R E, Lee E Y-H P, Bradley A, Li W, Bertino J R, Wahl G M. Deficiency of retinoblastoma protein leads to inappropriate S-phase entry, activation of E2F-responsive genes, and apoptosis. Proc Natl Acad Sci USA. 1995;92:5436–5440. doi: 10.1073/pnas.92.12.5436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alnemri E S, Livingston D J, Nicholson D W, Salvesen G, Thornberry N A, Wong W W, Yuan J. Human ICE/CED-3 protease nomenclature. Cell. 1996;87:171. doi: 10.1016/s0092-8674(00)81334-3. [DOI] [PubMed] [Google Scholar]
- 10.Bellacosa A, Franke T F, Gonzalez-Portal M E, Datta K, Taguchi T, Gardner J, Cheng J Q, Testa J R, Tsichlis P N. Structure, expression and chromosomal mapping of c-akt: relationship to v-akt and its implications. Oncogene. 1993;8:745–754. [PubMed] [Google Scholar]
- 11.Bellacosa A, Testa J R, Staal S P, Tsichlis P N. A retroviral oncogene, akt, encoding a serine-threonine kinase containing an SH2-like region. Science. 1991;254:274–277. doi: 10.1126/science.254.5029.274. [DOI] [PubMed] [Google Scholar]
- 12.Bottenstein J E. Growth and differentiation of neural cells in defined media. In: Bottenstein J E, Sato G, editors. Cell culture in the neurosciences. New York, N.Y: Plenum Press; 1985. pp. 3–43. [Google Scholar]
- 13.Burgering B M T, Coffer P J. Protein kinase B (c-Akt) in phosphatidylinositol-3-OH kinase signal transduction. Nature. 1995;376:599–602. doi: 10.1038/376599a0. [DOI] [PubMed] [Google Scholar]
- 14.Chan, T. O., A. Bellacosa, and P. N. Tsichlis. 1997. Unpublished data.
- 15.Clarke A R, Purdie C A, Harrison D J, Morris R G, Bird C C, Hooper M L, Wyllie A H. Thymocyte apoptosis induced by p53-dependent and independent pathways. Nature. 1993;362:849–852. doi: 10.1038/362849a0. [DOI] [PubMed] [Google Scholar]
- 16.Cohen P, Alessi D R, Cross D A E. PDK1, one of the missing lines in insulin signal transduction? FEBS Lett. 1997;410:3–10. doi: 10.1016/s0014-5793(97)00490-0. [DOI] [PubMed] [Google Scholar]
- 17.Datta K, Bellacosa A, Chan T O, Tsichlis P N. Akt is a direct target of the PI3-K: activation by growth factors, v-src and v-Ha-ras in Sf9 and mammalian cells. J Biol Chem. 1996;271:30835–30839. doi: 10.1074/jbc.271.48.30835. [DOI] [PubMed] [Google Scholar]
- 18.Datta S R, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg M E. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- 19.del Peso L, González-García M, Page C, Herrera R, Nuñez G. Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt. Science. 1997;278:687–689. doi: 10.1126/science.278.5338.687. [DOI] [PubMed] [Google Scholar]
- 20.de Luca A, Weller M, Frei K, Fontana A. Maturation-dependent modulation of apoptosis in cultured cerebellar granule neurons by cytokines and neurotrophins. Eur J Neurosci. 1996;8:1994–2005. doi: 10.1111/j.1460-9568.1996.tb01343.x. [DOI] [PubMed] [Google Scholar]
- 21.Diaz-Meco M T, Municio M M, Frutos S, Sanchez P, Lozano J, Sanz L, Moscat J. The product of par-4, a gene induced during apoptosis, interacts selectively with the atypical isoforms of protein kinase C. Cell. 1996;86:777–786. doi: 10.1016/s0092-8674(00)80152-x. [DOI] [PubMed] [Google Scholar]
- 22.Downward J. Regulating S6 kinase. Nature. 1994;371:378–379. doi: 10.1038/371378a0. [DOI] [PubMed] [Google Scholar]
- 23.Dudek H, Datta S R, Franke T F, Birnbaum M J, Yao R, Cooper G M, Segal R A, Kaplan D R, Greenberg M E. Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science. 1997;275:661–665. doi: 10.1126/science.275.5300.661. [DOI] [PubMed] [Google Scholar]
- 24.Dudley D T, Pang L, Decker S J, Bridges A J, Saltiel A R. A synthetic inhibitor of the mitogen-activated protein kinase cascade. Proc Natl Acad Sci USA. 1995;92:7686–7689. doi: 10.1073/pnas.92.17.7686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Earnshaw W C. Apoptosis: lessons from in vitro systems. Trends Cell Biol. 1995;5:217–220. doi: 10.1016/s0962-8924(00)89006-6. [DOI] [PubMed] [Google Scholar]
- 26.Eves E M, Boise L H, Thompson C B, Wagner A J, Hay N, Rosner M R. Apoptosis induced by differentiation or serum-deprivation in an immortalized central nervous system neuronal cell line. J Neurochem. 1996;67:1908–1920. doi: 10.1046/j.1471-4159.1996.67051908.x. [DOI] [PubMed] [Google Scholar]
- 27.Eves, E. M., D. Hanck, and M. R. Rosner. 1997. Unpublished data.
- 28.Eves E M, Kwon J, Downen M, Tucker M S, Wainer B H, Rosner M R. Conditional immortalization of neuronal cells from postmitotic cultures and adult CNS. Brain Res. 1994;656:396–404. doi: 10.1016/0006-8993(94)91484-2. [DOI] [PubMed] [Google Scholar]
- 29.Eves, E. M., and M. R. Rosner. 1997. Unpublished data.
- 30.Eves E M, Tucker M S, Roback J D, Downen M, Rosner M R, Wainer B H. Immortal rat hippocampal cell lines exhibit neuronal and glial lineages and neurotrophin gene expression. Proc Natl Acad Sci USA. 1992;89:4373–4377. doi: 10.1073/pnas.89.10.4373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Franke T F, Kaplan D R, Cantley L C, Toker A. Direct regulation of the Akt proto-oncogene product by phosphatidylinositol-3,4-biphosphate. Science. 1997;275:665–668. doi: 10.1126/science.275.5300.665. [DOI] [PubMed] [Google Scholar]
- 32.Franke T F, Yang S-I, Chan T O, Datta K, Kazlauskas A, Morrison D K, Kaplan D R, Tsichlis P N. The protein kinase encoded by the Akt proto-oncogene is a target of the PDGF-activated phosphatidylinositol 3-kinase. Cell. 1995;81:727–736. doi: 10.1016/0092-8674(95)90534-0. [DOI] [PubMed] [Google Scholar]
- 33.Galli-Resta L, Ensini M. An intrinsic time limit between genesis and death of individual neurons in the developing retinal ganglion cell layer. J Neurosci. 1996;16:2318–2324. doi: 10.1523/JNEUROSCI.16-07-02318.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Garcia I, Martinou I, Tsujimoto Y, Martinou J-C. Prevention of programmed cell death of sympathetic neurons by the bcl-2 proto-oncogene. Science. 1992;258:302–304. doi: 10.1126/science.1411528. [DOI] [PubMed] [Google Scholar]
- 35.Gonzalez-Garcia M, Garcia I, Ding L, O’Shea S, Boise L H, Thompson C B, Nunez G. bcl-x is expressed in embryonic and postnatal neural tissues and functions to prevent neuronal cell death. Proc Natl Acad Sci USA. 1995;92:4304–4308. doi: 10.1073/pnas.92.10.4304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Greene L A, Tischler A S. Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells that respond to nerve growth factor. Proc Natl Acad Sci USA. 1976;73:2424–2428. doi: 10.1073/pnas.73.7.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Greenlund L J S, Korsmeyer S J, Johnson E M., Jr Role of BCL-2 in the survival and function of developing and mature sympathetic neurons. Neuron. 1995;15:649–661. doi: 10.1016/0896-6273(95)90153-1. [DOI] [PubMed] [Google Scholar]
- 38.Hawes B E, Luttrell L M, van Biesen T, Lefkowitz R J. Phosphatidylinositol 3-kinase is an early intermediate in the Gβγ-mediated mitogen-activated protein kinase signaling pathway. J Biol Chem. 1996;271:12133–12136. doi: 10.1074/jbc.271.21.12133. [DOI] [PubMed] [Google Scholar]
- 39.Hawkins P T, Eguinoa A, Qiu R G, Stokoe D, Cooke F T, Walters R, Wennstrom S, Claesson W L, Evans T, Symons M, Stephens L. PDGF stimulates an increase in Rac-GTP via the activation of phosphoinositide 3-kinase. Curr Biol. 1995;5:393–403. doi: 10.1016/s0960-9822(95)00080-7. [DOI] [PubMed] [Google Scholar]
- 40.Hibi M, Lin A, Smeal T, Minden A, Karin M. Identification of an oncoprotein- and UV-responsive protein kinase that binds and potentiates the c-Jun activation domain. Genes Dev. 1993;7:2135–2148. doi: 10.1101/gad.7.11.2135. [DOI] [PubMed] [Google Scholar]
- 41.Kapeller R, Cantley L C. Phosphatidylinositol 3-kinase. BioEssays. 1994;16:565–576. doi: 10.1002/bies.950160810. [DOI] [PubMed] [Google Scholar]
- 42.Kauffmann-Zeh A, Rodriguez-Viciana P, Ulrich E, Gilbert C, Coffer P, Downward J, Evan G. Suppression of c-Myc-induced apoptosis by Ras signalling through PI(3)K and PKB. Nature. 1997;385:544–584. doi: 10.1038/385544a0. [DOI] [PubMed] [Google Scholar]
- 43.Kennedy S G, Wagner A J, Conzen S D, Jordan J, Bellacosa A, Tsichlis P N, Hay N. The PI 3-kinase/Akt signaling pathway delivers an anti-apoptotic signal. Genes Dev. 1997;11:701–713. doi: 10.1101/gad.11.6.701. [DOI] [PubMed] [Google Scholar]
- 44.Khwaja A, Rodriguez-Viciana P, Wennstrom S, Warne P H, Downward J. Matrix adhesion and Ras transformation both activate a phosphoinositide 3-OH kinase and protein kinase B/Akt cellular survival pathway. EMBO J. 1997;16:2783–2793. doi: 10.1093/emboj/16.10.2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kohn A D, Kovacina K S, Roth R A. Insulin stimulates the kinase activity of RAC-PK, a pleckstrin homology domain containing Ser/Thr kinase. EMBO J. 1995;14:4288–4295. doi: 10.1002/j.1460-2075.1995.tb00103.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kulik G, Klippel A, Weber M J. Antiapoptotic signalling by the insulin-like growth factor I receptor, phosphatidylinositol 3-kinase, and Akt. Mol Cell Biol. 1997;17:1595–1606. doi: 10.1128/mcb.17.3.1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kuo W-L, Abe M, Rhee J, Eves E M, McCarthy S A, Yan M, Templeton D J, McMahon M, Rosner M R. Raf, but not MEK or ERK, is sufficient for differentiation of hippocampal neuronal cells. Mol Cell Biol. 1996;16:1458–1470. doi: 10.1128/mcb.16.4.1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kuo W-L, Chung K-C, Rosner M R. Differentiation of central nervous system neuronal cells by fibroblast-derived growth factor requires at least two signaling pathways: roles for Ras and Src. Mol Cell Biol. 1997;17:4633–4643. doi: 10.1128/mcb.17.8.4633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lowe S W, Schmitt E M, Smith S W, Osborne B A, Jacks T. p53 is required for radiation-induced apoptosis in mouse thymocytes. Nature. 1993;362:847–849. doi: 10.1038/362847a0. [DOI] [PubMed] [Google Scholar]
- 50.Mah S, Zhong L, Liu Y, Roghani A, Edwards R, Bredesen D. The proto-oncogene bcl-2 inhibits apoptosis in PC12 cells. J Neurochem. 1993;60:1183–1186. doi: 10.1111/j.1471-4159.1993.tb03275.x. [DOI] [PubMed] [Google Scholar]
- 51.Martin S J, Green D R. Protease activation during apoptosis: death by a thousand cuts. Cell. 1995;82:349–352. doi: 10.1016/0092-8674(95)90422-0. [DOI] [PubMed] [Google Scholar]
- 52.Morgenstern J P, Land H. A series of mammalian expression vectors and characterisation of their expression of a reporter gene in stably and transiently transfected cells. Nucleic Acids Res. 1990;18:1068–1070. doi: 10.1093/nar/18.4.1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Moriya S, Kazlauskas A, Akimoto K, Hirai S-I, Mizuno K, Takenawa T, Fukui Y, Watanabe Y, Ozaki S, Ohno S. Platelet-derived growth factor activates protein kinase Cɛ through redundant and independent signaling pathways involving phospholipase Cγ or phosphatidylinositol 3-kinase. Proc Natl Acad Sci USA. 1996;93:151–155. doi: 10.1073/pnas.93.1.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nakanishi H, Brewer K A, Exton J H. Activation of the ζ isozyme of protein kinase C by phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem. 1993;268:13–16. [PubMed] [Google Scholar]
- 55.Oppenheim R W. Cell death during development of the nervous system. Annu Rev Neurosci. 1991;14:453–501. doi: 10.1146/annurev.ne.14.030191.002321. [DOI] [PubMed] [Google Scholar]
- 56.Palmer R, Dekker L V, Woscholski R, Le Good J A, Gigg R, Parker P. Activation of PRK1 by phosphatidylinositol 4,5-bisphosphate and phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem. 1995;270:22412–22416. doi: 10.1074/jbc.270.38.22412. [DOI] [PubMed] [Google Scholar]
- 57.Pan H, Griep A E. Temporally distinct patterns of p53-dependent and p53-independent apoptosis during mouse lens development. Genes Dev. 1995;9:2157–2169. doi: 10.1101/gad.9.17.2157. [DOI] [PubMed] [Google Scholar]
- 58.Pang L, Sawada T, Decker S, Saltiel A. Inhibition of MAP kinase kinase blocks the differentiation of PC-12 cells induced by nerve growth factor. J Biol Chem. 1995;270:13585–13588. doi: 10.1074/jbc.270.23.13585. [DOI] [PubMed] [Google Scholar]
- 59.Raff M C, Barres B A, Burne J F, Coles H S, Ishizaki Y, Jacobson M D. Programmed cell death and the control of cell survival: lessons from the nervous system. Science. 1993;262:695–700. doi: 10.1126/science.8235590. [DOI] [PubMed] [Google Scholar]
- 60.Ray J, Peterson D A, Schinstine M, Gage F H. Proliferation, differentiation, and long-term culture of primary hippocampal neurons. Proc Natl Acad Sci USA. 1993;90:3602–3606. doi: 10.1073/pnas.90.8.3602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sambrook J, Fritsch E F, Maniatis T. Molecular cloning: a laboratory manual. 2nd ed. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 62.Sato N, Hotta K, Waguri S, Nitatori T, Tohyama K, Tsujimoto Y, Uchiyama Y. Neuronal differentiation of PC12 cells as a result of prevention of cell death by bcl-2. J Neurobiol. 1994;25:1227–1234. doi: 10.1002/neu.480251005. [DOI] [PubMed] [Google Scholar]
- 63.Stokoe D, Stephens L R, Copeland T, Gaffney P R J, Reese C B, Painter G F, Holmes A B, McCormick F, Hawkins P T. Dual role of phosphatidylinositol-3,4,5-trisphosphate in the activation of protein kinase B. Science. 1997;277:567–570. doi: 10.1126/science.277.5325.567. [DOI] [PubMed] [Google Scholar]
- 64.Toker A, Meyer M, Reddy K K, Falck J R, Aneja R, Aneja S, Burns A P D J, Ballas L M, Cantley L C. Activation of protein kinase C family members by the novel polyphosphoinositides Ptdlns-3,4-P2 and Ptdlns-3,4,5-P3. J Biol Chem. 1994;269:32358–32367. [PubMed] [Google Scholar]
- 65.Ui M, Okada T, Hazeki K, Hazeki O. Wortmannin as a unique probe for an intracellular signalling protein, phosphoinositide 3-kinase. Trends Biochem Sci. 1995;20:303–307. doi: 10.1016/s0968-0004(00)89056-8. [DOI] [PubMed] [Google Scholar]
- 66.Voyvodic J T. Cell death in cortical development: how much? why? so what? Neuron. 1996;16:693–696. doi: 10.1016/s0896-6273(00)80089-6. [DOI] [PubMed] [Google Scholar]
- 67.Wagner A J, Kokontis J M, Hay N. Myc-mediated apoptosis requires wild-type p53 in a manner independent of cell cycle arrest and the ability of p53 to induce p21waf1/cip1. Genes Dev. 1994;8:2817–2830. doi: 10.1101/gad.8.23.2817. [DOI] [PubMed] [Google Scholar]
- 68.Whyte M, Evan G. The last cut is the deepest. Nature. 1995;376:17–18. doi: 10.1038/376017a0. [DOI] [PubMed] [Google Scholar]
- 69.Wood K A, Youle R J. The role of free radicals and p53 in neuron apoptosis in vivo. J Neurosci. 1995;15:5851–5857. doi: 10.1523/JNEUROSCI.15-08-05851.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Xia Z, Dickens M, Raingeaud J, Davis R J, Greenberg M E. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- 71.Yaginuma H, Tomita M, Takashita N, McKay S E, Cardwell C, Yin Q-W, Oppenheim R W. A novel type of programmed neuronal death in the cervical spinal cord of the chick embryo. J Neurosci. 1996;16:3685–3703. doi: 10.1523/JNEUROSCI.16-11-03685.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yao R, Cooper G M. Requirement for phosphatidylinositol-3 kinase in the prevention of apoptosis by nerve growth factor. Science. 1995;267:2003–2006. doi: 10.1126/science.7701324. [DOI] [PubMed] [Google Scholar]
- 73.Zhong L-T, Sarafian T, Kane D J, Charles A C, Mah S P, Edwards R H, Bredesen D E. bcl-2 inhibits death of central neural cells induced by multiple agents. Proc Natl Acad Sci USA. 1993;90:4533–4537. doi: 10.1073/pnas.90.10.4533. [DOI] [PMC free article] [PubMed] [Google Scholar]