

Abstract
Knowledge of the distribution of growth times from individual spores and quantification of this biovariability are important if predictions of growth in food are to be improved, particularly when, as for Clostridium botulinum, growth is likely to initiate from low numbers of spores. In this study we made a novel attempt to determine the distributions of times associated with the various stages of germination and subsequent growth from spores and the relationships between these stages. The time to germination (tgerm), time to emergence (temerg), and times to reach the lengths of one (tC1) and two (tC2) mature cells were quantified for individual spores of nonproteolytic C. botulinum Eklund 17B using phase-contrast microscopy and image analysis. The times to detection for wells inoculated with individual spores were recorded using a Bioscreen C automated turbidity reader and were compatible with the data obtained microscopically. The distributions of times to events during germination and subsequent growth showed considerable variability, and all stages contributed to the overall variability in the lag time. The times for germination (tgerm), emergence (temerg − tgerm), cell maturation (tC1 − temerg), and doubling (tC2 − tC1) were not found to be correlated. Consequently, it was not possible to predict the total duration of the lag phase from information for just one of the stages, such as germination. As the variability in postgermination stages is relatively large, the first spore to germinate will not necessarily be the first spore to produce actively dividing cells and start neurotoxin production. This information can make a substantial contribution to improved predictive modeling and better quantitative microbiological risk assessment.
Food microbiologists have devoted considerable research to creating mathematical models that allow the behavior of microorganisms to be predicted in defined environments. This predictive modeling has successfully reduced the numbers of growth studies and challenge tests required, but refinements in modeling techniques are ongoing. One area where improvements are being sought is lag phase modeling. Current models are usually based on empirical descriptions of population kinetics, but this approach can be problematic when the initial population is very small. To produce improved mechanistic descriptions of lag, it is important to understand the underlying distributions of lag times of individuals (1). Recently, lag phase models have been created using stochastic processes that take into account the variability between individual vegetative cells (3, 16). Mechanistic models of lag for spore inocula may differ from those for vegetative cells as the period between entering an environment suitable for growth and the appearance of actively dividing daughter cells encompasses different activities for spores and vegetative cells.
For the sake of simplicity we will define the lag time for spores as the time from which a dormant spore is exposed to conditions suitable for growth to the start of exponential growth. Germination and outgrowth of spores follow an ordered sequence of steps (10). Initially, spores may be activated, which can increase their ability to germinate. Germination is an irreversible cascade of degradative steps using preformed enzymes in which a dormant spore is transformed to a metabolically active cell. During germination, spores rehydrate and irreversibly loose their extreme heat and chemical resistance. In suitable conditions, metabolism is initiated, and the germinated spores begin macromolecular synthesis and start to grow. Eventually, the spore coats are shed, and a young cell emerges. Synthesis continues until cells obtain the full complement of molecules found in adult cells. Mature cells continue to increase in size and proceed to cell division.
To date, kinetic studies have been concentrated on only one part of the lag time, germination. For example, Billon et al. (6) examined germination of individual proteolytic Clostridium botulinum spores. However, germination is only part of the lag time; the lag also includes outgrowth. Knowledge of all stages in lag and the relationships between these stages is necessary if mechanistic models are to be produced. The distributions of times associated with the various stages and the relationships between the stages have not been determined previously.
C. botulinum is a group of spore-forming anaerobic bacteria that produce the extremely potent botulinum neurotoxin, the causative agent of botulism (7). While food-borne botulism is rare, the severity of the disease means that the prevention of outbreaks is a major aim of regulators and industry. Nonproteolytic C. botulinum is a particular concern for the safety of mildly heat-treated, refrigerated foods (11). C. botulinum spores are widespread in the environment but are usually present at low concentrations (7). Consequently, any growth in food packs is likely to initiate from just a few spores. It has been shown that decreasing the number of spores increased the uncertainty of estimation of time to observable turbidity in broth inoculated with spores of nonproteolytic C. botulinum (17). The authors suggested that this resulted from spores having a distribution of germination times, with larger populations more likely to have at least one spore with a shorter germination time present to initiate growth. Quantifying lag times from individual spores is important not only for estimating times to growth from small initial numbers but also for estimating uncertainty. It has also been shown that variability in lag times of individual spores is an important component in risk assessment for products contaminated at low levels (5). Knowledge of the distribution of lag times for individual spores should help refine risk assessment for such foods. Such distributions cannot be derived from observations made at the population level; instead, they must be determined from studies of individual spores.
The aim of this work was to determine both the biovariability of individual spores of nonproteolytic C. botulinum strain Eklund 17B and the relationships between different stages in germination and subsequent growth. Previous studies of single spores have focused only on germination. In this study we examined the time to germination (tgerm), the time to emergence (temerg), the times to growth to the length of one (tC1) and two (tC2) mature cells, and the time to turbidity, as well as the relationships between these stages of C. botulinum spore outgrowth.
MATERIALS AND METHODS
Spore preparation.
Spores of nonproteolytic C. botulinum type B strain Eklund 17B were produced using a two-phase medium consisting of distilled water over cooked meat agar as described previously (12). After incubation for 7 days at 30°C, spores were harvested from the liquid phase by centrifugation (6,000 × g, 4°C, 15 min). The resulting pellet was washed five times by centrifugation (as described above) in 0.85% (wt/vol) sterile saline (20 ml). After each wash the spore suspension was sonicated for 5 min by immersion in an ultrasonic cleaning bath to disperse any spore clumps. Free spores were separated from mother cells, germinated spores, and cell debris by discontinuous density gradient centrifugation. The spore pellet was suspended in 1 ml of 0.85% (wt/vol) saline, layered on top of 20 ml of 50% (vol/vol) Urografin 370 (Schering Health Care Ltd., West Sussex, United Kingdom), and centrifuged (6,000 × g, 4°C, 1 h). The top layer, containing debris and vegetative cells, and the Urografin layer were removed using a Pasteur pipette. The pellet of phase-bright spores was washed four times by centrifugation (6,000 × g, 4°C, 15 min) in 0.85% (wt/vol) saline (20 ml) that had been passed through a 0.2-μm filter. Cleaned spores were resuspended in 1 ml of 0.85% (wt/vol) saline, exposed to 5 min of sonication in an ultrasonic cleaning bath to disperse clumps, and then stored at 1°C until they were required. The viable count was determined on peptone, yeast extract, glucose, starch (PYGS) agar incubated for 2 days at 30°C under a headspace consisting of 10% carbon dioxide and 90% hydrogen. Culture purity and the absence of high levels of proteolytic activity were checked on VL blood agar and reinforced clostridial medium containing 5% (wt/vol) skim milk, respectively, incubated in the same conditions.
Time to turbidity measurement.
The time to turbidity was studied using a Bioscreen C automated turbidity reader (Lab Systems, Finland) installed in an anaerobic cabinet. All manipulations were performed in the anaerobic cabinet under a headspace consisting of 5% carbon dioxide, 10% hydrogen, and 85% nitrogen. Spores were diluted to 20 ml−1 in PYGS broth, and then 50 μl of the spore suspension and 350 μl of PYGS broth were dispensed into each of the 200 wells of the Bioscreen plates. Filled plates were placed into the Bioscreen C reader and incubated at 22°C for 2 days. Optical density at 600 nm (OD600) was measured, and readings for each well were taken every 10 min for 48 h. The time to detectable turbidity (tdet) was defined as the time taken for the measured OD600 to reach 0.14 U. Blank medium had an OD600 of around 0.10 U. The detection limit was 3 × 105 CFU ml−1, as determined from a calibration curve of cell number versus OD600. The specific growth rate of nonproteolytic C. botulinum 17B in PYGS broth at 22°C was determined from plate count measurements obtained during exponential growth. Plate counts were determined from appropriate dilutions of cells spread on PYGS agar and incubated under a headspace consisting of 90% hydrogen and 10% carbon dioxide for 2 days at 30°C.
Slide preparation for microscopic observation.
Slides were prepared by placing a suspension of spores in a defined area on the surface of an electrostatically charged Superfrost Plus microscope slide to obtain a concentration between 1,200 and 3,000 spores mm−2. After 30 min of contact time in a humid chamber at 1°C, each slide was washed in distilled water to remove any unattached spores, air dried in a fan-powered incubator at 30°C, and stored in an anaerobic cabinet for at least 16 h to remove any traces of oxygen from the slide. At time zero, the deoxygenated spores attached to the slide were overlaid with 8 μl of molten anaerobic PYGS medium containing 0.5% (wt/vol) agar. A coverslip (18 by 18 mm) was pressed onto the agar to create a thin film and was sealed in place with aluminum tape to prevent desiccation and oxygen ingress. The slide was transferred from the anaerobic cabinet to the microscope stage, and measurement was started.
Microscopy.
Spore outgrowth was observed by phase-contrast microscopy at a magnification of ×40 (Leica ×40/0.70 numerical aperture, PL FLUOTAR objective) with a Leica DMRB optical microscope. Samples were maintained at 22°C using a stage-mounted peltier device (Linkham Scientific Instruments, Tadworth, United Kingdom) with a temperature controller (Linkham PE60). The microscope was fitted with an XYZ stage (Marzhauser, Wetzlar-Steindorf, Germany) with an H128 motor controller (Prior Scientific Instruments, Cambridge, United Kingdom) controlled by Image-Pro Plus image analysis software (Media Cybernetics, Silver Spring, MD) so that the stage could be returned repeatedly to multiple set positions during each experiment. Images of each field of view were captured every 5 min for 15 h using a JVC KY-F70 three-charge-coupled device color digital camera. Individual images were compiled to give a sequence of frames for each field of view. This allowed the same spore to be monitored throughout the entire experiment. Spore data were collected from 11 replicate experiments.
Image processing.
The image sequences followed individual spores through dormancy, germination, emergence, elongation, and eventually cell division. The individual images were analyzed using Image Pro Plus image analysis software to determine the maximum pixel intensity and length of each spore or cell in each captured field of view. Images were sharpened to enhance intensity differences between neighboring pixels using two passes at strength 10 for a 7 × 7 kernel size and then outlined as objects with pixel intensities less than the mean background intensity minus three times the standard deviation. Each outline was checked by eye, and alterations, such as separation of touching objects or addition of nonoutlined areas, were made before the outline was saved. The outline was then overlaid on the raw image, and the maximum pixel intensity and the length of the object were measured.
Quantification of germination and outgrowth events.
For all measurements, time zero was defined as the time of nutrient addition. Germination was determined from when a spore changed from phase bright to phase dark, as measured by a decrease in pixel intensity. Maximum pixel intensity was plotted versus time and fitted with a linear triphasic model using Dmfit (www.ifr.ac.uk/safety/DMfit). The three lines represented the spore during periods of high (phase-bright), decreasing, and low (phase-dark) pixel intensity. Time to germination was defined as the time at the midpoint of the line representing decreasing pixel intensity. Time to emergence was defined as the time when a new cell was first observed emerging from the spore coats. Newly emerged spores do not contain the full complement of macromolecules found in adult cells and thus are smaller. The time to a mature cell was measured by determining the time that it took a cell to reach the length of a single mid-exponential cell in broth culture (found to be 5.5 μm in PYGS at 22°C). It was difficult to determine the time to cell division as septation and cell constriction were not always obvious microscopically; cells often appeared to elongate to many times their initial length before separation into multiple cells. It was decided that doubling time would be measured by determining the time that it took a cell to increase from the length of one mid-exponential cell to the length of two mid-exponential cells. DMfit was used to generate a best-fit curve for cell length versus time, and the times that it took to reach 5.5 μm and 11 μm were recorded as the times that it took to reach the lengths of one (tC1) and two (tC2) mature cells, respectively.
Statistical analysis.
The data sets were analyzed using an in-house program (Varifit) written in Visual Basic. The homogeneity of distributions was analyzed using a chi-square test, and a Bartlett test was used to compare the variances of the distributions. Pearson coefficients were calculated to study the correlation between growth event times and intervals during germination and subsequent growth.
RESULTS
The results described here are based on microscopic observations of 1,739 spores. The numbers of spores that could be followed to germination, emergence, and growth to lengths of 5.5 μm and 11 μm were 1,441, 1,169, 872, and 656, respectively. In the turbidity experiments in which a Bioscreen C automated turbidity reader was used, 800 filled wells were observed, and growth was detected in 557 wells. The log growth rate during the exponential phase was 0.3 h−1, which is equivalent to a population doubling time of 0.99 h. The distributions of time to germination, time to emergence, time to the length of one mature cell, time to the length of two mature cells, and time to detectable turbidity are shown in Fig. 1. Chi-square tests indicated that the only distributions between which homogeneity could be accepted were tC1 and tC2. The mean times and standard deviations for events during germination and subsequent growth are shown in Table 1. There was considerable heterogeneity in the population at all stages of growth. The variability was greatest for tgerm and then fell and was similar for tC1, tC2, and tdet. The distributions of times for germination (tgerm), times for outgrowth (tC1 − tgerm), and times for a mature cell to double in length (tC2 − tC1) are shown in Fig. 2. Under the good growth conditions tested in this experiment, the mean time for outgrowth (6.01 h) was much longer than the mean time for germination (2.56 h) or doubling (1.50 h).
FIG. 1.
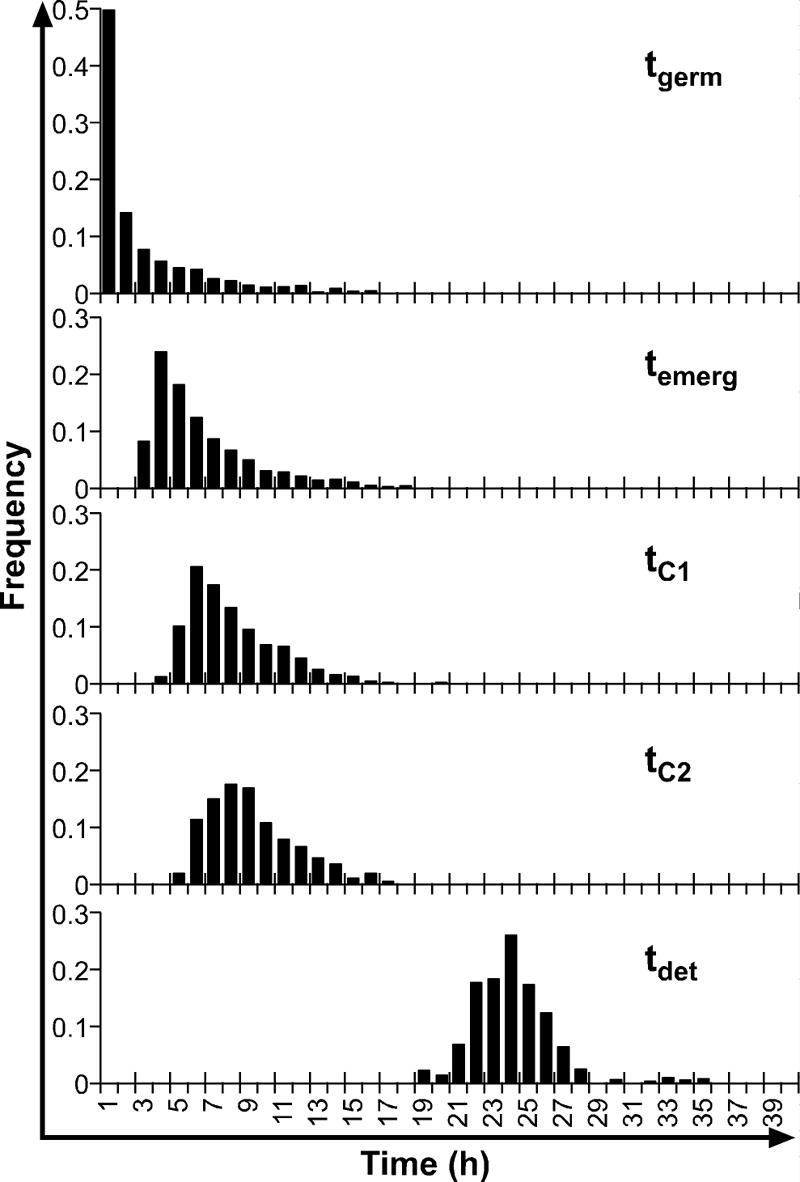
Frequency distributions for tgerm, temerg, tC1, tC2, and tdet for single spores of nonproteolytic C. botulinum Eklund 17B in PYGS medium at 22°C.
TABLE 1.
Average times and standard deviations for events occurring during germination of, and subsequent growth from, spores of nonproteolytic C. botulinum Eklund 17B in PYGS medium at 22°C
| Event | Mean (h) | SD (h) |
|---|---|---|
| tgerm | 2.56 | 3.19 |
| temerg | 5.82 | 2.92 |
| tC1 | 7.60 | 2.61 |
| tC2 | 8.66 | 2.51 |
| tdet | 23.64 | 2.54 |
| tC1 − tgerm | 6.01 | 1.75 |
| tC2 − tC1 | 1.50 | 0.73 |
FIG. 2.
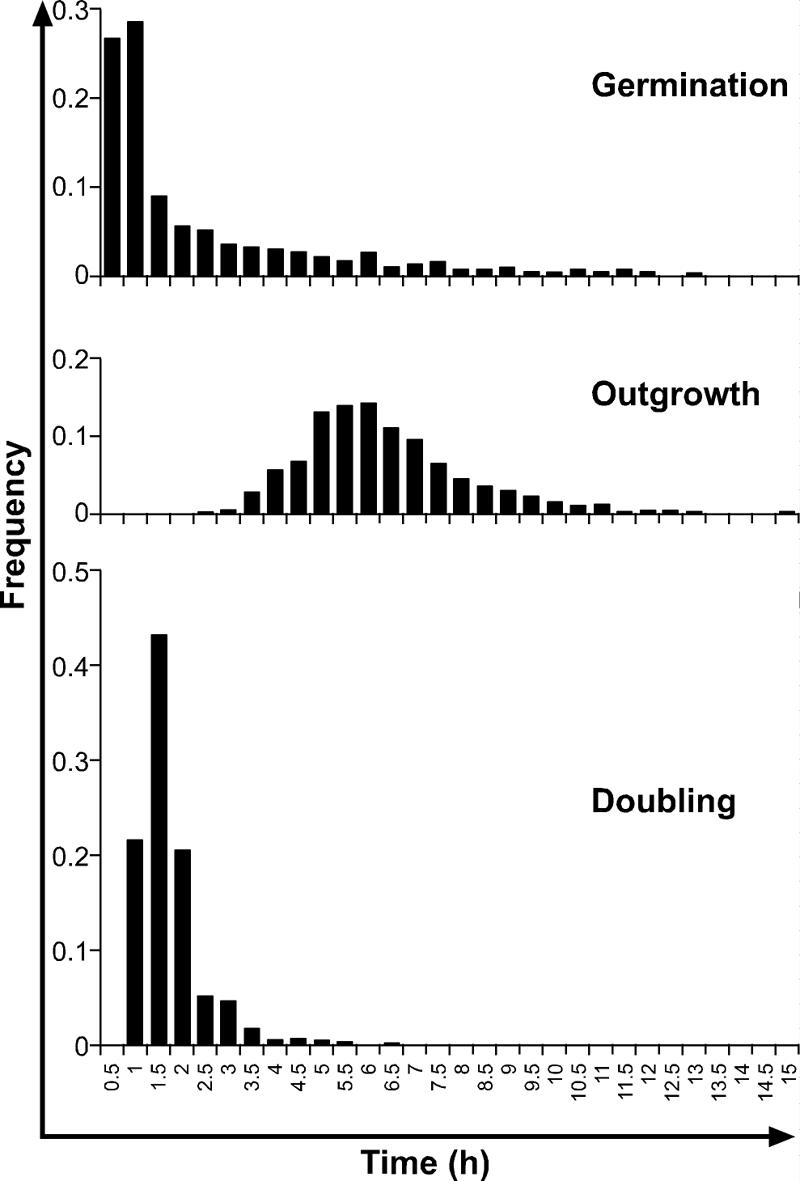
Frequency distributions for times for germination (tgerm), outgrowth (tC1 − tgerm), and first doubling (tC2 − tC1) for spores of nonproteolytic C. botulinum Eklund 17B in PYGS medium at 22°C.
The relationships between times for events and intervals during germination and subsequent growth are show in Table 2. The total times for germination (tgerm), emergence (temerg − tgerm), cell maturation (tC1 − temerg), and doubling (tC2 − tC1) were not correlated. Correlations between times to events in, and stages of, germination and subsequent growth were found only when one of the times was a component of the other period. In these cases the strength of the correlation reflected the size of the overlap between the periods. For example, the time to one cell formed a larger component of the time to two cells than did the time to germination and consequently showed a stronger correlation to tC2 than tgerm. Although there was a strong correlation between the time to germination and the total time to two cells, there was a large scatter in times to growth from spores at each germination time, and the first spore to germinate was not the first spore to reach the length of two mature cells (Fig. 3). Spores that germinated within 30 min of nutrient addition took between 4.5 and 14.4 h to reach the length of two cells. Spores that germinated after 30 min frequently reached two cell lengths before spores that germinated earlier (Fig. 3).
TABLE 2.
Pearson product moment correlations between pairs of variables representing times for events during germination of and subsequent outgrowth from spores of nonproteolytic C. botulinum Eklund 17B
| Event | Pearson product moment coefficientsa
|
||||||||
|---|---|---|---|---|---|---|---|---|---|
| temerg | tC1 | tC2 | temerg − tgerm | tC1 − tgerm | tC2 − tgerm | tC1 − temerg | tC2 − temerg | tC2 − tC1 | |
| tgerm | 0.838b | 0.735b | 0.720b | 0.053 | 0.128 | 0.115 | 0.215 | 0.188 | 0.005 |
| temerg | 0.889b | 0.862b | 0.589 | 0.511 | 0.459 | 0.274 | 0.246 | 0.117 | |
| tC1 | 0.959b | 0.474 | 0.767b | 0.649 | 0.684 | 0.554 | 0.173 | ||
| tC2 | 0.443 | 0.731b | 0.773b | 0.619 | 0.703 | 0.447 | |||
| temerg − tgerm | 0.727b | 0.675 | 0.164 | 0.156 | 0.211 | ||||
| tC1 − tgerm | 0.917b | 0.797b | 0.682 | 0.274 | |||||
| tC2 − tgerm | 0.656 | 0.834b | 0.634 | ||||||
| tC1 − temerg | 0.839b | 0.186 | |||||||
| tC2 − temerg | 0.691 | ||||||||
Boldface type indicates independent variables. Normal type indicates nested variables, where one outgrowth event is a component of the other.
Pairs of events found to be strongly correlated (>0.7).
FIG. 3.
Scatter diagram showing tgerm versus tC2 for spores of nonproteolytic C. botulinum Eklund 17B in PYGS agar at 22°C.
DISCUSSION
It is often said that the first spore to germinate is the important one as it will produce the first cell to grow and start toxin production. The results of this experiment show that this is not necessarily true. A microscope and image analysis system was used to measure the durations of germination (tgerm), emergence (temerg), growth to one cell length (tC1), and growth to two cell lengths (tC2) for single spores of nonproteolytic C. botulinum. The time intervals for germination, emergence, outgrowth, and doubling were all independent of each other and showed considerable variability between individuals. A consequence of the lack of correlation between, and high variability in, the different stages of the lag phase is that under the good growth conditions used in present tests, it is not possible to predict the total duration of the lag phase from just one stage. Thus, it is not possible to predict the total duration of the lag phase from the germination time.
The independence of different stages of lag phase indicates that the time for each stage is not related to a single factor but depends on many diverse factors. The lag phase for spores of nonproteolytic C. botulinum is a multistage process. Germination differs from other growth stages in that it is a series of degradative reactions in which preformed enzymes are used, while the later stages require macromolecular synthesis (10). It is likely that different metabolic processes are required at different times during emergence, outgrowth, and cell multiplication and also that these stages depend on nonmetabolic factors, such as the strength of the spore coat or the initial size or chemical complement of the spore. Adverse conditions or processes could add further complexity as they may affect each stage differently. For example, heat treatment is known to damage the spore germination system (13), and while the presence of oxygen does not affect germination of C. botulinum (15), it adversely affects subsequent stages that require active metabolism. This complexity ensures that mechanistic models have to be created specifically for spore inocula.
Strong correlations were found between the total times to different stages of the lag phase when the time for one stage formed a substantial component of the time for another stage. Correlation would be expected in these circumstances but does not necessarily allow one event to be predicted from the other. For example, tgerm was found to correlate strongly with tC2. The time to two cells depends on both tgerm and the time for growth from germination to two cells (tC2 − tgerm). These intervals encompass different growth events and were shown to be independent of each other. The correlation between tgerm and tC2 depends on their relative magnitudes and occurs only when tgerm is a large proportion of tC2. Figure 3 shows that the correlation between tgerm and tC2 was strong only for slowly germinating spores, when an extended tgerm contributed a large proportion of tC2. The majority of spores (>50%) germinated within 1 h. For these rapidly germinating spores, tC1 was highly variable, ranging from 4.5 to 14.4 h, and correlated poorly with tgerm. Despite overall strong correlations, it is unwise to estimate time to late growth events from early growth events, such as germination, unless there is a mechanistic dependence.
Two independent methods were used to determine the shape of lag distributions in this study. Early stages in germination and outgrowth were measured using microscopy, and lag distributions were derived from times to detection in a Bioscreen automated turbidity reader. A previous study suggested that microscopy provided a more accurate estimate of Listeria monocytogenes cell lag times than the Bioscreen method (18). Using microscopy, individual lag times are determined directly. However, all spores are in a single chamber so they may interfere with each other, either spatially or chemically. With the Bioscreen method, spores are physically separated and thus unable to affect each other's lag times, but single lag times are not measured directly; the Bioscreen reader records detection times for populations originating from about one spore. Another difference between the methods was that turbidity measurements were made in broth media, whereas agar was added to the medium used in microscopic studies to keep individuals in a fixed location that allowed identification at multiple observation times. The time to a mature cell could be longer for attached spores in agar than for free spores in broth. Despite the differences between the two methods, the Bioscreen and microscopic methods produced distributions with very similar estimates of lag variability.
To predict individual lag times from the tdet measured by the Bioscreen method, it is assumed that growth in each well arises from a single spore, the time from lag to detection is constant in identical conditions, and the growth conditions are identical in all wells. If these assumptions are true, the measured tdet distribution should be the same as the desired lag time distribution, simply shifted in time (8). One assumption made in the consideration described above is that growth originates from a single spore. As samples were prepared by dilution, the number of spores per Bioscreen well would have followed a Poisson distribution, so not all wells would have contained a single spore. The size of the inoculum used in the Bioscreen analysis was a compromise between the percentage of wells with a single spore and the number of wells in which growth occurred giving a distribution of the detection times. The dilution used resulted in growth in only 70% of the observed wells, so according to the Poisson distribution ca. 30% of the wells contained no spores, 36% of the wells contained one spore, 22% of the wells contained two spores, and the rest of the wells contained more than two spores. Computer simulations showed that this composition of inocula would still allow estimation of the distribution of single cell parameters (results not shown). It is therefore a reasonable assumption that the measured distribution approximated the distribution that would have been obtained using single spores. The agreement was also demonstrated by the finding that the variance in the distribution of lag times measured by the Bioscreen method was the same as that obtained microscopically. If the lag distributions measured microscopically and by the Bioscreen method were identical and growth after lag occurs at a constant rate, then the tdet distribution should be the same as the tC1 distribution simply shifted by the time required for 18.2 doublings. The mean observed tdet was 23.6 h, compared to 25.7 h estimated from the mean time to one cell plus time for multiplication from one cell to a detectable population calculated using a population doubling time of 0.99 h. Although there is a discrepancy of 2.1 h, this value is small considering that the doubling time was extrapolated over 18.2 generations.
The population doubling time of 0.99 h measured in broth culture was much shorter than the mean time required for a new cell to double in length (tC2 − tC1) (1.50 h). The relationship between the doubling time of a population (td) and the mean generation time of the single cells within that population (tg) is not straightforward and depends on the distribution of single cell generation times (4). In the special case of all cells having identical generation times, td is equal to tg. When single cell generation times are exponentially distributed, the population doubling time is much shorter than the mean generation time, and td = ln2 × tg (4). In most cases, populations have distributions somewhere between these two special cases, and td has a value between ln2tg and tg. Using tC2 − tC1 as a measure of average single cell generation time and assuming that the observed distribution of generation times is maintained for all generations throughout growth, it is possible to simulate a growth curve and estimate the population doubling time. The estimated td in this experiment was 1.30 h, which gives an estimate for tdet of 31.2 h. The difference between the population doubling time measured during exponential growth and that calculated from cells observed immediately after lag could arise from differences in the measurement techniques, as mentioned above, but it could also be because the assumption that the generation time distribution remains constant throughout growth does not hold. Recent work has suggested that the average and spread of generation times decrease during the first three generations after lag (9, 14). Further examination of growth kinetics for generations soon after lag is required if the real shape of growth curves originating from single cells is to be determined and times for small increases in cell numbers are to be predicted.
Knowledge of individual spore lag time distributions is an important component in quantitative risk assessment and process risk modeling of products containing low numbers of spores (5) and also is necessary for improved prediction of population lag (2). Population lag is traditionally determined as the breakpoint from the stationary phase of log cell count against time and can thus depend on the initial count. The relationship between population lag and the average lag of the individual cells in the population is not simple and again depends on the distribution of single cell lag times. This distribution can be determined only from observations of times to growth from individual cells as population lag is a stochastic process from which the underlying distributions cannot be determined (3). Determination of lag phase distributions as observed in this study should help the development of more mechanistic models to predict bacterial growth.
The distribution curves determined in this study highlight the finding that there is considerable heterogeneity within spore populations. The shape of the distribution of germination times was skewed with a long tail and was similar to the shape of distributions previously reported for proteolytic C. botulinum (6). The asymmetry of the distribution implies that mean germination time is not a good parameter to use in risk assessment work as it underestimates the number of spores germinating at times shorter than the mean time. In the present study, the mean germination time was 2.6 h, but 50% of the spores germinated within 1 h. Information on the shape of the distribution curve should be useful in refining estimates of germination.
Food-borne botulism is an intoxication. Consequently, the time to toxin is of paramount importance in food safety. In the absence of direct toxin measurements, safety is usually estimated from the time to growth. Comparison of the times for germination, outgrowth, and doubling show that the time between germination and outgrowth to a full-sized cell is a large part of the lag in good growth conditions. Using germination time as an estimate of lag time underestimates times to growth. This study also showed the distribution curves for germination, and subsequent stages of growth were not simply projections of each other shifted in time, so later events cannot be predicted by adding a constant value to earlier values. Knowledge of spore germination kinetics alone cannot be used to estimate time to growth or time to toxin.
Acknowledgments
This research was carried out with financial support from the EU program Quality of Life and Management of Living Resources (QLK1-CT-2001-01145; BACANOVA) and from a competitive strategic grant from the BBSRC.
We thank József Baranyi for useful discussions.
REFERENCES
- 1.Baranyi, J. 1997. Simple is good as long as it is enough. Food Microbiol. 14:189-192. [Google Scholar]
- 2.Baranyi, J. 1998. Comparison of stochastic and deterministic concepts of bacterial lag. J. Theor. Biol. 192:403-408. [DOI] [PubMed] [Google Scholar]
- 3.Baranyi, J. 2002. Stochastic modelling of bacterial lag phase. Int. J. Food Microbiol. 73:203-206. [DOI] [PubMed] [Google Scholar]
- 4.Baranyi, J., and C. Pin. 2001. A parallel study on bacterial growth and inactivation. J. Theor. Biol. 210:327-336. [DOI] [PubMed] [Google Scholar]
- 5.Barker, G. C., P. K. Malakar, and M. W. Peck. 2005. Germination and growth from spores: variability and uncertainty in the assessment of food borne hazards. Int. J. Food Microbiol. 100:67-76. [DOI] [PubMed] [Google Scholar]
- 6.Billon, C. M. P., C. J. McKirgan, P. J. McClure, and C. Adair. 1997. The effect of temperature on the germination of single spores of Clostridium botulinum 62A. J. Appl. Microbiol. 82:48-56. [DOI] [PubMed] [Google Scholar]
- 7.Lund, B. M., and M. W. Peck. 2000. Clostridium botulinum, p. 1057-1109. In B. M. Lund, T. C. Baird-Parker, and G. W. Gould (ed.), The microbiological safety and quality of food. Aspen Publishers, Gaithersburg, Md.
- 8.Metris, A., S. M. George, M. W. Peck, and J. Baranyi. 2003. Distribution of turbidity detection times produced by single cell-generated bacterial populations. J. Microbiol. Methods 55:821-827. [DOI] [PubMed] [Google Scholar]
- 9.Metris, A., Y. Le Marc, A. Elfwing, A. Ballagi, and J. Baranyi. 2003. Modelling the variability of lag times and the first generation times of single cells of E. coli, p. 37-39. In J. F. M. Van Impe, A. H. Geeraerd, I. Leguerinel, and P. Malfart (ed.), Predictive modelling in foods—conference proceedings. Katholieke Universiteil, Leuven, Belgium. [DOI] [PubMed]
- 10.Paidhungat, M., and P. Setlow. 2002. Spore germination and outgrowth, p. 537-548. In A. L. Sonenshein, J. A. Hoch, and R. Losick (ed.), Bacillus subtilis and its closest relatives: from genes to cells. ASM Press, Washington, D.C.
- 11.Peck, M. W. 1997. Clostridium botulinum and the safety of refrigerated processed foods of extended durability. Trends Food Sci. Technol. 8:186-192. [Google Scholar]
- 12.Peck, M. W., D. A. Fairbairn, and B. M. Lund. 1992. The effect of recovery medium on the estimated heat-inactivation of spores of non-proteolytic Clostridium botulinum. Lett. Appl. Microbiol. 15:146-151. [DOI] [PubMed] [Google Scholar]
- 13.Peck, M. W., D. A. Fairbairn, and B. M. Lund. 1992. Factors affecting growth from heat-treated spores of non-proteolytic Clostridium botulinum. Lett. Appl. Microbiol. 15:152-155. [DOI] [PubMed] [Google Scholar]
- 14.Pin, C., and J. Baranyi. Distribution of the lag times of individual cells as a function of the age of the cells in the inoculum. Int. J. Food Microbiol., in press.
- 15.Plowman, J., and M. W. Peck. 2002. Use of a novel method to characterize the response of spores of non-proteolytic Clostridium botulinum types B, E and F to a wide range of germinants and conditions. J. Appl. Microbiol. 92:681-694. [DOI] [PubMed] [Google Scholar]
- 16.Swinnen, I. A. M., K. Bernaerts, E. J. J. Dens, A. H. Geeraerd, and J. F. Van Impe. 2004. Predictive modelling of the microbial lag phase: a review. Int. J. Food Microbiol. 94:137-159. [DOI] [PubMed] [Google Scholar]
- 17.Whiting, R. C., and J. C. Oriente. 1997. Time-to-turbidity model for non-proteolytic type B Clostridium botulinum. Int. J. Food Microbiol. 35:49-60. [DOI] [PubMed] [Google Scholar]
- 18.Wu, Y., M. W. Griffiths, and R. C. McKellar. 2000. A comparison of the Bioscreen method and microscopy for the determination of lag times of individual cells of Listeria monocytogenes. Lett. Appl, Microbiol. 30:468-472. [DOI] [PubMed] [Google Scholar]