

Abstract
Many actions of the proinflammatory cytokines tumor necrosis factor (TNF) and interleukin-1 (IL-1) on gene expression are mediated by the transcription factor NF-κB. Activation of NF-κB by TNF and IL-1 is initiated by the phosphorylation of the inhibitory subunit, IκB, which targets IκB for degradation and leads to the release of active NF-κB. The nonsteroidal anti-inflammatory drug sodium salicylate (NaSal) interferes with TNF-induced NF-κB activation by inhibiting phosphorylation and subsequent degradation of the IκBα protein. Recent evidence indicated that NaSal activates the p38 mitogen-activated protein kinase (MAPK), raising the possibility that inhibition of NF-κB activation by NaSal is mediated by p38 MAPK. We now show that inhibition of TNF-induced IκBα phosphorylation and degradation by NaSal is prevented by treatment of cells with SB203580, a highly specific p38 MAPK inhibitor. Both p38 activation and inhibition of TNF-induced IκBα degradation were seen after only 30 s to 1 min of NaSal treatment. Induction of p38 MAPK activation and inhibition of TNF-induced IκBα degradation were demonstrated with pharmacologically achievable doses of NaSal. These findings provide evidence for a role of NaSal-induced p38 MAPK activation in the inhibition of TNF signaling and suggest a possible role for the p38 MAPK in the anti-inflammatory actions of salicylates. In addition, these results implicate the p38 MAPK as a possible negative regulator of TNF signaling that leads to NF-κB activation.
Mitogen-activated protein kinases (MAPKs) are proline-directed serine-threonine kinases that have important functions as mediators of cellular responses to a variety of extracellular stimuli (10, 34, 45). Three major subfamilies of structurally related MAPKs have been identified in mammalian cells: the extracellular signal-regulated kinases (ERKs), the c-Jun N-terminal kinases/stress-activated protein kinases (JNK/SAPKs), and the p38 kinases. Members of all three MAPK subfamilies are activated by upstream dual specificity kinases (MAPK kinases, or MKKs) which produce a simultaneous phosphorylation on threonine and tyrosine residues that are separated by one other amino acid. MAPK kinases, in turn, are activated by a family of serine-threonine kinases termed MAPK kinase kinases, or MEKKs. ERKs are characteristically activated by various growth factors and by phorbol esters. Members of the JNK/SAPK and p38 MAPK subfamilies are strongly activated in response to stress stimuli such as UV radiation, heat shock, and hyperosmolarity (27, 36, 43). JNK/SAPKs and p38s are also characteristically activated by the major proinflammatory cytokines tumor necrosis factor (TNF) and interleukin-1 (IL-1). The specificity of activating stimuli for the three subfamilies of MAPKs is not absolute; for instance, TNF and IL-1 are known to activate ERKs in many cell lines, and some growth factors can produce a weak activation of the JNK/SAPKs and p38 kinases (35, 46, 48, 53, 54). Whereas ERKs are characteristically associated with cell proliferation and protection from apoptosis, JNK/SAPKs and p38 kinases can promote apoptosis in many systems (17, 23, 24, 59). Ten isoforms of JNK/SAPKs (19) and four isoforms of p38 kinases (13, 21, 26, 28, 30) have been identified in mammalian cells. Among the identified substrates of MAPKs are a variety of transcription factors that become activated upon their phosphorylation (10, 34, 45, 56).
TNF and IL-1 are potent activators of gene expression, and many actions of these cytokines, including those that characteristically occur during inflammation, can be ascribed to their ability to activate the transcription factor NF-κB (2–4). In most vertebrate cells, NF-κB proteins are present in a latent form, sequestered in the cytoplasm by members of the IκB family of inhibitory proteins. The two major forms of IκB are IκBα and IκBβ (39). The release of active NF-κB proteins from the inactive complex and their translocation to the nucleus are initiated by site-specific phosphorylation of serine residues on IκB proteins (serines 32 and 36 on IκBα and serines 19 and 23 on IκBβ), which provides a signal for the ubiquitination and degradation of IκB proteins by a proteasome-dependent pathway. The pathway leading to TNF-induced IκBα phosphorylation has been recently elucidated. Cross-linking of the p55 TNF receptor (TNF-RI) by its ligand leads to the association of several intracellular adapter proteins with the death domain region of TNF-RI (20). One of the proteins present in the complex, termed TRAF2, then interacts with, and activates, a kinase termed NIK (NF-κB-inducing kinase), a member of the MAPK kinase kinase family (33). NIK in turn produces phosphorylation and activation of a kinase termed CHUK (12) or IKK-α, the enzyme capable of phosphorylating serines 32 and 36 on IκBα (44). CHUK/IKK-α may also be responsible for phosphorylating IκBβ, which provides an alternative route of TNF-induced NF-κB activation in some types of cells (44).
We have recently demonstrated the activation of p38 MAPK in cells after their treatment with sodium salicylate (NaSal), a nonsteroidal anti-inflammatory drug (NSAID) (47). To learn about the possible functional significance of NaSal-induced p38 kinase activation, we recently examined the effect of the pyridinyl imidazole compound SB203580, a selective p38 MAPK inhibitor (14, 29), on NaSal-induced apoptosis in cultured normal human diploid fibroblasts (47). Treatment with SB203580 protected human fibroblasts from programmed cell death, indicating that p38 MAPK activation was essential for NaSal-induced apoptosis in this cell system. The latter finding raised the possibility that p38 MAPK played a role in some other actions produced by NaSal or other NSAIDs. One recently discovered, extensively documented action of NaSal and its acetylated form, aspirin, is the inhibition of NF-κB activation (7, 9, 15, 16, 18, 25, 40, 42). In TNF-treated cells, NaSal was shown to inhibit NF-κB activation by preventing IκBα phosphorylation and subsequent IκBα degradation, but the pathway responsible for this inhibitory action has not been elucidated (25, 42). In the present study, we demonstrate that p38 kinase activation is required for the inhibitory action of NaSal on TNF-induced IκBα phosphorylation and degradation. Our findings shed light on the pathway whereby NaSal inhibits TNF signaling leading to NF-κB activation. In addition, these results suggest that TNF-induced p38 kinase activation may exert a negative regulatory influence on the process of NF-κB activation by this cytokine.
MATERIALS AND METHODS
Cell culture.
COS-1 African green monkey kidney cells were cultured at 37°C in the presence of 5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM) with 10% fetal bovine serum (FBS). Before use in experiments, COS-1 cells were serum starved for 18 h in DMEM with 0.5% FBS. HT-29 human colon adenocarcinoma cells were cultured in DMEM with 8% FBS and before use in experiments were serum starved for 24 h in DMEM with 0.25% FBS.
Materials.
The rabbit polyclonal anti-IκBα antibody was purchased from Santa Cruz Biotechnology. The rabbit polyclonal anti-phospho-IκBα, anti-phospho-p38 MAPK, and anti-p38 MAPK antibodies were obtained from New England Biolabs. The anti-phospho-IκBα antibody is specific for phosphorylated serine 32 of IκBα, and the anti-phospho-p38 antibody is specific for phosphorylated tyrosine 182 of p38 MAPK. Antibody specific for IκBβ was purchased from Santa Cruz Biotechnology. Recombinant human TNF-α was supplied by Masafumi Tsujimoto, Suntory Institute for Biomedical Research, Osaka, Japan. Recombinant human IL-1α was obtained from the National Cancer Institute, Bethesda, Md. NaSal was purchased from Sigma and dissolved in distilled water. The p38 MAPK inhibitor SB203580 (29) was kindly supplied by John C. Lee and Peter Young (SmithKline Beecham, King of Prussia, Pa.) and was solubilized in dimethyl sulfoxide. Control experiments demonstrated that treatment with the same concentration of dimethyl sulfoxide alone had no effect on the ability of NaSal to inhibit TNF-induced IκBα phosphorylation and degradation. At the concentration used (10 μM), SB203580 did not inhibit JNK/SAPK activity in an in vitro kinase assay using glutathione S-transferase–c-Jun as the substrate (data not shown).
Immunoblotting.
Western blot analysis was performed essentially as described previously (47, 48). Briefly, whole-cell lysates were generated by using a buffer consisting of 1% Nonidet P-40, 50 mM HEPES (pH 7.5), 100 mM NaCl, 2 mM EDTA, 1 mM pyrophosphate, 10 mM sodium orthovanadate, 1 mM phenylmethylsulfonyl fluoride, and 100 mM sodium fluoride. Equal amounts of lysates were subjected to sodium dodecyl sulfate–10% polyacrylamide gel electrophoresis and then transferred to Immobilon-P membranes (Millipore) in transfer buffer (25 mM Tris, 192 mM glycine, 20% [vol/vol] methanol). Membranes were first rinsed in Tris-buffered saline (TBS; 10 mM Tris [pH 7.4], 150 mM NaCl) and then blocked overnight at room temperature in TBS–5% bovine serum albumin (BSA). The anti-IκBα antibody was used at a dilution of 1:200 in TBS–5% BSA. The anti-phospho-IκBα, anti-phospho-p38 MAPK, anti-p38 MAPK, and anti-IκBβ antibodies were each used at a dilution of 1:1,000 in TBS–5% BSA. Antibody-antigen complexes were detected with the aid of horseradish peroxidase-conjugated protein A or horseradish peroxidase-conjugated goat anti-rabbit immunoglobulin G (Bio-Rad) and a chemiluminescent substrate development kit (Kirkegaard & Perry Laboratories). For IκBα blots, equal loading was ascertained by the presence of an ∼70-kDa nonspecific band recognized by the anti-IκBα antibody (not shown).
RESULTS
NaSal strongly inhibits TNF-induced, but not IL-1-induced, IκBα phosphorylation and degradation.
To analyze the effect of NaSal on the TNF-induced degradation of IκBα, COS-1 cells were treated for different time periods with TNF alone or with TNF in the presence of NaSal (Fig. 1A, upper panel). Treatment with TNF for 5 min resulted in the appearance of a slower-migrating IκBα band, corresponding to the phosphorylated form of IκBα (6, 38, 49). Disappearance of the IκBα protein, as a consequence of its proteolytic degradation (6, 38, 49), was apparent at 10 and 15 min after TNF addition. In agreement with earlier reports (25, 42), treatment with NaSal completely inhibited TNF-induced degradation of IκBα. Appearance of the phosphorylated form of IκBα, visualized with the aid of an antibody specific for phospho-IκBα, peaked at 5 min after TNF addition and was inhibited in the presence of NaSal (Fig. 1A, lower panel). Thus, in agreement with earlier findings (25, 42), our results indicate that NaSal inhibits IκBα degradation by interfering with IκBα phosphorylation, which is required for targeting IκBα for degradation by the ubiquitin-proteasome pathway (41). Figure 1B shows that in the absence of NaSal, IκBα protein begins to reappear in COS-1 cells by 30 min following TNF addition, and by 1 h after the onset of TNF treatment the levels approached those seen in untreated control cells. No change in the levels of IκBα protein was seen in NaSal-treated cells at any of the examined time periods after TNF addition.
FIG. 1.
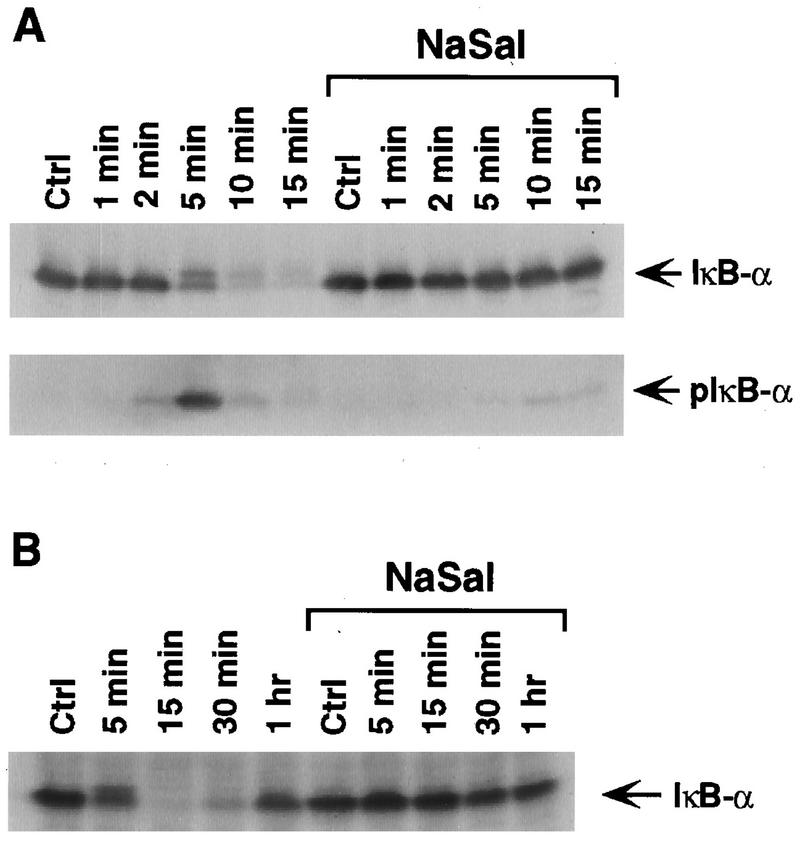
NaSal inhibits TNF-induced IκBα phosphorylation and degradation. (A) COS-1 cells were either treated for 1 h with NaSal (20 mM) or left untreated. They were then either left unstimulated (Ctrl) or stimulated for the indicated times with TNF (20 ng/ml). Lysates were blotted with antibodies against IκBα (top panel) or with antibodies to phosphorylated IκBα (pIκB-α; bottom panel). (B) COS-1 cells were treated as described above, and lysates were blotted with an anti-IκBα antibody.
Our earlier studies indicated that NaSal produced a selective inhibition of TNF-induced signaling, as evidenced by the fact that NaSal suppressed the activation of ERK and JNK MAPKs by TNF much more strongly than the activation of the same kinases by other cytokines or growth factors (47, 48). Therefore, we compared the effects of NaSal on IκBα degradation induced by TNF and by IL-1 (Fig. 2). IκBα degradation induced by TNF was completely inhibited, whereas IκBα degradation elicited by treatment with IL-1 was much less affected. The selectivity of NaSal’s inhibitory action on TNF-induced IκBα degradation is supported by the finding that prior treatment of cells with NaSal and TNF did not prevent the ability of IL-1 to induce IκBα degradation, as shown in the last lane of Fig. 2. The latter finding shows that treatment with NaSal and TNF does not render IκBα refractory to phosphorylation by the IL-1-triggered pathway.
FIG. 2.
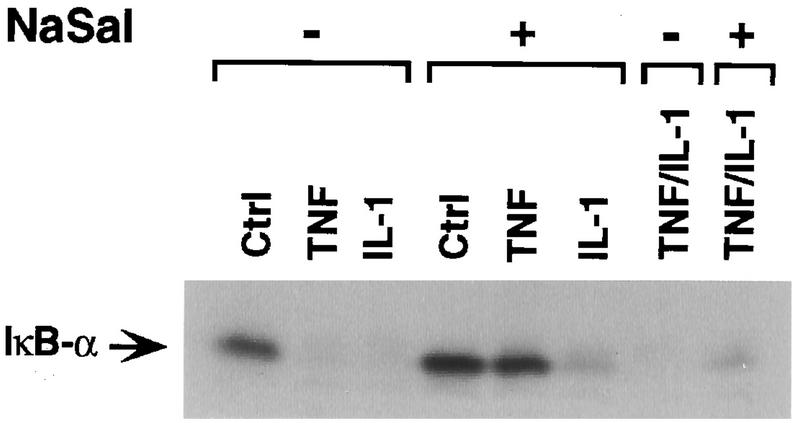
Selective inhibition of TNF-induced IκBα degradation by NaSal. COS-1 cells were preincubated for 1 h in the presence (+) or absence (−) of NaSal (20 mM). They were then left untreated (Ctrl) or treated with TNF (20 ng/ml) or IL-1 (4 ng/ml) for 15 min. In the last two lanes, an initial 15-min TNF treatment was immediately followed by a 15-min IL-1 treatment. Lysates were blotted with anti-IκBα antibody.
The inhibitory action of NaSal on TNF-induced IκBα phosphorylation and degradation is prevented by selective inhibition of p38 MAPK activity.
We have recently demonstrated that NaSal treatment of normal human fibroblasts or COS-1 cells leads to an activation of the p38 MAPK and that NaSal-induced p38 MAPK activation is required for apoptosis induced by NaSal in human fibroblasts (47). In view of the demonstrated inverse relationship between apoptosis and NF-κB activity (5, 32, 52, 55), we considered the possibility that p38 promotes NaSal-induced apoptosis by inhibiting NF-κB function. To determine whether NaSal-induced inhibition of IκBα phosphorylation and degradation was mediated by p38, we used a highly specific p38 kinase inhibitor, the pyridinyl imidazole compound SB203580 (14, 29, 58, 60). The ability of NaSal to inhibit TNF-induced IκBα phosphorylation was largely prevented in both COS-1 and HT-29 cells that were treated with SB203580 before their exposure to TNF (Fig. 3A, upper panel). In both types of cells, a 5-min treatment with TNF induced the appearance of the phosphorylated form of IκBα (lane 2), which was inhibited by the addition of NaSal (lane 4). Whereas treatment of cells with SB203580 in the absence of NaSal did not affect TNF-induced IκBα phosphorylation (lane 6), the inhibitory activity of NaSal on IκBα phosphorylation was largely prevented when cells were treated with SB203580 before their exposure to NaSal and TNF (lane 8). In a similar type of experiment, we also examined the effect of SB203580 on the ability of NaSal to inhibit TNF-induced IκBα degradation (Fig. 3B). A 15-min treatment of either COS-1 or HT-29 cells with TNF led to the disappearance of the IκBα band from the cell extracts, indicating that IκBα had been intracellularly degraded (lane 2). This TNF-induced IκBα degradation was inhibited by treatment with NaSal (lane 4). Whereas SB203580 did not affect TNF-induced IκBα degradation in the absence of NaSal (lane 6), treatment with the p38 kinase inhibitor significantly prevented the ability of NaSal to suppress TNF-induced IκBα degradation (lane 8). Similar results were obtained when we examined the effect of SB203580 on the ability of NaSal to inhibit IκBβ degradation (50). Treatment with NaSal blocked TNF-induced degradation of IκBβ in HT-29 cells, and this blockage was prevented upon treatment with SB203580 (data not shown).
FIG. 3.

Inhibition of TNF-induced IκBα phosphorylation and degradation by NaSal is prevented by SB203580. (A) COS-1 and HT-29 cells were preincubated for 1.5 h in the presence (+) or absence (−) of SB203580 (10 μM). The cells were then incubated for 1 h in the presence or absence of NaSal (20 mM) and subsequently incubated for 5 min in the presence or absence of TNF (20 ng/ml). Lysates were blotted with antibodies against phosphorylated IκBα (pIκB-α; top panel) and with antibodies to IκBα (bottom panel). (B) COS-1 and HT-29 cells were treated as described above except that the duration of TNF treatment was 15 min instead of 5 min. Lysates were blotted with anti-IκBα antibody.
Induction of p38 MAPK by NaSal correlates with the inhibitory action on TNF-induced IκBα phosphorylation and degradation.
The finding that inhibition of TNF-induced IκBα phosphorylation and degradation by NaSal was prevented in the presence of SB203580 strongly suggested that the earlier demonstrated induction of p38 MAPK activation by NaSal (47) plays a role in this process. To further analyze the role of p38, we compared the kinetics of the inhibition of TNF-induced IκBα phosphorylation with the kinetics of p38 activation by NaSal. COS-1 cells were treated with NaSal for periods ranging from 30 s to 1 h before their exposure to TNF, and the extent of IκBα degradation was determined at 15 min after TNF addition (Fig. 4A). Treatment with NaSal for 30 s was sufficient to produce a detectable inhibition of IκBα degradation, although the degree of inhibition increased with longer periods of NaSal pretreatment. An analysis of the kinetics of p38 activation (determined by the appearance of a band specifically recognized by antibody to phosphorylated p38) revealed that phospho-p38 was detectable within 30 s and reached a plateau at 5 min after NaSal addition (Fig. 4B). We (reference 47 and data not shown) and others (43) have shown that p38 phosphorylation correlates with kinase activity. Thus, p38 activation by NaSal is extremely rapid, as is the establishment of the inhibitory action of NaSal on IκBα degradation.
FIG. 4.
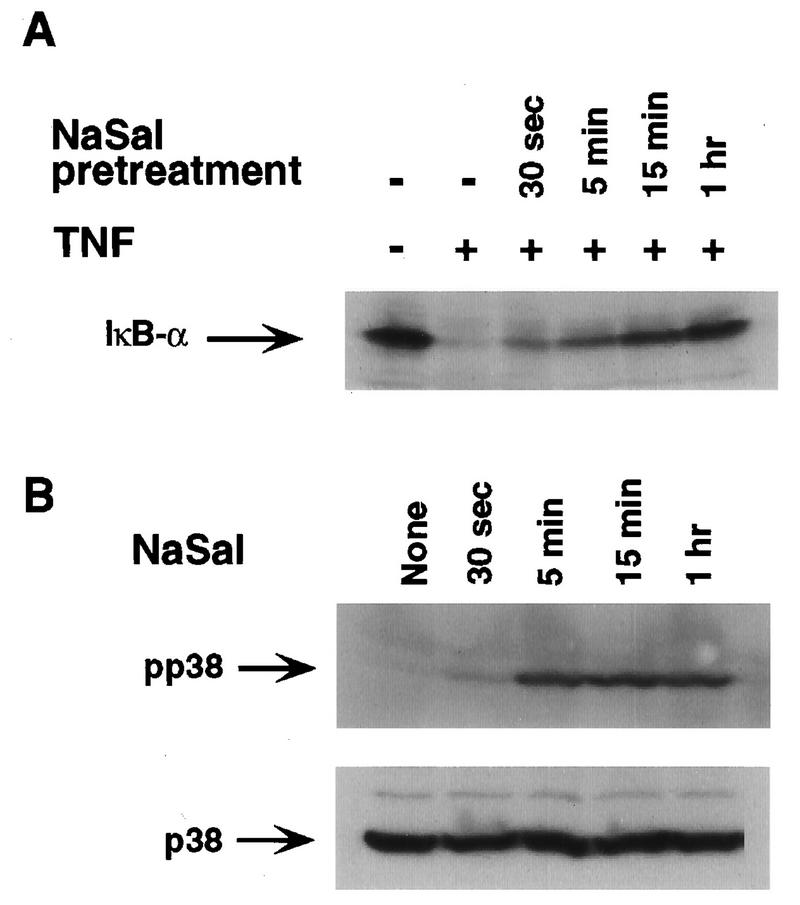
Kinetics of inhibition of IκBα degradation and of p38 MAPK activation by NaSal. (A) COS-1 cells were either left untreated (−) or treated for the indicated times with NaSal (20 mM) and then stimulated for 15 min with TNF (20 ng/ml). Lysates were blotted with anti-IκBα antibody. (B) COS-1 cells were treated for the indicated times with NaSal (20 mM) alone, lysed, and blotted with anti-phospho-p38 MAPK antibody (pp38; top panel) and with anti-p38 MAPK antibody (p38; bottom panel).
To further analyze the correlation between p38 MAPK activation and the inhibition of TNF-induced IκBα phosphorylation and degradation by NaSal, we examined the abilities of different doses of NaSal to inhibit TNF-induced IκBα degradation and to activate p38 MAPK. A very strong inhibition of TNF-induced IκBα degradation was seen in COS-1 cells with 20 and 10 mM NaSal, while 5 mM produced a strong, and 2 mM NaSal produced a slight, inhibition (Fig. 5A). Similarly, although 20 mM NaSal was required for an optimal activation of p38 (as measured by the appearance of phospho-p38), 2 mM was sufficient to produce a detectable activation. Thus, the dose responses of p38 activation and inhibition of IκBα phosphorylation and degradation by NaSal are very similar. Taken together, these findings support the conclusion that p38 activation by NaSal, either alone or in conjunction with some other action, is responsible for the inhibition of TNF-induced IκBα phosphorylation and subsequent degradation.
FIG. 5.
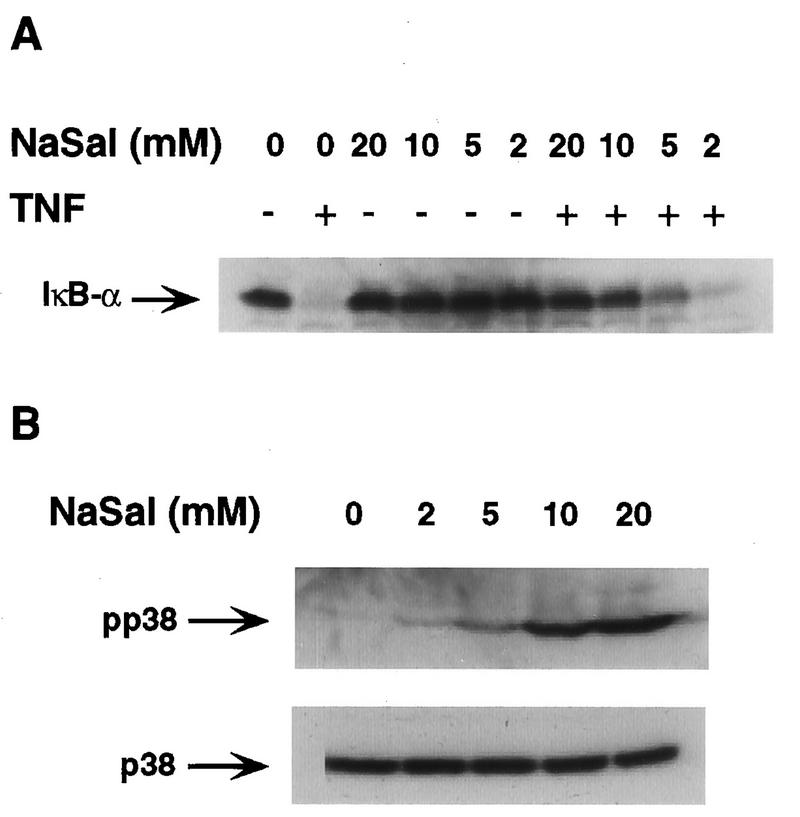
Inhibition of IκBα degradation and induction of p38 MAPK activation by different doses of NaSal. (A) COS-1 cells were treated for 15 min with the indicated doses of NaSal and then left unstimulated (−) or stimulated (+) for 15 min with TNF (20 ng/ml). Lysates were blotted with anti-IκBα antibody. (B) COS-1 cells were treated for 15 min with the indicated doses of NaSal alone, lysed, and blotted with anti-phospho-p38 MAPK antibody (pp38; top panel) and with anti-p38 MAPK antibody (p38; bottom panel).
NaSal activates p38 MAPK more rapidly than TNF.
TNF itself is known to be a potent activator of p38 MAPK (31, 43), raising the question of what the role of TNF-induced p38 activation is in the process of IκBα phosphorylation and degradation. If p38 activation leads to an inhibition of IκBα phosphorylation, how is TNF able to induce both IκBα phosphorylation and p38 activation? To resolve this paradox, we compared the kinetics of p38 activation in COS-1 cells treated with either TNF or NaSal (Fig. 6). An increase in p38 phosphorylation was detected 5 min after TNF addition, whereas incubation of cells in the presence of TNF for 1 or 2 min did not result in a demonstrable p38 activation. In contrast, a marked activation of p38 was demonstrable within 1 min of NaSal addition. The same results were obtained when the kinetics of TNF- and NaSal-induced p38 activation were compared in HT-29 cells (data not shown). Other experiments showed that p38 activation in NaSal-treated cells remained strong for at least 8 h (data not shown). The fact that it takes about 5 min for TNF to produce p38 MAPK activation may explain why p38 activation by TNF fails to prevent TNF-induced IκBα phosphorylation and degradation. Since IκB-α phosphorylation also peaks at about 5 min after TNF addition (Fig. 1), TNF-induced p38 activation probably does not occur early enough to prevent IκBα phosphorylation and its subsequent degradation leading to NF-κB activation.
FIG. 6.
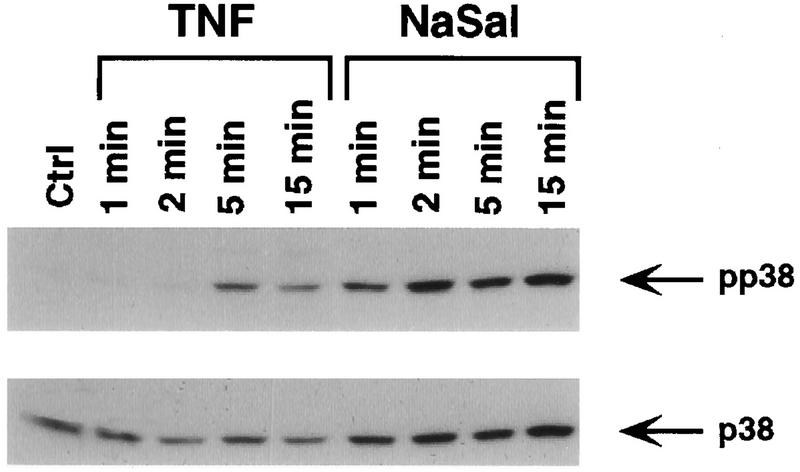
Kinetics of p38 MAPK activation by NaSal and TNF. COS-1 cells were either left untreated (Ctrl) or treated for the indicated times with TNF (20 ng/ml) or NaSal (20 mM). Lysates were blotted with anti-phospho-p38 MAPK antibody (pp38; top panel) and with anti-p38 MAPK antibody (p38; bottom panel).
DISCUSSION
The inhibition of NF-κB activation by NaSal and aspirin has been widely documented in a variety of cell systems (16, 18, 25, 42). NaSal was shown to inhibit the phosphorylation of IκBα that precedes IκBα degradation by the ubiquitin-proteasome pathway and leads to the release of activated NF-κB (25, 42). However, the pathway responsible for the inhibition of IκBα phosphorylation by NaSal has not been elucidated. A recent study from our laboratory showed that treatment of cells with NaSal results in p38 MAPK activation, as demonstrated in a direct kinase assay with glutathione S-transferase–ATF2 as the substrate and by a demonstration of increased p38 phosphorylation (47). The latter demonstration raised the possibility that p38 MAPK activation by NaSal may play a role in some of the anti-inflammatory actions of NaSal, including its inhibition of TNF-induced IκBα phosphorylation and subsequent degradation. The present study provides evidence that p38 MAPK activity is required for the inhibitory action of NaSal on TNF-induced IκBα phosphorylation and degradation, based on the demonstration that treatment of cells with a highly selective p38 kinase inhibitor, the pyridinyl imidazole compound SB203580, prevented the ability of NaSal to suppress both the phosphorylation and degradation of IκBα induced by TNF (Fig. 3). SB203580 binds with a high affinity to p38 near the ATP binding site, thus rendering p38 inactive (60). At the concentration of 10 μM used in our experiments, SB203580 fails to affect other kinases, including other MAPKs in intact cells (14, 58). To further substantiate the conclusion that p38 activation by NaSal is required for the inhibitory action of NaSal on TNF-induced IκBα phosphorylation, we also compared the kinetics (Fig. 4) and the dose response (Fig. 5) of p38 activation and of the inhibition of TNF-induced IκBα phosphorylation and degradation by NaSal. The results of the latter experiments show that p38 activation by NaSal correlates with its inhibitory activity on TNF-induced IκBα phosphorylation and subsequent degradation.
NaSal was much less effective in inhibiting IL-1-induced IκBα degradation than IκBα degradation induced by TNF (Fig. 2). Earlier, we showed that in normal human fibroblasts, NaSal strongly inhibited ERK and JNK/SAPK activation by TNF but was much less effective in inhibiting activation of these MAPKs by other agents, e.g., epidermal growth factor or IL-1 (47, 48). Thus, our earlier and present findings support the notion that a principal target of NaSal action is an early event in TNF-triggered signaling that leads to IκBα phosphorylation as well as to ERK and JNK/SAPK activation. Among the possible targets are TRAF2, a signaling protein implicated in both NF-κB and JNK/SAPK activation by TNF (20, 32, 44), and RIP, found to be essential for TNF-induced NF-κB activation (51). Others, however, have reported that NaSal efficiently inhibited NF-κB activation induced by bacterial lipopolysaccharide (40), insulin (7), human immunodeficiency virus Tat protein (15), or respiratory syncytial virus infection (9), suggesting that at least under some circumstances, the inhibitory effect of NaSal is not specific for TNF-induced NF-κB activation.
Although our experiments demonstrate that p38 MAPK is required for the inhibitory effect on TNF-induced IκBα phosphorylation, it is not known whether p38 activation is sufficient for the establishment of the inhibition or whether some other NaSal-induced action is also necessary. Many stimuli that lead to p38 kinase activation also result in the activation of the JNK/SAPK subfamily of MAPKs (43). In fact, we have seen that treatment with NaSal leads to an activation of JNK/SAPK in both COS-1 and HT-29 cells (data not shown). Thus, we cannot rule out the possibility that activation of both p38 and JNK/SAPK is required for NaSal-induced inhibition of TNF-induced IκBα phosphorylation and degradation. Alternatively, it might be possible that some other unknown function activated by NaSal, together with p38 activation, is required for the inhibitory action.
Actions of NSAIDs may be divided into two categories: those that are mediated by inhibition of prostaglandin biosynthesis and those that are independent of effects on prostaglandin synthesis (57). For a number of reasons, the activation of p38 by NaSal very likely falls within the latter category. First, unlike other NSAIDs, NaSal is known to be a weak inhibitor of both cyclooxygenase isoforms (COX1 and COX2) and, hence, of prostaglandin synthesis (37). Even more importantly, our demonstration that p38 kinase activation was evident within 30 s to 1 min of NaSal addition (Fig. 4 and 6) makes it very unlikely that this action was secondary to an inhibition of prostaglandin synthesis.
Recent evidence indicates the existence of four distinct isoforms of p38 MAPK, termed p38α, p38β, p38γ/SAPK3, and p38δ/SAPK4 (13, 21, 26, 28, 30). The four isoforms are similar in size (360 to 372 amino acids in length), show about 60 to 75% sequence homology, and are all activated by TNF, IL-1, UV radiation, and hyperosmolar medium, but they differ in substrate specificity and in susceptibility to inhibition by SB203580. Since one study found that p38α and p38β, but not p38γ and p38δ, were inhibited by SB203580 (26), it seems less likely that either the γ or the δ isoform is responsible for the inhibition of IκBα phosphorylation by NaSal. Availability of expression vectors encoding the individual tagged p38 isoforms should make it possible to test directly which of the four isoforms can be activated by NaSal and which is able to inhibit IκBα phosphorylation.
Since TNF itself potently activates p38 MAPK (31, 43) as well as inducing IκBα phosphorylation, our finding that p38 mediates the inhibition of the latter process at first seemed difficult to reconcile. However, a careful comparison of the kinetics of p38 activation and IκBα phosphorylation in TNF-treated cells may help to explain these seemingly paradoxical observations. Both IκBα phosphorylation (Fig. 1) and p38 activation (Fig. 6) are first demonstrable at about 5 min after the exposure of cells to TNF. Thus, p38 activation by TNF probably occurs too late to prevent IκBα phosphorylation. On the other hand, our findings raise the distinct possibility that TNF-induced p38 activation plays a negative autoregulatory role in the process of IκBα phosphorylation and degradation. It is known that TNF-induced IκBα degradation is quite transient and that the IκBα protein rapidly reappears even when cells are kept in the continuous presence of TNF (6, 49). In both TNF-treated COS-1 cells (Fig. 1B) and HT-29 cells (data not shown), reappearance of IκBα was apparent as soon as 30 min after TNF addition. IκBα reappearance is the result of rapid IκBα resynthesis, a process that is promoted by the NF-κB transcription factor (39). In view of our findings, it is plausible that another factor aiding in the rapid reappearance of IκBα is p38 activation by TNF, which may serve to prevent IκBα phosphorylation and resulting degradation of the newly synthesized IκBα protein.
There is evidence that under some circumstances, p38 MAPK activation promotes apoptosis (22, 24, 47, 59). There is also extensive evidence showing that NF-κB activation suppresses apoptosis, most likely because NF-κB promotes the synthesis of proteins that can protect cells from apoptosis (5, 32, 52, 55). Our findings suggest that the propensity of p38 MAPK to promote apoptosis could be at least partly due to the inhibition of NF-κB activation by p38. However, other factors are likely to be involved in these processes because, in two different cell systems, we and others were unable to demonstrate a significant protection from TNF-induced apoptosis by incubating cells in the presence of the p38 kinase inhibitor SB203580 (8, 47).
Earlier we demonstrated that SB203580 inhibits apoptosis induced by NaSal in cultured diploid human fibroblasts, indicating that p38 activation by NaSal was essential for this process (47). Based on this earlier work, we concluded that the activation of p38 MAPK and the resulting induction of apoptosis may be important in the widely demonstrated antineoplastic actions of aspirin and other NSAIDs. Our present findings suggest that p38 activation and the resulting inhibition of TNF-induced NF-κB activation are of some significance in the anti-inflammatory actions of NSAIDs. To inhibit NF-κB activation and NF-κB-mediated actions in cultured cells, many investigators relied on high doses of NaSal (usually 20 mM) that are unlikely to be achievable systemically in the intact organism without producing severe toxic side effects (11). However, concentrations of salicylates that are locally available may be increased in the mildly acidic environments that prevail at inflammatory sites (1, 57). Moreover, our present study shows that a significant activation of p38 kinase and inhibition of IκBα phosphorylation and degradation can be achieved with 2 to 5 mM NaSal (Fig. 5), which is a dose range achievable upon systemic administration of salicylates during anti-inflammatory therapy (1). These findings support the notion that the observations reported in the present study are relevant to some of the anti-inflammatory actions of NSAIDs in the intact organism.
ACKNOWLEDGMENTS
We thank John C. Lee, Peter Young, Jiahuai Han, Gerald Weissmann, and Bruce Cronstein for reagents and helpful discussions and Ilene Totillo for preparing the manuscript.
This work was supported by NIH grant R35CA42568. P.S. was supported by a predoctoral fellowship from NIH training grant 5T32-CA09161. D.A. was supported by an MSTP fellowship from NIH training grant 5T32-GM07308.
REFERENCES
- 1.Abramson S B, Weissmann G. The mechanisms of action of nonsteroidal antiinflammatory drugs. Arthritis Rheum. 1989;32:1–9. doi: 10.1002/anr.1780320102. [DOI] [PubMed] [Google Scholar]
- 2.Baeuerle P A, Baltimore D. NF-κB: ten years after. Cell. 1996;87:13–20. doi: 10.1016/s0092-8674(00)81318-5. [DOI] [PubMed] [Google Scholar]
- 3.Baldwin A S., Jr The NF-κB and IκB proteins: new discoveries and insights. Annu Rev Immunol. 1996;14:649–683. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- 4.Beauparlant P, Hiscott J. Biological and biochemical inhibitors of the NF-κB/Rel proteins and cytokine synthesis. Cytokine Growth Factor Rev. 1996;7:175–190. doi: 10.1016/1359-6101(96)00020-2. [DOI] [PubMed] [Google Scholar]
- 5.Beg A A, Baltimore D. An essential role for NF-κB in preventing TNF-α-induced cell death. Science. 1996;274:782–784. doi: 10.1126/science.274.5288.782. [DOI] [PubMed] [Google Scholar]
- 6.Beg A A, Finco T S, Nantermet P V, Baldwin A S., Jr Tumor necrosis factor and interleukin-1 lead to phosphorylation and loss of IκBα: a mechanism for NF-κB activation. Mol Cell Biol. 1993;13:3301–3310. doi: 10.1128/mcb.13.6.3301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bertrand F, Philippe C, Antoine P J, Baud L, Groyer A, Capeau J, Cherqui G. Insulin activates nuclear factor kappa B in mammalian cells through a Raf-1-mediated pathway. J Biol Chem. 1995;270:24435–24441. doi: 10.1074/jbc.270.41.24435. [DOI] [PubMed] [Google Scholar]
- 8.Beyaert R, Cuenda A, Vanden Berghe W, Plaisance S, Lee J C, Haegeman G, Cohen P, Fiers W. The p38/RK mitogen-activated protein kinase pathway regulates interleukin-6 synthesis in response to tumour necrosis factor. EMBO J. 1996;15:1914–1923. [PMC free article] [PubMed] [Google Scholar]
- 9.Bitko V, Velazquez A, Yang L, Yang Y C, Barik S. Transcriptional induction of multiple cytokines by human respiratory syncytial virus requires activation of NF-κB and is inhibited by sodium salicylate and aspirin. Virology. 1997;232:369–378. doi: 10.1006/viro.1997.8582. [DOI] [PubMed] [Google Scholar]
- 10.Cano E, Mahadevan L C. Parallel signal processing among mammalian MAPKs. Trends Biochem Sci. 1995;20:117–122. doi: 10.1016/s0968-0004(00)88978-1. [DOI] [PubMed] [Google Scholar]
- 11.Chalasani N, Roman J, Jurado R L. Systemic inflammatory response syndrome caused by chronic salicylate intoxication. South Med J. 1996;89:479–482. doi: 10.1097/00007611-199605000-00005. [DOI] [PubMed] [Google Scholar]
- 12.Connelly M A, Marcu K B. CHUK, a new member of the helix-loop-helix and leucine zipper families of interacting proteins, contains a serine-threonine kinase catalytic domain. Cell Mol Biol Res. 1995;41:537–549. [PubMed] [Google Scholar]
- 13.Cuenda A, Cohen P, Buee-Scherrer V, Goedert M. Activation of stress-activated protein kinase-3 (SAPK3) by cytokines and cellular stresses is mediated via SAPKK3 (MKK6); comparison of the specificities of SAPK3 and SAPK2 (RK/p38) EMBO J. 1997;16:295–305. doi: 10.1093/emboj/16.2.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cuenda A, Rouse J, Doza Y N, Meier R, Cohen P, Gallagher T F, Young P R, Lee J C. SB 203580 is a specific inhibitor of a MAP kinase homologue which is stimulated by cellular stresses and interleukin-1. FEBS Lett. 1995;364:229–233. doi: 10.1016/0014-5793(95)00357-f. [DOI] [PubMed] [Google Scholar]
- 15.Demarchi F, d’Adda di Fagagna F, Falaschi A, Giacca M. Activation of transcription factor NF-κB by the Tat protein of human immunodeficiency virus type 1. J Virol. 1996;70:4427–4437. doi: 10.1128/jvi.70.7.4427-4437.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gautam S C, Pindolia K R, Noth C J, Janakiraman N, Xu Y X, Chapman R A. Chemokine gene expression in bone marrow stromal cells: downregulation with sodium salicylate. Blood. 1995;86:2541–2550. [PubMed] [Google Scholar]
- 17.Graves J D, Draves K E, Craxton A, Saklatvala J, Krebs E G, Clark E A. Involvement of stress-activated protein kinase and p38 mitogen-activated protein kinase in mlgM-induced apoptosis of human B lymphocytes. Proc Natl Acad Sci USA. 1996;93:13814–13818. doi: 10.1073/pnas.93.24.13814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grilli M, Pizzi M, Memo M, Spano P. Neuroprotection by aspirin and sodium salicylate through blockade of NF-κB activation. Science. 1996;274:1383–1385. doi: 10.1126/science.274.5291.1383. [DOI] [PubMed] [Google Scholar]
- 19.Gupta S, Barrett T, Whitmarsh A J, Cavanagh J, Sluss H K, Dérijard B, Davis R J. Selective interaction of JNK protein kinase isoforms with transcription factors. EMBO J. 1996;15:2760–2770. [PMC free article] [PubMed] [Google Scholar]
- 20.Hsu H, Shu H-B, Pan M-G, Goeddel D V. TRADD-TRAF2 and TRADD-FADD interactions define two distinct TNF receptor 1 signal transduction pathways. Cell. 1996;84:299–308. doi: 10.1016/s0092-8674(00)80984-8. [DOI] [PubMed] [Google Scholar]
- 21.Jiang Y, Chen C, Li Z, Guo W, Gegner J A, Lin S, Han J. Characterization of the structure and function of a new mitogen-activated protein kinase (p38β) J Biol Chem. 1996;271:17920–17926. doi: 10.1074/jbc.271.30.17920. [DOI] [PubMed] [Google Scholar]
- 22.Juo P, Kuo C J, Reynolds S E, Konz R F, Raingeaud J, Davis R J, Biemann H P, Blenis J. Fas activation of the p38 mitogen-activated protein kinase signalling pathway requires ICE/CED-3 family proteases. Mol Cell Biol. 1997;17:24–35. doi: 10.1128/mcb.17.1.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kawasaki H, Morooka T, Shimohama S, Kimura J, Hirano T, Gotoh Y, Nishida E. Activation and involvement of p38 mitogen-activated protein kinase in glutamate-induced apoptosis in rat cerebellar granule cells. J Biol Chem. 1997;272:18518–18521. doi: 10.1074/jbc.272.30.18518. [DOI] [PubMed] [Google Scholar]
- 24.Kinoshita T, Yokota T, Arai K, Miyajima A. Suppression of apoptotic death in hematopoietic cells by signalling through the IL-3/GM-CSF receptors. EMBO J. 1995;14:266–275. doi: 10.1002/j.1460-2075.1995.tb07000.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kopp E, Ghosh S. Inhibition of NF-κB by sodium salicylate and aspirin. Science. 1994;265:956–959. doi: 10.1126/science.8052854. [DOI] [PubMed] [Google Scholar]
- 26.Kumar S, McDonnell P C, Gum R J, Hand A T, Lee J C, Young P R. Novel homologues of CSBP/p38 MAP kinase: activation, substrate specificity and sensitivity to inhibition by pyridinyl imidazoles. Biochem Biophys Res Commun. 1997;235:533–538. doi: 10.1006/bbrc.1997.6849. [DOI] [PubMed] [Google Scholar]
- 27.Kyriakis J M, Banerjee P, Nikolakaki E, Dai T, Rubie E A, Ahmad M F, Avruch J, Woodgett J R. The stress-activated protein kinase subfamily of c-Jun kinases. Nature. 1994;369:156–160. doi: 10.1038/369156a0. [DOI] [PubMed] [Google Scholar]
- 28.Lechner C, Zahalka M A, Giot J F, Moller N P, Ullrich A. ERK6, a mitogen-activated protein kinase involved in C2C12 myoblast differentiation. Proc Natl Acad Sci USA. 1996;93:4355–4359. doi: 10.1073/pnas.93.9.4355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee J C, Laydon J T, McDonnell P C, Gallagher T F, Kumar S, Green D, McNulty D, Blumenthal M J, Heys J R, Landvatter S W, Strickler J E, McLaughlin M M, Siemens I R, Fisher S M, Livi G P, White J R, Adams J L, Young P R. A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature. 1994;372:739–746. doi: 10.1038/372739a0. [DOI] [PubMed] [Google Scholar]
- 30.Li Z, Jiang Y, Ulevitch R J, Han J. The primary structure of p38 gamma: a new member of p38 group of MAP kinases. Biochem Biophys Res Commun. 1996;228:334–340. doi: 10.1006/bbrc.1996.1662. [DOI] [PubMed] [Google Scholar]
- 31.Lin A, Minden A, Martinetto H, Claret F X, Lange-Carter C, Mercurio F, Johnson G L, Karin M. Identification of a dual specificity kinase that activates the Jun kinases and p38-Mpk2. Science. 1995;268:286–290. doi: 10.1126/science.7716521. [DOI] [PubMed] [Google Scholar]
- 32.Liu Z-G, Hsu H, Goeddel D V, Karin M. Dissection of TNF receptor 1 effector functions: JNK activation is not linked to apoptosis while NF-κB activation prevents cell death. Cell. 1996;87:565–576. doi: 10.1016/s0092-8674(00)81375-6. [DOI] [PubMed] [Google Scholar]
- 33.Malinin N L, Boldin M P, Kovalenko A V, Wallach D. MAP3K-related kinase involved in NF-κB induction by TNF, CD95 and IL-1. Nature. 1997;385:540–544. doi: 10.1038/385540a0. [DOI] [PubMed] [Google Scholar]
- 34.Marshall C J. Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell. 1995;80:179–185. doi: 10.1016/0092-8674(95)90401-8. [DOI] [PubMed] [Google Scholar]
- 35.Minden A, Lin A, McMahon M, Lange-Carter C, Dérijard B, Davis R J, Johnson G L, Karin M. Differential activation of ERK and JNK mitogen-activated protein kinases by Raf-1 and MEKK. Science. 1994;266:1719–1723. doi: 10.1126/science.7992057. [DOI] [PubMed] [Google Scholar]
- 36.Minden A, Lin A, Smeal T, Dérijard B, Cobb M, Davis R, Karin M. c-Jun N-terminal phosphorylation correlates with activation of the JNK subgroup but not the ERK subgroup of mitogen-activated protein kinases. Mol Cell Biol. 1994;14:6683–6688. doi: 10.1128/mcb.14.10.6683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mitchell J A, Akarasereenont P, Thiemermann C, Flower R J, Vane J R. Selectivity of nonsteroidal antiinflammatory drugs as inhibitors of constitutive and inducible cyclooxygenase. Proc Natl Acad Sci USA. 1993;90:11693–11697. doi: 10.1073/pnas.90.24.11693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miyamoto S, Maki M, Schmitt M J, Hatanaka M, Verma I M. Tumor necrosis factor α-induced phosphorylation of IκBα is a signal for its degradation but not dissociation from NF-κB. Proc Natl Acad Sci USA. 1994;91:12740–12744. doi: 10.1073/pnas.91.26.12740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Miyamoto S, Verma I M. Rel/NF-κB/IκB story. Adv Cancer Res. 1995;66:255–292. [PubMed] [Google Scholar]
- 40.Oeth P, Mackman N. Salicylates inhibit lipopolysaccharide-induced transcriptional activation of the tissue factor gene in human monocytic cells. Blood. 1995;86:4144–4152. [PubMed] [Google Scholar]
- 41.Palombella V J, Rando O J, Goldberg A L, Maniatis T. The ubiquitin-proteasome pathway is required for processing the NF-κB1 precursor protein and the activation of NF-κB. Cell. 1994;78:773–785. doi: 10.1016/s0092-8674(94)90482-0. [DOI] [PubMed] [Google Scholar]
- 42.Pierce J W, Read M A, Ding H, Luscinskas F W, Collins T. Salicylates inhibit IκB-α phosphorylation, endothelial-leukocyte adhesion molecule expression, and neutrophil transmigration. J Immunol. 1996;156:3961–3969. [PubMed] [Google Scholar]
- 43.Raingeaud J, Gupta S, Rogers J S, Dickens M, Han J, Ulevitch R J, Davis R J. Pro-inflammatory cytokines and environmental stress cause p38 mitogen-activated protein kinase activation by dual phosphorylation on tyrosine and threonine. J Biol Chem. 1995;270:7420–7426. doi: 10.1074/jbc.270.13.7420. [DOI] [PubMed] [Google Scholar]
- 44.Regnier C H, Song H Y, Gao X, Goeddel D V, Cao Z, Rothe M. Identification and characterization of an IκB kinase. Cell. 1997;90:373–383. doi: 10.1016/s0092-8674(00)80344-x. [DOI] [PubMed] [Google Scholar]
- 45.Robinson M J, Cobb M H. Mitogen-activated protein kinase pathways. Curr Opin Cell Biol. 1997;9:180–186. doi: 10.1016/s0955-0674(97)80061-0. [DOI] [PubMed] [Google Scholar]
- 46.Sanchez I, Hughes R T, Mayer B J, Yee K, Woodgett J R, Avruch J, Kyriakis J M, Zon L I. Role of SAPK/ERK kinase-1 in the stress-activated pathway regulating transcription factor c-Jun. Nature. 1994;372:794–798. doi: 10.1038/372794a0. [DOI] [PubMed] [Google Scholar]
- 47.Schwenger P, Bellosta P, Vietor I, Basilico C, Skolnik E Y, Vilcek J. Sodium salicylate induces apoptosis via p38 mitogen-activated protein kinase but inhibits tumor necrosis factor-induced c-Jun N-terminal kinase/stress-activated protein kinase activation. Proc Natl Acad Sci USA. 1997;94:2869–2873. doi: 10.1073/pnas.94.7.2869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schwenger P, Skolnik E Y, Vilcek J. Inhibition of tumor necrosis factor-induced p42/p44 mitogen-activated protein kinase by sodium salicylate. J Biol Chem. 1996;271:8089–8094. doi: 10.1074/jbc.271.14.8089. [DOI] [PubMed] [Google Scholar]
- 49.Sun S C, Ganchi P A, Beraud C, Ballard D W, Greene W C. Autoregulation of the NF-κB transactivator RelA (p65) by multiple cytoplasmic inhibitors containing ankyrin motifs. Proc Natl Acad Sci USA. 1994;91:1346–1350. doi: 10.1073/pnas.91.4.1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Thompson J E, Phillips R J, Erdjument-Bromage H, Tempst P, Ghosh S. IκB-β regulates the persistent response in a biphasic activation of NF-κB. Cell. 1995;80:573–582. doi: 10.1016/0092-8674(95)90511-1. [DOI] [PubMed] [Google Scholar]
- 51.Ting A T, Pimentel-Muinos F X, Seed B. RIP mediates tumor necrosis factor receptor 1 activation of NF-κB but not Fas/APO-1-initiated apoptosis. EMBO J. 1996;15:6189–6196. [PMC free article] [PubMed] [Google Scholar]
- 52.Van Antwerp D J, Martin S J, Kafri T, Green D R, Verma I M. Suppression of TNF-α-induced apoptosis by NF-κB. Science. 1996;274:787–789. doi: 10.1126/science.274.5288.787. [DOI] [PubMed] [Google Scholar]
- 53.Van Lint J, Agostinis P, Vandevoorde V, Haegeman G, Fiers W, Merlevede W, Vandenheede J R. Tumor necrosis factor stimulates multiple serine/threonine protein kinases in Swiss 3T3 and L929 cells. Implication of casein kinase-2 and extracellular signal-regulated kinases in the tumor necrosis factor signal transduction pathway. J Biol Chem. 1992;267:25916–25921. [PubMed] [Google Scholar]
- 54.Vietor I, Schwenger P, Li W, Schlessinger J, Vilcek J. Tumor necrosis factor-induced activation and increased tyrosine phosphorylation of mitogen-activated protein (MAP) kinase in human fibroblasts. J Biol Chem. 1993;268:18994–18999. [PubMed] [Google Scholar]
- 55.Wang C-Y, Mayo M W, Baldwin A S., Jr TNF- and cancer therapy-induced apoptosis: potentiation by inhibition of NF-κB. Science. 1996;274:784–787. doi: 10.1126/science.274.5288.784. [DOI] [PubMed] [Google Scholar]
- 56.Wang X Z, Ron D. Stress-induced phosphorylation and activation of the transcription factor CHOP (GADD153) by p38 MAP kinase. Science. 1996;272:1347–1349. doi: 10.1126/science.272.5266.1347. [DOI] [PubMed] [Google Scholar]
- 57.Weissmann G. Aspirin. Sci Am. 1991;264:84–90. doi: 10.1038/scientificamerican0191-84. [DOI] [PubMed] [Google Scholar]
- 58.Whitmarsh A J, Yang S-H, Su M S-S, Sharrocks A D, Davis R J. Role of p38 and JNK mitogen-activated protein kinases in the activation of ternary complex factors. Mol Cell Biol. 1997;17:2360–2371. doi: 10.1128/mcb.17.5.2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xia Z, Dickens M, Raingeaud J, Davis R J, Greenberg M E. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- 60.Young P R, McLaughlin M M, Kumar S, Kassis S, Doyle M L, McNulty D, Gallagher T F, Fisher S, McDonnell P C, Carr S A, Huddleston M J, Seibel G, Porter T G, Livi G P, Adams J L, Lee J C. Pyridinyl imidazole inhibitors of p38 mitogen-activated protein kinase bind in the ATP site. J Biol Chem. 1997;272:12116–12121. doi: 10.1074/jbc.272.18.12116. [DOI] [PubMed] [Google Scholar]