

Abstract
Most strains of Candida glabrata switch spontaneously between a number of phenotypes distinguishable by graded brown coloration on agar containing 1 mM CuSO4, a phenomenon referred to as “core switching.” C. glabrata also switches spontaneously and reversibly from core phenotypes to an irregular wrinkle (IWr) phenotype, a phenomenon referred to as “irregular wrinkle switching.” To identify genes differentially expressed in the core phenotypes white (Wh) and dark brown (DB), a cDNA subtraction strategy was employed. Twenty-three genes were identified as up-regulated in DB, four in Wh, and six in IWr. Up-regulation was verified in two unrelated strains, one a and one α strain. The functions of these genes were deduced from the functions of their Saccharomyces cerevisiae orthologs. The majority of genes up-regulated in DB (78%) played deduced roles in copper assimilation, sulfur assimilation, and stress responses. These genes were differentially up-regulated in DB even though the conditions of growth for Wh and DB, including CuSO4 concentration, were identical. Hence, the regulation of these genes, normally regulated by environmental cues, has been usurped by switching, presumably as an adaptation to the challenging host environment. These results are consistent with the suggestion that switching provides colonizing populations with a minority of cells expressing a phenotype that allows them to enrich in response to an environmental challenge, a form of rapid adaptation. However, DB is the most commonly expressed phenotype at sites of host colonization, in the apparent absence of elevated copper levels. Hence, up-regulation of these genes by switching suggests that in some cases they may play roles in colonization and virulence not immediately obvious from the roles played by their orthologs in S. cerevisiae.
Candida glabrata is the second most prevalent Candida species colonizing humans (13, 17, 33, 43, 46). Because C. glabrata is carried in the same anatomical niches as Candida albicans and results in similar infections, it has been assumed that its basic biology would be similar to that of C. albicans. However, sequencing studies revealed that C. glabrata is more closely related to Saccharomyces cerevisiae than to C. albicans (3, 7, 8, 25, 67), suggesting that it could employ mechanisms different from those of C. albicans to generate phenotypic plasticity during pathogenesis. This indeed appears to be the case for spontaneous phenotypic switching. C. glabrata switches in a unique fashion between a number of phenotypes distinguishable by graded colony coloration on agar containing 1 mM CuSO4, a phenomenon referred to as “core switching” (28, 29). Core switching has not been observed in C. albicans. Core phenotypes include white (Wh), light brown (LB), dark brown (DB), and very dark brown (vDB) (28). Additionally, cells expressing any one of the core phenotypes can spontaneously and reversibly switch to an irregular wrinkle (IWr) phenotype, a transition referred to as “irregular wrinkle switching” (28). It is assumed that the graded brown coloration of the core phenotypes is the result of graded levels of conversions of Cu2+ to Cu1+ and the associated reduction of SO42− to S1−. Although growth on CuSO4-containing agar originally revealed the core switching system (29), CuSO4 does not appear to induce switching. Switching occurs at the same frequencies on agar lacking CuSO4 and can be identified by adding phloxine B to the supporting agar (28). Phloxine B stains Wh, LB, DB, and vDB colonies graded shades of red, but in an intensity order reversed from the brown graded colors on CuSO4-containing agar. The coloration of the IWr phenotype is white on CuSO4-containing agar, regardless of the core phenotype from which the IWr strain arose (28). However, IWr exhibits a propensity for switching back to the core phenotype from which it arose, suggesting that although it expresses characteristics of Wh, it retains, or “remembers,” its core phenotype of origin.
The graded differences between the core phenotypes in copper-based coloration suggested that core switching involved the regulation of genes that played roles in copper homeostasis and detoxification. Lachke et al. (29) originally demonstrated that the transcript levels of the metallothionein genes MT-I and MT-II were lower in Wh than in DB and that these differences were expressed in the absence as well as the presence of CuSO4. Prior studies had revealed that MT-I and MT-II were up-regulated upon exposure to CuSO4 (36, 38, 71) through the metalloregulatory transcription factor Amt1p (72). Furthermore, Mehra et al. (37) demonstrated that repeated exposure to increasing concentrations of CuSO4 resulted in concomitant increases in resistance to CuSO4 and associated amplification of the MT-II gene. However, Lachke et al. (29) demonstrated that amplification of MT-II was not responsible for the differential expression of MT-II genes during core switching. Rather, spontaneous switching directly regulated expression of these genes, regardless of extracellular CuSO4 levels, presumably by phase-specific trans-acting factors. Lachke et al. (29) demonstrated that genes other than those involved in copper assimilation, such as HLP1, were also regulated by core switching. Furthermore, Srikantha et al. (59) demonstrated that MTLα1 was regulated by core switching, but in reverse order, with expression highest in Wh and lowest in DB.
Here we have performed a cDNA subtraction screen of phase-specific libraries in order to identify additional genes regulated by core switching. Genes enriched in cDNA pools of Wh or DB cells subtracted with excess driver cDNA from the alternative phenotype were then tested by Northern blot hybridization for levels of expression in Wh, DB, and IWr cells derived from either an a or an α strain. To determine if the differential expression of phase-regulated genes was mediated by upstream promoter regions, the promoters of select genes were fused with the reporter gene expressing Renilla reniformis luciferase (RLUC), and reporter activity was assessed in Wh and DB cells. The results indicate that more genes are up-regulated in DB than in Wh, that the majority of these genes are related to copper detoxification and stress responses, and that these genes are regulated by core switching at the level of the promoter. However, because DB is a common colonizing phenotype, and because copper toxicity does not appear to be an environmental factor contributing to colonization, we propose that some of these differentially expressed genes may play roles other than those deduced from their S. cerevisiae orthologs.
MATERIALS AND METHODS
Strains and culture conditions.
The origins of the strains used in this study are provided in Table 1. All strains were stored in 20% glycerol at −80°C. In the course of an experiment, cells of different switch phenotypes were isolated and plated onto YPD agar (2% [wt/vol] Bacto peptone, 2% [wt/vol] glucose, 1% [wt/vol] yeast extract, 1 mM CuSO4, 1.5% [wt/vol] agar). For experiments, cells were obtained from colonies grown at 25°C for 4 to 6 days. The ura3− strains were maintained on defined synthetic dextrose agar (2 g of a complete amino acid mixture, 20 g dextrose, 5 g ammonium sulfate, 1.45 g yeast nitrogen base, and 50 mg uracil per liter [54]). Only colonies exhibiting a homogeneous switch phenotype were used for experiments.
TABLE 1.
Strains used in this study
| Strain | Parent straina | Genotypeb | Mating type | Reference |
|---|---|---|---|---|
| 35B11 | — | — | MTLa | 59 |
| PB921 | — | — | MTLα | 59 |
| 1480.49 | — | — | MTLα | 59 |
| 1480.47 | — | — | MTLα | 59 |
| 1480.50 | — | — | MTLa | 59 |
| 40F1 | 35B11 | ura3Δ(−85 + 932)::Tn903NeoR | MTLa | This study |
| 12F1 | PB921 | ura3Δ(−85 + 932)::Tn903NeoR | MTLα | This study |
—, strain is natural and therefore has no identifiable parent.
—, wild type.
cDNA subtraction.
The generation of cDNA libraries enriched for mRNA species of the switch phenotypes Wh and DB was based on the subtraction strategy of Hubank and Schatz (18). The Oligotex mRNA kit (QIAGEN, Valencia, CA) was used to isolate poly(A)+ mRNA individually from the Wh and DB phenotypes of strains 35B11 (MTLa) and 1480.49 (MTLα). This RNA was used to synthesize double-stranded cDNA, employing the PCR-Select cDNA subtraction kit (BD Sciences, Palo Alto, CA) with minor modifications. Subtractions were performed reciprocally between Wh and DB for both the a and α strains. The cDNA sample from each cell phenotype was digested with RsaI to create blunt ends and then split into three aliquots, one to be used as a driver in subtraction and the other two, ligated at both ends to adaptors 1 and 2R, respectively, to be used as “targets” (Table 2). Two sequential subtraction hybridizations were performed, the first for the duplicate Wh targets versus the DB driver and the second for the duplicate DB targets versus the Wh driver, for both the a and α strains. The eight subtracted cDNA pools, two from Wh and two from DB of each of the two strains, were then selectively amplified, first by using a PCR strategy to eliminate unsubtracted cDNAs and second by a PCR enrichment strategy employing nested primers 1 and 2 (Table 2). The secondary PCR-amplified cDNA pools were purified with the Wizard-PCR Clean kit and ligated to the pGEM-T-Easy plasmid (both from Promega, Madison, WI). Ligation products were transformed into Escherichia coli strain DH10B by electroporation. Approximately 1,500 transformants from each ligation were screened by colony hybridization (16) with MT-II and MT-I, which were presumed to be overrepresented cDNAs in DB (29), and with MTLα1, which was assumed to be an overrepresented cDNA in α cells (59). Clones that hybridized to these probes were excluded from further analysis. Two hundred fifty recombinant clones were chosen for further study. Extracted plasmid DNAs were digested with EcoRI and analyzed for the presence of inserts. All positive plasmid clones were sequenced.
TABLE 2.
Primers and adaptors used in this study
| Primer or adaptor | Gene or purposea | Sequence |
|---|---|---|
| Adaptor 1 | cDNA subtraction | 5′-CTA ATA CGA CTC ACT ATA GGG CTC GAG CGG CCG CCC GGG CAG GT-3′ |
| Adaptor 2R | cDNA subtraction | 5′-CTA ATA CGA CTC ACT ATA GGG CAG CGT GGT CGC GGC CGA GGT-3′ |
| PCR primer 1 | cDNA subtraction | 5′-CTA ATA CGA CTC ACT ATA GGGC-3′ |
| Nested primer 1 | cDNA subtraction | 5′-TCG AGC GGC CGC CCG GGC AGGT-3′ |
| Nested primer 2R | cDNA subtraction | 5′-AGC GTG GTC GCG GCC GAGGT-3′ |
| REFV | RLUC ORF | 5′-TAA CTC GAG TCG CGA CCG CGG CTG CAG ATG ACT TCG AAA GTT TAT GAT-3′ |
| RERV | RLUC ORF | 5′-TAA GAG CTC TTA TTG TTC ATT TTT GAG AACT-3′ |
| ADFV | 3′ end ADE2 | 5′-TAA GAG CTC TAA TAG AGC ACC ATC TAT AAA ACCA-3′ |
| ADRV | 3′ end ADE2 | 5′-TAA ATC GAT TGG CGT TGA TAT TTG CGC CT-3′ |
| TCMF1 | SCTCM1 | 5′-TCG CCC GGG CAG CTG CTA AGC CGG GAA GTT-3′ |
| TCMR1 | 5′-TCG CCC GGG ATA TGT ACA TGT AGTC-3′ | |
| TCIn-R1 | 5′-TCG TCG CGA GTC GAC GAA GTT TTG TTA GAA AAT AAATC-3′ | |
| TCIn-F1 | 5′-TCG TCG CGA CCG CGG GGT ACC GAT TGA TTG ATT GTT GTA GTA ACTG-3′ | |
| NATSacF1 | NAT1 ORF | 5′-ATT GAG CTC ATG ACC ACT CTT GAC GAC ACG GC-3′ |
| NATSacR1 | NAT1 ORF | 5′-ATT GAG CTC TCA GGG GCA GGG CAT GCT CAT GTA GAGC-3′ |
| cghof1 | HO | 5′-ATT ATG CAT CTC GAG AAA TGT AAT CCT GTA TTT TCT GG-3′ |
| cghor1 | HO | 5′-TAA CTG CAG TGT ACT CTA TTG TAC CGC CTG TTTC-3′ |
| PMPF1 | PMP1 | 5′-TCG CTG CAG TTA CAA ATA GGA CTA GAC TAC-3′ |
| PMPR1 | Promoter | 5′-TCG CTG CAG GTC GTC AAC ACA ACG TAC-3′ |
| TARF2 | TAR | 5′-TTA CTC GAG GTG TAT TCC GGC ACC TTA AC-3′ |
| TARR1 | Promoter | 5′-TCG CTG CAG CAC TGG GCC AGC ATC GGT TTT GGC-3′ |
| ECM17F1 | ECM17 | 5′-TAA CTG CAG GCT TAA AAA AAG GTG ATC ATA GAT TC-3′ |
| ECM17R1 | Promoter | 5′-TAA CTG CAG GGT GAA TAA CAA AAA ATCG-3′ |
| ACPF1 | ACP9 | 5′-TCG CTG CAG TGT GGC AGG TCT TGC TCC CC-3′ |
| APCR1 | Promoter | 5′-TCG CTG CAG TCA ATC AAG CCA GAA ACA TT-3′ |
| HSPF1 | HSP104 | 5′-TAA CTG CAG ATG AAC GAC GAA ACA CAG TT-3′ |
| HSPR1 | Promoter | 5′-TAA CTG CAG CAT GGG TTA GAC TTT ATTC-3′ |
| PGKF1 | PGK1 | 5′-TCG CTG CAG AAC TGT GAG ATT ACC ATG AC-3′ |
| PGKR1 | Promoter | 5′-TCG CTG CAG TAT CGA ATA GAT GTA TGT ATGC-3′ |
| TEFF1 | TEF4 | 5′-TCG CTG CAG ATG TTA TCA TCT ATT TTG-3′ |
| TEFR1 | Promoter | 5′-TCG CTG CAG TTT TTT CTT GGT ATG CTT GC-3′ |
| SUTF1 | SUT1 | 5′-TCG CTG CAG AGC ACA ATG TGT GGA AAT TTAC-3′ |
| SUTR1 | Promoter | 5′-TCG CTG CAG TTT GCT GTA TTG AAA TAA ATC-3′ |
| M14F1 | ECM14 | 5′-GAT AGA CAT CGA CCA CTC-3′ |
| M14R1 | 5′-TGG GTC GAG TAA GAA ATC-3′ | |
| M25F1 | ECM25 | 5′-CAG AAG CGG ATC TGT ATC-3′ |
| M25R1 | 5′-CTA GAC TGA ATT CTG ATGC-3′ | |
| R8F1 | FAR8 | 5′-ATT GTT GAT GTT GTA CTC-3′ |
| R8R1 | 5′-TTA TTT AGA GTT TAC TAAC-3′ | |
| ZFF1 | FZF1 | 5′-ATA TCG TCT GAA CAA CAC-3′ |
| ZFR1 | 5′-TAG TAC ACG CTC CTG GAC-3′ | |
| P10F1 | HSP104 | 5′-TCA TTA TGA CTT CCA ACC-3′ |
| P10R1 | 5′-TTA ATC AAC ATC AAC AGC-3′ | |
| AXF1 | RAX2 | 5′-ATG ACA TAT ATA CAG CCTC-3′ |
| AXR1 | 5′-AAG TTC AAC AAG TGT GGC-3′ | |
| OXF1 | ROX1 | 5′-TTG TCT TCT CTG AGC AGC-3′ |
| OXR1 | 5′-TAG TGG TGA AGA GTT TTGC-3′ | |
| GOF1 | SGO1 | 5′-TAG TTG ATG CTA CTA CAAC-3′ |
| GOR1 | 5′-CTT TAT TCT TTC TGG ATC-3′ | |
| ILF1 | SIL1 | 5′-ACG CAT TGA AGC AAG GCTC-3′ |
| ILR1 | 5′-TCA ACA ATA TAT GGA TCC | |
| PCF1 | SPC98 | 5′-GCT CAA TTC TTC TGA CGC-3′ |
| PCR1 | 5′-TCA ACG TAA ATG CCT ACTC-3′ | |
| CTV2f1 | CTR2 | 5′-ATG AAG ATG GAT CAC TCA GGC-3′ |
| CTV2V1 | 5′-TCA GTG GCA TGC TAG TGT TC-3′ | |
| CCC2f1 | CCC2 | 5′-CGT TCC GTA GAG TGA AAC-3′ |
| CCC2r1 | 5′-GTC CTT TGT GAG GAT AAAC-3′ | |
| Zrtf1 | ZRT1 | CAT GTT CAC AAC ACT ACC-3′ |
| Zrtr1 | 5′-GAT CCG ATA GCG ATA GC-3′ | |
| Ctrf1 | CTR1 | 5′-CTT CAT GCC TTC TTT CAC-3′ |
| Ctrr1 | 5′-TTC TCG TCG TAC TCT TCC-3′ | |
| fre6f1 | FRE6 | 5′-AAT ACC CAG ATT CTT CTG GC-3′ |
| fre6r1 | 5′-GAT ACA AAG TTA CGG TAA CC-3′ | |
| AQYf1 | AQY1 | 5′-ATG GAA ACA GAA CAT CAG GC-3′ |
| AQYr1 | 5′-TTA GGG TTC AAC CAT TACC-3′ | |
| cbff1 | CBF1 | 5′-CCA AAG CCA GAG TAC AGC-3′ |
| cbfr1 | 5′-AGA CTC CAT CTT CGC CAGC-3′ | |
| Ecm17f1 | ECM17 | 5′-ACC AAC ATG TGG TCT AGC-3′ |
| Ecm17r1 | 5′-GCA TCT TCA GAG ATA TCC-3′ | |
| Met31f1 | MET31 | 5′-ATG TCT ACG GGA GAT GATC-3′ |
| Met31r1 | 5′-GGC TTC ATT TAT GAG TTC-3′ | |
| Mscf1 | MSC1 | 5′-CAC TGA CGA CTT TGT TAGC-3′ |
| Mscr1 | 5′-GTA GCA CAC CAT TGT ATA CC-3′ | |
| RFXf1 | RFX1 | 5′-TGA CAA GTA TTT CGG AGC-3′ |
| RFXr1 | 5′-TCC GAT AAA TAA CCA GTC-3′ | |
| Yapf1 | YAP1 | 5′-AAA CGA CTA CTT TAC AAC-3′ |
| Yapr1 | 5′-AAC CTG TAC ATC ATC GGC-3′ | |
| met4f1 | MET4 | 5′-ACA GAG CTC AGA CGA CAC-3′ |
| met4r1 | 5′-TCA ACG TTT AAC CTT CGC-3′ | |
| TSAF1 | TSA1 | 5′-AGT TCA AAA GCC TGC TCC-3′ |
| TSAR1 | 5′-GTA CTC CTT GGA GTC CTC-3′ |
ORF, open reading frame.
Northern blot analysis.
A minority of cDNA clones contained 250 to 400 nucleotides. They were amplified by PCR, using nested primers 1 and 2R, to generate probes for Northern blot analysis. A majority of clones contained 100 to 150 nucleotides. They were amplified by gene-specific primer pairs (Table 2) determined from the sequences of the C. glabrata genome database (http://cbi.labri.fr/Genolevures/elt/CAGL). Northern blot analyses were performed by methods previously described (58, 59).
Construction of RLUC reporter plasmids.
To test whether genes were up-regulated by switching at the level of their promoters, 450- to 500-base-pair regions upstream of such genes were synthesized, fused to a luciferase reporter, transformed into cells, and tested for expression in Wh and DB cells by measuring luciferase activity. The basic plasmid pT2.1, containing the S. cerevisiae URA3 gene, the C. glabrata centromere sequence CEN, an autonomously replicating sequence (ARS), and the Renilla reniformis luciferase reporter gene RLUC, was constructed as follows. A 1,235-bp AatII-MspA1I fragment of the S. cerevisiae URA3 gene from plasmid p112-8XM (23) was end repaired and subcloned at the AatII site of plasmid pSP72 (Promega, Madison, WI) to generate plasmid pE4.20. The CEN-based plasmid derivative pE11.3 was derived by subcloning an 850-bp end-repaired XhoI-Eco109I fragment from plasmid pCGAct14 (23) into pE4.20 at the HpaI site. The reporter module, containing a 936-bp fragment of the RLUC coding region (57) and a 270-bp fragment spanning the 3′ end of the C. glabrata ADE2 coding region, was subcloned in one step between the XhoI and ClaI sites of pE11.3 to generate pT2.1. The RLUC coding region was derived by PCR using the primer pair REFV-RERV (Table 2) and was digested with XhoI and SacI. The 3′ end of ADE2 was also derived by PCR using the primer pair ADFV-ADRV (Table 2) and was digested with SacI and ClaI. The nourseothricin resistance (NATr) module, flanking the S. cerevisiae TCM1 promoter at the 5′ end and the TCM1 transcription- and translation-termination regulatory sequences at the 3′ untranslated region (UTR) (http://www.duke.edu/web/microlabs/mccusker/), was inserted at the SspI site to generate pG5-T6. This module was created by the following steps. First, the full-length TEM1 gene (http://www.yeastgenome.org/) of 1,849 bp was amplified by PCR using primers TEMF1 and TCMR1 (Table 2) and then subcloned into the pGEM-T-Easy vector. The 5′ promoter and 3′ UTR of TEM1 were fused by inverse PCR using the primer pair TCInF1-TCInR1 (Table 2), followed by digestion with NruI. The NATr coding region was derived by PCR using pAG35 (http://www.duke.edu/web/microlabs/mccusker/) as a template and the primer pair NATSacF1-NATSacR1 (Table 2), followed by digestion with SacI and end repair using T4 DNA polymerase (New England Biolabs, Beverly, MA). This was inserted at the NruI site of the inverse PCR fragment to derive the NATr-dominant marker module. The module was amplified from the derivative plasmid, end repaired, phosphorylated using T4 polynucleotide kinase, and subcloned into pT2.1 at the SspI site to derive pG5-T6. To produce the C. glabrata HO gene-specific targeting plasmid pH12.7, a 1,200-bp PCR fragment of HO (7) was generated using C. glabrata genomic DNA as a template and primer pair CgHOF1-CgHOR1 (Table 2), followed by digestion with NsiI and PstI, and subcloning of pG5-T6 at the PstI site. The plasmid derivative containing the intact PstI site abutting the ATG start codon of the RLUC coding sequence was identified and used for promoter analysis. DNA fragments of approximately 450 to 500 base pairs encompassing the 5′ upstream regions of the PMP3, TAR1, ECM17, APC9, HSP104, PGK1, SUT1, and TEF1 genes were obtained by PCR using gene-specific primer pairs (Table 2) and C. glabrata genomic DNA as a template. The PCR products were digested with PstI and inserted at the PstI site of pH12.7. The derived plasmids were designated pH95.2 (CgPMP3), pH94.1 (CgPGK1), pH96.1 (CgSUT1), pH99.1 (CgTEF1), pI22.6 (CgTAR1), pI.3 (CgECM17), pI2.7 (CgHSP104), and pH91.2 (CgAPC9). The correct orientation of the promoters was verified by DNA sequencing.
Construction of URA3 deletion strains for promoter analysis.
For transformation with plasmids containing reporter constructs, URA3 deletion strains were first generated. The plasmid containing the C. glabrata URA3 deletion construct, pBC39.1, was a generous gift from Brendan Cormack, Johns Hopkins School of Medicine, Baltimore, MD. The deletion cassette contained the 5′ and 3′ untranslated regions of URA3 flanking the neo gene from Tn903 (12). The deletion cassette used for transformation was isolated from pBC39.1 by digestion with PstI, followed by separation from the plasmid backbone using agarose gel electrophoresis.
To replace wild-type URA3, approximately 2 μg of the purified deletion cassette was used to transform C. glabrata strains 35B11 (MTLa) and pB921 (MTLα) (59). To isolate a URA3 deletion derivative of strain 35B11, the primary transformants were selected on synthetic complete medium supplemented with a complete amino acid mixture (2 g/liter), 250 to 500 μg/ml of G418 for neomycin resistance, and 50 μg/ml of uracil for URA3 auxotrophy. G418-resistant colonies were tested for 5-fluoroorotic acid (5-FOA) resistance to identify putative uracil auxotrophs. Since strain PB921 exhibited high levels of intrinsic resistance to G418, URA3 auxotrophs were selected by direct plating of the primary transformants on 5-FOA plates. Southern blot analyses of FOAR transformants verified that in a transformant clone of each strain, the URA3 coding region had been replaced by the deleted copy of the URA3 gene harboring the neomycin resistance cassette. The URA3 deletion derivatives of 35B11 and PB921 were designated 40F1 and 12F1, respectively.
Integrative transformation of C. glabrata.
For all integrative transformations, 2 μg of a particular plasmid was linearized by digestion with the restriction enzyme Bsu36I, a unique site in the HO gene (7). The linearized plasmid DNA was targeted to the HO gene by transformation as follows. The ura3− strains were grown overnight to saturation phase. Cells from this primary culture were then diluted into fresh YPD medium plus 50 μg/ml uracil and grown for 4 h. Cells were spun down and washed once with 10 ml of water and once with 10 ml of LET solution (0.1 M lithium acetate, 1 mM EDTA, and 10 mM Tris, pH 7.5) (14). The cells were then resuspended in 2 ml of LET solution. For each transformation, 200 μl of cells was mixed with 5 μl of linearized plasmid DNA and 20 μl of denatured salmon sperm DNA (200 μg) and then incubated for 30 min at 30°C in an orbital water bath shaker. Then 1.2 ml of LET solution containing 40% polyethylene glycol was added, and the mixture was incubated for an additional 30 min. Twelve percent dimethyl sulfoxide was added, and the mixture was heat shocked for 20 min at 42°C. Cells were collected by centrifugation and spread on synthetic dextrose agar medium plates lacking uracil. Six to eight transformants were analyzed by PCR and Southern blot hybridization to select clones that were targeted to the HO locus and that were present as a single copy in the genome.
Measurement of RLUC activity.
Transformants were streaked onto YPD agar supplemented with 1 mM CuSO4 and allowed to grow at 25°C for 4 days, when the colonies could be discriminated by the intensity of coloration. Cells from three Wh or three DB colonies were inoculated into 2 ml of YPD broth supplemented with 1 mM CuSO4. Duplicate cultures were grown for either 15 to 16 h (mid-log phase) or 35 to 40 h (saturation phase) at 30°C prior to measurement of RLUC activity. Cell-free protein extracts were prepared as previously described (57). RLUC activity was measured for 30 seconds at 480 nm in the integration mode with a Monolight 2001 luminometer (Analytical Luminescence, San Diego, CA). RLUC activity is expressed as relative luminescence per 30 s per μg of protein. Protein was measured using the Coomassie Plus protein assay reagent (Pierce Labs, Rockford, IL) in a 96-well titer plate format with a VERSAmax plate reader (Molecular Devices Corp., Sunnyvale, CA).
RESULTS
Cell preparations.
Cells from homogeneous Wh colonies of the MTLa strain 35B11 (Fig. 1A) and the MTLα strain 1480.49 (Fig. 1G) were plated at low density, and spontaneous DB offspring were isolated from each strain (Fig. 1C and I, respectively). DB cells were plated, and spontaneous IWr offspring were isolated from each strain (Fig. 1E and K). When cells from the Wh, DB, and IWr colonies were replated, they formed colonies of the respective phenotypes (Fig. 1B and H, D and J, and F and L, respectively). Wh and DB cells exhibited phenotype-specific characteristics, the former staining dark red and the latter pink on agar containing phloxine B (28) (data not shown). On agar containing CuSO4, IWr colonies of both the a and α strains were white and were composed of 70 to 80% pseudohyphal cells or cells with tubes (data not shown), both characteristics of IWr (28). In addition, IWr isolates exhibited a propensity to switch back to the parental DB phenotype (data not shown), an additional characteristic of this phenotype (28).
FIG. 1.

Colony phenotypes of the two major strains employed in the screen to identify switch phenotype-regulated genes.
cDNA subtraction.
To identify genes regulated by the core switching system, a cDNA subtraction strategy (18) was applied. In the first step of this strategy, two different cDNA pools were generated for Wh and two for DB of both the a strain 35B11 and the α strain 1480.49. The first of each pair contained primer sequences for cloning into the pGEM-T-Easy plasmid and represented the target cDNA pool. The second of each pair lacked these sequences and represented the driver pool. Each target cDNA pool was hybridized with an excess of the driver cDNA pool of the alternative phenotype of that strain (i.e., target DB cDNA and driver Wh cDNA, or target Wh cDNA and driver DB cDNA). After two successive hybridizations, those target cDNAs that had not hybridized with excess driver were cloned into the pGEM-T-Easy plasmid to generate phase-enriched subpools.
Approximately 1,500 clones of each of the four subpools (35B11-Wh, 35B11-DB, 1480.49-Wh, 1480.49-DB) were screened for hybridization to MT-II and MT-I, which were presumed to be expressed at high levels in DB cells (29). Approximately 80% of the clones from the DB cDNA subpools of both the a and α strains hybridized with the MT-II and MT-I probes, while only 5% of the clones from the Wh cDNA subpools of both strains hybridized to these probes, indicating that the subtractions resulted in phenotype-specific enrichment. The identified MT-II and MT-I clones were excluded from further analysis. Clones from the α strain 1480.49 were also screened with MTLα1, which was presumed to be overexpressed in α cells (59), and the clones thus identified were excluded from further analysis. The total number of putative recombinant clones from the Wh pools was approximately 6,000 and that from the Db pools 12,000. Restriction enzyme analysis and sequencing of 85 clones from the former and 165 from the latter subpools revealed that 87% (218) contained recombinant sequences and that 40% of these were represented once or twice, while 60% were represented three or more times. Forty-five unique clones were subsequently selected for Northern blot analysis, 13 from the Wh subpools and 32 from the DB subpools, which represented the approximate proportions of putative unique sequences from the respective core phenotypes. Each of the 45 clones was used to probe Northern blots containing total-cell RNA from Wh, DB, and IWr cells from each of the a (35B11) and α (1480.49) strains. Comparisons of the intensities of hybridization of each gene probe (Table 2) with Wh, DB, and IWr RNAs were made within each strain, either a or α, not between strains (i.e., not between a and α strains). The patterns of relative expression between the switch phenotypes held true within both strains for all genes tested (Tables 3 and 4).
TABLE 3.
Genes up-regulated in DB
| General category | Gene | Identification in screena | Relative transcript levelb
|
Relative transcript abundancec | Deduced functiond | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Strain 35B11 (a)
|
Strain 1480.49 (α)
|
|||||||||
| Wh | DB | IWr | Wh | DB | IWr | |||||
| Sulfur metabolism | ECM17 | √ | ++ | ++++ | ++ | − | ++++ | + | Low | Sulfite reductase subunit (β subunit) |
| CBF1 | √ | ++ | ++++ | ++ | ++ | ++++ | ++ | Low | Transcription factor, methionine biosynthesis | |
| MET31 | √ | + | ++++ | ++++ | + | ++++ | ++++ | Low | Transcription factor, methionine biosynthesis | |
| MET10 | √ | ++ | ++++ | ++ | + | ++++ | + | Low | Sulfite reductase subunit (α subunit) | |
| Copper | FRE6 | √ | ++ | ++++ | ++ | ++ | ++++ | ++ | Low | Copper-iron reductase |
| metabolism | CTR1 | √ | + | ++++ | ++++ | − | ++++ | ++ | Low | Copper transporter, high affinity |
| MAC1 | √ | ++ | ++++ | ++ | + | ++++ | + | Low | Copper-sensing transcription factor | |
| CCC2 | √ | +/− | ++++ | +/− | ++ | ++++ | ++ | Low | Copper transport, P-type ATPase | |
| MT-II | √ | +/− | ++++ | +/− | +/− | ++++ | +/− | Very high | Metallothionein IIa and IIb | |
| FET3 | √ | ++ | ++++ | +++ | ++ | ++++ | +++ | Medium | Multicopper oxidoreductase | |
| FTR1 | √ | ++ | ++++ | ++ | + | ++++ | ++ | Low | High-affinity copper and iron permease | |
| AMT1 | X | ++ | ++++ | ++ | ++ | ++++ | ++ | High | Transcriptional activator of MT-II, MT-I | |
| MT-1 | √ | ++ | ++++ | ++ | ++ | ++++ | ++ | High | Metallothionein I | |
| Stress response | ROX1 | √ | + | ++++ | +++ | + | +++ | ++++ | Low | Repressor of hypoxic genes |
| TSA1 | √ | ++ | ++++ | ++++ | + | ++++ | +++ | Medium | Thioredoxin peroxidase, redox homeostasis | |
| PMP3 | √ | ++ | ++++ | ++ | +++ | ++++ | +++ | Low | Salt tolerance | |
| HSP104 | √ | + | ++++ | +++ | + | ++++ | ++++ | Medium | Chaperone, stress response | |
| RB12 | √ | ++ | ++++ | ++ | + | ++++ | + | High | Vacuole fusion, endopeptidase inhibitor | |
| Other | ECM14 | √ | ++ | ++++ | ++ | ++ | ++++ | ++ | Medium | Zinc carboxypeptidase |
| ECM25 | √ | ++ | ++++ | ++ | − | ++++ | + | Low | Cell wall organization | |
| TAR1 | √ | + | ++++ | ++ | + | ++++ | ++ | Medium | Mitochondrial RNA, Pol-associated | |
| MSC1 | √ | ++ | ++++ | ++ | ++ | ++++ | ++ | Low (a)/high (α) | Meiotic recombination | |
| SGO1 | √ | + | ++++ | ++++ | ++ | ++++ | ++++ | Low (a)/high (α) | Chromosome segregation | |
Checks indicate genes identified in screen. X indicates gene not identified in screen but analyzed by Northern blot hybridization.
Symbols: ++++, maximum; +++, slightly reduced; ++, very reduced; +, extremely reduced; −, not detectable. Luminescence measurements of bands in Northern blot hybridization patterns indicate that the difference between − and ++++ is between 6- and 35-fold, the difference between + or ++ and ++++ is at least 4-fold, and the difference between +++ and ++++ is approximately 2-fold. These fold differences are underestimates because of pixel saturation artifacts at the high end of band intensity.
Level of maximum expression relative to levels of expression of other messages.
Deduced from the demonstrated functions of S. cerevisiae orthologs.
TABLE 4.
Genes up-regulated in Wh, up-regulated or down-regulated in IWr, or constitutively expressed
| General category | Gene | Identification in screena | Relative transcript levelb
|
Relative transcript abundancec | Deduced functiond | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Strain 35B11 (a)
|
Strain 1480.47 (α)
|
|||||||||
| Wh | DB | IWr | Wh | DB | IWr | |||||
| Up-regulated in Wh | APC9 | √ | +++ | + | ++++ | ++++ | + | ++++ | Medium | Ubiquitin protein ligase, cell cycle |
| FAR8 | √ | ++++ | + | ++++ | ++++ | − | ++++ | Low | Cell cycle, pheromone response | |
| CTR2 | √ | ++++ | ++ | ++++ | ++++ | ++ | ++++ | Low | Copper transporter, low affinity | |
| YAP1 | √ | ++++ | ++ | ++++ | ++++ | + | ++++ | Low | Transcription factor, oxidative response | |
| Up-regulated in IWr | TEF4 | √ | + | + | ++++ | + | + | ++++ | Low | Elongation factor, translation |
| RPS23A | √ | ++ | ++ | ++++ | + | + | ++++ | High | 40S ribosome subunit, translation | |
| SIL1 | √ | ++ | + | ++++ | ++ | + | ++++ | Medium | Protein translocation into ER | |
| SUT1 | √ | ++ | + | ++++ | + | + | ++++ | Medium | RNA Pol II regulator, sterol transport | |
| PUT1 | √ | + | + | ++++ | ++ | ++ | ++++ | Low | Proline dehydrogenase, mitochondria | |
| HEM4 | √ | ++ | + | ++++ | ++ | + | ++++ | Low | Uroporphyrogen III synthase | |
| Down-regulated in IWr | EFT1 | √ | ++++ | ++++ | ++ | ++++ | +++ | ++ | Medium | Elongation factor, translation |
| MET4 | √ | ++++ | ++++ | + | +++ | ++++ | + | Low | Transcription factor, methionine biosynthesis | |
| S32 | √ | ++++ | ++++ | ++ | ++ | ++++ | + | Low | Regulator of sulfur aa biosynthesis | |
| Constitutive | SPC98 | √ | ++++ | ++++ | ++++ | ++++ | ++++ | ++++ | High | Microtubule nucleation |
| RAX2 | √ | ++++ | ++++ | ++++ | ++++ | ++++ | ++++ | High | Bud site selection | |
| RFX1 | √ | ++++ | ++++ | ++++ | ++++ | ++++ | ++++ | High | DNA replication checkpoint | |
| CYT1 | √ | ++++ | ++++ | ++++ | ++++ | ++++ | ++++ | High | Cytochrome electron transport | |
| PST2 | √ | ++++ | ++++ | ++++ | ++++ | ++++ | ++++ | High | Flavodoxin-like protein | |
| ZRT1 | √ | ++++ | ++++ | ++++ | ++++ | ++++ | ++++ | High | Zinc transporter, high affinity | |
| TIR1 | √ | ++++ | ++++ | ++++ | ++++ | ++++ | ++++ | High | Cold shock induced, cell wall | |
| ENO2 | √ | ++++ | ++++ | ++++ | ++++ | ++++ | ++++ | High | Enolase | |
| SOD2 | √ | ++++ | ++++ | ++++ | ++++ | ++++ | ++++ | High | Mn2+-containing superoxide dismutase | |
Checks indicate genes identified in screen, X indicates gene not identified in screen but analyzed by Northern blot hybridization.
Symbols: ++++, maximum; +++, slightly reduced; ++, very reduced; +, extremely reduced; −, not detectable. Luminescence measurements of bands in Northern blot hybridization patterns indicate that the difference between − and ++++ is between 6- and 35-fold, the difference between + or ++ and ++++ is at least 4-fold, and the difference between +++ and ++++ is approximately 2-fold. These fold differences are underestimates because of pixel saturation artifacts at the high end of band intensity.
Level of maximum expression relative to levels of expression of other messages.
Deduced from the demonstrated functions of S. cerevisiae orthologs. ER, endoplasmic reticulum; aa, amino acid.
Elevated expression in DB.
Of the 32 putative DB-enriched genes analyzed, 22 (69%) proved by Northern blot analysis to be expressed at higher levels in DB than in Wh (Table 3; Fig. 2). In deducing the putative functions of these up-regulated genes from the functions of their S. cerevisiae orthologs, it became apparent that 18 of them, representing the majority (77%), were involved in either sulfur assimilation, copper assimilation, or stress responses (Table 3).
FIG. 2.
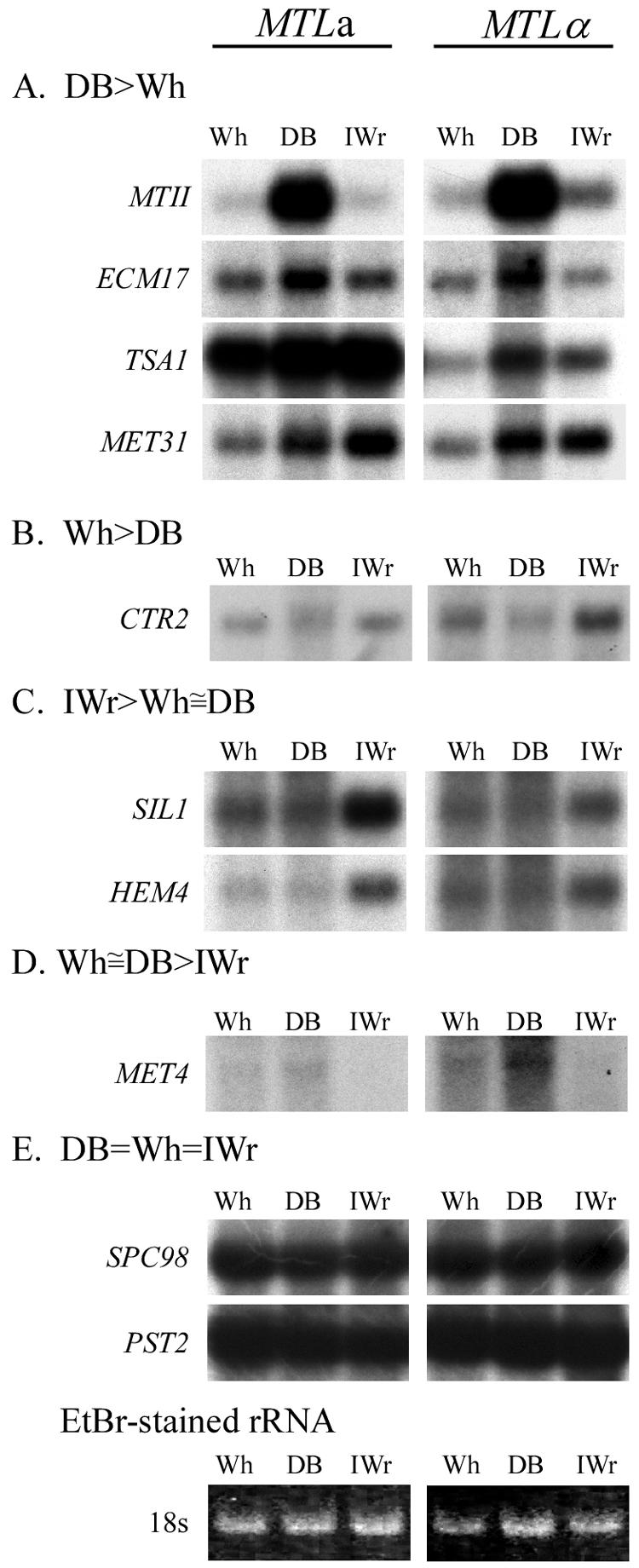
Examples of the different Northern blot hybridization patterns of genes in the Wh, DB, and IWr phenotypes.
Sulfur assimilation.
Studies with both S. cerevisiae and Mucor rouxii have demonstrated that copper-induced brown coloration results from the reduction of CuSO4 to CuS (2, 62, 66). In S. cerevisiae, this reaction is catalyzed primarily by sulfite reductase, which is a heterotetramer of the gene products of ECM17 and MET10 (62, 66). Both of these genes were identified in the subtracted DB cDNA pool and demonstrated by Northern blot analysis to be up-regulated in DB cells (Table 3; Fig. 2). In S. cerevisiae, the promoters of both ECM17 and MET10 contain the binding site CACGTG for the trans-activator Cbf1p, which has been demonstrated to be involved in the regulation of both centromere function and sulfur assimilation in S. cerevisiae (9). In C. glabrata, the promoter of MET10 contained two Cbf1p binding sites and the promoter of ECM17 contained one site (http://cbi.labri.fr/Genolevures/elt/CAGL). CBF1 was also identified in the subtracted DB cDNA pool and demonstrated by Northern blot analysis to be up-regulated in DB cells (Table 3). These results suggest that up-regulation of ECM17 and MET10 in DB cells may be mediated by Cbf1p. The ECM17 promoter also contained a binding site for Amt1p, a transactivator of genes in the copper-sequestering pathway of S. cerevisiae (15, 45). Although we did not identify AMT1 in the subtraction screen, we tested its expression by Northern blot analysis and found it to be up-regulated in DB cells of both the a and α strains, like CBF1 (Table 3). Hence, it is possible that Amt1p may also play a role in up-regulating ECM17 in DB cells.
In S. cerevisiae, Cbf1p is a member of the transcription complexes Cbf1p/Met4p/Met28p (24) and Met31p/Met32p (4). MET31 was also identified in the subtracted DB cDNA pool and demonstrated by Northern blot analysis to be up-regulated in DB (Table 3). Hence, the two genes encoding the subunits of sulfite reductase and three genes encoding trans-acting factors that regulate their expression in S. cerevisiae were up-regulated in DB cells.
Copper assimilation.
C. glabrata, like other microorganisms, has evolved intricate molecular mechanisms to deal with both limiting and toxic concentrations of copper (15, 48, 65). Several genes that either were previously demonstrated to be involved in copper assimilation or were orthologs of S. cerevisiae genes involved in copper assimilation were identified in the subtracted DB cDNA pool and demonstrated by Northern blot analysis to be up-regulated in DB cells (Table 3; Fig. 2). They included the two metallothionein genes MT-II and MT-I, originally observed by Lachke et al. (29) to be up-regulated by core switching in DB cells; a copper and iron reductase gene, FRE6 (50); two copper transporter genes, CTR1 (27) and CCC2 (69); a copper-sensing transcription factor gene, MAC1 (49); a multicopper oxidoreductase gene, FET3 (50); and a high-affinity copper/iron permease gene, FTR1 (50) (Table 3). As previously noted, Northern blot analysis revealed that AMT1, which regulates MT-I and MT-II expression in response to extracellular copper levels (72), was also up-regulated in DB cells (Table 3). While the promoters of MT-II and MT-I contained putative Amt1p binding sites, the promoters of FRE6, FET3, and FTR1, which were similarly up-regulated in DB cells, did not contain binding sites for Amt1p (http://cbi.labri.fr/Genolevures/elt/CAGL).
Stress response pathways.
A surprisingly high proportion of the remaining nine genes identified in the screen as up-regulated in DB had deduced functions in stress response pathways. ROX1, which encodes a transcription factor that represses hypoxic genes in response to oxidative stress in S. cerevisiae (52, 61), was identified in our screen and demonstrated by Northern blot analysis to be up-regulated in DB cells (Table 3). CCC2, a copper chaperone and a Rox1p target gene in S. cerevisiae (61, 69), was also identified in the subtraction screen and demonstrated by Northern blot analysis to be up-regulated in DB (Table 3). However, Northern blot analyses revealed that orthologs of four additional S. cerevisiae target genes of ROX1 (CYT1, SOD2, T1R1, and SUT1) (61) were not similarly up-regulated in DB (data not shown). TSA1, which encodes thioredoxin peroxidase, a component of the oxidative response (10), PMP3, which is regulated by alkalinity (40), HSP104, which is regulated by heat shock (51), and PBI2, which encodes a protein involved in redox homeostasis (68), were also identified in the subtraction screen and demonstrated by Northern blot analysis to be up-regulated in DB cells (Table 3).
Genes with miscellaneous functions.
Of the 23 genes identified in the subtraction screen to be up-regulated in DB, only 5 were not implicated in sulfur assimilation, copper assimilation, or a stress response. These included ECM14, a zinc carboxypeptidase involved in cell wall organization in S. cerevisiae (34); ECM25, also involved in S. cerevisiae cell wall organization (34); TAR1, a suppressor of the S. cerevisiae mitochondrial RNA polymerase mutation rpo41(R129D) (11); MSC1, involved in meiotic recombination in S. cerevisiae (63); and SGO1, involved in chromosome segregation in S. cerevisiae (20) (Table 3).
Elevated expression in Wh.
Of the 13 putative Wh enriched genes analyzed, only 4 (31%) proved by Northern blot analysis to be expressed at higher levels in Wh than in DB (Table 4; Fig. 2). One of these genes, CTR2 (47), functions as a low-affinity copper transporter, and another, FAR8 (22), as a key regulator of cell cycle arrest in the pheromone response in S. cerevisiae. The third gene, YAP1 (42), functions as a b-zip transcription factor involved in the oxidative response, and the fourth gene, APC9 (70), is a ubiquitin protein ligase involved in the cell cycle of S. cerevisiae. It seems no coincidence that three of these four genes are involved in copper assimilation, a stress response, or a pheromone response.
Elevated expression in IWr.
In this study, genes enriched in subtracted Wh or DB cDNA pools were tested for relative expression by Northern blot analyses not only in Wh and DB cells but also in IWr cells. Northern blot analysis revealed five patterns that involved differential gene expression in IWr: (i) DB > Wh ≅ IWr, (ii) DB ≅ IWr > Wh, (iii) Wh ≅ IWr > DB, (iv) Wh ≅ DB > IWr, and (v) IWr > Wh ≅ DB (Tables 3 and 4). IWr exhibits the coloration of Wh, independently of the core phenotype of origin (28). Hence, similarities between the gene expression patterns of IWr and Wh (Table 3) may reflect this commonality. The patterns of expression of genes involved in sulfur and copper assimilation appeared to reflect this. Three out of the four genes involved in sulfur assimilation and all nine genes involved in copper assimilation that were expressed at higher levels in DB and lower levels in Wh were also expressed at lower levels in IWr (Table 3). In addition, all of the genes expressed at higher levels in Wh than in DB were up-regulated in IWr as well (Table 4). However, IWr has a propensity to switch back to the core phenotype from which it emerged, suggesting that even though the coloration is that of Wh, IWr maintains, or “remembers,” its original core phenotype (28). The patterns of expression of genes involved in stress responses appeared to reflect this. Three out of the five stress response-related genes that were expressed at higher levels in DB than in Wh were also expressed at higher levels in IWr (Table 3).
Six genes were selectively up-regulated and three down-regulated in IWr, but not in either Wh or DB (Table 4). Interestingly, the deduced functions of several of these genes (TEF4, RPS23A, SIL1, SUT1, EFT1, and MET4) involved RNA synthesis, protein synthesis, or protein translocation in S. cerevisiae (5, 21, 35, 41, 60, 64). None of the genes selectively up-regulated in IWr were related to copper detoxification or stress. In addition, none were involved in pseudohypha formation, an involvement one might have expected given the high proportion of cells in IWr colonies that express this phenotype (28). However, both Wh and DB colonies also contain pseudohyphae, although at lower proportions, and furthermore, the screens were not designed for the enrichment of IWr-specific transcripts.
Constitutively expressed genes.
In our screen, we serendipitously identified nine genes that were enriched in either Wh or DB cDNA pools but proved to be constitutively expressed by Northern blot analysis (Fig. 2; Table 4). All of these genes exhibited high transcript abundance in the three switch phenotypes tested, while only 17% of these genes in the regulated categories (6 of 36) exhibited high abundance, which may explain why the former may have slipped through the cDNA screening protocol. It is also noteworthy that none of these genes were involved in copper detoxification or stress responses.
Verification of regulation of select genes in additional strains.
To verify regulation patterns, we analyzed the expression of PMP3, TAR1, CBF1, ECM17, CTR1, and CTR2 in Wh and DB in two additional, unrelated strains, the a strain 1480.50 and the α strain 1480.47. As was the case for the a strain 35B11 and the α strain 1480.49 (Tables 3 and 4), expression of the first five of these genes was up-regulated in DB cells, while that of CTR2 was up-regulated in Wh cells (data not shown).
Promoter activity reflects phenotype-specific expression.
To test whether the patterns of differential gene expression among the switch phenotypes reflected promoter activity, the promoter regions of select genes from different categories were fused to the coding region of the RLUC reporter gene (57). The plasmid constructs were then targeted to the HO locus of C. glabrata (7), which plays a specific role only in mating type switching and hence was considered a neutral, nonessential gene for growth, core switching, and IWr switching. Two unrelated strains, one MTLa (35B11) and one MTLα (PB921) strain, were transformed with each of eight genes representing different categories of gene regulation. Two independent transformants were selected for each promoter and strain combination. Cells of each transformant were then plated, and three Wh colonies and three DB colonies were separately pooled in a growth medium containing 1 mM CuSO4. Cells were assayed at late-exponential phase. Luciferase activities in Table 5 are presented as the means (± standard deviations) of six measurements, which included three from each of the two independent transformants. For every targeted gene, the results of the promoter comparison were similar to the results of the Northern blot comparison (Table 5), indicating that the regulation of gene expression by switching occurs at the level of the promoter.
TABLE 5.
Phenotypic regulation of gene expression assessed by Northern blot analysis reflects promoter activity assessed in cells transformed with promoter-luciferase fusions
| Gene | Northern blotting result | RLUC sp act (104 RLUC units/30 s/μg protein)a
|
RLUC result | Fold difference of RLUC comparison
|
|||||
|---|---|---|---|---|---|---|---|---|---|
| Strain 40F1 (a)
|
Strain 12F1 (α)
|
||||||||
| Wh | DB | Wh | DB | Comparison | MTLa | MTLα | |||
| PMP3 | DB > Wh | 2.8 ± 1.6 | 21.1 ± 7.9 | 1.1 ± 0.5 | 4.8 ± 1.8 | DB > Wh | DB/Wh | 7.5 | 4.3 |
| TAR1 | DB > Wh | 1.6 ± 0.8 | 18.6 ± 8.2 | 1.3 ± 0.8 | 12.4 ± 5.9 | DB > Wh | DB/Wh | 11.6 | 9.5 |
| ECM17 | DB > Wh | 1.2 ± 0.6 | 6.9 ± 2.6 | 1.2 ± 0.6 | 6.5 ± 2.8 | DB > Wh | DB/Wh | 5.7 | 6.1 |
| HSP104 | DB > Wh | 1.9 ± 0.9 | 17.0 ± 8.4 | 1.2 ± 0.7 | 7.3 ± 4.6 | DB > Wh | DB/Wh | 8.9 | 6.1 |
| APC9 | Wh > DB | 13.1 ± 5.1 | 2.2 ± 1.1 | 14.0 ± 6.3 | 2.3 ± 1.5 | Wh > DB | Wh/DB | 5.9 | 6.1 |
| SUT1 | Wh ≅ DB | 4.3 ± 2.4 | 3.5 ± 2.0 | 2.6 ± 1.7 | 2.9 ± 1.8 | Wh ≅ DB | Wh/DB | 1.2 | 0.9 |
| TEF4 | Wh ≅ DB | 1.9 ± 1.1 | 1.5 ± 0.8 | 1.5 ± 0.5 | 1.9 ± 0.9 | Wh ≅ DB | Wh/DB | 1.3 | 0.8 |
The basal RLUC activity of strains harboring the promoterless vector pH12.7 targeted to the HO gene ranged between 5.9 × 103 and 6.2 × 103 units. This was subtracted from all RLUC activities obtained with promoter-containing constructs. Each measurement shown is the mean (± standard deviation) of six measurements of relative luminescence, three from each of two independent transformants.
To test whether growth phase affected phenotype-specific promoter activity, we compared luciferase activities between exponential- and saturation-phase cells grown in liquid culture for a third strain, 35B1, which is MTLa. For every tested category of gene expression, regulation between Wh and DB was similar in the two growth phases (Table 6). Hence, regulation of promoter activity by switching was independent of growth phase. It should also be noted that the reproducibility of Wh and DB regulation in the three unrelated test strains was remarkably high (compare the PMP3, TAR1, HSP104, APC9, and SUT1 genes in Tables 5 and 6).
TABLE 6.
Phenotypic regulation of promoter activity is independent of the growth phase in liquid culturea
| Gene | Northern blotting result | RLUC sp act (104 RLUC units/30 s/μg protein)b
|
RLUC result
|
Fold difference of comparison
|
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Log phase
|
Saturation phase
|
|||||||||
| Wh | DB | Wh | DB | Log phase | Saturation phase | Comparison | Log phase | Saturation phase | ||
| PMP3 | DB > Wh | 3.5 ± 2.1 | 23.1 ± 9.8 | 4.9 ± 2.7 | 24.7 ± 10.6 | DB > Wh | DB > Wh | DB/Wh | 6.6 | 5.0 |
| TAR1 | DB > Wh | 1.8 ± 0.9 | 21.4 ± 8.2 | 2.7 ± 1.5 | 24.6 ± 8.8 | DB > Wh | DB > Wh | DB/Wh | 11.9 | 9.1 |
| HSP104 | DB > Wh | 1.9 ± 0.9 | 18.5 ± 7.4 | 2.9 ± 1.8 | 21.9 ± 9.7 | DB > Wh | DB > Wh | DB/Wh | 9.7 | 7.6 |
| APC9 | Wh > DB | 17.4 ± 7.2 | 2.3 ± 1.7 | 23.0 ± 10.4 | 2.7 ± 1.6 | Wh > DB | Wh > DB | Wh/DB | 7.6 | 8.5 |
| SUT1 | IWr > Wh ≅ DB | 4.1 ± 2.2 | 3.6 ± 2.4 | 4.9 ± 2.6 | 4.5 ± 2.4 | Wh ≅ DB | Wh ≅ DB | Wh/DB | 1.1 | 1.1 |
| PGK1 | W ≅ DB | 1.7 ± 0.6 | 1.6 ± 0.6 | 2.7 ± 1.3 | 2.3 ± 1.2 | Wh ≅ DB | Wh ≅ DB | Wh/DB | 1.1 | 1.0 |
The strain used in this analysis was 35B11, which is MTLa.
See Table 5, footnote a.
DISCUSSION
A majority of C. glabrata strains switch spontaneously between core phenotypes and to the irregular wrinkle phenotype (6, 28, 29). It has been suggested that switching in the infectious fungi provides variants in natural colonizing populations which may be enriched in response to rapid environmental challenges, a mechanism for rapid adaptation (55, 56). White-opaque switching in C. albicans has been demonstrated to facilitate skin colonization (26) and to be essential for mating (32, 39). One way of deducing the roles these complex phenotypic transitions play in host colonization and pathogenesis is to identify the genes regulated by them (30). Here we have used a subtraction strategy to identify genes differentially regulated by the core switching system. This screen identified 35 such genes, the majority of which were up-regulated in DB. Regulation of these genes was verified in two unrelated strains, one a and one α, and select genes were verified in two additional strains. In the white-opaque transition in C. albicans, more genes appear to be up-regulated in the white-to-opaque direction than in the opaque-to-white direction (30), suggesting a more specialized role for the opaque-phase phenotype and, by inference, a more specialized role for the DB phenotype in C. glabrata.
The S. cerevisiae orthologs of 17 of the 22 genes (77%) identified in the screen as up-regulated in DB function in S. cerevisiae in sulfur assimilation, copper assimilation, and stress responses. They included reductases, transporters, and permeases, as well as trans-acting factors that regulate these genes. In several organisms including S. cerevisiae, the genes encoding proteins involved in copper homeostasis and detoxification, as well as in stress responses, are up-regulated in response to environmental cues such as toxic levels of CuSO4, increases in temperature, or changes in oxygen tension (1, 19, 31, 53). In C. glabrata, the orthologs of some of the same genes have also been demonstrated to be regulated by environmental cues. MT-I and MT-II in C. glabrata have been demonstrated to be up-regulated by high levels of extracellular CuSO4 (71, 72). However, in our comparison of gene expression, both Wh and DB cells were grown in media containing the same concentration of CuSO4. They were also grown in the same nutrient medium at the same temperature and were harvested for comparison at the same growth phase. Therefore, in C. glabrata these genes are also regulated by spontaneous phenotypic switching and, as we have demonstrated here, at the level of promoter activation. We found that the differences in promoter activity between Wh and DB were similar in mid-log- and saturation-phase cells. Lachke et al. (29) further demonstrated that at least in the case of MT-II, graded expression (i.e., Wh < LB < DB) was similar in cells grown in the presence and in the absence of 1 mM CuSO4. They also demonstrated this to be the case for the hemolysin-like protein gene HLP1, which is expressed in a similar graded fashion (i.e., Wh < LB < DB) (29). Therefore, core switching in C. glabrata regulates a number of genes normally under the regulation of environmental cues in other organisms.
Our results would appear to be consistent with the hypothesis that switching provides populations with a minority of cells expressing variant phenotypes that can be rapidly enriched in response to an environmental challenge (44, 55, 56). However, this explanation does not appear to be sufficient in the case of C. glabrata, since DB may represent the common core phenotype expressed at sites of colonization (6; S. Lachke and D. R. Soll, unpublished observations). The up-regulation of genes associated with sulfur homeostasis, copper homeostasis, and stress responses, therefore, may not be associated with a rare phenotype, but possibly with the most common colonizing phenotype of C. glabrata. However, Brockert et al. (6) also observed that for one patient, while DB was the predominant phenotype in cheek and tongue samples, Wh of the same strain was the predominant phenotype in the vaginal canal, indicating a specialization that may be based on the differences in gene expression patterns demonstrated here.
While only four genes were identified as up-regulated in Wh, three were orthologs of S. cerevisiae genes regulated by environmental cues. These three included a low-affinity copper transporter, a protein involved in the pheromone response, and a transcription factor involved in the oxidative response. In marked contrast, only one of eight constitutively expressed genes that were picked up in the screen due to their high abundance had a deduced function in copper or sulfur assimilation or a stress response. Hence, the functional bias of genes up-regulated in DB or Wh toward copper detoxification and stress responses cannot be due to chance. This conclusion is further supported by the deduced functions of the six genes identified in the screen as up-regulated in IWr. None of them had a deduced role in copper assimilation, sulfur assimilation, or a stress response.
In addition to the 6 genes identified as up-regulated in IWr, 5 of the 18 genes up-regulated in DB were also up-regulated in IWr, which may reflect the DB origin of the IWr isolates analyzed here. More interestingly, all four of the genes up-regulated in Wh were also up-regulated in IWr. This is consistent with our original assessment (28) that IWr seemed to exhibit Wh characteristics, regardless of the core phenotype from which it arose. These characteristics included white color on agar containing 1 mM CuSO4, red color on agar containing phloxine B, a high switching frequency, and low levels of MT-II transcript (28). Our results further suggest that although the core and IWr switching systems appear to be distinct, there may be overlap in the genes that are regulated by the two programs.
Our results, therefore, indicate that core switching in C. glabrata regulates a subset of genes that have been implicated in copper detoxification and stress responses in S. cerevisiae. The majority of these genes are up-regulated in the DB phenotype, which may represent the prevalent phenotype at sites of infection. Although we have suggested that up-regulation of these genes in vitro in the transition from Wh to DB is a result of switching and not environmental cues, it may be that for this pathogen, the regulation of such genes has been usurped by the spontaneous core-switching system in adaptation to the challenging host environment. As such, switching may represent a supervirulence factor regulating a number of genes, the combined expression of which facilitates pathogenesis. Because the host environment does not include high levels of CuSO4, we further suggest that the functions of these and perhaps other genes up-regulated in DB may not be the same as the functions in S. cerevisiae, which has not similarly evolved as a pathogen.
Acknowledgments
This research was supported by National Institutes of Health grant DE014219.
REFERENCES
- 1.Agell, G., M. J. Uriz, E. Cebrian, and R. Marti. 2001. Does stress protein induction by copper modify natural toxicity in sponges? Environ. Toxicol. Chem. 20:2588-2593. [PubMed] [Google Scholar]
- 2.Ashida, J., N. Higashi, and T. Kikuchi. 1963. An electron microscopic study on copper precipitation by copper-resistant yeast cells. Protoplasma 57:27-32. [Google Scholar]
- 3.Barns, S. M., D. J. Lane, M. L. Sogin, C. Bibeau, and W. G. Weisburg. 1991. Evolutionary relationships among pathogenic Candida species and relatives. J. Bacteriol. 173:2250-2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blaiseau, P. L., A. D. Isnard, Y. Surdin-Kerjan, and D. Thomas. 1997. Met31p and Met32p, two related zinc finger proteins, are involved in transcriptional regulation of yeast sulfur amino acid metabolism. Mol. Cell. Biol. 17:3640-3648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blaiseau, P. L., and D. Thomas. 1998. Multiple transcriptional activation complexes tether the yeast activator Met4 to DNA. EMBO J. 17:6327-6336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brockert, P. J., S. A. Lachke, T. Srikantha, C. Pujol, R. Galask, and D. R. Soll. 2003. Phenotypic switching and mating type switching of Candida glabrata at sites of colonization. Infect. Immun. 71:7109-7118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Butler, G., C. Kenny, A. Fagan, C. Kurischko, C. Gaillardin, and K. H. Wolfe. 2004. Evolution of the MAT locus and its Ho endonuclease in yeast species. Proc. Natl. Acad. Sci. USA 101:1632-1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cai, J., I. N. Roberts, and M. D. Collins. 1996. Phylogenetic relationships among members of the ascomycetous yeast genera Brettanomyces, Debaryomyces, Dekkera, and Kluyveromyces deduced by small-subunit rRNA gene sequences. Int. J. Syst. Bacteriol. 46:542-549. [DOI] [PubMed] [Google Scholar]
- 9.Cai, M., and R. W. Davis. 1990. Yeast centromere binding protein CBF1, of the helix-loop-helix protein family, is required for chromosome stability and methionine prototrophy. Cell 61:437-446. [DOI] [PubMed] [Google Scholar]
- 10.Chae, H. Z., I. H. Kim, K. Kim, and S. G. Rhee. 1993. Cloning, sequencing, and mutation of thiol-specific antioxidant gene of Saccharomyces cerevisiae. J. Biol. Chem. 268:16815-16821. [PubMed] [Google Scholar]
- 11.Coelho, P. S., A. C. Bryan, A. Kumar, G. S. Shadel, and M. Snyder. 2002. A novel mitochondrial protein, Tar1p, is encoded on the antisense strand of the nuclear 25S rDNA. Genes Dev. 16:2755-2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cormack, B. P., and S. Falkow. 1999. Efficient homologous and illegitimate recombination in the opportunistic yeast pathogen Candida glabrata. Genetics 151:979-987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fidel, P. L., Jr., J. A. Vazquez, and J. D. Sobel. 1999. Candida glabrata: review of epidemiology, pathogenesis, and clinical disease with comparison to C. albicans. Clin. Microbiol. Rev. 12:80-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gietz, R. D., and R. A. Woods. 2002. Transformation of yeast by lithium acetate/single-stranded carrier DNA/polyethylene glycol method. Methods Enzymol. 350:87-96. [DOI] [PubMed] [Google Scholar]
- 15.Gross, C., M. Kelleher, V. R. Iyer, P. O. Brown, and D. R. Winge. 2000. Identification of the copper regulon in Saccharomyces cerevisiae by DNA microarrays. J. Biol. Chem. 275:32310-32316. [DOI] [PubMed] [Google Scholar]
- 16.Grunstein, M., and D. S. Hogness. 1992. Colony hybridization: a method for the isolation of cloned DNAs that contain a specific gene. Bio/Technology 24:117-121. [PubMed] [Google Scholar]
- 17.Hazen, K. C. 1995. New and emerging yeast pathogens. Clin. Microbiol. Rev. 8:462-478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hubank, M., and D. G. Schatz. 1994. Identifying differences in mRNA expression by representational difference analysis of cDNA. Nucleic Acids Res. 22:5640-5648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ikeda, K., H. Nakayashiki, M. Takagi, Y. Tosa, and S. Mayama. 2001. Heat shock, copper sulfate and oxidative stress activate the retrotransposon MAGGY resident in the plant pathogenic fungus Magnaporthe grisea. Mol. Genet. Genomics 266:318-325. [DOI] [PubMed] [Google Scholar]
- 20.Indjeian, V. B., B. M. Stern, and A. W. Murray. 2005. The centromeric protein Sgo1 is required to sense lack of tension on mitotic chromosomes. Science 307:130-133. [DOI] [PubMed] [Google Scholar]
- 21.Justice, M. C., M. J. Hsu, B. Tse, T. Ku, J. Balkovec, D. Schmatz, and J. Nielsen. 1998. Elongation factor 2 as a novel target for selective inhibition of fungal protein synthesis. J. Biol. Chem. 273:3148-3151. [DOI] [PubMed] [Google Scholar]
- 22.Kemp, H. A., and G. F. Sprague, Jr. 2003. Far3 and five interacting proteins prevent premature recovery from pheromone arrest in the budding yeast Saccharomyces cerevisiae. Mol. Cell. Biol. 23:1750-1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kitada, K., E. Yamaguchi, and M. Arisawa. 1996. Isolation of a Candida glabrata centromere and its use in construction of plasmid vectors. Gene 175:105-108. [DOI] [PubMed] [Google Scholar]
- 24.Kuras. L., R. Barbey, and D. Thomas. 1997. Assembly of a bZIP-bHLH transcription activation complex: formation of the yeast Cbf1-Met4-Met28 complex is regulated through Met28 stimulation of Cbf1 DNA binding. EMBO J. 16:2441-2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kurtzman, C. P., and C. J. Robnett. 1998. Identification and phylogeny of ascomycetous yeasts from analysis of nuclear large subunit (26S) ribosomal DNA partial sequences. Antonie Leeuwenhoek 73:331-371. [DOI] [PubMed] [Google Scholar]
- 26.Kvaal, C., S. A. Lachke, T. Srikantha, K. Daniels, J. McCoy, and D. R. Soll. 1999. Misexpression of the opaque-phase-specific gene PEP1 (SAP1) in the white phase of Candida albicans confers increased virulence in a mouse model of cutaneous infection. Infect. Immun. 67:6652-6662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Labbe, S., Z. Zhu, and D. J. Thiele. 1997. Copper-specific transcriptional repression of yeast genes encoding critical components in the copper transport pathway. J. Biol. Chem. 272:15951-15958. [DOI] [PubMed] [Google Scholar]
- 28.Lachke, S. A., S. Joly, K. Daniels, and D. R. Soll. 2002. Phenotypic switching and filamentation in Candida glabrata. Microbiology 148:2661-2674. [DOI] [PubMed] [Google Scholar]
- 29.Lachke, S. A., T. Srikantha, L. K. Tsai, K. Daniels, and D. R. Soll. 2000. Phenotypic switching in Candida glabrata involves phase-specific regulation of the metallothionein gene MT-II and the newly discovered hemolysin gene HLP. Infect. Immun. 68:884-895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lan, C. Y., G. Newport, L. A. Murillo, T. Jones, S. Scherer, R. W. Davis, and N. Agabian. 2002. Metabolic specialization associated with phenotypic switching in Candida albicans. Proc. Natl. Acad. Sci. USA 99:14907-14912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu, X. D., and D. J. Thiele. 1997. Yeast metallothionein gene expression in response to metals and oxidative stress. Methods 11:289-299. [DOI] [PubMed] [Google Scholar]
- 32.Lockhart, S. R., K. J. Daniels, R. Zhao, D. Wessels, and D. R. Soll. 2003. Cell biology of mating in Candida albicans. Eukaryot. Cell 2:49-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lockhart, S. R., S. Joly, C. Pujol, J. D. Sobel, M. A. Pfaller, and D. R. Soll. 1999. Development and verification of fingerprinting probes for Candida glabrata. Microbiology 143:3733-3746. [DOI] [PubMed] [Google Scholar]
- 34.Lussier, M., A. M. White, J. Sheraton, T. di Paolo, J. Treadwell, S. B. Southard, C. I. Horenstein, J. Chen-Weiner, A. F. Ram, J. C. Kapteyn, T. W. Roemer, D. H. Vo, D. C. Bondoc, J. Hall, W. W. Zhong, A. M. Sdicu, J. Davies, F. M. Klis, P. W. Robbins, and H. Bussey. 1997. Large scale identification of genes involved in cell surface biosynthesis and architecture in Saccharomyces cerevisiae. Genetics 147:435-450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McCarthy, J. E. 1998. Posttranscriptional control of gene expression in yeast. Microbiol. Mol. Biol. Rev. 62:1492-1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mehra, R. K., J. L. Thorvaldsen, I. G. Macreadie, and D. R. Winge. 1992. Disruption analysis of metallothionein-encoding genes in Candida glabrata. Gene 114:75-80. [DOI] [PubMed] [Google Scholar]
- 37.Mehra, R. K., J. R. Garey, and D. R. Winge. 1990. Selective and tandem amplification of a member of the metallothionein gene family in Candida glabrata. J. Biol. Chem. 265:6369-6375. [PubMed] [Google Scholar]
- 38.Mehra, R. K., J. R. Garey, T. R. Butt, W. R. Gray, and D. R. Winge. 1989. Candida glabrata metallothioneins. Cloning and sequence of the genes and characterization of proteins. J. Biol. Chem. 264:19747-19753. [PubMed] [Google Scholar]
- 39.Miller, M. G., and A. D. Johnson. 2002. White-opaque switching in Candida albicans is controlled by mating-type locus homeodomain proteins and allows efficient mating. Cell 110:293-302. [DOI] [PubMed] [Google Scholar]
- 40.Navarre, C., and A. Goffeau. 2000. Membrane hyperpolarization and salt sensitivity induced by deletion of PMP3, a highly conserved small protein of yeast plasma membrane. EMBO J. 19:2515-2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ness, F., S. Bourot, M. Regnacq, R. Spagnoli, T. Berges, and F. Karst. 2001. SUT1 is a putative Zn[II]2Cys6-transcription factor whose up-regulation enhances both sterol uptake and synthesis in aerobically growing Saccharomyces cerevisiae cells. Eur. J. Biochem. 268:1585-1595. [PubMed] [Google Scholar]
- 42.Nguyen, D. T., A. M. Alarco, and M. Raymond. 2001. Multiple Yap1p-binding sites mediate induction of the yeast major facilitator FLR1 gene in response to drugs, oxidants, and alkylating agents. J. Biol. Chem. 276:1138-1145. [DOI] [PubMed] [Google Scholar]
- 43.Odds, F. C. 1988. Candida and candidiasis. Baillière Tindall, London, United Kingdom.
- 44.Odds, F. C., and L. A. Mercon-Davies. 1989. Colony variation in Candida species. Mycoses 32:275-282. [DOI] [PubMed] [Google Scholar]
- 45.Pena, M. M., K. A. Koch, and D. J. Thiele. 1998. Dynamic regulation of copper uptake and detoxification genes in Saccharomyces cerevisiae. Mol. Cell. Biol. 18:2514-2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pfaller, M. A., R. N. Jones, S. A. Messer, M. B. Edmond, R. P. Wenzel, et al. 1998. National surveillance of nosocomial blood stream infection due to species of Candida other than Candida albicans: frequency of occurrence and antifungal susceptibility in the SCOPE Program. Diagn. Microbiol. Infect. Dis. 30:121-129. [DOI] [PubMed] [Google Scholar]
- 47.Portnoy, M. E., P. J. Schmidt, R. S. Rogers, and V. C. Culotta. 2001. Metal transporters that contribute copper to metallochaperones in Saccharomyces cerevisiae. Mol. Genet. Genomics 265:873-882. [DOI] [PubMed] [Google Scholar]
- 48.Puig, S., and D. J. Thiele. 2002. Molecular mechanisms of copper uptake and distribution. Curr. Opin. Chem. Biol. 6:171-180. [DOI] [PubMed] [Google Scholar]
- 49.Rees, E. M., and D. J. Thiele. 2004. From aging to virulence: forging connections through the study of copper homeostasis in eukaryotic microorganisms. Curr. Opin. Microbiol. 7:175-184. [DOI] [PubMed] [Google Scholar]
- 50.Rutherford, J. C., and A. J. Bird. 2004. Metal-responsive transcription factors that regulate iron, zinc, and copper homeostasis in eukaryotic cells. Eukaryot. Cell 3:1-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sanchez, Y., J. Taulien, K. A. Borkovich, and S. Lindquist. 1992. Hsp104 is required for tolerance to many forms of stress. EMBO J. 11:2357-2364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sertil, O., R. Kapoor, B. D. Cohen, N. Abramova, and C. V. Lowry. 2003. Synergistic repression of anaerobic genes by Mot3 and Rox1 in Saccharomyces cerevisiae. Nucleic Acids Res. 31:5831-5837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shanmuganathan, A., S. V. Avery, S. A. Willetts, and J. E. Houghton. 2004. Copper-induced oxidative stress in Saccharomyces cerevisiae targets enzymes of the glycolytic pathway. FEBS Lett. 556:253-259. [DOI] [PubMed] [Google Scholar]
- 54.Sherman, F., G. R. Fink, and J. B. Hicks. 1986. Laboratory course manual for methods in yeast genetics. Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y.
- 55.Soll, D. R. 2003. Candida albicans, p. 165-201. In A. Craig and A. Scherf (ed.), Antigenic variation. Academic Press, London, United Kingdom.
- 56.Soll, D. R. 1992. High-frequency switching in Candida albicans. Clin. Microbiol. Rev. 5:183-203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Srikantha, T., A. Klapach, W. W. Lorenz, L. K. Tsai, L. A. Laughlin, J. A. Gorman, and D. R. Soll. 1996. The sea pansy Renilla reniformis luciferase serves as a sensitive bioluminescent reporter for differential gene expression in Candida albicans. J. Bacteriol. 178:121-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Srikantha, T., L. Tsai, K. Daniels, A. J. Klar, and D. R. Soll. 2001. The histone deacetylase genes HDA1 and RPD3 play distinct roles in regulation of high-frequency phenotypic switching in Candida albicans. J. Bacteriol. 183:4614-4625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Srikantha, T., S. A. Lachke, and D. R. Soll. 2003. Three mating type-like loci in Candida glabrata. Eukaryot. Cell 2:328-340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Synetos, D., C. P. Frantziou, and L. E. Alksne. 1996. Mutations in yeast ribosomal proteins S28 and S4 affect the accuracy of translation and alter the sensitivity of the ribosomes to paromomycin. Biochim. Biophys. Acta 1309:156-166. [DOI] [PubMed] [Google Scholar]
- 61.Ter Linde, J. J., and H. Y. Steensma. 2002. A microarray-assisted screen for potential Hap1 and Rox1 target genes in Saccharomyces cerevisiae. Yeast 19:825-840. [DOI] [PubMed] [Google Scholar]
- 62.Thomas, D., and Y. Surdin-Kerjan. 1997. Metabolism of sulfur amino acids in Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 61:503-532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Thompson, D. A., and F. W. Stahl. 1999. Genetic control of recombination partner preference in yeast meiosis. Isolation and characterization of mutants elevated for meiotic unequal sister-chromatid recombination. Genetics 153:621-641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tyson, J. R., and C. J. Stirling. 2000. LHS1 and SIL1 provide a lumenal function that is essential for protein translocation into the endoplasmic reticulum. EMBO J. 19:6440-6452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Van Ho, A., D. M. Ward, and J. Kaplan. 2002. Transition metal transport in yeast. Annu. Rev. Microbiol. 56:237-261. [DOI] [PubMed] [Google Scholar]
- 66.Vido, K., D. Spector, G. Lagniel, S. Lopez, M. B. Toledano, and J. Labarre. 2001. A proteome analysis of the cadmium response in Saccharomyces cerevisiae. J. Biol. Chem. 276:8469-8474. [DOI] [PubMed] [Google Scholar]
- 67.Wong, S., M. A. Fares, W. Zimmermann, G. Butler, and K. H. Wolfe. 2003. Evidence from comparative genomics for a complete sexual cycle in the ‘asexual’ pathogenic yeast Candida glabrata. Genome Biol. 4:R10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Xu, Z., K. Sato, and W. Wickner. 1998. LMA1 binds to vacuoles at Sec18p (NSF), transfers upon ATP hydrolysis to a t-SNARE (Vam3p) complex, and is released during fusion. Cell 93:1125-1134. [DOI] [PubMed] [Google Scholar]
- 69.Yuan, D. S., R. Stearman, A. Dancis, T. Dunn, T. Beeler, and R. D. Klausner. 1995. The Menkes/Wilson disease gene homologue in yeast provides copper to a ceruloplasmin-like oxidase required for iron uptake. Proc. Natl. Acad. Sci. USA 92:2632-2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zachariae, W., and K. Nasmyth. 1999. Whose end is destruction: cell division and the anaphase-promoting complex. Genes Dev. 13:2039-2058. [DOI] [PubMed] [Google Scholar]
- 71.Zhou, P., and D. J. Thiele. 1993. Copper and gene regulation in yeast. Bioessays 4:105-115. [PubMed] [Google Scholar]
- 72.Zhou, P., M. S. Szczypka, T. Sosinowski, and D. J. Thiele. 1992. Expression of a yeast metallothionein gene family is activated by a single metalloregulatory transcription factor. Mol. Cell. Biol. 12:3766-3775. [DOI] [PMC free article] [PubMed] [Google Scholar]