

Abstract
Cell stress, viral infection, and translational inhibition increase the abundance of human Alu RNA, suggesting that the level of these transcripts is sensitive to the translational state of the cell. To determine whether Alu RNA functions in translational homeostasis, we investigated its role in the regulation of double-stranded RNA-activated kinase PKR. We found that overexpression of Alu RNA by cotransient transfection increased the expression of a reporter construct, which is consistent with an inhibitory effect on PKR. Alu RNA formed stable, discrete complexes with PKR in vitro, bound PKR in vivo, and antagonized PKR activation both in vitro and in vivo. Alu RNAs produced by either overexpression or exposure of cells to heat shock bound PKR, whereas transiently overexpressed Alu RNA antagonized virus-induced activation of PKR in vivo. Cycloheximide treatment of cells decreased PKR activity, coincident with an increase in Alu RNA. These observations suggest that the increased levels of Alu RNAs caused by cellular exposure to different stresses regulate protein synthesis by antagonizing PKR activation. This provides a functional role for mammalian short interspersed elements, prototypical junk DNA.
Selfish DNA sequences are hypothesized to be neutral or nearly neutral parasites. However, repetitive, human Alu elements cause deleterious mutations both by retrotranspositional insertion and by unequal, homologous recombination (40). Accordingly, there may be some compensating selective advantage, if not a normal physiological role, for maintaining Alus within the human lineage. Among various possibilities, Alu transcripts may provide this selective advantage.
Despite the presence of nearly one million Alus, most of which have internal promoter elements for RNA polymerase III (Pol III), Alu RNA transcripts are very scarce in cultured human cells (29, 43). Pol III proceeds through an Alu template until a fortuitous terminator, consisting of four or more T residues, is encountered within the 3′ flanking sequence. These primary full-length Alu (flAlu) RNAs contain the dimeric repeat structure that is typical of consensus Alu elements and have a cytoplasmic lifetime of about 30 min (6, 39, 44). A fraction of flAlu RNA is processed into more stable, small cytoplasmic Alu (scAlu) RNA, which corresponds to the left monomer of the dimeric structure (32). There are about 500 copies of flAlu and scAlu RNAs in uninduced HeLa cells (29).
An infection of human cells by either adenovirus, human immunodeficiency virus, or herpes simplex virus dramatically increases the abundance of flAlu RNA (19, 20, 36). In the case of adenovirus, virus-encoded gene products increase the rate of Alu transcription (36). In addition, cell stress (whether by heat shock or other treatments) and translational inhibition by cycloheximide cause transient increases in the abundance of flAlu RNA (28). Cell stress and cycloheximide also increase the abundance of mouse and rabbit short interspersed element (SINE) transcripts, indicating that this response is evolutionarily conserved (28). Although these treatments increase the level of flAlu RNA, they increase only slightly the abundance of scAlu RNA, which is posttranscriptionally regulated (28).
One common feature of cell stress, viral infection, and translational inhibition is altered protein synthesis. Considering heat shock as one example of cell stress, protein synthesis is first inhibited, subsequently restored during cell recovery to engage expression of newly synthesized heat shock protein mRNAs, and later switched to restore expression of the pre-heat shock mRNA cohort (12). Mechanisms that regulate translational initiation are the primary effectors of these changes.
Viral infection affects translational initiation by several different pathways, including activation of double-stranded RNA (dsRNA)-regulated protein kinase PKR (8, 33, 41). Viral dsRNAs induce PKR autophosphorylation and kinase activation, leading to phosphorylation of the α subunit of eukaryotic initiation factor 2 (eIF-2α) and subsequent repression of host cell protein synthesis (9, 34). As a counter measure, small highly structured virus-encoded RNAs, such as adenovirus VAI RNA, competitively bind PKR, blocking its autophosphorylation and autoactivation (22, 41). High concentrations of activator dsRNAs, such as human immunodeficiency virus type 1 TAR RNA, can inhibit PKR activity (17, 30) as can a number of virus-encoded proteins (21).
The effects of cell stress, particularly heat shock, upon translational initiation are complex. Upon heat shock, eIF-2α is initially phosphorylated to block protein synthesis by hemin-regulated initiation factor-2 kinase, which is unresponsive to dsRNA (8, 10). The role of PKR during heat shock is unknown, but it first becomes associated with an insoluble cytosolic fraction and subsequently reenters the soluble fraction during recovery (11). During heat shock recovery, the level of eIF-2α phosphorylation decreases as protein synthesis is restored (12).
Given the increase in Alu RNA caused by cell stress, inhibition of protein synthesis with cycloheximide, and viral infection and the agonistic and antagonistic effects of dsRNAs upon PKR activity, we investigated the effects of Alu RNA on the expression of a reporter gene and PKR activation as well as the changes in PKR activity caused by these cellular treatments.
MATERIALS AND METHODS
Expression and induction of Alu RNAs.
Human 293 cells were cultured and transfected by the calcium phosphate method as previously described and harvested either 24 or 48 h later for analysis (6, 18). Clones for transfection included a luciferase reporter; a previously described flAlu RNA-overexpressing clone, XAT (6); and a clone, XAL, which overproduces left Alu RNA monomers (i.e., scAlu-like RNA). This clone contains the 7SL RNA promoter elements and the left Alu monomer from clone XAT, followed by five T residues, beginning at Alu consensus position 119 (39). Equivalent amounts of DNA were transfected in all experiments by adding additional pUC DNA. Cloned genes expressing VAI RNA, 5S rRNA, and 7SL RNA were also used in transfection assays (3, 24, 27).
RNA was analyzed by primer extension with reverse transcriptase and primers to detect 7SL RNA, flAlu RNA, 5S rRNA, and scAlu RNAs as previously described (6). The 7SL RNA primer (ATGCCGAACTTAGTGCGG) gives a 128-nucleotide (nt) primer extension product. Primer Alu 71 (GGTTTCACCGTGTTAGCCA) is directed toward the left Alu monomer, giving a 107-nt primer extension product for both scAlu and flAlu RNAs. Primer Alu 21-mer (GCGATCTCGGCTCACTGCAAG) gives a 238-nt product for flAlu RNA and two additional products. Alu 21-mer also misprimes transcripts from clone XAT, giving a shorter (210-nt) product that is diagnostic for XAT transcripts. Additionally, this primer gives a longer (350-nt) primer extension product, corresponding to an endogenous mRNA that evidently contains an Alu element (29). Primers for VAI RNA (AAAAGGAGCGCTCCCCCGTT) and 5S rRNA (AAAAAGCCTACAGCACCCGGTA) give 159- and 120-nt primer extension products, respectively. Heat shock, cycloheximide addition, and adenovirus infection of 293 cells were performed according to previous protocols, with exceptional details described in the text (28, 36). In particular, we employed a significantly lower multiplicity of infection (1 to 5 PFU per cell) in this study.
Preparation of recombinant PKR.
PKR was cloned into expression vector pET-15b (Novagen) and bacterially expressed as a hexahistidine-tagged fusion protein as described previously (33). Cell culture, cell lysis and extraction, and protein purification by nickel-agarose affinity column chromatography and by gel filtration fast-performance liquid chromatography were carried out as previously described (34) with the following modifications. The gel filtration chromatograph was developed in 20 mM sodium phosphate buffer (pH 7.0) containing 200 mM NaCl and 1 mM EDTA. The PKR fraction was loaded on a 2-ml bed volume SP Sepharose FF ion-exchange column (Pharmacia), washed with the same buffer, and eluted in buffer containing 300 mM NaCl. The eluent was concentrated to approximately 0.3 mg/ml with a disposable ultrafiltration device (Ultrafree-CL; Millipore) and stored in small aliquots at −80°C until used. Protein concentrations were estimated by Coomassie staining of sodium dodecyl sulfate (SDS) polyacrylamide gel electrophoresis gels and by spectrophotometric absorbance with a protein extinction coefficient derived from a purified enzymatically inactive mutant form of PKR, PKR[K296R] (4, 5). The final protein preparation was judged to be free of nucleic acid contamination by UV spectrophotometry.
Coimmunoprecipitation assays of PKR-Alu RNA complexes.
293 cells (treated with either heat shock or cycloheximide, infected with adenovirus, or transiently cotransfected with Alu expression vectors) were harvested at the indicated times. Cell extracts were reacted with either HL 71/10 monoclonal antibody against PKR (15) or normal mouse immunoglobulin G in buffer I (0.15 M NaCl, 0.4% Nonidet P-40 [NP-40], 20 mM VRC [Bethesda Research Laboratories], 10 mM Tris-HCl [pH 7.6], 0.2 mM phenylmethylsulfonyl fluoride, 1 μg [each] of aprotinin, leupeptin, and pepstatin per ml) on ice for 30 min. Protein A agarose (20 μl; Santa Cruz Biotechnology) was added and incubated with gentle shaking at 4°C for another 3 h (23). After being washed in buffer II (0.15 M KCl, 10 mM Tris-HCl [pH 7.6], 0.2 mM EDTA, 20% glycerol, 0.125% NP-40, 0.2 mM phenylmethylsulfonyl fluoride, 1 μg [each] of aprotinin, leupeptin, and pepstatin per ml), immunoprecipitates were resuspended in RNA lysis buffer (0.14 M NaCl, 0.5% NP-40, 0.5 mM EDTA, 10 mM Tris-HCl [pH 7.6]), extracted with phenol-chloroform, and precipitated with ethanol. RNAs were assayed by primer extension as described above.
RNA electrophoretic mobility shift assays.
Plasmids containing Alu, tRNA, and VAI sequences driven by the T7 promoter (pT7flAlu, pT7tRNA, and pT7VAI, respectively) were constructed by PCR amplification from clone XAT (6), the Xenopus tRNAMet gene (24), and the VAI gene (2), respectively, followed by directional cloning into pGEM3Z (Promega). The sequences of the primers used to create each construct were as follows: forward primer 628 (5′-AAAGAATTCGGCCGGGCGCGGTGGT-3′) and reverse primer 629 (5′-AAAGGATCCGAGACGGAGTCTCGCT-3′) for pT7fluAlu, forward primer XltRNAmeteco.f (5′-TCCGAATTCAGCAGAGTGGCGCAGC-3′) and reverse primer XltRNAbam.r (5′-TCTGGATCCTAGCAGAGGATGGTTTCG-3′) for pT7tRNA, and forward primer VAIeco.f (5′-GCAGAATTCGGGCACTCTTCCGTGGTCTG-3′) and reverse primer VAIxba.r (5′-GCATCTAGAGGAGCGCTCCCCCGTTGTCT-3′) for pT7VAI. The identities of these constructs were verified by sequencing. After purification by banding in CsCl-ethidium bromide gradients, plasmids were linearized by cleavage with BamHI (pT7flAlu and pT7tRNA) or XbaI (pT7VAI), isolated as a band from an agarose gel, and purified with Gene Clean (Bio 101). After transcription with T7 RNA polymerase and with [α-32P]UTP as the labeled substrate, RNA was purified by DNase treatment, extraction with phenol-chloroform-isoamyl alcohol, and precipitation with ethanol. The RNA concentration was determined by scintillation counting.
For binding assays, PKR[K296R] at the indicated concentrations was incubated with 0.2 ng of labeled transcript in a reaction buffer consisting of 50 mM KCl, 5 mM MgCl2, 15 mM HEPES-KOH (pH 7.4), 1 mM dithiothreitol, 1 mM Pefabloc SC (Boehringer Mannheim), 0.1 mg of bovine serum albumin per ml, 6 U of RNAguard (Pharmacia) per ml, and 1.0 μg (each) of pepstatin, leupeptin, and aprotinin per ml. After binding for 20 min at room temperature, dye-glycerol was added and reactions were analyzed by electrophoresis through nondenaturing 5% polyacrylamide gels (bisacrylamide/acrylamide ratio, 1:29) run in 1× Tris-borate-EDTA. Quantitation was performed by using a Fuji Bas 1000 phosphorimager and MacBas software.
For competition binding assays, labeled transcripts were preincubated with PKR for 5 min at room temperature. After preincubation, unlabeled competitor RNA was added, and samples were incubated for an additional 15 min. Dye-glycerol was added, and samples were analyzed as described above.
PKR autophosphorylation assays.
Recombinant PKR (0.33 μg) was preincubated at 30°C with various RNAs for 6 min in reaction buffer (55 mM KCl, 5 mM Tris-HCl [pH 7.6], 10% glycerol, 2 mM MgCl2, 1 mM MnCl2, 0.1 mM EDTA) (23). Transcripts synthesized by T7 RNA polymerase as described above were subjected to an additional step of gel purification for use in these assays. Poly(I) · poly(C) (3 ng/μl [final concentration]) and 5 μCi of [γ-32P]ATP with unlabeled ATP to a 1.5 μM final concentration were added in a final reaction volume of 20 μl, and the mixture was incubated for 25 min at 30°C. Twenty microliters of Laemmli’s buffer was added, and samples were boiled for 5 min, resolved on an SDS–8% polyacrylamide gel, and analyzed by autoradiography and by phosphorimager analysis.
Assay of PKR activity.
Human 293 cells were transiently transfected for 48 h prior to being harvested with genes for various small RNAs or equivalent amounts of plasmid DNA. In addition, cells were subjected to either heat shock, cycloheximide treatment, or viral infection as indicated in the text. In studies of viral infection, cells were infected with 1 to 5 PFU of either wild-type adenovirus or adenovirus dl720 (Ad720) or were mock infected 20 h prior to being harvested. Ad720 has defective VAI and VAII RNA genes (9). At 17 h prior to being harvested, cells were treated with 1,000 U of interferon (Sigma) per ml. PKR was purified with either a commercial PKR antibody (Santa Cruz) or a previously described PKR antibody (4) by the immunoprecipitation procedure described above and resuspended in buffer III (10 mM Tris-HCl [pH 7.6], 100 mM KCl, 20% glycerol, 0.2 mM EDTA). The authenticity of PKR precipitated by the commercial antibody was verified by Western analysis with a well-characterized polyclonal antibody (4). PKR activity was assayed as described above, except that 75 mM KCl was employed. The level of PKR was measured by Western blotting.
RESULTS
Overexpression of Alu RNAs increases protein synthesis.
Previously, we developed a flAlu RNA overexpression vector (clone XAT) consisting of the 5′ flanking sequence of the 7SL RNA gene linked to an Alu element with an efficient terminator (6). Subsequently, we developed a system (clone XAL) to overexpress left monomers of this Alu element (scAlu-like RNA) by placing an efficient terminator, consisting of five consecutive T residues, immediately adjacent to the left Alu monomer in clone XAT (29a). Upon transient transfection into 293 cells, clone XAL produces very high levels of long-lived 118-nt left monomer scAlu RNA transcripts.
flAlu and scAlu-like RNA overexpression vectors were transiently cotransfected with a luciferase reporter construct to test the effects of Alu transcripts on protein synthesis (Fig. 1A). As controls, equivalent amounts of plasmid pUC, a cloned VAI RNA gene, and a cloned 7SL RNA gene were also transiently cotransfected with the reporter. Compared to the pUC control, overexpression of scAlu-like RNA and 7SL RNA led to modest but consistent increases in luciferase activity. In contrast, flAlu RNA and VAI RNA provided significantly greater stimulation of luciferase activity (ca. 10- to 16-fold in replicate trials for cells transfected with 10 μg of the flAlu overexpression clone) (Fig. 1A and data not shown). The very different effects of scAlu-like and flAlu RNAs on luciferase activity provided a negative control for this experiment. We routinely observed much higher levels of scAlu-like RNA compared to those of flAlu RNA in overexpression assays (data not shown) (see Fig. 7). However, scAlu-like RNA had relatively modest effects upon luciferase activity. scAlu-like RNA effectively served as a defective Alu mutant, providing a negative control for this and subsequent experiments.
FIG. 1.

Effects of Alu RNAs on a transiently coexpressed reporter gene. (A) 293 cells were transiently cotransfected with a luciferase reporter gene (0.1 μg) and the indicated quantity of a clone overexpressing 7SL, scAlu-like, flAlu (clone XAT), or VAI RNA or plasmid pUC for a 293 cell control and with additional pUC DNA to provide a total of 40 μg of DNA for each sample. Luciferase activities were assayed at 24 h after transfection and are reported in arbitrary luminosity units. (B) The amounts of luciferase mRNA were assayed by primer extension in cells transfected with 0.1, 2.5, and 10 μg of clones overexpressing the indicated RNA.
FIG. 7.

flAlu RNA inhibits virus-induced PKR activation in vivo. Human 293 cells were transiently transfected for 48 h with 5 or 15 μg of cloned genes to express various small RNA genes (lanes 4 through 13) or with plasmid DNA (lanes 1 through 3). The type of small-RNA gene employed in each transfection is noted above the corresponding lane. Twenty hours prior to being harvested, cells were mock infected (293; lane 2) or infected with 1 to 5 PFU of either adenovirus (adv; lane 1) or mutant Ad720 (720; lanes 3 through 13). Seventeen hours prior to being harvested, cells were treated with interferon (1,000 U/ml). (A) PKR activity was assayed by in vitro autophosphorylation. (B) PKR protein levels were determined by Western analysis. No PKR activity or PKR protein was detected in a mock precipitation control (lane not shown). (C) Primer extension analysis was employed to assay the levels of expression of various small RNAs in control cells (293), cells infected with mutant Ad720 (720), and cells transfected with either 5 or 15 μg of cloned genes for various small RNAs. The relative amounts of RNA (2 μg is 1×) used in these primer extension assays are indicated.
Differences in luciferase activity may result from differences in either transcriptional or translational activity. As assayed by primer extension, luciferase mRNA accumulated to similar levels in these transfection assays (Fig. 1B). By phosphorimager analysis, the levels of luciferase mRNA in control 293 cells and cells overexpressing flAlu RNA were virtually identical. We therefore conclude that flAlu RNA, like VAI, increases luciferase activity by increasing the translational activity of its mRNA.
While the results are reproducible, such transient-overexpression experiments must be cautiously interpreted. As reported below, PKR is implicated in the effects that Alu RNA has upon protein expression. Since PKR negatively regulates its own expression (2), complex changes in cell physiology may occur during the time span employed for transient-transfection assays.
The effects of flAlu RNA on the luciferase reporter may be an artifact of its gross overexpression. In this event, the level of overexpressed flAlu RNA required to increase expression of the reporter gene would greatly exceed naturally occurring levels of endogenous flAlu RNA. We therefore compared the levels of exogenous and endogenous flAlu RNAs (Fig. 2). In agreement with the results shown in Fig. 1, overexpression of flAlu RNA increased expression of the luciferase reporter gene nearly fivefold over the range of transfected XAT DNA investigated (Fig. 2). The level of overexpressed flAlu RNA was determined by primer extension relative to the abundance of 7SL RNA to control for RNA loading. The abundance of flAlu RNA increased about fivefold over the experimental range investigated (Fig. 2). Moreover, there was an excellent correlation between increasing levels of flAlu RNA and luciferase activity (Fig. 2).
FIG. 2.

Cotransfection of the luciferase reporter gene and the flAlu RNA-overexpressing clone XAT was performed as described in the legend to Fig. 1, except that cells were harvested at 48 h after transfection. The level of luciferase activity (expressed in arbitrary luminosity units) is shown. Primer extension (insert) was used to assay for the abundances of both Alu RNA and 7SL RNA for 293 control cells (lane 1); cells transfected with 0.5, 2.5, 5.0, and 1.25 μg of clone XAT (lanes 2 through 5, respectively); cells infected with adenovirus for 24 h (lane 6); cells treated with 100 μg of cycloheximide per ml for 4 h (lane 7); and cells recovering for 2 h from heat shock (lane 8). The primers for flAlu and 7SL RNAs were mixed at a ratio of 30 to 1. The abundance of flAlu RNA (circles), relative to that of 7SL RNA, was determined by phosphorimager analysis of the corresponding primer extension products. The positions of 7SL and Alu primer extension products are indicated; the diffuse intermediate band is an artifact that is occasionally observed for the 7SL primer.
We also observed two- to threefold increases of flAlu RNA in cells that either were treated with cycloheximide, were recovering from heat shock, or were infected with adenovirus (Fig. 2). The endogenous levels of flAlu RNA induced by these cellular treatments were similar to the levels that increased expression of the reporter gene. In interpreting this experiment, the remarkable efficiency with which Pol III-directed templates are expressed in a majority of transfected cells should be noted (1). Apparently, endogenous flAlu RNA can achieve the levels required to stimulate translational expression. Since overexpression of flAlu RNA and VAI RNA, a known PKR antagonist (22), had similar effects upon luciferase activity (Fig. 1), these RNAs may increase translation through a similar mechanism.
PKR binds flAlu RNA in vivo.
To determine whether Alu RNAs interact with PKR in vivo, the association of overexpressed flAlu RNA with PKR was assayed by immunoaffinity binding of PKR and by primer extension of Alu RNAs with reverse transcriptase (Fig. 3A). As negative and positive controls, we tested the binding of PKR to 7SL RNA and overexpressed VAI RNA, respectively. As a further control for possible effects due to the transfection procedure, we also employed cells which had been transfected with the 7SL RNA gene, although we did not attempt to distinguish between endogenous and overexpressed exogenous 7SL RNAs. None of these three RNAs precipitated in a mock control (Fig. 3A, lanes 6 through 8). Both overexpressed VAI and flAlu RNAs immunoprecipitated with PKR, but there was no detectable interaction between 7SL RNA and PKR (Fig. 3A, lanes 9 through 11). Although we have not examined whether our procedures result in quantitative precipitation of PKR, we calculate that 0.4% of flAlu RNA coprecipitated with PKR in this experiment.
FIG. 3.

Coimmunoprecipitation of Alu RNAs with PKR. PKR was immunoprecipitated with monoclonal antibody against PKR or normal mouse immunoglobulin G by absorption upon protein A-Sepharose, resulting in supernatant (Sup), immunoprecipitation (IP), and control (Mock) fractions, which were assayed by primer extension. (A) Cells were transiently transfected with clones (10 μg) XAT (lanes 1, 5, 8, and 11), 7SL (lanes 2, 3, 6, and 9) and VAI (lanes 4, 7, and 10). The resulting samples were assayed with the specific primer directed toward the corresponding RNA, except for lane 1, in which we employed a mixture of Alu, 7SL, and VAI RNA primers. The relative amounts of primers employed for the supernatant fractions were adjusted in the Alu/VAI/7SL ratio of 30:3:1 to provide comparable cDNA products, except for lane 3, in which the 7SL primer was used at the same concentration as was the Alu primer. Equal amounts of the three primers were used to assay the mock and immunoprecipitation fractions. Equal amounts of RNA (20 μg) were assayed in supernatant fractions, except for lane 3, in which 0.66 μg of RNA was assayed. The total mock and immunoprecipitation fractions were assayed. (B) Using 7SL/Alu primer mixture ratios of 1:30 for supernatants (Sup) and 1:1 for immunoprecipitates, primer extension was performed with control 293 cells (lanes 1 and 9) and cells infected with adenovirus mutant Ad720 at 1 to 5 PFU per cell (lanes 2 and 10), treated with 100 μg of cycloheximide per ml for 4 h (lane 3 and 11), or recovering for 2 h from heat shock (lane 4 and 12), as well as cells transfected with pUC alone (lanes 5 and 13) and XAT (2.5 [lanes 6 and 14], 5 [lanes 7 and 15], and 10 [lanes 8 and 16] μg). All transfections were adjusted to 10 μg with pUC. For supernatant fractions (lanes 1 through 8), 10 μg of RNA was assayed. The entire immunoprecipitate was assayed for lanes 9 through 12, but the immunoprecipitates for transfected samples (lanes 13 through 16) were divided for primer extension analysis (70%) and PKR assays (30%) as described in the legend to Fig. 8. Because of this difference and the number of cells employed, the immunoprecipitates for transfected samples (lanes 13 through 16) corresponded to 35% of the material examined for endogenous samples (lanes 9 through 12). Phosphorimager determinations of the relative abundances of flAlu RNA in supernatant fractions, with the intensities of 7SL RNA bands used as internal controls for RNA loading, are cited in the text.
Since overexpression of Alu RNAs from transiently transfected templates may artifactually cause PKR to bind Alu RNA, we tested the binding of PKR to endogenous flAlu RNA (Fig. 3B). To compare the effect of flAlu RNA overexpression on PKR activity, we also repeated the immunoprecipitation of overexpressed flAlu RNA with part of the resulting immunoprecipitate for primer extension analysis (Fig. 3B, lanes 13 through 16) and part for assays of PKR activity (see Fig. 8). As previously reported, the abundance of flAlu RNA increased in cells infected with virus (4.2-fold; Fig. 3B, lane 2), treated with cycloheximide (2.4-fold; lane 3), or recovering from heat shock (4.3-fold; lane 4). We also observed a 2.0- to 4.3-fold increase in flAlu RNA upon overexpression (Fig. 3B, lanes 5 through 8); therefore, the range of overexpression examined here is comparable to the increases induced by these cellular insults. In contrast to 7SL RNA, endogenous flAlu RNAs coimmunoprecipitated with PKR (Fig. 3B, lanes 9 through 12). The low signals of bound Alu RNA (Fig. 3B, lanes 9 through 16) cannot be accurately quantitated against the lane-specific background. However, we qualitatively observe that higher levels of either endogenous or exogenous flAlu RNA generally resulted in higher levels of association with PKR (Fig. 3B; compare lanes 2 and 10, lanes 4 and 12, and lanes 8 and 16). The slight increase in flAlu caused by cycloheximide in this particular experiment (Fig. 3B, lanes 1 and 3) did not cause a convincing increase in PKR binding relative to that of the control (lanes 9 and 11).
FIG. 8.
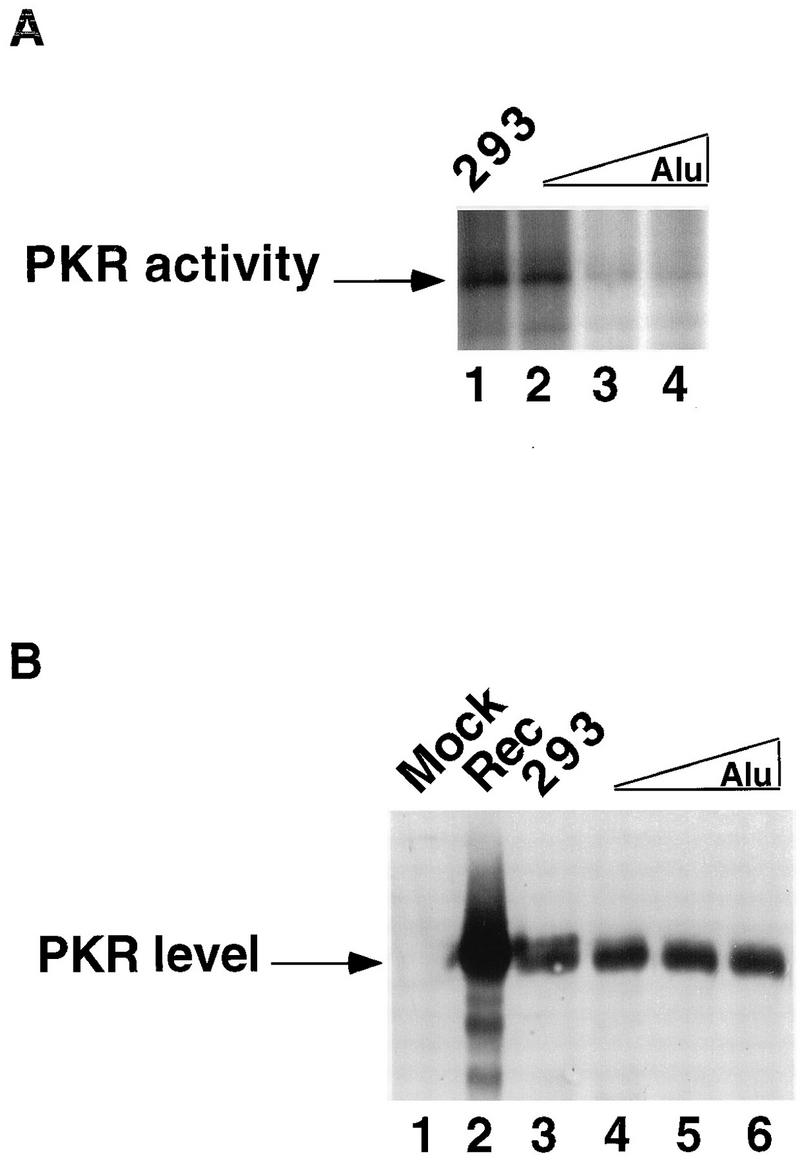
Effects of flAlu RNA overexpression on PKR activity. (A) Aliquots of immunoprecipitates from cells transfected with clone XAT as described in the legend to Fig. 3B were tested for PKR autophosphorylation. Immunoprecipitated samples were from 293 cells transfected with pUC (293; lane 1) and with 2.5, 5, and 10 μg of clone XAT (Alu; lanes 2 through 4, respectively). By phosphorimager analysis, the levels of PKR activity were 82% (lane 2) and 26% (lanes 3 and 4) of the activity in the control (lane 1). (B) The levels of PKR in the same immunoprecipitates were tested by Western analysis with a previously described antibody (4). The samples were from 293 cells transfected with pUC as a control (293; lane 3) and with 2.5, 5, and 10 μg of clone XAT (Alu; lanes 4 through 6, respectively). As additional controls, cells overexpressing recombinant PKR (Rec; lane 2) and a mock immunoprecipitation (Mock; lane 1) were examined. By using a lower percentage of acrylamide and running the gel longer, a doublet band of PKR was revealed as discussed in the text.
FlAlu RNA forms stable PKR complexes.
The association of flAlu RNA with PKR may result from direct interaction or may be mediated by other factors. Gel mobility shift assays were employed to detect possible complexes between PKR and flAlu RNA. PKR forms two discrete complexes, C1 and C2, with flAlu RNA (Fig. 4). As the PKR concentration increased, the abundance of the lower-mobility complex, C2, increased relative to that of the higher-mobility complex, C1 (Fig. 4).
FIG. 4.

Gel mobility shift assays of PKR-flAlu RNA complexes. Labeled flAlu RNA was incubated with the indicated amounts of PKR[K296R] and assayed by gel electrophoresis. The positions of free probe, high- (C1) and low (C2)-mobility complexes, and wells are indicated. Phosphorimager analysis was used to measure the amounts of free RNA and complexes; the dissociation constant of complex C1 was 0.26 μM. As discussed in the text, the dissociation constant for C2 was model dependent. Assuming that C2 results from occupancy of two PKR binding sites within flAlu RNA, the estimated dissociation constant for the PKR complexes associated with each site was 0.24 μM.
The observed mobility shift patterns for flAlu RNA can be interpreted as resulting from either protein oligomerization, the binding of protein to multiple sites in the dimeric sequence of flAlu RNA, or a combination of both. If we assumed that C1 and C2 consist of one and two PKRs bound to flAlu RNA, respectively, then the dissociation constant for each of these two sites was estimated to be 0.25 μM. This suggests that the dimeric sequence within flAlu RNA contains two indistinguishable and independent PKR binding sites (Fig. 4), a very attractive possibility. However, PKR is in equilibrium between monomeric and dimeric forms, and the addition of dsRNA shifts this equilibrium toward the protein dimeric state, presumably by stabilizing the dimer (4). Thus, the C2 complex may instead result from the stabilization of the PKR dimer at higher protein concentrations.
VAI RNA and tRNA also formed discrete complexes with PKR (Fig. 5, lanes 6 and 11). The T7-synthesized VAI RNA used in this study contains additional polylinker sequence, which may alter its structure and association with PKR. However, in excellent agreement with a previous determination (38), the dissociation constant for the major PKR-VAI RNA complex was 0.3 μM (Fig. 5, lane 11, and data not shown). Thus, the stabilities of the Alu RNA and VAI RNA mobility shift complexes with PKR were virtually identical. Like flAlu RNA, VAI RNA also formed higher-order PKR complexes (Fig. 5). The PKR-tRNA complex was only slightly less stable, having a dissociation constant of 0.6 μM (Fig. 5, lane 6, and data not shown).
FIG. 5.

Competition by VAI RNA against PKR-RNA gel shift complexes. Labeled RNAs (ca. 0.2 ng or about 0.1 nM [final concentration]) were preincubated with 250 nM PKR [K296R] for 5 min and then incubated for an additional 15 min with different concentrations of unlabeled VAI RNA (2.5 nM [lanes 2, 7, and 12], 25 nM [lanes 3, 8, and 13], 250 nM [lanes 4, 9, and 14], and 2.5 mM [lanes 5, 10, and 15]) or without VAI RNA competitor (−; lanes 1, 6, and 11). Mobility shift complexes were analyzed on 5% nondenaturing polyacrylamide gels. The positions of wells (W), free probe (F), and complexes (C) are indicated. The mobilities of the three probes in the absence of PKR were examined in separate control experiments.
Although the dissociation constants for complexes with these three RNAs were very similar, competition assays with preformed complexes revealed a significant difference between the binding of PKR to flAlu RNA and binding to either tRNA or VAI RNA (Fig. 5). Complexes with a radiolabeled RNA tracer were preformed for 5 min before the addition of unlabeled competitor RNA (VAI RNA in this example), after which the mixture was incubated for an additional 15 min. Preliminary gel mobility shift assays indicated that 5 min is sufficient to completely preform these complexes, as the observed dissociation constant remained unchanged during incubation for 45 min. In addition, competition occurred within 5 min after the addition of unlabeled competitor so the amount of complex was not changed by competitive incubation for at least 0.5 h. VAI RNA at a concentration of 25 nM was sufficient to displace both tRNA and labeled VAI RNA from preformed PKR complexes (Fig. 5, lanes 8 and 13). In contrast, the PKR-flAlu RNA complex resisted competition by 2,500 nM VAI RNA (Fig. 5, lane 5). In otherwise identical experiments with either flAlu RNA, B1 RNA, or poly(I) · poly(C) as the unlabeled competitor, we obtained results similar to those shown in Fig. 5 (data not shown). Thus, the PKR-flAlu complex is stable at competitor concentrations which eliminate tRNA and VAI RNA complexes.
Since the dissociation constants observed for the PKR complexes formed with these RNAs were rather similar, these results are surprising. Among several possibilities, the flAlu-PKR complex may be kinetically more stable than are complexes formed with tRNA and VAI RNAs. As precedence for this possibility, preformed VAI RNA-PKR and poly(I) · poly(C)-PKR complexes are kinetically resistant to competition by poly(I) · poly(C) (15). We strongly favor this kinetic interpretation since we also observed that preformed flAlu-PKR complexes resisted competition by unlabeled flAlu RNA. However, PKR has high- and low-affinity binding sites (15); therefore, complex formation and competition for these RNAs may occur at different sites. While the difficult question of whether this difference is kinetically or thermodynamically determined remains to be decisively resolved, PKR-flAlu RNA complexes were more stable than were the complexes that PKR formed with either tRNA or VAI RNA (Fig. 5).
Alu RNA antagonizes PKR autophosphorylation.
Since flAlu RNA forms complexes with PKR, we tested its effects on PKR autophosphorylation in vitro. flAlu RNA at concentrations of 50 fg/μl to 15 ng/μl did not stimulate PKR autophosphorylation in vitro (data not shown). Therefore, we next examined its possible antagonism of PKR activation in the presence of dsRNA.
Low concentrations of poly(I) · poly(C) induce PKR autophosphorylation, whereas high concentrations are inhibitory (14, 23). In preliminary experiments, the optimal concentration of poly(I) · poly(C) was found to be 3 ng/μl (Fig. 6, lanes 7 and 10). In the absence of dsRNA, PKR exhibited no kinase activity (Fig. 6, lanes 8 and 9). Very low concentrations (e.g., 0.1 ng/μl [corresponding to about 1 nM]) of either flAlu RNA or VAI RNA partially inhibited (60% inhibition) PKR autophosphorylation in the presence of poly(I) · poly(C) (Fig. 6, lanes 1 and 4). Either of these RNAs at a concentration of 3 ng/μl (ca. 30 nM) caused a fourfold decrease in kinase activity (Fig. 6, lanes 3 and 6). Thus, VAI RNA and flAlu RNA were equally potent PKR antagonists in vitro (Fig. 6, lanes 1 through 6). Consistent with these results, 10 nM VAI RNA has been previously observed to inhibit PKR activation (15); therefore, our results for both the inhibitory activity of VAI RNA and the dissociation constant of its PKR gel shift complexes agree with previous determinations. tRNA does not antagonize PKR activation (35). Discounting 30% fluctuations in activity, we also observed that tRNA was a relatively ineffective PKR antagonist (Fig. 6, lanes 9 through 14). For example, the level of PKR activity in the presence of 50 ng of tRNA per ml (Fig. 6, lane 14) was 76% of the control level (lane 10).
FIG. 6.

flAlu RNA inhibits PKR autophosphorylation in vitro. Recombinant PKR (16 ng/μl) was preincubated with either flAlu RNA (0.1, 0.5, and 3 ng/μl [lanes 1 through 3, respectively]), VAI RNA (0.1, 0.5, and 3 ng/μl [lanes 4 through 6, respectively]), yeast tRNA (0.1, 0.5, 3, and 50 ng/μl [lanes 11 through 14, respectively]), or buffer (lanes 7 through 10) at 30°C for 6 min. Subsequently, poly(I) · poly(C) (poly I.C.) at 3 ng/μl (lanes 1 through 7 and 10 through 14) and labeled ATP (all lanes) were added to initiate phosphorylation. Samples were then incubated for 25 min and assayed by SDS-polyacrylamide gel electrophoresis.
The concentration of PKR employed in these autophosphorylation experiments (240 nM) was comparable to the dissociation constants observed for gel shift complexes. However, the inhibitory effect provided by 1 to 30 nM either flAlu RNA or VAI RNA was significantly greater than would be expected from the formation of discrete complexes with a stoichiometry of only one or two PKR molecules for each RNA, as observed in gel shift assays. PKR has at least two RNA binding sites, and it has previously been suggested that one site is involved in activation and the other is involved in inhibition of PKR kinase activity (15). PKR also binds unstructured, synthetic single-stranded RNAs (15). Consequently, small RNAs, such as Alu and VAI, which contain highly structured regions interspersed with relatively less structured regions may form higher-order PKR complexes in addition to discrete gel shift complexes. Aggregates form when highly purified PKR is titrated with saturating amounts of dsRNA, leading to the suggestion that this phenomenon represents a mechanism for inhibiting PKR with small structured RNAs (4). A second possibility is that flAlu and VAI RNAs exert their inhibitory effects through direct competition with poly(I) · poly(C) binding to the dsRNA binding domains of PKR and that both the activating and inhibitory concentrations of dsRNA are much lower than the PKR concentration.
flAlu RNA antagonizes PKR activity in vivo.
Presumably, any RNA with a sufficient secondary structure may antagonize PKR activation in vitro (31). We therefore tested the effects of flAlu RNA on virus-induced PKR activation in vivo (Fig. 7). Adenovirus infection of 293 cells decreased the activity of PKR (Fig. 7A, lanes 1 and 2). As assayed by Western blotting, the level of PKR in this and subsequent experiments was relatively constant and thus not responsible for differences in its activity (Fig. 7B). In contrast to wild-type virus, infection with Ad720 (which has a deleted VAI RNA gene [9]) increased PKR activity (Fig. 7A, lanes 2 and 3). As previously observed, virus-induced activation of PKR can be inhibited by transient coexpression of VAI RNA (1) (Fig. 7A, lanes 4 and 5). VAI RNA overexpression in this experiment was confirmed by primer extension analysis (Fig. 7C).
flAlu RNA overexpression also inhibited virus-induced activation of PKR (Fig. 7A, lanes 6 and 7). Presumably, endogenous levels of flAlu stimulated by viral infection (Fig. 7C) are insufficient to completely counter virus-induced activation of PKR. In a comparison of the levels of VAI RNA and flAlu RNA overexpression (Fig. 7C), relatively low levels of Alu RNA antagonized PKR to almost the same degree as did much higher levels (ca. 50-fold) of the positive control, VAI RNA. As judged by this comparison, flAlu RNA is a potent PKR antagonist in vivo. Overexpression of scAlu, 7SL, and 5S RNAs provided negative controls for this experiment. scAlu RNA had little or no effect on PKR activity, even when it was expressed at very high levels (approximately the levels of VAI RNA overexpression employed in these experiments) (Fig. 7A, lanes 8 and 9, and C). This result was consistent with the previous observation that scAlu overexpression had little effect on the expression of the cotransfected luciferase reporter gene (Fig. 1). Again, scAlu essentially provided a defective Alu mutant control for possible nonspecific effects of flAlu RNA overexpression. The transfection of either the 7SL RNA or 5S RNA gene had no effect on PKR activity in this assay (Fig. 7A, lanes 10 through 13). The expression of these two RNAs from their exogenous genes is difficult to document against their very high levels of endogenous expression (Fig. 7C). Nonetheless, we observed less 7SL RNA and 5S RNA in Ad720-infected cells than in control 293 cells; upon transient transfection, these levels increased. Thus, in contrast to the positive results for VAI and flAlu RNAs, overexpression of scAlu RNA or expression of exogenous 5S and 7SL RNAs did not significantly antagonize virus-induced PKR activation.
The effects of flAlu RNA upon PKR activity may result from unnaturally high levels of overexpression. Previously, we compared the expression levels and PKR binding of endogenous and exogenous flAlu RNAs (Fig. 3B). That comparison provided an opportunity to observe the effects of physiologically relevant levels of flAlu RNA expression and PKR binding on PKR activity in vivo. PKR activity decreased in a dose-dependent manner with the level of flAlu overexpression (Fig. 8A). Higher levels of overexpression caused a three- to fourfold decrease in PKR activity (Fig. 8A, lanes 3 and 4), but even the lowest level of overexpression tested caused a slight decrease in PKR activity (lane 2). As previously discussed, the amount of flAlu RNA associated with PKR apparently increased with the level of flAlu RNA expression (Fig. 3B). Thus, these data suggest a correlation among the level of flAlu RNA, its association with PKR, and its effects on PKR activity. Of particular significance, this decrease in PKR activity (Fig. 8A) was observed at overexpression levels (two- to fourfold) corresponding to the two- to fourfold increases in endogenous levels of flAlu RNA that resulted from various cellular insults (Fig. 3B). These observations for PKR activity (Fig. 8A) complemented the earlier observation that physiologically relevant levels of flAlu RNA overexpression also increased luciferase reporter expression in a dose-dependent manner (Fig. 2).
Western analysis showed that the level of PKR was approximately constant (Fig. 8B, lanes 3 through 6), indicating again that the effect of flAlu RNA on PKR activity results from a decrease in its activation. The higher-resolution conditions used in this experiment resolved a closely spaced doublet of PKR bands (Fig. 8B, lane 3). Presumably, this doublet resulted from different degrees of phosphorylation; interestingly, the slower-mobility band decreased in intensity (Fig. 8B, lanes 4 through 6) in parallel with the decrease in PKR activity caused by flAlu RNA overexpression (Fig. 8A).
Cycloheximide treatment decreases PKR activity.
The role of PKR and its activation in response to viral infection is well understood. Since exposing cells to cycloheximide and cell stress also increases the accumulation of flAlu RNA, we investigated whether these treatments, like viral infection, affect PKR activity.
The addition of cycloheximide caused a decrease in PKR activity and, in agreement with our previous results (6), an increase in the abundance of flAlu RNA (Fig. 9). The level of PKR protein was unchanged during the time course of this experiment (Fig. 9B), indicating that kinase deactivation was responsible for the decrease in PKR activity. Both the increase in Alu RNA and the decrease in PKR activity began within 20 min of cycloheximide addition (Fig. 9A). Since cycloheximide-induced flAlu transcripts bound PKR (Fig. 3B) and since overexpressed flAlu RNA antagonized PKR activation (Fig. 7), we hypothesize that flAlu RNA is at least partially responsible for this initial decrease in PKR activity. However, 4 h after the addition of cycloheximide, the level of PKR activity was still low, but short-lived flAlu RNA returned to basal levels (Fig. 9). While factors other than flAlu RNA may be responsible for this continuing repression of PKR activity, as discussed above, PKR may form kinetically stable complexes with RNA antagonists, thereby inhibiting its kinase activity even after the RNA returns to basal level.
FIG. 9.
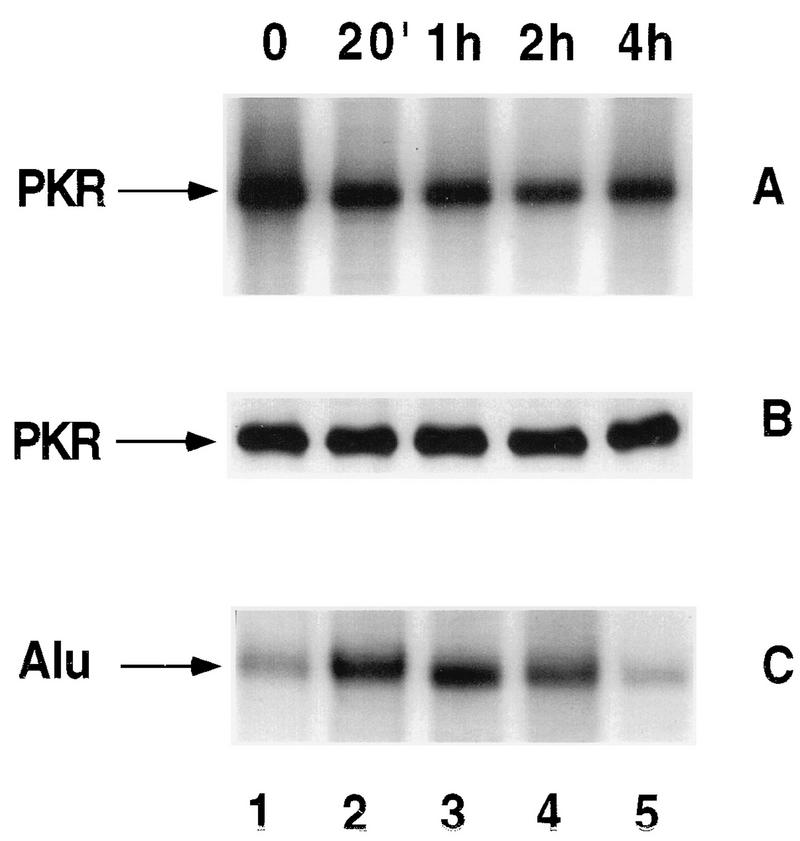
Effects of cycloheximide on PKR activity and flAlu RNA. 293 cells incubated for 17 h with 1,000 U of interferon per ml were exposed to cycloheximide (100 μg/ml) for the indicated times and then assayed for PKR autophosphorylation activity (A) and the level of PKR protein by Western analysis (B). (C) The abundances of flAlu RNA in the same samples were analyzed by primer extension at the indicated times. 20′, 20 min.
DISCUSSION
The rapid, dramatic increases in flAlu RNA in response to viral infection, cell stress, and translational inhibition have raised the possibility that these transcripts serve a physiological role (19, 20, 28, 36). Since each of these treatments also affects protein synthesis, we tested the effects of flAlu RNA on translation by using the expression of a reporter gene. Overexpression of flAlu RNA significantly increased the expression of a luciferase reporter. Negative controls, particularly one employing scAlu RNA, indicated that the stimulated expression of this reporter was not an artifact of RNA overexpression but was attributable to the specific activity of flAlu RNA. Furthermore, the levels of Alu overexpression required to cause an increase in luciferase were comparable to the levels of flAlu RNA induced by cell stress and viral infection, suggesting that endogenous levels of flAlu RNA have similar effects on translational expression. Similarly, levels of flAlu RNA overexpression comparable to those induced by cell stress and viral infection also caused a decrease in PKR activity, suggesting a mechanism for the effects of flAlu RNA on translational expression.
Viral infection and cell stress alter translation by multiple and redundant pathways, including pathways which initially increase and subsequently decrease eIF-2α phosphorylation, thereby first repressing and then derepressing translational initiation (12, 41). Significantly, in both cases, dephosphorylation of eIF-2α to reactivate translational initiation parallels the appearance of entirely new mRNA cohorts encoding viral or heat shock proteins. Inhibition of translational elongation has previously been reported to decrease eIF2α phosphorylation (42). In agreement with that result, we observed that cycloheximide treatment caused a very rapid decrease in PKR activity, which may lead to a decrease in eIF-2α phosphorylation. Presumably, an increase in translational initiation would be an initial, regulated cellular response to a decreased rate of translational elongation. In any event, PKR activity was altered by each of these three cellular treatments, which also increase the level of flAlu RNA.
We further observed that flAlu RNA bound PKR both in vitro and in vivo. In gel mobility shift assays, Alu RNA formed discrete complexes with PKR. The relationship of these complexes to either the binding of PKR to Alu RNA in vivo or the inhibition of PKR by Alu RNA is uncertain. However, under competitive binding conditions, preformed flAlu RNA complexes were relatively stable compared to those formed by VAI RNA, a well-studied PKR antagonist. Presumably, any RNA with a sufficient secondary structure could bind to PKR in vitro (7, 31), but many such RNAs, in the form of stable ribonucleoprotein structures, would not be available for PKR binding in vivo. In agreement with this suggestion, both VAI RNA and flAlu RNA bound PKR in vivo, but we did not observe any interaction between PKR and 7SL RNA, which is far more abundant than is flAlu RNA. As a component of fully formed, functional signal recognition particles (SRPs), 7SL RNA would be protected from PKR binding. The most active PKR inhibitors, as exemplified by VAI RNA, may be highly structured RNAs which do not serve functions that require being tightly sequestered in ribonucleoprotein structures. flAlu RNA is known to bind only the two smallest SRP proteins, presumably making it accessible to PKR in vivo.
flAlu RNA was equally as effective as was VAI RNA in antagonizing the activation of PKR in vitro; more importantly, overexpressed flAlu RNA also antagonized virus-induced activation of PKR in vivo. Interestingly, overexpressed flAlu RNA antagonized PKR activation to almost the same degree as did far higher concentrations of VAI RNA, leading us to conclude that flAlu RNA is indeed a potent PKR inhibitor. Higher levels of overexpressed scAlu RNA did not cause this inhibition, showing that the antagonism of PKR by flAlu RNA is not an artifact of gross overexpression but depends upon the structure of flAlu RNA.
These observations suggest that endogenous flAlu RNA affects translational expression by inhibiting PKR activation. We have not yet tested this possibility directly. However, cell stress, cycloheximide treatment, and viral infection changed PKR activity, which presumably modulates changes in protein synthesis caused by these same treatments. We also observed a rather simple dose-response relationship between the abundance of exogenously expressed flAlu RNA and the levels of both transiently coexpressed luciferase activity and PKR activity. Furthermore, the increased levels of flAlu RNA caused by viral infection, translational inhibition, and cell stress were similar to the levels of flAlu RNA overexpression that stimulated expression of the luciferase reporter and concomitantly decreased PKR activity. Thus, we consider it a possibility that increases in flAlu RNA caused by these three cellular treatments affect PKR activity and consequently protein synthesis.
As a host defense against viral infection, PKR is activated by dsRNA to phosphorylate eIF-2α, thereby blocking protein synthesis (8, 41). PKR is also an important signal transducer for interferon and cytokine induction of gene expression through regulation of transcription factors NF-κB and IRF-1 (25, 26). Viral counterstrategies to block PKR activation include the synthesis of massive quantities of small RNAs, such as VAI RNA in the case of adenovirus, to antagonize PKR’s activation (8, 22, 41). In the most thoroughly studied case of adenovirus, viral gene products direct the increase in Alu transcription after infection (36). By binding PKR and antagonizing its activation, these induced flAlu transcripts may provide another viral defense against PKR activation by the host. There are already many known viral strategies to counter host defenses, and the existence of yet another is not surprising (41). Of course, VAI RNA encoded by adenovirus accumulates to a much higher level than does short-lived flAlu RNA and would therefore be expected to serve as a far more effective PKR antagonist. However, virus-encoded pathways are often deleterious to the host cell. A cellular PKR antagonist would not need to achieve the level of PKR antagonism provided by VAI RNA. It is noteworthy that VAI deletion mutants, although impaired, remain viable, indicating that there are redundant pathways to overcome host defenses (41). Although the physiological function of flAlu RNA cannot be to enhance viral infectivity, viruses typically co-opt normal cellular regulatory mechanisms.
As discussed above, a decrease in PKR activity is plausibly an initial cellular response to the inhibition of translational elongation caused by drugs such as cycloheximide. More than 20 years ago, Reichman and Penman identified a factor, termed an activator, which stimulates translational initiation (37). They demonstrated that this activator is an RNA with a half-life of about 1 h, which is approximately the short lifetime of flAlu RNA (6, 16). Like flAlu RNA, activator RNA is induced by both heat shock and cycloheximide treatment of cells. Comparing the results of Goldstein et al. (16) to those of Liu et al. (28), the transient increases in both activator RNA and flAlu RNA levels in response to cycloheximide are virtually identical. We suspect that flAlu RNA is this translational activator. Retrospectively, the antagonism of PKR activation by the increased levels of flAlu RNA caused by cycloheximide treatment provides a mechanistic explanation for the observed biochemical activity of activator RNA upon translational initiation.
Hemin-regulated initiation factor-2 kinase, a PKR homolog, phosphorylates eIF-2α in response to heat shock, but PKR itself has no known role in the heat shock response (10). During long-term heat shock, PKR changes from a soluble form to an insoluble form (11). We observed transient increases in PKR activity during heat shock recovery. However, the kinetic relationship of these changes in PKR activity to transient increases in flAlu RNA abundance during heat shock recovery is not evident. At present, any proposed function that PKR may serve during the heat shock response would be entirely speculative; therefore, identifying the effects that flAlu RNA may cause by acting on PKR during this period is even more problematic. However, complex changes in translational expression occur during both heat shock and heat shock recovery and, in part, these changes are regulated by changes in eIF-2α phosphorylation (12). Consequently, PKR and its regulators are almost certainly involved in the heat shock response. As suggested above, activator RNA may be a PKR regulator which is induced by heat shock (6, 16).
Ideally, the possibility that Alu RNA fulfills a physiological role would be tested by genetics. Because of their extraordinary copy number, Alu RNAs are not amenable to classical genetic tests. In Tetrahymena spp., a small, Pol III-transcribed RNA rapidly accumulates after heat shock and is required for the establishment of thermal tolerance (13). Interestingly, this transcript, like human Alu RNAs, is related to 7SL RNA. We do not know whether there is any relationship between the function of this gene and the present observations concerning flAlu RNA. However, there are several intriguing parallels between these two systems and certainly the Tetrahymena results provide strong precedence for the functionality of small heat shock-induced RNAs.
This proposed role for human Alu RNA potentially explains some unusual evolutionary features of mammalian SINEs. (i) There is no homology between human Alu RNAs and many other mammalian SINEs, such as rabbit SINEs (39, 44). Either SINEs are functionless or their function(s) does not depend upon sequence but upon some other, higher-order structure. However, entirely nonhomologous mammalian SINE RNAs could antagonize PKR since the binding of PKR to duplex RNA regions does not depend strongly on the sequence but rather requires a minimum number of base pairs of sufficient base pair fidelity (8, 31). Interestingly, the dimeric structure of human Alu elements may make their transcripts unusually potent PKR antagonists since each flAlu RNA potentially binds two molecules of PKR. The results from gel shift assays support this intriguing possibility. (ii) Individual mammalian SINEs are weakly promoted; therefore, their expression is easily repressed (6, 39). Nevertheless, the extremely large number of SINEs guarantees that many elements are always in active chromatin domains, thereby permitting a robust transcriptional response in all cell types despite the weakness of their individual promoters (6, 28). (iii) The short lifetime of SINE transcripts, which lack other normal cellular functions, makes them ideally suited to signal PKR. The abundance of a long-lived RNA serving an essential constitutive function could not be rapidly and significantly increased without disrupting that function.
Although an exact physiological role for Alu RNA and more generally mammalian SINE RNAs remains to be determined, overexpressed Alu transcripts stimulate translation and almost certainly do so by antagonizing PKR activation. Adaptation of these effects to regulate translational expression could provide a significant selective advantage for the maintenance of SINEs within the mammalian genome. The induction of Alu RNAs by cell stress and other treatments and the association between Alu RNA and PKR suggest that this potential selective advantage is being exploited.
ACKNOWLEDGMENTS
We thank John Hershey for critical interest and advice. We also thank Wen-Man Liu for designing, testing, and providing clone XAL.
This research was supported in part by USPHS grant GM21346 and the Agriculture Experiment Station, University of California, Davis (C.W.S.) and USPHS grant AI 34039 (B.R.G.W.).
REFERENCES
- 1.Akusjarvi G, Svensson C, Nygard O. A mechanism by which adenovirus virus-associated RNAI controls translation in a transient-expression assay. Mol Cell Biol. 1987;7:549–551. doi: 10.1128/mcb.7.1.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barber G N, Wambach M, Wong M L, Dever T E, Hinnebusch A G, Katze M G. Translational regulation by the interferon-induced double-stranded-RNA-activated 68-kDa protein kinase. Proc Natl Acad Sci USA. 1993;90:4621–4625. doi: 10.1073/pnas.90.10.4621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bredow S, Surig D, Muller J, Kleinert H, Benecke B J. Activating-transcription-factor (ATF) regulates human 7SL RNA transcription by RNA polymerase III in vivo and in vitro. Nucleic Acids Res. 1990;18:6779–6784. doi: 10.1093/nar/18.23.6779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carpick B W, Graziano V, Schneider D, Maitra R K, Lee X, Williams B R G. Characterization of the solution complex between the interferon-induced double-stranded RNA-activated protein kinase and HIV-1 trans-activating region RNA. J Biol Chem. 1997;272:9510–9516. doi: 10.1074/jbc.272.14.9510. [DOI] [PubMed] [Google Scholar]
- 5.Chong K, Schappert K, Friesen J, Donahue T, Meurs A, Hovanessian A G, Williams B R G. Expression of the human p68 kinase in yeast reveals a growth inhibiting phenotype and functional homology to the translational regulator GCN2. EMBO J. 1992;11:1553–1562. doi: 10.1002/j.1460-2075.1992.tb05200.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chu W-M, Liu W-M, Schmid C W. RNA polymerase III promoter and terminator elements affect Alu transcription. Nucleic Acids Res. 1995;23:1750–1757. doi: 10.1093/nar/23.10.1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Clarke P A, Schwemmle M, Schickinger J, Hilse K, Clemens M. Binding of Epstein-Barr virus small RNA EBER-1 to the double-stranded RNA-activated protein kinase DAI. Nucleic Acids Res. 1991;19:243–248. doi: 10.1093/nar/19.2.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Clemens M J. Protein kinases that phosphorylate eIF2 and eIF2B and their role in eukaryotic cell translational control. In: Hershey J W B, Mathews M B, Sonenburg N, editors. Translational control. Plainview, N.Y: Cold Spring Harbor Laboratory Press; 1996. pp. 139–172. [Google Scholar]
- 9.Davies M V, Futrado M, Hershey J N, Thimmappaya B, Kaufman R J. Complementation of adenovirus-associated RNA I gene deletion by expression of a mutant eukaryotic translation initiation factor. Proc Natl Acad Sci USA. 1989;86:9163–9167. doi: 10.1073/pnas.86.23.9163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.De Benedetti A, Baglioni C. Activation of hemin-regulated initiation factor-2 kinase in heat-shocked HeLa cells. J Biol Chem. 1986;261:338–342. [PubMed] [Google Scholar]
- 11.Dubois M F, Galabru J, Lebon P, Safer B, Hovanessian A G. Reduced activity of the interferon-induced double-stranded RNA-dependent protein kinase during a heat shock stress. J Biol Chem. 1989;264:12165–12171. [PubMed] [Google Scholar]
- 12.Duncan R F. Translational control during heat shock. In: Hershey J W B, Mathews M B, Sonenburg N, editors. Translational control. Plainview, N.Y: Cold Spring Harbor Laboratory Press; 1996. pp. 271–293. [Google Scholar]
- 13.Fung P A, Gaertig J, Gorovsky M A, Hallberg R L. Requirement of a small cytoplasmic RNA for the establishment of thermotolerance. Science. 1995;268:1036–1039. doi: 10.1126/science.7754381. [DOI] [PubMed] [Google Scholar]
- 14.Galabru J, Hovanessian A. Autophosphorylation of the protein kinase dependent on double stranded RNA. J Biol Chem. 1987;262:15538–15544. [PubMed] [Google Scholar]
- 15.Galabru J, Katze M G, Robert N, Hovanessian A G. The binding of double-stranded RNA and adenovirus VAI RNA to the interferon-induced kinase. Eur J Biochem. 1989;178:581–589. doi: 10.1111/j.1432-1033.1989.tb14485.x. [DOI] [PubMed] [Google Scholar]
- 16.Goldstein E S, Reichman M E, Penman S. The regulation of protein synthesis in mammalian cells. VI. Soluble and polyribosome associated components in controlling in vitro polypeptide initiation in HeLa cells. Proc Natl Acad Sci USA. 1974;71:4752–4756. doi: 10.1073/pnas.71.12.4752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gunnery S A, Rice A P, Robertson H D, Mathews M B. Tat-responsive region RNA of human immunodeficiency virus 1 can prevent activation of the double-stranded-RNA-activated protein kinase. Proc Natl Acad Sci USA. 1990;87:8687–8691. doi: 10.1073/pnas.87.22.8687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hickman M A, Malone R W, Lehman-Bruinsma K, Sih T, Knoell D, Szoka F C, Walzem R, Carlson D M, Powell J S. Gene expression following direct injection of DNA into liver. Hum Gene Ther. 1994;5:1477–1483. doi: 10.1089/hum.1994.5.12-1477. [DOI] [PubMed] [Google Scholar]
- 19.Jang K L, Latchman D S. The herpes simplex virus immediate-early protein ICP27 stimulates the transcription of cellular Alu repeated sequences by increasing the activity of transcription factor TFIIIC. Biochem J. 1992;284:667–673. doi: 10.1042/bj2840667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jang K L, Collins M K, Latchman D S. The human immunodeficiency virus tat protein increases the transcription of human Alu repeated sequences by increasing the activity of the cellular transcription factor TFIIIC. J Acquired Immune Defic Syndr. 1992;5:1142–1147. [PubMed] [Google Scholar]
- 21.Katze M G. The war against the interferon-induced dsRNA-activated protein kinase: can viruses win? J Interferon Res. 1992;12:241–248. doi: 10.1089/jir.1992.12.241. [DOI] [PubMed] [Google Scholar]
- 22.Katze M G, DeCorato D, Safer B, Galabru J, Hovanessian A G. Adenovirus VAI RNA complexes with the 68,000 Mr protein kinase to regulate its autophosphorylation and activity. EMBO J. 1987;6:689–697. doi: 10.1002/j.1460-2075.1987.tb04809.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Katze M G, Wambach M, Wong M-L, Garfinkel M, Meurs E, Chong K, Williams B R G, Hovanessian A G, Barber G N. Functional expression and RNA binding analysis of the interferon-induced, double-stranded RNA-activated, 68,000-Mr protein kinase in a cell-free system. Mol Cell Biol. 1991;11:5497–5505. doi: 10.1128/mcb.11.11.5497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Koski R A, Clarkson S G. Synthesis and mutation of Xenopus laevis methionine tRNA gene transcription homologous cell-free extracts. J Biol Chem. 1982;257:4514. [PubMed] [Google Scholar]
- 25.Kumar A, Hague J, Lacaste J, Hiscott J, Williams B R G. The dsRNA-dependent protein kinase, PKR, activates transcription factor NF-κB by binding IkB. Proc Natl Acad Sci USA. 1994;9:6288–6292. doi: 10.1073/pnas.91.14.6288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kumar A, Yang Y-L, Flati V, Der S, Kadereit S, Hague J, Deb A, Reis L, Weismann C, Williams B R G. Deficient cytokine signaling in mouse embryo fibroblasts with a targeted deletion in the PKR gene: role of IRF-1 and NFκB. EMBO J. 1997;16:406–416. doi: 10.1093/emboj/16.2.406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Little R, Bratten M. Genomic organization of human 5S rDNA and sequence of one tandem repeat. Genomics. 1989;4:376–383. doi: 10.1016/0888-7543(89)90345-5. [DOI] [PubMed] [Google Scholar]
- 28.Liu W-M, Chu W-M, Choudary P V, Schmid C W. Cell stress and translational inhibitors transiently increase the abundance of mammalian SINE transcripts. Nucleic Acids Res. 1995;23:1758–1765. doi: 10.1093/nar/23.10.1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu W-M, Maraia R J, Rubin C M, Schmid C W. Alu transcripts: cytoplasmic localization and regulation of DNA methylation. Nucleic Acids Res. 1994;22:1087–1095. doi: 10.1093/nar/22.6.1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29a.Liu, W.-M., and C. W. Schmid. Unpublished data.
- 30.Maitra R K, McMillan N A J, Desai S, McSwiggen J, Hovanessian A G, Sen G, Williams B R G, Silverman R H. HIV-1 TAR RNA has an intrinsic ability to activate interferon-inducible enzymes. Virology. 1994;204:823–827. doi: 10.1006/viro.1994.1601. [DOI] [PubMed] [Google Scholar]
- 31.Manche L, Green S R, Schmedt C, Mathews M B. Interactions between double-stranded RNA regulators and the protein kinase DAI. Mol Cell Biol. 1992;12:5238–5248. doi: 10.1128/mcb.12.11.5238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maraia R J, Driscoll C T, Bilyeu T, Hsu K, Darlington G J. Multiple dispersed loci produce small cytoplasmic Alu RNA. Mol Cell Biol. 1993;13:4233–4241. doi: 10.1128/mcb.13.7.4233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McMillan N A J, Williams B R G. Structure and function of the interferon-induced protein kinase PKR and related enzymes. In: Clemens M J, editor. Structure and function of the interferon-induced protein kinase, PKR, and related enzymes in protein phosphorylation in cell growth regulation. Amsterdam, The Netherlands: Harwood Academic Publishers; 1996. pp. 225–253. [Google Scholar]
- 34.Murtha-Riel P, Davies M V, Scherer B J, Choi S Y, Hershey J W, Kaufman R J. Expression of a phosphorylation-resistant eukaryotic initiation factor 2 alpha-subunit mitigates heat shock inhibition of protein synthesis. J Biol Chem. 1993;268:12946–12951. [PubMed] [Google Scholar]
- 35.O’Malley R P, Mariano T M, Siekierka J, Mathews M M. A mechanism for the control of protein synthesis by adenovirus VA RNAI. Cell. 1986;44:391–400. doi: 10.1016/0092-8674(86)90460-5. [DOI] [PubMed] [Google Scholar]
- 36.Panning B, Smiley J R. Activation of RNA polymerase III transcription of human Alu repetitive elements by adenovirus type 5: requirement for the E1b 58-kilodaton protein and the products of E4 open reading frames 3 and 6. Mol Cell Biol. 1993;13:3231–3244. doi: 10.1128/mcb.13.6.3231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reichman M, Penman S. Stimulation of polypeptide initiation in vitro after protein synthesis inhibition in vivo in HeLa cells. Proc Natl Acad Sci USA. 1973;70:2678–2682. doi: 10.1073/pnas.70.9.2678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schmedt C, Gree S R, Manche L, Taylor D R, Ma Y, Mathews M B. Functional characterization of the RNA-binding domain and motif of the double stranded RNA-dependent protein kinase DAI (PKR) J Mol Biol. 1995;249:29–44. doi: 10.1006/jmbi.1995.0278. [DOI] [PubMed] [Google Scholar]
- 39.Schmid C W, Maraia R. Transcriptional regulation and transpositional selection of active SINE sequences. Curr Opin Genet Dev. 1992;2:874–882. doi: 10.1016/s0959-437x(05)80110-8. [DOI] [PubMed] [Google Scholar]
- 40.Schmid C W. Alu: structure, origin, evolution, significance and function of one-tenth of human DNA. Prog Nucleic Acid Res Mol Biol. 1996;53:283–319. doi: 10.1016/s0079-6603(08)60148-8. [DOI] [PubMed] [Google Scholar]
- 41.Schneider R J. Adenovirus and vaccinia virus translational control. In: Hershey J W B, Mathews M B, Sonenburg N, editors. Translational control. Plainview, N.Y: Cold Spring Harbor Laboratory Press; 1996. pp. 575–606. [Google Scholar]
- 42.Scorsone K A, Panniers R, Rowlands A G, Henshaw E C. Phosphorylation of eukaryotic initiation factor 2 during physiological stresses which affect protein synthesis. J Biol Chem. 1987;262:14538–14541. [PubMed] [Google Scholar]
- 43.Sinnett D, Richer C, Deragon J M, Labuda D. Alu RNA transcripts in human embryonal carcinoma cells. Model of post-transcriptional selection of master sequences. J Mol Biol. 1992;226:689–706. doi: 10.1016/0022-2836(92)90626-u. [DOI] [PubMed] [Google Scholar]
- 44.Weiner A M, Deininger P L, Efstratiadis A. Nonviral retroposons: genes, pseudogenes, and transposable elements generated by the reverse flow of genetic information. Annu Rev Biochem. 1986;55:631–661. doi: 10.1146/annurev.bi.55.070186.003215. [DOI] [PubMed] [Google Scholar]