

Abstract
Activation of heterodimeric E2F-DP transcription factors can drive the G1-S transition. Mutation of the Drosophila melanogaster dE2F gene eliminates transcriptional activation of several replication factors at the G1-S transition and compromises DNA replication. Here we describe a mutation in the Drosophila dDP gene. As expected for a defect in the dE2F partner, this mutation blocks G1-S transcription of DmRNR2 and cyclin E as previously described for mutations of dE2F. Mutation of dDP also causes an incomplete block of DNA replication. When S phase is compromised by reducing the activity of dE2F-dDP by either a dE2F or dDP mutation, the first phenotype detected is a reduction in the intensity of BrdU incorporation and a prolongation of the labeling. Notably, in many cells, there was no detected delay in entry into this compromised S phase. In contrast, when cyclin E function was reduced by a hypomorphic allele combination, BrdU incorporation was robust but the timing of S-phase entry was delayed. We suggest that dE2F-dDP contributes to the expression of two classes of gene products: replication factors, whose abundance has a graded effect on replication, and cyclin E, which triggers an all-or-nothing transition from G1 to S phase.
One of the key points of cell cycle regulation is the transition from G1 into S phase (50, 58). Despite the identification of a number of the regulators that influence this transition, we do not fully understand how it is controlled. Detailed analyses of the phenotypes of mutations in the genes that regulate the transition might provide more insight into the nature of the decisive events that trigger the onset of DNA replication. Such analyses of Saccharomyces cerevisiae have demonstrated two types of phenotypes when entry into S phase is partially compromised (40, 55). Defects in some genes do not delay the transition into S phase but prolong S phase, apparently by reducing replication efficiency. Defects in other genes appear to have an all-or-nothing effect on a decisive event that is presumed to represent a switch from the G1 to the S-phase states. Analysis of the behavior of cells in other organisms (26) and of nuclei replicating in Xenopus laevis in vitro extracts (39) suggests that an all-or-nothing switch is generally associated with the G1-to-S transition.
Several findings argue for the importance of the activation of transcription of specific genes that accompanies the G1-S transition. Chief among these is the finding that activation of specific transcription factors appears to occur immediately upstream of the decisive transition into S phase in yeast (1, 5), Drosophila melanogaster (15, 17), and mammalian cells (37, 44). These factors stimulate a program of expression of genes whose products are required for DNA replication. However, because at least some of the required products are stable and are present as a result of earlier expression, the activation of this gene expression does not necessarily provide direct coupling between replication factor transcription and entry into S phase. The role of triggering entry into S phase may instead be mediated by a second type of gene product induced as part of the S-phase transcription program. In yeast (43, 45), Drosophila (16), and vertebrates (4, 23, 47), G1 cyclins are part of this transcription program (e.g., CLN1 and CLN2 in S. cerevisiae and cyclin E in metazoans). It is not entirely clear what roles each of these two classes of induced gene products, replication factors and G1 cyclins, plays in the G1-to-S transition.
In mammals and Drosophila, the G1-S-phase transcription program is controlled by the E2F-DP family of heterodimeric transcription factors (17, 37, 44, 59). Five related E2F genes and two related DP genes have been identified in mammals (59). Mice containing a homozygous knockout mutation of the E2F-1 gene develop normally and are viable, indicating that E2F-1 is not essential for cell cycle progression (22, 72). While E2F-1 knockout mice do display several abnormal phenotypes, most strikingly the eventual acquisition of a variety of tumors in older individuals, the absence of a more dramatic phenotype due to loss of E2F-1 function could be explained by redundancies among the five known E2Fs. In Drosophila, only a single E2F gene (dE2F) and DP gene (dDP) have yet been reported (18, 27, 48), although an E2F-related sequence has recently been identified as an EST from a Drosophila embryonic cDNA library (28). As in mice, therefore, there exists a potential for functional redundancy among Drosophila E2F genes. Nevertheless, mutations of dE2F cause lethality (6, 17). In embryos, dE2F mutations block the activation of transcription that usually occurs at the G1-to-S transition and inhibit DNA replication (17). Thus, dE2F plays an essential role, likely as a requirement for normal cell cycle progression.
Here we explore further the role of the activation of G1-S phase transcription by identifying mutations of the dDP gene and comparing embryonic phenotypes caused by genetic reductions in the function of dDP, dE2F, or cyclin E, a downstream target of dE2F-dDP that is required for S phase (16, 33, 51). We find that both dDP and dE2F are required for the activation of transcription of the same genes at the G1-to-S transition, consistent with dE2F and dDP acting as a heterodimer in vivo. If cyclin E and dE2F-dDP are jointly required to trigger a common step needed for the initiation of DNA replication, then reductions in the functions of these gene products should lead to similar phenotypes. In contrast to this, we find distinct behaviors. Reducing dE2F-dDP function first diminishes the rate of DNA replication, and only with more severe reductions in functions do we detect delays or defects in the transition into S phase. Reduction of cyclin E function leads to a stochastic increase in the length of G1 without compromising replication in those cells that initiate DNA synthesis. We discuss these results in terms of two distinct roles for dE2F-dDP and cyclin E in the pulse of transcription at the G1-to-S transition: one role is a direct and dose-dependent participation in DNA replication and the other is a regulatory role that triggers an all-or-nothing change required for S phase.
MATERIALS AND METHODS
Genetics and fly stocks.
The overlapping Df(2R)vg56 and Df(2R)vg33 deficiencies (breakpoints 49D;49F and 49D;50A, respectively) and the recessive-lethal EMS-induced mutations from the 49F region were isolated in a previously described mutagenesis screen (36). Single mutant stocks representing a subset of the 18 complementation groups were screened with the DmRNR2 probe (vr-3, -4, -6, -8, -9, -10, -11, -13, -15, -17, -18, and -19). These were graciously provided to us by Ting Wu of Harvard University. The dDPvr10 mutation we refer to throughout the text was originally designated vr10-13 (36) and contained the recessive markers al, b, c, and sp. We separated three other lethal mutations from the original dDPvr10 chromosome by two successive meiotic recombinations with a wild-type Sevelen second chromosome. The possibility that this new recombinant chromosome (designated dDPvr10-5D) only contains the dDPvr10 semilethal allele is supported by the rare (<2%) appearance of dDPvr10/dDPvr10 homozygous flies in the stock. The presence of these three additional lethal mutations did not significantly affect the phenotype in comparisons of DmRNR2 staining of dDPvr10/dDPvr10, dDPvr10/Df(2R)vg56, dDPvr10-5D/dDPvr10-5D, and dDPvr10-5D/Df(2R)vg56 mutant embryos. The cyclin EP28 and cyclin E05206 alleles are described in references 2 and 32 and 33, respectively. The dE2F164 P-element allele is described in reference 56; all other dE2F alleles are described in reference 17. All second chromosome mutant stocks were maintained with a CyO P[w+; wg-lacZ] balancer, allowing unambiguous identification of mutant progeny embryos, which fail to express lacZ.
Molecular analysis of the dDP locus and dDP cDNA.
Polytene chromosome hybridizations were performed with digoxigenin-labeled dDP cDNA (encoding amino acids 76 to 322) exactly as described previously (15). To identify the dDPvr10 mutation, dDP genomic DNA was PCR amplified in three overlapping regions with primers designed from the sequence of the full-length cDNA (see below). Template DNA was prepared from single adult dDPvr10/Df(2R)vg56 or Canton S control flies as described previously (25). For each genotype, products from two independent PCR amplifications with Thermococcus litoralis DNA polymerase (New England Biolabs) were subcloned into pBluescript and sequenced. The missense mutation was found in both dDPvr10 PCR amplifications but not in the wild-type amplifications. Several pieces of evidence suggested that our original dDP cDNA (18) was not full length. First, immunoblot analysis revealed that heat shock expression of this cDNA produces a polypeptide that by sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis migrates with an apparent molecular mass ∼8 kDa smaller than that of the endogenous dDP (14). Second, Hao et al. (27) reported the amino acid sequence of a Drosophila dDP cDNA that was nearly identical to the previously reported dDP cDNA translation product (18) from residues 8 to 377 but contained an amino-terminal extension of 74 amino acids. Third, the Dynlacht et al. (18) sequence does not contain an in-frame stop codon upstream of the assigned initiator Met. We PCR amplified the 5′ region from a probable full-length dDP cDNA with nested primers and template DNA prepared from a 0- to 4-h embryonic cDNA library (7). In the first PCR, a vector primer that hybridizes 5′ to the cloning site and contains a HindIII recognition sequence was paired with a primer (5′ ACAGTTGTTGTCGTACGCAT) that hybridizes at the unique BsiWI site in the dDP cDNA. A 1:1,000 dilution of the first reaction mixture was used as the template in a second PCR that paired the same 5′ vector primer with a dDP primer (5′ TTGCCCGATGCCGCTAGCAC) that hybridizes at the unique NheI site just upstream of the initiator Met assigned by Dynlacht et al. (18). The single product from this reaction was cut with HindIII/NheI and subcloned in frame with the Dynlacht et al. (18) cDNA contained in pBluescript SK.
Embryo in situ hybridization and BrdU labeling.
Digoxigenin-labeled RNA probes from DmRNR2 genomic DNA (15) and cyclin E type I cDNA (51) were used for embryo in situ hybridizations as described previously (15, 38, 65). Embryos were pulse labeled for 15 min in Schneider’s medium containing 1 mg of BrdU/ml as described previously (19, 54). Incorporated BrdU was detected with anti-BrdU mouse monoclonal primary antibody (Becton Dickinson) and rhodamine-conjugated goat anti-mouse secondary antibody (Jackson). For heat shock experiments, eggs from dDPvr10/CyO P[w+; wg-lacZ]; hsp70-dDP/hsp70-dDP flies were collected on grape juice agar plates, aged at 18°C, and heat shocked by floating the collection plate on the surface of a 37°C water bath for 30 min. After a 70-min recovery at room temperature, the embryos were fixed and stained for DmRNR2 expression. The hsp70-dDP P-element construct is described in reference 14.
Immunoblot analysis.
Eggs were collected for 1 h at room temperature from w; Df(2R)vg56/CyO P[w+; wg-lacZ] flies and aged 23 h at 17°C. The eggs were dechorionated in 2.5% sodium hypochlorite, and the embryos were devitellinized and fixed by shaking in a 1:1 mixture of methanol and heptane. The embryos were washed twice with MeOH, transferred to an aqueous buffer containing protease inhibitors (10 mM Tris [pH 7.8], 150 mM NaCl, 1 mM EDTA, 1 mM phenylmethylsulfonyl fluoride, 2 mM benzamidine, 0.05% Tween 20), and stained with the DNA binding dye Hoechst 33258. Ten Df(2R)vg56/Df(2R)vg56 or 10 Df(2R)vg56/CyO P[w+; wg-lacZ] methanol-fixed embryos were hand selected under UV light with a fluorescence microscope. The three types of progeny from this stock {Df(2R)vg56/Df(2R)vg56, Df(2R)vg56/CyO P[w+; wg-lacZ], and CyO P[w+; wg-lacZ]/CyO P[w+; wg-lacZ]} could easily be distinguished from each other at germ band-retracted stages due to differences between the aberrant morphology of the deficiency homozygotes and the CyO/CyO balancer homozygotes, which have a wg mutant phenotype. Selected embryos were dissociated by boiling them for 3 min in sample buffer (62.5 mM Tris [pH 6.8], 2% SDS, 5% glycerol, 5% β-mercaptoethanol) and subjected to SDS-polyacrylamide gel electrophoresis. Separated proteins were electrophoretically transferred to nitrocellulose, and dDP polypeptides were detected with a 1:10 dilution of monoclonal antibody supernatant Yun1 (13).
Nucleotide sequence accession number.
The additional dDP cDNA sequence discussed above has been submitted to GenBank (accession no. AF031700).
RESULTS
Identification of a dDP mutation.
Drosophila dDP cDNA was cloned previously and shown to encode a functional binding partner for dE2F (18, 27). We took a reverse genetic approach to identify mutations of dDP in order to test its role in transcription and cell cycle control. Hybridizations of a dDP cDNA probe to salivary gland polytene chromosomes (Fig. 1A) allowed us to map the dDP locus to position 49F on the right arm of the second chromosome (not shown). A series of overlapping deletion mutations that uncover region 49F were generated by Lasko and Pardue (36). In situ analysis of Df(2R)/+ polytene chromosomes revealed that the Df(2R)vg56 deletion chromosome failed to hybridize with our dDP cDNA probe (Fig. 1A). We conclude from this result that dDP coding sequences are missing in the Df(2R)vg56 deletion. Hao et al. previously reported that dDP mapped outside this particular deficiency (27). While we cannot readily explain this discrepancy, two other observations confirm our mapping of dDP within the Df(2R)vg56 deletion. First, in embryos that had progressed to cellularization or beyond, in situ hybridization detected zygotic dDP expression in control but not in Df(2R)vg56/Df(2R)vg56 embryos, consistent with the elimination of maternal dDP RNA by cell cycle 14 (data not shown). Second, Western blotting (see Materials and Methods) of total protein extracted from germ band-retracted Df(2R)vg56/Df(2R)vg56 embryos with anti-dDP monoclonal antibodies indicated a severe reduction in the amount of dDP protein compared to that of Df(2R)vg56/+ sibling embryos (Fig. 1B). Taken together these data indicate that the Df(2R)vg56 chromosome lacks a functional dDP gene.
FIG. 1.

Identification of dDP mutations. (A) Hybridization of a Df(2R)vg56/+ salivary gland polytene chromosome with a dDP cDNA probe. The hybridization signal (arrow) is lost in the Df(2R)vg56 deletion and consequently appears over half the width of the heterozygous polytene chromosome. (B) Western blot of extract of germ band-retracted embryos collected from the Df(2R)vg56/CyO stock probed with anti-dDP monoclonal antibody. Since Df(2R)vg56 deletes the dDP gene, any dDP protein present in Df(2R)vg56/Df(2R)vg56 embryos must be of maternal origin. (C) Germ band-extended control (+/?) and Df(2R)vg56/Df(2R)vg56 mutant stage 11 embryos subjected to in situ hybridization with a digoxigenin-labeled probe derived from the DmRNR2 gene. DmRNR2 expression is failing in the developing ventral (toward the bottom) nerve cord of the mutant embryo. (D) Intron and exon structure of the transcribed region of the dDP gene. Exons are represented by shaded boxes, and introns are represented by a straight line. ATG and TGA indicate the beginning and end of the dDP open reading frame, respectively. The Arg-to-His missense mutation in exon 5 of the dDPvr10 allele is indicated. (E) Alignment of a highly conserved region of the heterodimerization domain of DPs from Drosophila (this paper), human (hum) (30, 70), mouse (mus) (30, 49), and Xenopus (xen) (24). The numbers indicate the position in each primary amino acid sequence. The position of the Arg-to-His change in the dDPvr10 allele is indicated at the top.
Mutations of dE2F reduce the zygotic expression of several genes involved in DNA replication, including those encoding the small subunit of ribonucleotide reductase (DmRNR2) and PCNA (17). This phenotype is first apparent in the central nervous system (CNS) of germ band-extended embryos during late stage 11. In germ band-extended Df(2R)vg56/Df(2R)vg56 embryos the amount of DmRNR2 and PCNA mRNA in the CNS was significantly reduced relative to that of the wild type (Fig. 1C). An essentially identical gene expression defect was observed in embryos homozygous for Df(2R)vg33 (not shown), another deletion mutation isolated by Lasko and Pardue that removes polytene region 49F (36). Therefore, loss of the dDP locus is associated with a phenotype that closely resembles the phenotype associated with the loss of dE2F function.
Embryos homozygous for the Df(2R)vg56 and Df(2R)vg33 deletions deviate dramatically from wild-type morphology during and after germ band retraction, presumably because of the loss of multiple gene functions. This precluded an accurate analysis of gene expression patterns in the deficiencies at stages when we usually analyze the G1-S transcription program in wild-type embryos. We therefore examined existing mutations that mapped to 49F for perturbations in embryonic DmRNR2 expression. Mutations causing defective DmRNR2 expression would be candidates for alleles of dDP. This approach had proven successful in identifying dE2F mutations (17). A collection of EMS-induced mutations representing 18 lethal complementation groups that fail to complement Df(2R)vg56, Df(2R)vg33, and other deletions in the 49F region were generated by Lasko and Pardue (36). Embryos were collected from extant stocks carrying representative alleles from most of these complementation groups (see Materials and Methods) and probed for DmRNR2 expression by in situ hybridization. In one lethal complementation group, designated dDPvr10, DmRNR2 expression was greatly reduced compared to that of wild-type embryos (for germ band-retracted stages, cf. Fig. 2A and B). No obvious change in the expression of DmRNR2 was observed in any of the other mutants tested. At germ band-extended stages, the dDPvr10 DmRNR2 expression phenotype was similar to that caused by Df(2R)vg56 and Df(2R)vg33 (not shown), suggesting that the dDPvr10 chromosome contained a mutation in the relevant gene deleted in these deficiencies.
FIG. 2.

dDP is required for expression of DmRNR2. Each panel shows a stage 13 embryo (anterior at the left and dorsal at the top) subjected to in situ hybridization with a digoxigenin-labeled probe derived from the DmRNR2 gene. (A) Wild type (WT). The pattern of DmRNR2 expression is coincident with the pattern of DNA replication (Fig. 5A). This includes internal endoreduplicating cells (e.g., midgut cells) and proliferating cells in the CNS. (B) dDPvr10/dDPvr10 (dDP−). DmRNR2 expression is not activated in the dDP mutant. (C) A dDPvr10/dDPvr10; hsp70-dDP/hsp70-dDP (dDP− hs-dDP) embryo subjected to a 30-min 37°C heat shock followed by a 70-min recovery at room temperature. Expression of dDP cDNA in the mutant restores the normal pattern of DmRNR2 expression. Each of the images has an internal focal plane in order to observe the anterior midgut (arrows).
The original complementation mapping by Lasko and Pardue (36) placed the dDPvr10 lethal mutation outside of the Df(2R)vg56 deletion; i.e., dDPvr10 and Df(2R)vg56 were scored as complementing mutations in their test crosses. Since this was inconsistent with our interpretation of the DmRNR2 expression phenotype, we repeated the complementation tests with our dDPvr10 stock (Table 1). When crosses with Df(2R)vg56 flies were reared at 25°C, less than 2% of the adult progeny (n = 683) were of the dDPvr10/Df(2R)vg56 class (complementing mutations should comprise 33% of adult progeny). Similarly, in crosses with Df(2R)vg33 flies less than 1% of the adult progeny (n = 531) were of the dDPvr10/Df(2R)vg33 class. These rare “escapers” were not phenotypically normal: many flies had notched wings, some had rough eyes, and both males and females were sterile (the females failed to lay eggs). Thus, the dDPvr10 chromosome is semilethal at 25°C over both deficiencies. Because this semilethality gives occasional survivors, it could be scored as complementation when smaller numbers of progeny are scored: presumably this explains the mapping results of Lasko and Pardue. When we reared our crosses at 29°C, no Df(2R)/dDPvr10 flies eclosed (Table 1). A single copy of a hsp70-dDP construct contained in a P-element vector was sufficient to induce dDPvr10/Df(2R)vg56 or dDPvr10/Df(2R)vg33 flies to eclose at 29°C in the absence of a 37°C heat shock treatment (Table 1). We conclude that the dDPvr10 mutation fails to complement deficiencies that delete the dDP locus and that low-level expression of a dDP cDNA can rescue dDPvr10/Df(2R) lethality. Additionally, the incomplete lethality of the dDPvr10/Df(2R) heteroallelic combination at lower temperatures indicates either that dDP is not absolutely essential or that dDPvr10 is not null for the locus.
TABLE 1.
Results of Df(2R)/dDPvr10 complementation tests
| Cross | No. of adult progeny when reared ata:
|
|
|---|---|---|
| 25°C | 29°C | |
| Df(2R)vg56/CyO wg-lacZ × dDPvr10/CyO wg-lacZ | 12/683 (2%) | 0/487 (0%) |
| Df(2R)vg56/CyO wg-lacZ × dDPvr10/CyO wg-lacZ; hsp70-dDP/hsp70-dDP | 20/265 (8%) | 46/475 (10%) |
| Df(2R)vg33/CyO wg-lacZ × dDPvr10/CyO wg-lacZ | 5/531 (1%) | 0/467 (0%) |
| Df(2R)vg33/CyO wg-lacZ × dDPvr10/CyO wg-lacZ; hsp70-dDP/hsp70-dDP | 21/388 (5%) | 43/468 (9%) |
Data are expressed as the number of Df(2R)/dDPvr10 adult progeny/total number of adult progeny. Full complementation is 33% (CyO wg-lacZ/CyO wg-lacZ progeny do not survive until adulthood).
These data suggested that the dDPvr10 chromosome contained a mutation in the dDP gene. To confirm this, we molecularly characterized the dDP locus of wild-type and dDPvr10 chromosomes. The full-length dDP transcription unit spans approximately 4 kb of genomic DNA and contains eight introns (Fig. 1D). Our analysis predicts that the wild-type dDP translation product consists of 446 residues with a predicted molecular mass of 49,718 Da. This is in contrast to previously published reports (see Materials and Methods). Southern blotting indicated that there was no gross rearrangement of the dDP locus in the dDPvr10 chromosome (data not shown). PCR amplification and sequencing of the dDP locus from individual dDPvr10/Df(2R)vg56 or dDPvr10/dDPvr10 adult flies revealed a single nucleotide change encoding an Arg-to-His substitution at residue 217 (Fig. 1E). This nucleotide change destroys a unique AatII restriction site present in our dDP cDNA. This consequently provided a means of readily assessing whether the change is simply a wild-type polymorphism. PCR amplification products of dDP genomic DNA isolated from dDPvr10/CyO flies could be partially digested by AatII, indicating that the CyO balancer chromosome contains the AatII site (not shown). In contrast, similar dDP genomic PCR products derived from a stock (vr18/CyO) containing a mutant chromosome (vr18) independently isolated in the original Lasko and Pardue (36) mutagenesis screen could be completely digested by AatII, strongly suggesting that the vr18 chromosome contains the AatII site (not shown). Furthermore, vr18 complements dDPvr10 and does not cause defects in DmRNR2 expression (not shown). We conclude from this data that the Arg-to-His amino acid change present in dDPvr10 is not a polymorphism that was present in the flies originally mutagenized by Lasko and Pardue (36).
Arg 217 is in a region of the dDP protein that is highly conserved among Xenopus, mouse, human, and Drosophila DPs (Fig. 1E). This region of mammalian DP-1 has been shown to be required for dimerization with E2F-1 (3, 30, 34, 69). Thus, the Arg-to-His mutation may reduce the ability of the dDPvr10 polypeptide to bind to dE2F. Dimerization of dDP with dE2F is required for efficient DNA binding activity in vitro (18). Moreover, coexpression of dDP and dE2F is needed for effective transactivation of promoters containing dE2F-dDP binding sites in cultured cells (13, 18) and for stimulating transcription of endogenous target genes in embryos (14). We conclude that the dDPvr10 chromosome contains an alteration of the dDP gene and suggest that this change impairs the ability of dDP protein to act as a transcription factor, perhaps due to a reduced capacity to dimerize with dE2F. Royzman et al. (52) have also recently reported the characterization of a dDP mutant allele (a1) encoding an Arg-to-Cys change at residue 217.
Since dDPvr10 encodes a point mutation of dDP, we cannot be sure that the phenotypes we describe below represent the null situation. Moreover, we could still detect a small amount of maternal dDP protein in germ band-retracted Df(2R)vg56/Df(2R)vg56 embryos (Fig. 1B). Nevertheless, we did not observe obvious differences in the transcription and DNA replication phenotypes in dDPvr10/dDPvr10 and dDPvr10/Df(2R)vg56 embryos. This indicates that a twofold reduction in the zygotic dose of dDPvr10 does not detectably enhance the phenotypes we are scoring and suggests that dDPvr10 retains very little function.
dDP is required for the expression of replication genes at the G1-S transition.
The first 16 cell cycles of Drosophila embryogenesis lack a G1 phase. Following the introduction of a G1 phase in cell cycle 17, subsequent entry into S phase is accompanied by coordinate dE2F-dependent transcriptional activation of replication factor genes (15). We used probes derived from DmRNR2 or cyclin E as representatives of this group of genes (cf. Fig. 2A and 4A). DmRNR2 transcripts are present continuously and nearly uniformly prior to cycle 17 in dDPvr10 mutants, as they are in the wild type (not shown). By cycle 17, DmRNR2 is not expressed in its usual stereotypic pattern in endoreduplicating tissues in the mutants (cf. Fig. 2A and B). The dDPvr10 mutation also dramatically reduces DmRNR2 expression in proliferating cells of the developing CNS (brain lobes and ventral nerve cord). Heat shock expression of a dDP cDNA contained in a P-element transgene restored the wild-type pattern of DmRNR2 expression (Fig. 2C). We conclude that dDP function is required for the activation of DmRNR2 transcription in both endoreduplicating and proliferating cells.
FIG. 4.

Mutation of dDP and dE2F causes distinct phenotypes. Each panel shows a stage 14 embryo (anterior at the left) subjected to in situ hybridization with a digoxigenin-labeled probe derived from dMCM3 cDNA. The perspective is ventral to view the nerve cord (VNC) of the CNS (arrowheads). (A) Wild type (WT). Expression occurs coincident with replication, generating the observed pattern. (B) dDPvr10/dDPvr10 mutant. dMCM3 expression continues in the VNC, but occurs in an expanded domain of cells. This suggests that dDP functions to prevent dMCM3 from becoming derepressed in this tissue. (C) dE2F91/dE2F91 null mutant. Very little if any dMCM3 expression can be detected in the VNC, indicating that dE2F is required for normal dMCM3 expression. No ectopic dMCM3 expression is observed, perhaps because dE2F does not perform an essential repression function. Residual dMCM3 expression in the brain accounts for the out-of-focus staining at the left.
The pattern of cyclin E expression resembles that of genes encoding other replication factors during cycle 17 (16, 33). However, while genes encoding other factors (e.g., DmRNR2) require dE2F function in all domains of their expression (16), cyclin E expression relies on dE2F in all domains except the CNS (16). Similarly, expression of cyclin E relies on dDP in all domains but the CNS (cf. Fig. 3A and B). The factors that control cyclin E expression in the CNS have not yet been identified.
FIG. 3.

dDP is required for cyclin E expression at the G1-S transition in endoreduplicating cells. Each panel shows a stage 13 embryo (anterior at the left and dorsal at the top) subjected to in situ hybridization with a digoxigenin-labeled probe derived from cyclin E cDNA. (A) Wild type (WT). The arrows indicate the midgut. The arrowhead indicates the ventral nerve cord. (B) dDPvr10/Df(2R)vg56 (dDP−). The cells of the midgut normally express cyclin E at this stage but fail to do so in the dDP mutant. CNS expression of cyclin E is normal in the dDP mutant. (C) cyclin E5206/cyclin E5206 mutant embryo. This allele contains a P-element insertion into the upstream regulatory region of the cyclin E gene (33). cyclin E is not expressed in the midgut or other cells that normally endoreduplicate but is expressed in the CNS. (D) cyclin E5206/cyclin EP28 transheterozygote. Expression of cyclin E, presumably from the cyclin EP28 homolog, initiates at the correct developmental time in the midgut. However, transcription is not down regulated as usual, leading to persistently high levels of cyclin E mRNA.
Our results indicate that the dDP mutant phenotype is very similar to that of dE2F mutants with respect to activation of transcription of DmRNR2 and cyclin E. This is consistent with a requirement for dE2F-dDP heterodimers in the transcription of replication genes in vivo. However, if dE2F and dDP perform any functions independent of each other, then we would also expect to observe differences in their respective phenotypes. This possibility is further suggested by the presence of at least one other E2F-like gene, and therefore at least one other potential dDP binding partner, in Drosophila (28). We detected a phenotypic difference in the embryonic CNS with a probe derived from the Drosophila dMCM3 gene (22a, 64). dMCM3 is a member of the MCM family of proteins, which act at origins of replication and are required for S phase (8, 61, 67). dMCM genes are expressed in replicating cells in a pattern similar to that of genes like DmRNR2 (21, 66). In dE2F mutant embryos, there is substantially less dMCM3 RNA in the CNS than in the wild-type CNS, indicating that dE2F is required for dMCM3 expression in this tissue (cf. Fig. 4A and C). The dDPvr10 mutation also alters dMCM3 expression, but in very different way. The intense staining of particular cells seen in wild-type embryos is absent in the mutant, suggesting that this high-level expression requires dDP (Fig. 4B). However, other cells of the CNS that normally have extremely low levels of dMCM3 RNA exhibit increased staining (Fig. 4B). This derepression of dMCM3 as a result of mutation of dDP suggests that dDP normally plays a role in the repression of this gene, at least in some cells of the CNS.
Mutation of dDP attenuates DNA replication and lengthens S phase.
Replicating nuclei can be readily observed in situ by pulse labeling live embryos with BrdU and subsequently detecting BrdU incorporation immunologically after fixation. The wild-type profile of endocycle S phases during embryonic stages 12 to 15 in the midgut has been well described (15, 60). The first of these S phases begins from G1 of cycle 17 (Fig. 5A). dDPvr10/dDPvr10 and dDPvr10/Df(2R)vg56 mutant embryos incorporated much less BrdU in the midgut during a 15-min pulse labeling than did wild-type embryos (for dDPvr10/dDPvr10, cf. Fig. 5A and B). Reduced BrdU incorporation is also seen in other endocycling tissues, such as the hindgut. This decrease in BrdU incorporation was not restricted to endocycles, as mitotic cells in the CNS also labeled poorly relative to the wild type (cf. Fig. 5A and B). We conclude that dDP function is required for wild-type levels of DNA synthesis in endocycling and dividing cells.
FIG. 5.
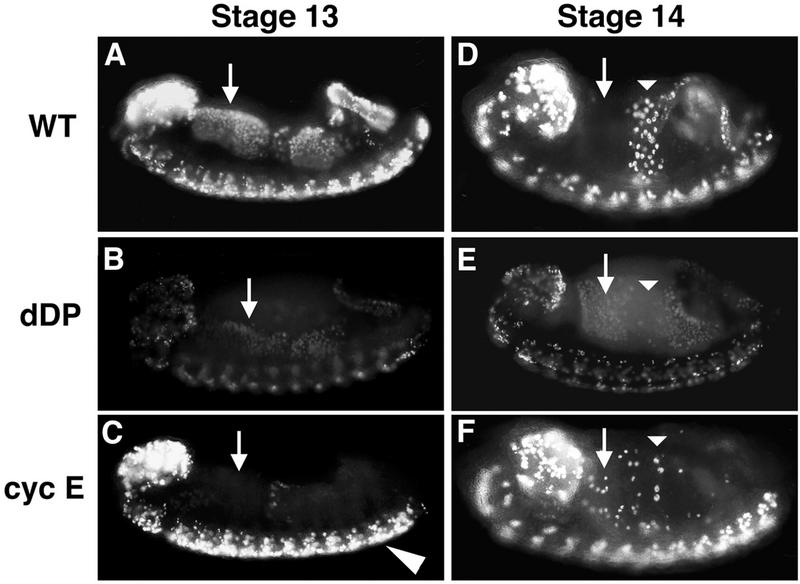
Different cell cycle roles for cyclin E and dDP. Embryos were collected and aged until stage 13 (A to C) or stage 14 (D to F), pulse labeled for 15 min with BrdU, and then immediately fixed. BrdU that incorporated into replicating nuclei during the pulse was subsequently detected by indirect immunofluorescence. The orientation of each embryo is the same as in Fig. 2. (A and D) Wild type (WT). (B and E) dDPvr10/dDPvr10 (dDP). (C and F) cyclin EP28/cyclin E05206 (cyc E). The arrows indicate the cells of the anterior midgut (AMG). These cells begin S phase 17 during stage 12, continue replication into stage 13 (A), and exit S phase by stage 14 (D). In the dDP mutant, all or most of the AMG cells enter S phase at the correct time but incorporate much less BrdU (B). By stage 14 these cells are still replicating (E), indicating that S phase is longer than it is in the wild type. In the hypomorphic cyclin E mutant, the AMG cells do not begin S phase 17 on schedule (C) but eventually enter S phase in a random fashion beginning in stage 14 (F). Thus, G1 of cycle 17 is longer than usual in these cells. The actual length of G1 depends upon when the cells reach the critical level of cyclin E activity that triggers S phase. Also note that replication in the CNS appears normal, consistent with wild-type cyclin E expression in the CNS of cyclin E05206 mutants.
Analysis of BrdU incorporation in an embryo allows one to score the timing of entry into S phase and the duration of S phase in addition to scoring the level of labeled nucleotide incorporated during the pulse. As described above, the dDPvr10 mutant embryos exhibit an obvious reduction in BrdU incorporation, but the pattern of BrdU incorporation reveals that the programming of the G1-to-S transition still operates. The tissues that enter S phase 17 relatively early in the wild type, such as the anterior midgut, posterior midgut, and hindgut, appear to begin their compromised S phase at a similar embryonic stage in the dDP mutant (Fig. 5B). This demonstrated that reduction of dDP function first affects the level of incorporation rather than the timing of the transition from G1 to S phase. Analysis of the incorporation pattern in successively older embryos, whose age is determined both by time from egg deposition and by staging according to morphological criteria, defines the duration of S phase. Whereas the tissues that begin S phase 17 early in wild-type embryos discontinue incorporation by stage 14, in the mutant incorporation continues at this stage. This changes the pattern of BrdU incorporation such that cells which no longer label in the wild type are still labeled in the mutant (e.g., anterior midgut cells [Fig. 5D and E]). The prolongation of S phase combined with decreased BrdU incorporation suggests that the replication rate is reduced. Since the dDPvr10 mutant compromises the expression of genes such as DmRNR2, we suggest that the mutant exhibits a reduction in DNA synthesis as a result of decreases in the levels of replication functions.
Our original analysis of the replication phenotype in dE2F mutant embryos led us to the conclusion that the mutation blocked S phase (17), whereas the analysis of the dDP mutation suggests a more quantitative defect. We thus conducted more detailed analyses of dE2F mutants. The results suggest that reductions in the function of dE2F also have a quantitative effect on replication. dE2F164 is a homozygous, viable, hypomorphic P-element-induced allele of dE2F that expresses nearly undetectable levels of dE2F mRNA (56). Nonetheless, dE2F164 fully complements a deletion [Df(3R)eD7] of the dE2F gene: adult flies are viable and fertile. Despite the absence of an effect on viability, we detected a change in the pattern of BrdU incorporation in Df(3R)eD7/dE2F164 mutant embryos. As with the dDPvr10 mutant, this pattern is consistent with a prolongation of S phase 17 in the midgut (cf. Fig. 5E and 6A). This suggests that the requirement for dE2F can be satisfied by relatively low levels of activity and that a reduction in the efficiency of S phase is not lethal. We therefore reexamined embryos homozygous for lethal null alleles of dE2F. We could detect a low level of BrdU incorporation in the midgut, as well as in the CNS (Fig. 6B). This incorporation can be more readily detected when longer BrdU-labeling periods are used (e.g., 40 vs. 15 min). Again, the pattern of BrdU incorporation was altered in a way that suggests the prolongation of S phase 17 in the midgut (cf. Fig. 5E and 6B). We conclude that reductions in function of dE2F/dDP activity over a large range result in a graded reduction in S phase.
FIG. 6.
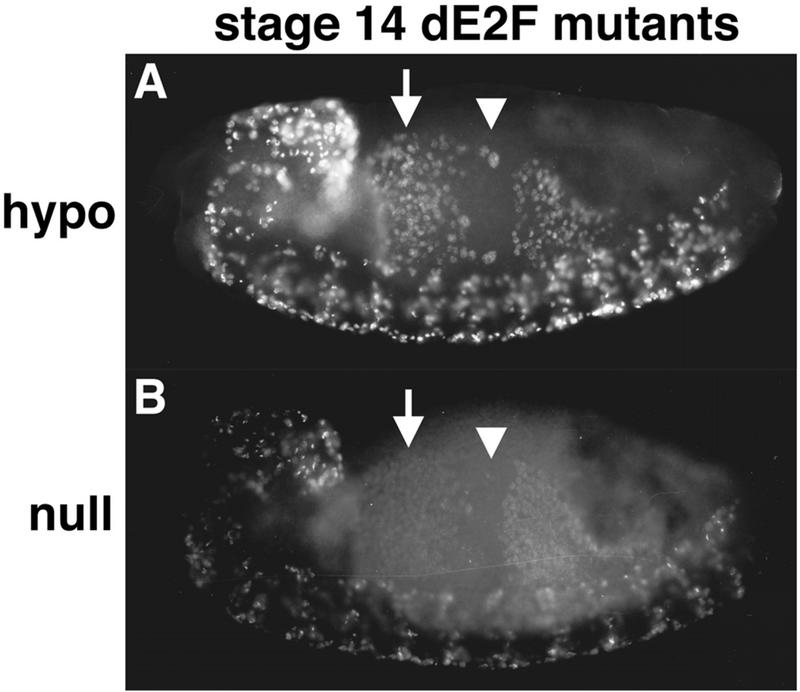
Prolonged S phase in dE2F mutants. Stage 14 dE2F164/Def(3R)eD7 (hypo) (A) and dE2F7172/dE2F7172 (null) (B) embryos pulse labeled for 15 min with BrdU. S phase 17 continues in the anterior midgut (arrows) when in the wild type it has already finished (Fig. 5D). This is an indication that S phase is prolonged in the mutants. In the central portion of the midgut (arrowheads) S phase 18 has not started on schedule for most of the cells (Fig. 5D). This indicates that dE2F also contributes to correct timing of the G1-S transition.
Cyclin E triggers S phase in an all-or-nothing manner.
During G1 phase, eukaryotic cells are thought to respond to growth-promoting signals by making a commitment to enter the cell cycle and begin S phase. A typical feature of the process of commitment is that it is an all-or-nothing step. Reductions in function of a component contributing to such an all-or-nothing step ought to exhibit a sharp threshold, with reductions below a critical level blocking the ability to progress beyond the decisive step. As described above, mutations that compromise the activity of dE2F-dDP suggest that this transcription factor makes a graded contribution to the efficiency of S phase, the antithesis of the expectation for a regulator of a decisive all-or-nothing transition.
Cyclin E mutations on the other hand show the behavior expected for a contribution to an all-or-nothing transition. Cyclin E null mutations complete embryonic cell cycle 16 and cause an arrest in G1 of cell cycle 17 (33). Our immunofluorescent methods cannot detect any BrdU incorporation in these mutants. Thus, in this instance the cyclin E threshold is never reached. To test the consequence to cell cycle progression of reduced but significant levels of cyclin E function, we used two hypomorphic mutations of cyclin E: cyclin E05206, a P-element insertion mutation that eliminates cyclin E expression in endocycling cells but not in the CNS (Fig. 3C), and cyclin EP28, an EMS-induced mutation. We examined BrdU incorporation in the anterior midgut cells of cyclin E05206/cyclin EP28 transheterozygous mutant embryos. At stage 13, no BrdU incorporation was detected (Fig. 5C). These cells are therefore still in G1 of cycle 17 at a time when wild-type cells have already begun S phase 17 (Fig. 5A). However, at stage 14 we could detect BrdU incorporation in some of the anterior midgut cells (Fig. 5F). The number of BrdU-positive cells varied from embryo to embryo, and the distribution of cells within the tissue was random. In sharp contrast to the results with the dDP and dE2F mutations, the level of BrdU staining in those nuclei that had made the transition to S phase was just as intense as in the wild type. We interpret this phenotype as a delay of the G1-S transition, or a reduction of the probability of undergoing this transition, and suggest that it reflects a partial defect in the execution of an all-or-nothing event leading to S phase (Fig. 7).
FIG. 7.

Model of dE2F-dDP and cyclin E function at the G1-S transition and schematic representation of the G1-S transition and BrdU incorporation in the embryonic midgut. (A) Wild type. Components of the prereplicative complex (open symbols) assemble on chromosomes (parallel lines) at origins of DNA replication (solid rectangles). Since E2F-DP is required for the transcription of genes encoding known components of prereplicative complexes like dMCM3 in Drosophila and HsOrc1 in humans (46), we propose that dE2F-dDP contributes to prereplicative complex assembly. Cells remain in G1 until they are induced to progress through the cell cycle by a developmental signal. DNA synthesis is then triggered at the beginning of S phase via cyclin E action, and bidirectional replication forks appear in the chromosome (ovals). After BrdU pulse labeling and in situ immunofluorescent detection of incorporated BrdU, the nuclei in a field of cells that synchronously entered S phase stain brightly and uniformly (nine open circles on the black background). (B) Reduced cyclin E function. Prereplicative complexes form normally at a frequency identical to that of the wild type, because dE2F-dDP is activated independently of cyclin E (16). If cyclin E function is eliminated, replication is not triggered and cells remain in G1. If the cyclin E function is reduced but not eliminated, S phase will still be triggered provided cyclin E function can eventually achieve the critical threshold. In the latter situation, G1 is prolonged and DNA replication is normal once S phase begins. In a field of cells of this type, the length of G1 might vary stochastically as cells approach the S phase threshold. The pattern of BrdU-labeled nuclei would appear random, and each nucleus would have the wild-type intensity of staining (three open circles instead of nine on the black background). (C) Reduced dE2F-dDP function. Decreased provision of components causes limited assembly of prereplicative complexes. If those that assemble are triggered at the usual time in development, the length of G1 does not change. However, fewer prereplicative complexes initiate fewer bidirectional replication forks. Consequently, S phase is prolonged and BrdU incorporation during pulse labeling is significantly reduced. A field of cells of this type appears uniformly and weakly labeled (nine shaded circles on the black background). (Note that the symbols designating components of the prereplicative complex are not shown on the DNA after bidirectional DNA synthesis has begun for the purpose of clarity. Some components of this complex [e.g., ORC] are thought to remain bound to the DNA throughout the entire cell cycle [11].)
Strikingly, a small number of cyclin E05206/cyclin EP28 progeny eclosed as adult flies (12 out of 317 total progeny [3.7%]). These flies were sterile, had rough eyes, and had incomplete formation of the fifth wing vein. Therefore, this combination of cyclin E alleles is semilethal. This result indicates that dramatic alterations to the timing of the G1-S transition at embryonic stages is still compatible with a substantial amount of development.
Drosophila cyclin E inhibits its own expression in endocycles by one or more negative feedback loops (16, 41, 53), and mammalian cyclin E has been shown to promote its own destruction (9, 68). As a result, cyclin E is ordinarily expressed only transiently at the G1-S transition. We examined the behavior of cyclin E expression in cyclin E05206/cyclin EP28 embryos and found that these embryos activate cyclin E transcription (presumably from the mutant cyclin EP28 chromosome) at the correct time in cycle 17 in the midgut but fail to terminate transcription at the normal time (Fig. 3D). This, and similar results with cyclin E null mutants (16, 53), suggests that the normal down regulation of cyclin E synthesis involves negative feedback. There appear to be multiple components to this negative feedback. Cyclin E acts particularly effectively as a repressor of its own expression (53), and in a separate step it plays a role in the down regulation of dE2F-dDP-dependent transcription in general (16). This mode of regulation buffers the effect of mutation on cyclin E function in that mutant alleles of cyclin E are expressed for a longer time and mRNA (and presumably the mutant protein it encodes) can accumulate to higher levels (Fig. 3D). Therefore, we propose that part of the delay in progress to S phase in the cyclin E05206/cyclin EP28 mutant is due to an extension in the time required for mutant cyclin E protein to achieve a critical threshold of cyclin E function (e.g., activating cdk2).
dE2F/dDP contributes to the all-or-nothing transition into S phase.
Based on the characteristic defects seen in hypomorphic conditions for cyclin E and dE2F, and in the dDPvr10 mutant, it appears that cyclin E has important inputs into an all-or-nothing event required for transition to S phase while dE2F-dDP makes a quantitative contribution to functions required for S phase. However, our work has demonstrated that cyclin E expression requires dE2F and dDP at the G1-S transition of cycle 17. We consequently expected that dE2F-dDP should also make a contribution to the G1-S phase trigger. Such a contribution might be masked if a relatively high level of dE2F-dDP activity were required to fully satisfy the quantitative contribution to S phase while a very low level of activity of dE2F-dDP provided sufficient cyclin E to satisfy the threshold of the all-or-nothing event. Indeed, further characterization of the dDP and dE2F mutant phenotypes suggests that this is the case.
As we described above, cells that enter S phase 17 relatively early (e.g., anterior midgut cells) in the wild type do so at approximately the normal time in the dDPvr10 mutant. However, the ensuing S phase is prolonged in the mutant and displays a reduced rate of BrdU incorporation. If a low level of maternal dDP persists until the time that tissues begin to enter S phase 17, one might expect that, as a result of continued decay, cells which enter S phase early would benefit from a higher level of this maternal dDP than cells which enter S phase at a later stage. Consistent with this, S phase 18 in the central midgut (Fig. 5D), which begins about 2 h after the beginning of S phase 17 in the anterior midgut, seems to be more dramatically affected in the dDPvr10 mutant embryos (Fig. 5E). Importantly, the timing of entry into S phase 18 is affected as well as the efficiency of replication. In stage 14, only a few cells in the mutant’s central midgut have begun to incorporate BrdU (Fig. 5E) compared to the number in wild-type embryos (Fig. 5D). These cells have either been arrested or they initiate S phase at a much later time than the wild type (i.e., beyond the stages we have examined). Moreover, the few cells that do incorporate BrdU are stained weakly, again consistent with a reduced rate of DNA replication once S phase is initiated. A very similar phenotype is seen in the same cells of dE2F mutant embryos (Fig. 6). These data suggest that dE2F-dDP contributes both to the correct timing of the G1-S transition and to the efficiency of S phase.
DISCUSSION
In this report we describe the identification and characterization of mutations of the Drosophila dDP gene, which encodes a component of the E2F-DP transcription factor. dDP is required for activation of expression of replication genes at the G1-S transition in embryonic cells, and mutants have an attenuated and sometimes delayed S phase. Characterization of BrdU incorporation in different mutant backgrounds and at different embryonic stages suggests that reductions in dE2F-dDP over a large range results in graded attenuation of DNA replication and that only severe reductions in dE2F-dDP function delay or block entry into S phase. In contrast, partial loss of function of cyclin E results in a delay of S-phase entry without detectable diminution of BrdU incorporation. We suggest that dE2F-dDP makes two different kinds of contributions to DNA replication. It drives the expression of numerous replication functions, some of which are limiting for the rate of DNA replication. Additionally, it drives the expression of at least one product, cyclin E, that is required to trigger an all-or-nothing transition from G1 into S phase.
dE2F and dDP collaborate to activate G1-S transcription.
A substantial amount of biochemical evidence indicates that E2F-DP heterodimers function as a DNA binding transcription factor that contributes to the activation of transcription at the G1-S transition (reviewed in references 37 and 59). Our previous genetic analysis had demonstrated that the Drosophila dE2F gene is essential for transcriptional activation of several replication genes at the G1-S transition in embryos. The present work demonstrates that mutation of the Drosophila dDP gene also eliminates this activation of expression for the DmRNR2 and cyclin E genes. While this article was undergoing review, Royzman et al. reported similar transcriptional phenotypes due to mutation of dDP (52). The parallel defects in the transcription of several genes are consistent with the joint action of dE2F and dDP in this aspect of their function. Several experiments imply that transcriptional activation is likely a result of a direct interaction between dE2F-dDP and cis-acting elements in the promoters of replication genes. Expression of the dE2F cDNA stimulates the DNA polymerase α promoter in cultured Drosophila cells (48). Furthermore, dE2F-dDP binding sites found upstream of the PCNA transcriptional start site are required for reporter gene expression in cell culture and β-galactosidase activity in embryo extracts prepared from flies carrying PCNA-promoter-lacZ fusion transgenes (71).
Is normal regulation of S phase essential?
The vr10 allele of dDP produced some adult flies when it was heterozygous to a deficiency for the locus. While it is possible that this result is in part due to residual function of this mutant allele, it is clear that this same allele reduces G1-S transcription of various replication genes to levels below detection and substantially compromises replication in the embryonic S phases that we have examined. Similarly, a dE2F hypomorphic allele that alters embryonic S phases is viable, and a combination of cyclin E alleles that greatly altered the embryonic S-phase program (Fig. 5F) nonetheless gave viable flies. Based on these observations, we suggest that most aspects of patterning, growth, and differentiation are regulated in ways that are not easily disturbed by alterations in the G1-S program. Note that adult dDPvr10 mutant flies were rare, defective, and infertile. Hence the activity of the gene and presumably the control of S phase is clearly of some relevance to development, but it is nonetheless surprising that an intact fly can be produced when defects in S phase can be scored in midembryogenesis.
Is dE2F-dDP activity essential to S phase?
We previously reported that the compromised embryonic S phases in a dE2F mutant indicated that dE2F function was required for the G1-S transition (17). However, here we have shown that the available dE2F and dDP mutations have their primary effect on the efficiency of S phase. If dE2F-dDP is an integral part of the normal cascade of signaling that triggers S phase, we might expect that the mutations would result in an absolute block to S phase rather than a reduction in its efficiency. However, we find that many cells enter a compromised S phase 17 according to a normal schedule. Nonetheless, we cannot conclude that dE2F-dDP function is dispensable for the G1-S transition, because it is possible that the threshold of dE2F-dDP activity required to drive the transition is lower than the levels of dE2F-dDP required for maximal efficiency of DNA replication.
There are three possible sources of dE2F-dDP activity in the mutant embryos. First, it appears that a small amount of maternally expressed protein persists to late embryonic stages (Fig. 1B). Second, the mutations might not remove all activity of the gene. Third, there may be homologs that provide a comparable activity. Indeed, sequences of random cDNAs (ESTs) by the Berkeley Drosophila Genome Project uncovered the existence of a second dE2F homolog (28). This and perhaps other undetected homologs of dE2F and/or dDP might contribute to dE2F-dDP activity in the mutant backgrounds. Hence, while our experiments allow us to say that mutations that reduce dE2F-dDP activity affect S phase, it is more difficult to address whether DNA replication seen in these mutant backgrounds depends on a low level of residual dE2F-dDP activity from one or more of these sources.
One important observation leads us to suggest that dE2F-dDP contributes to the transition from G1 to S as well as to the efficiency of DNA replication. Analysis of a later S phase (S phase 18 in the central midgut), when maternal functions have further decayed, shows that entry into S phase is delayed and variable. Additionally, an involvement of dE2F-dDP in the G1-S transition is also suggested by the involvement of the dE2F-dDP-stimulated gene cyclin E in triggering the G1-S transition (see below) and by the extraordinary correlation between the schedule of activation of dE2F-dDP and the developmental program of S phases. We consequently favor the interpretation that dE2F-dDP drives expression of cyclin E at the G1-S transition and that cyclin E is essential for this transition.
Our favored interpretation might seem strained because it requires that undetectable levels of cyclin E RNA trigger S phase. However, the genetics indicates that zygotic expression of cyclin E is absolutely required for S phase 17 (33). Hence, regardless of our failure to detect cyclin E RNA in the dE2F and dDP mutant embryos, sufficient cyclin E protein must accumulate to allow S phase. How can undetectable levels of cyclin E mRNA produce sufficient cyclin E protein to trigger S phase? We suggest that particular aspects of cyclin E regulation might allow gradual accumulation of cyclin E protein at very low levels of mRNA. Cyclin E inhibits its own dE2F-dDP-mediated transcription in embryos (16, 53). Moreover, mammalian cyclin E protein appears to drive its own destruction through ubiquitin-mediated proteolysis (9, 68). Hence, once expression begins cyclin E will accumulate until it reaches levels that activate these negative feedback loops. As the level of cyclin E RNA is reduced, the rate of accumulation of cyclin E protein will decline, but it will accumulate over a longer period and ultimately reach its threshold levels. Accordingly, we suggest that severe reductions in dE2F-dDP activity would be required before one could detect a substantial defect in the G1-S transition. In addition, it is also possible that in the complete absence of dE2F-dDP, low basal expression of cyclin E might allow accumulation of cyclin E protein over a very long time. In this case, entry into S phase would not be timed properly, but could still occur. Such dE2F-dDP-independent expression of cyclin E might sustain the growth of dE2F and dDP mutant larvae, as reported by Royzman et al. (52).
The dE2F-dDP contribution to the efficiency of DNA replication.
In S. cerevisiae, limitation of components that bind replication origins (e.g., ORC and MCM proteins) causes a reduced rate of replication that is associated with reduced origin utilization (reviewed in reference 11). In Drosophila, reduction in the function of dMCM2 results in a prolongation of S phase consistent with such a reduction in origin firing (66). We suggest an analogous interpretation of the phenotype caused by reduction of dE2F-dDP function (Fig. 7). In both the dDP and dE2F mutant embryos the first defect in DNA replication that appears is a reduction in the intensity of BrdU incorporation. An increase in the length of S phase 17 observed in the midgut argues that this decreased incorporation is associated with a reduced rate of DNA replication. Although we have not identified a specific factor whose expression is rate limiting for these embryonic S phases, we have demonstrated that dE2F and dDP are required for the expression of several genes encoding replication factors. We propose that one or more replication factors become limiting when dE2F-dDP function is compromised and that the consequence is a reduction in the number of forks that successfully initiate replication.
Cyclin E provides an S-phase threshold.
Evidence from many different systems indicates that cyclin E-cdk2 activity is required for cell cycle progression (reviewed in reference 29). In what way is cyclin E-cdk2 activity required for S phase? Here we have shown that Drosophila cyclin E provides a threshold for entry into S phase. By reducing but not eliminating cyclin E function by a hypomorphic combination of alleles, we showed that endoreduplicating embryonic midgut cells fail to execute a developmentally controlled G1-S transition on schedule in cell cycle 17. But rather than remaining in G1 arrest indefinitely, these cells subsequently enter S phase. In this delayed S phase, BrdU incorporation appears normal, probably because dE2F-dDP is independently activated in these cells and the replication machinery other than cyclin E is not limiting (17). The midgut cells do not all enter S phase at the same time, however, but rather at random times. We interpret this stochastic entry into S phase in cycle 17 as evidence for the participation of cyclin E in a molecular threshold. That is, when cyclin E, and by extension cyclin E-cdk2 activity, reaches a critical level S phase is triggered in an all-or-nothing manner. This will only be true if cyclin E function can continue to accumulate during the extended G1 period. Consistent with this, the mutant cyclin E gene is activated at the normal developmental time and continues to be expressed during the extended G1 period due to a failure to activate its own down regulation (see above).
The molecular mechanism behind such an all-or-nothing threshold is not clear. Positive feedback amplification has been a popular proposal for controlling all-or-nothing cell cycle transitions. The biological activities of mammalian and Drosophila cyclin E and E2F-DP can formally be placed into a positive feedback loop: overexpression of cyclin E activates E2F-DP, probably due to cyclin E-cdk2 phosphorylation of pRB, and E2F-DP activates cyclin E transcription. This type of positive feedback loop makes an attractive model for control of the restriction point (10, 23, 57), the time during G1 that mammalian cells commit to S-phase entry (50). Similar types of interactions between the S. cerevisiae Swi4/6 transcription factor and G1 cyclin genes (CLNs) can be demonstrated (43, 45). However, during START, the yeast equivalent of the restriction point, careful genetic experiments have ruled out positive feedback amplification between CLN1 or CLN2 and the Swi4/6 transcription factor (12, 63). Similarly, in Drosophila endocycling cells the genetic evidence indicates that there is no positive feedback amplification between cyclin E and dE2F-dDP at the G1-S transition, although each can induce the activity of the other when overexpressed (14, 16). The lack of positive feedback appears to be the consequence of negative feedback mechanisms involving cyclin E, as described above (16, 41, 53). These overcome any contribution of cyclin E to positive feedback. Thus, because cyclin E does not make a positive contribution to its own expression in our experimental context, cyclin E provides a threshold for entry into S phase by a mechanism that does not require positive feedback between cyclin E and dE2F-dDP.
What then is the function of cyclin E? After formation of prereplicative complexes during G1, and a commitment to progress into S phase, a poorly described mechanism exists that triggers the initiation of DNA replication. In vitro experiments suggest that cdk activity could comprise part of the trigger mechanism. In frog and human extracts, cyclin E-cdk2 and cyclin A-cdk2 are required for DNA synthesis (20, 31, 35, 62). These kinases presumably have substrates that must be phosphorylated in order for replication origins to fire. Moreover, cyclin E has an essential S-phase function that is independent of E2F-DP-mediated transcription (14, 42). Whatever the critical substrates are, our data suggest that cyclin E-cdk2 must be part of a mechanism that in vivo has an all-or-nothing response. Cyclin E could activate an amplifying kinase cascade, trigger changes in protein degradation, or affect DNA unwinding and initiation directly at origins of replication.
ACKNOWLEDGMENTS
We thank Nick Dyson for dDP cDNAs and anti-dDP monoclonal antibodies, Ting Wu for mutant stocks from the 49F region, Carole Seum and Pierre Spierer for the dE2F164 mutant stock, Shelagh Campbell and Tin Tin Su for library cDNA and primers, Peter Follette for dMCM3 cDNA, Terry Orr-Weaver for communicating results prior to publication, and Mark Peifer and Jenny Adam for comments on the manuscript.
This work was initiated while R.J.D. was supported by a fellowship from the Cancer Research Fund of the Damon Runyon-Walter Winchell Foundation (DRG-1161). This work was funded in part by a grant from the National Institutes of Health to P.H.O. (GM37193).
REFERENCES
- 1.Andrews B J, Herskowitz I. Regulation of cell cycle-dependent gene expression in yeast. J Biol Chem. 1990;265:14057–14060. [PubMed] [Google Scholar]
- 2.Ashburner M, Thompson P, Roote J, Lasko P F, Grau Y, El Messal M, Roth S, Simpson P. The genetics of a small autosomal region of Drosophila melanogaster containing the structural gene for alcohol dehydrogenase. VII. Characterization of the region around the snail and cactus loci. Genetics. 1990;126:679–694. doi: 10.1093/genetics/126.3.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bandara L R, Buck V M, Zamanian M, Johnston L H, La T N. Functional synergy between DP-1 and E2F-1 in the cell cycle-regulating transcription factor DRTF1/E2F. EMBO J. 1993;12:4317–4324. doi: 10.1002/j.1460-2075.1993.tb06116.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Botz J, Zerfass-Thome K, Spitkovsky D, Delius H, Vogt B, Eilers M, Hatzigeorgiou A, Jansen-Durr P. Cell cycle regulation of the murine cyclin E gene depends on an E2F binding site in the promoter. Mol Cell Biol. 1996;16:3401–3409. doi: 10.1128/mcb.16.7.3401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Breeden L. Start-specific transcription in yeast. Curr Top Microbiol Immunol. 1996;208:95–127. doi: 10.1007/978-3-642-79910-5_5. [DOI] [PubMed] [Google Scholar]
- 6.Brook A, Xie J E, Du W, Dyson N. Requirements for dE2F function in proliferating cells and in post-mitotic differentiating cells. EMBO J. 1996;15:3676–3683. [PMC free article] [PubMed] [Google Scholar]
- 7.Brown N H, Kafatos F C. Functional cDNA libraries from Drosophila embryos. J Mol Biol. 1988;203:425–437. doi: 10.1016/0022-2836(88)90010-1. [DOI] [PubMed] [Google Scholar]
- 8.Chong J P, Thommes P, Blow J J. The role of MCM/P1 proteins in the licensing of DNA replication. Trends Biochem Sci. 1996;21:102–106. [PubMed] [Google Scholar]
- 9.Clurman B E, Sheaff R J, Thress K, Groudine M, Roberts J M. Turnover of cyclin E by the ubiquitin-proteasome pathway is regulated by cdk2 binding and cyclin phosphorylation. Genes Dev. 1996;10:1979–1990. doi: 10.1101/gad.10.16.1979. [DOI] [PubMed] [Google Scholar]
- 10.DeGregori J, Kowalik T, Nevins J R. Cellular targets for activation by the E2F1 transcription factor include DNA synthesis- and G1/S-regulatory genes. Mol Cell Biol. 1995;15:4215–4224. doi: 10.1128/mcb.15.8.4215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Diffley J F. Once and only once upon a time: specifying and regulating origins of DNA replication in eukaryotic cells. Genes Dev. 1996;10:2819–2830. doi: 10.1101/gad.10.22.2819. [DOI] [PubMed] [Google Scholar]
- 12.Dirick L, Bohm T, Nasmyth K. Roles and regulation of Cln-Cdc28 kinases at the start of the cell cycle of Saccharomyces cerevisiae. EMBO J. 1995;14:4803–4813. doi: 10.1002/j.1460-2075.1995.tb00162.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Du W, Vidal M, Xie J E, Dyson N. RBF, a novel RB-related gene that regulates E2F activity and interacts with cyclin E in Drosophila. Genes Dev. 1996;10:1206–1218. doi: 10.1101/gad.10.10.1206. [DOI] [PubMed] [Google Scholar]
- 14.Duronio R J, Brook A, Dyson N, O’Farrell P H. E2F-induced S phase requires cyclin E. Genes Dev. 1996;10:2505–2513. doi: 10.1101/gad.10.19.2505. [DOI] [PubMed] [Google Scholar]
- 15.Duronio R J, O’Farrell P H. Developmental control of a G1-S transcriptional program in Drosophila. Development. 1994;120:1503–1515. doi: 10.1242/dev.120.6.1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Duronio R J, O’Farrell P H. Developmental control of the G1 to S transition in Drosophila: cyclin E is a limiting downstream target of E2F. Genes Dev. 1995;9:1456–1468. doi: 10.1101/gad.9.12.1456. [DOI] [PubMed] [Google Scholar]
- 17.Duronio R J, O’Farrell P H, Xie J E, Brook A, Dyson N. The transcription factor E2F is required for S phase during Drosophila embryogenesis. Genes Dev. 1995;9:1445–1455. doi: 10.1101/gad.9.12.1445. [DOI] [PubMed] [Google Scholar]
- 18.Dynlacht B D, Brook A, Dembski M, Yenush L, Dyson N. DNA-binding and trans-activation properties of Drosophila E2F and DP proteins. Proc Natl Acad Sci USA. 1994;91:6359–6363. doi: 10.1073/pnas.91.14.6359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Edgar B A, O’Farrell P H. The three postblastoderm cell cycles of Drosophila embryogenesis are regulated in G2 by string. Cell. 1990;62:469–480. doi: 10.1016/0092-8674(90)90012-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fang F, Newport J W. Evidence that the G1-S and G2-M transitions are controlled by different cdc2 proteins in higher eukaryotes. Cell. 1991;66:731–742. doi: 10.1016/0092-8674(91)90117-h. [DOI] [PubMed] [Google Scholar]
- 21.Feger G, Vaessin H, Su T T, Wolff E, Jan L Y, Jan Y N. dpa, a member of the MCM family, is required for mitotic DNA replication but not endoreplication in Drosophila. EMBO J. 1995;14:5387–5398. doi: 10.1002/j.1460-2075.1995.tb00223.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Field S J, Tsai F Y, Kuo F, Zubiaga A M, Kaelin W G, Jr, Livingston D M, Orkin S H, Greenberg M E. E2F-1 functions in mice to promote apoptosis and suppress proliferation. Cell. 1996;85:549–561. doi: 10.1016/s0092-8674(00)81255-6. [DOI] [PubMed] [Google Scholar]
- 22a.Follette, P., and P. H. O’Farrell. Unpublished data.
- 23.Geng Y, Eaton E N, Picon M, Roberts J M, Lundberg A S, Gifford A, Sardet C, Weinberg R A. Regulation of cyclin E transcription by E2Fs and retinoblastoma protein. Oncogene. 1996;12:1173–1180. [PubMed] [Google Scholar]
- 24.Girling R, Bandara L R, Ormondroyd E, Lam E W, Kotecha S, Mohun T, La Thangue N B. Molecular characterization of Xenopus laevis DP proteins. Mol Biol Cell. 1994;5:1081–1092. doi: 10.1091/mbc.5.10.1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gloor G B, Preston C R, Johnson-Schlitz D M, Nassif N A, Phillis R W, Benz W K, Robertson H M, Engels W R. Type I repressors of P element mobility. Genetics. 1993;135:81–95. doi: 10.1093/genetics/135.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gong J, Traganos F, Darzynkiewicz Z. Threshold expression of cyclin E but not D type cyclins characterizes normal and tumour cells entering S phase. Cell Prolif. 1995;28:337–346. doi: 10.1111/j.1365-2184.1995.tb00075.x. [DOI] [PubMed] [Google Scholar]
- 27.Hao X F, Alphey L, Bandara L R, Lam E W, Glover D, La Thangue N B. Functional conservation of the cell cycle-regulating transcription factor DRTF1/E2F and its pathway of control in Drosophila melanogaster. J Cell Sci. 1995;108:2945–2954. doi: 10.1242/jcs.108.9.2945. [DOI] [PubMed] [Google Scholar]
- 28.Harvey D, Hong L, Evans-Holm M, Pendleton J, Su C, Brokstein P, Lewis S, Rubin G M. Clone #LD02934. Berkeley, Calif: Berkeley Drosophila Genome Project; 1997. [Google Scholar]
- 29.Heichman K A, Roberts J M. Rules to replicate by. Cell. 1994;79:557–562. doi: 10.1016/0092-8674(94)90541-x. [DOI] [PubMed] [Google Scholar]
- 30.Helin K, Wu C L, Fattaey A R, Lees J A, Dynlacht B D, Ngwu C, Harlow E. Heterodimerization of the transcription factors E2F-1 and DP-1 leads to cooperative trans-activation. Genes Dev. 1993;7:1850–1861. doi: 10.1101/gad.7.10.1850. [DOI] [PubMed] [Google Scholar]
- 31.Jackson P K, Chevalier S, Philippe M, Kirschner M W. Early events in DNA replication require cyclin E and are blocked by p21CIP1. J Cell Biol. 1995;130:755–769. doi: 10.1083/jcb.130.4.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Karpen G H, Spradling A C. Analysis of subtelomeric heterochromatin in the Drosophila minichromosome DP1187 by single P-element insertional mutagenesis. Genetics. 1992;132:737–753. doi: 10.1093/genetics/132.3.737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Knoblich J A, Sauer K, Jones L, Richardson H, Saint R, Lehner C F. Cyclin E controls S phase progression and its down-regulation during Drosophila embryogenesis is required for the arrest of cell proliferation. Cell. 1994;77:107–120. doi: 10.1016/0092-8674(94)90239-9. [DOI] [PubMed] [Google Scholar]
- 34.Krek W, Livingston D M, Shirodkar S. Binding to DNA and the retinoblastoma gene product promoted by complex formation of different E2F family members. Science. 1993;262:1557–1560. doi: 10.1126/science.8248803. [DOI] [PubMed] [Google Scholar]
- 35.Krude T, Jackman M, Pines J, Laskey R A. Cyclin/Cdk-dependent initiation of DNA replication in a human cell-free system. Cell. 1997;88:109–119. doi: 10.1016/s0092-8674(00)81863-2. [DOI] [PubMed] [Google Scholar]
- 36.Lasko P F, Pardue M L. Studies of the genetic organization of the vestigial microregion of Drosophila melanogaster. Genetics. 1988;120:495–502. doi: 10.1093/genetics/120.2.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.La Thangue N B. DP and E2F proteins: components of a heterodimeric transcription factor implicated in cell cycle control. Curr Opin Cell Biol. 1994;6:443–450. doi: 10.1016/0955-0674(94)90038-8. [DOI] [PubMed] [Google Scholar]
- 38.Lehner C F, O’Farrell P H. The roles of Drosophila cyclins A and B in mitotic control. Cell. 1990;61:535–547. doi: 10.1016/0092-8674(90)90535-m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Leno G H, Laskey R A. The nuclear membrane determines the timing of DNA replication in Xenopus egg extracts. J Cell Biol. 1991;112:557–566. doi: 10.1083/jcb.112.4.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liang C, Weinreich M, Stillman B. ORC and Cdc6p interact and determine the frequency of initiation of DNA replication in the genome. Cell. 1995;81:667–676. doi: 10.1016/0092-8674(95)90528-6. [DOI] [PubMed] [Google Scholar]
- 41.Lilly M A, Spradling A C. The Drosophila endocycle is controlled by Cyclin E and lacks a checkpoint ensuring S-phase completion. Genes Dev. 1996;10:2514–2526. doi: 10.1101/gad.10.19.2514. [DOI] [PubMed] [Google Scholar]
- 42.Lukas J, Herzinger T, Hansen K, Moroni M C, Resnitzky D, Helin K, Reed S I, Bartek J. Cyclin E-induced S phase without activation of the pRb/E2F pathway. Genes Dev. 1997;11:1479–1492. doi: 10.1101/gad.11.11.1479. [DOI] [PubMed] [Google Scholar]
- 43.Nasmyth K, Dirick L. The role of SW14 and SW16 in the activity of G1 cyclins in yeast. Cell. 1991;66:995–1013. doi: 10.1016/0092-8674(91)90444-4. [DOI] [PubMed] [Google Scholar]
- 44.Nevins J R. E2F: a link between the Rb tumor suppressor protein and viral oncoproteins. Science. 1992;258:424–429. doi: 10.1126/science.1411535. [DOI] [PubMed] [Google Scholar]
- 45.Ogas J, Andrews B J, Herskowitz I. Transcriptional activation of CLN1, CLN2, and a putative new G1 cyclin (HCS26) by SWI4, a positive regulator of G1-specific transcription. Cell. 1991;66:1015–1026. doi: 10.1016/0092-8674(91)90445-5. [DOI] [PubMed] [Google Scholar]
- 46.Ohtani K, DeGregori J, Leone G, Herendeen D R, Kelly T J, Nevins J R. Expression of the HsOrc1 gene, a human ORC1 homolog, is regulated by cell proliferation via the E2F transcription factor. Mol Cell Biol. 1996;16:6977–6984. doi: 10.1128/mcb.16.12.6977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ohtani K, DeGregori J, Nevins J R. Regulation of the cyclin E gene by transcription factor E2F1. Proc Natl Acad Sci USA. 1995;92:12146–12150. doi: 10.1073/pnas.92.26.12146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ohtani K, Nevins J R. Functional properties of a Drosophila homolog of the E2F1 gene. Mol Cell Biol. 1994;14:1603–1612. doi: 10.1128/mcb.14.3.1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ormondroyd E, de la Luna S, La Thangue N B. A new member of the DP family, DP-3, with distinct protein products suggests a regulatory role for alternative splicing in the cell cycle transcription factor DRTF1/E2F. Oncogene. 1995;11:1437–1446. [PubMed] [Google Scholar]
- 50.Pardee A B. G1 events and regulation of cell proliferation. Science. 1989;246:603–608. doi: 10.1126/science.2683075. [DOI] [PubMed] [Google Scholar]
- 51.Richardson H E, O’Keefe L V, Reed S I, Saint R. A Drosophila G1-specific cyclin E homolog exhibits different modes of expression during embryogenesis. Development. 1993;119:673–690. doi: 10.1242/dev.119.3.673. [DOI] [PubMed] [Google Scholar]
- 52.Royzman I, Whittaker A J, Orr-Weaver T L. Mutations in Drosophila DP and E2F distinguish G1-S progression from an associated transcriptional program. Genes Dev. 1997;11:1999–2011. doi: 10.1101/gad.11.15.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sauer K, Knoblich J A, Richardson H, Lehner C F. Distinct modes of cyclin E/cdc2c kinase regulation and S-phase control in mitotic and endoreduplication cycles of Drosophila embryogenesis. Genes Dev. 1995;9:1327–1339. doi: 10.1101/gad.9.11.1327. [DOI] [PubMed] [Google Scholar]
- 54.Schubiger M, Palka J. Changing spatial patterns of DNA replication in the developing wing of Drosophila. Dev Biol. 1987;123:145–153. doi: 10.1016/0012-1606(87)90436-2. [DOI] [PubMed] [Google Scholar]
- 55.Schwob E, Nasmyth K. CLB5 and CLB6, a new pair of B cyclins involved in DNA replication in Saccharomyces cerevisiae. Genes Dev. 1993;7:1160–1175. doi: 10.1101/gad.7.7a.1160. [DOI] [PubMed] [Google Scholar]
- 56.Seum C, Spierer A, Pauli D, Szidonya J, Reuter G, Spierer P. Position-effect variegation in Drosophila depends on dose of the gene encoding the E2F transcriptional activator and cell cycle regulator. Development. 1996;122:1949–1956. doi: 10.1242/dev.122.6.1949. [DOI] [PubMed] [Google Scholar]
- 57.Sherr C J. Cancer cell cycles. Science. 1996;274:1672–1677. doi: 10.1126/science.274.5293.1672. [DOI] [PubMed] [Google Scholar]
- 58.Sherr C J. G1 phase progression: cycling on cue. Cell. 1994;79:551–555. doi: 10.1016/0092-8674(94)90540-1. [DOI] [PubMed] [Google Scholar]
- 59.Slansky J E, Farnham P J. Introduction to the E2F family: protein structure and gene regulation. Curr Top Microbiol Immunol. 1996;208:1–30. doi: 10.1007/978-3-642-79910-5_1. [DOI] [PubMed] [Google Scholar]
- 60.Smith A V, Orr-Weaver T L. The regulation of the cell cycle during Drosophila embryogenesis: the transition to polyteny. Development. 1991;112:997–1008. doi: 10.1242/dev.112.4.997. [DOI] [PubMed] [Google Scholar]
- 61.Stillman B. Cell cycle control of DNA replication. Science. 1996;274:1659–1664. doi: 10.1126/science.274.5293.1659. [DOI] [PubMed] [Google Scholar]
- 62.Strausfeld U P, Howell M, Descombes P, Chevalier S, Rempel R E, Adamczewski J, Maller J L, Hunt T, Blow J J. Both cyclin A and cyclin E have S-phase promoting (SPF) activity in Xenopus egg extracts. J Cell Sci. 1996;109:1555–1563. doi: 10.1242/jcs.109.6.1555. [DOI] [PubMed] [Google Scholar]
- 63.Stuart D, Wittenberg C. CLN3, not positive feedback, determines the timing of CLN2 transcription in cycling cells. Genes Dev. 1995;9:2780–2794. doi: 10.1101/gad.9.22.2780. [DOI] [PubMed] [Google Scholar]
- 64.Su T T, Yakubovich N, O’Farrell P H. Cloning of Drosophila MCM homologs and analysis of their requirement during embryogenesis. Gene. 1997;192:283–289. doi: 10.1016/s0378-1119(97)00107-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tautz D, Pfeifle C. A non-radioactive in situ hybridization method for the localization of specific RNAs in Drosophila embryos reveals translational control of the segmentation gene hunchback. Chromosoma. 1989;98:81–85. doi: 10.1007/BF00291041. [DOI] [PubMed] [Google Scholar]
- 66.Treisman J E, Follette P J, O’Farrell P H, Rubin G M. Cell proliferation and DNA replication defects in a Drosophila MCM2 mutant. Genes Dev. 1995;9:1709–1715. doi: 10.1101/gad.9.14.1709. [DOI] [PubMed] [Google Scholar]
- 67.Tye B-K. The MCM2-3-5 proteins: are they replication licensing factors? Trends Cell Biol. 1994;4:160–166. doi: 10.1016/0962-8924(94)90200-3. [DOI] [PubMed] [Google Scholar]
- 68.Won K A, Reed S I. Activation of cyclin E/CDK2 is coupled to site-specific autophosphorylation and ubiquitin-dependent degradation of cyclin E. EMBO J. 1996;15:4182–4193. [PMC free article] [PubMed] [Google Scholar]
- 69.Wu C-L, Classon M, Dyson N, Harlow E. Expression of dominant-negative mutant DP-1 blocks cell cycle progression in G1. Mol Cell Biol. 1996;16:3698–3706. doi: 10.1128/mcb.16.7.3698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wu C-L, Zukerberg L R, Ngwu C, Harlow E, Lees J A. In vivo association of E2F and DP family proteins. Mol Cell Biol. 1995;15:2536–2546. doi: 10.1128/mcb.15.5.2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yamaguchi M, Hayashi Y, Matsukage A. Essential role of E2F recognition sites in regulation of the proliferating cell nuclear antigen gene promoter during Drosophila development. J Biol Chem. 1995;270:25159–25165. doi: 10.1074/jbc.270.42.25159. [DOI] [PubMed] [Google Scholar]
- 72.Yamasaki L, Jacks T, Bronson R, Goillot E, Harlow E, Dyson N J. Tumor induction and tissue atrophy in mice lacking E2F-1. Cell. 1996;85:537–548. doi: 10.1016/s0092-8674(00)81254-4. [DOI] [PubMed] [Google Scholar]