

Significance
IECs mature and release IL-18 in response to barrier breach, which is subsequently sensed by underlying immune cells to initiate an inflammatory response. In addition to this epithelial-derived signaling network, IL-18 is also thought to have epithelial intrinsic function. However, the identity of IL18R1+ IECs is poorly defined. Here, we identify a subset of enterochromaffin cells (ECC) as an IL18R1+ population at homeostasis and implicate a plastic population of IL18R1+ revival stem cells (revSC) as the predominant epithelial-intrinsic IL-18 effector population in the recovering crypt.
Keywords: enterochromaffin cells, IL-18 receptor, intestinal injury, revival stem cells, inflammasomes
Abstract
Upon injury, epithelial-derived IL-18 is released and induces an inflammatory response in underlying IL18R1+ lamina propria cells. Notably, Il18r1 is also predicted to be expressed and functional in intestinal epithelial cells (IECs), since epithelial IL18R1 deficiency contributes to worsened outcomes upon inflammatory challenge. However, the nature of Il18r1+ IECs, and their subsequent role in epithelial-intrinsic IL-18 signaling is poorly characterized. Here, we show that, in the murine small intestine, the IL-18 receptor is expressed by rare IECs that we identified to be a subset of enterochromaffin cells (ECC). While these cells are the major producers of serotonin in the intestine, we found no evidence that IL-18 regulated serotonin metabolism or release. Rather, upon radiation-induced injury, Il18r1+ cells appeared in the crypt base and took on a revival stem cell (revSC) program, marked by mixed expression of YAP/TAZ and enteroendocrine genes signatures. Functionally, irradiated Il18−/− mice display reduced epithelial proliferation and altered differentiation in the small intestine, characterized by increased Paneth cells (PC) and elevated Wnt3 levels, which was partially recapitulated in Il18−/− ileal organoids. In sum, we identified an Il18r1+ population in the epithelium and revealed a role for IEC-intrinsic IL-18 signaling during injury.
The intestinal epithelium is a site of major immune regulation and must balance tolerance of commensal microbes with the need to rapidly respond to pathogens. Inflammasomes are cytosolic multiprotein complexes that regulate the release of IL-1 family cytokines and induction of proinflammatory pyroptotic cell death (1–4). Compellingly, inflammasome components are well expressed within the intestinal epithelium, and play a major role in preserving epithelial barrier integrity during enteric infection (5–7). Inflammasomes are generally composed of a proform of the cysteine protease, Caspase-1, the adaptor protein ASC (gene name Pycard), and a Nod-like receptor (NLR) pattern recognition protein (1–4). Once formed, inflammasomes recruit other pro-Caspase-1 molecules and process inactive pro-Caspase-1 into a cleaved, active form (1–4). Caspase-1 activation subsequently results in the cleavage of Gasdermin-D (GSDMD), and cleavage of both pro-IL-18 and pro-IL-1β into their mature, active forms (1–4). Once cleaved, GSDMD inserts itself into the cell’s membrane and forms pores, resulting in pyroptosis, and the release of active IL-1β and IL-18 into the extracellular space (1–4).
IL-18 is the dominant cytokine matured by inflammasomes in the intestinal epithelium (8–12), where it is detected by responsive cells expressing the alpha chain of the IL-18 receptor (IL18R1) as well as the IL-18 receptor-associated protein (IL18RAP) (13–16). IL-18 is thought to exert both proinflammatory and homeostatic functions at mucosal surfaces. In the intestine, IL-18 regulates T cell polarization and expression of IFNγ by CD4+ T cells (8, 17–19) and acts on Th17 (8, 10, 19) and regulatory CD4+ T cell (Treg) populations (8, 20). The notion that IL-18 may be essential not only for the inflammatory phase but also the healing and resolution phase of intestinal inflammation is reinforced by the observation that IL-18 regulates the signaling of IL-22, a key cytokine involved in the repair of the intestinal mucosal barrier and secretion of antimicrobial peptides (20–24).
Bioactive IL-18 levels are controlled at the transcriptional level, as is the case with IL-22-mediated induction of Il18 (23), at the protein level through inflammasome activation, and in the extracellular space, through competitive inhibition with IL-18 binding protein (IL-18 bp), a soluble decoy receptor (16, 25). Clinical studies have revealed that IL18 expression is increased in both epithelial and lamina propria cells from patients with inflammatory bowel disease (IBD) (26), suggesting that IL-18 may be either proinflammatory in the gut or induced as a consequence of inflammation. Likewise, genetic studies have identified mutations in the risk allele rs917996, which encodes IL18RAP as a risk factor for Crohn’s disease (CD) (27–29), suggesting that altered IL-18 signaling may be directly involved in CD pathogenesis.
Importantly, expression of the IL-18 receptor is not restricted to immune cells. However, little is known regarding the IEC-intrinsic function of IL-18 signaling in the intestine. Here, using a combination of single-cell RNA-seq (scRNA-seq), animal studies, and murine ileal organoids, we identify enterochromaffin cells (ECC) as the dominant IEC population expressing Il18r1 in the murine small intestine under homeostasis. Moreover, we demonstrate that Il18r1 is dynamically regulated during irradiation induced injury and additionally marks a population of Clu+ revival stem cells (revSC). Finally, we identify epithelial-intrinsic IL-18 signaling as a key regulator of IEC proliferation and differentiation in the recovering crypt.
Results
The IL-18 Receptor Is Discreetly Expressed in a Rare Intestinal Epithelial Cell (IECs) Population.
We began by exploring the location of inflammasome components in murine ileal and colonic tissues. In the small intestine, we found broad expression of Caspase1, Il18, Pycard, and Gsdmd in IECs (Fig. 1A and SI Appendix, Fig. S1A), which we validated orthogonally through reanalysis of an existing scRNA-seq dataset of murine epithelial cells (30) according to distinct spatially resolved gene signatures (31) labeling enterocyte populations from the crypt base, through the transit-amplifying (TA) zone and up the villus (Fig. 1B). Overall, inflammasome transcripts displayed the greatest expression in epithelial cells of the TA region (Fig. 1 A and B and SI Appendix, Fig. S1A). In contrast, while ileal Il18r1 transcripts were strongly expressed in lymphatic cells (Fig. 1A), expression in IECs was rare and restricted to cells that appeared as narrow and elongated (Fig. 1A, Inset). Likewise, in the colon, Il18r1 transcripts were abundantly expressed in immune cells (SI Appendix, Fig. S1B). Notably, Il18r1+ IECs in the colon were sporadic and had only weak foci (SI Appendix, Fig. S1B), whereas Il18r1+ IECs of the ileum had distinct staining and were consistently located in the mid-crypt to TA region (+4 to +10; Fig. 1A).
Fig. 1.

The IL-18 receptor is discreetly expressed in a rare intestinal epithelial cell (IECs) population. (A) Representative images of RNA in situ hybridization for stated inflammasome transcripts in the murine ileum. Enlarged areas indicated by red boxes. n > 2 per probe. (B) Heatmap of stated inflammasome transcripts within ileal enterocytes, clustered according to spatial-transcriptomic profiling of murine crypt–villus axis from intestinal stem cells (ISC) through TA (TA1-TA2) and villus (V1–V6) regions. Accessed from NCBI GEO: GSE195742.
ECC are the Predominant Il18r1+ Population in the Murine Small Intestine.
Next, we queried available scRNA-seq datasets for epithelial expression of Il18r1, which encodes the major chain of the IL-18 receptor. Unsupervised clustering of a murine small-intestinal dataset (32) revealed 17 distinct populations of IECs (Fig. 2 A and B). In support of our in situ hybridization data, we found abundant expression of Il18 across clusters (Fig. 2 C and D). Similarly, we observed discrete expression of Il18r1, with greatest abundance in cluster 12, followed by minimal expression in cluster 4 (Fig. 2 E and F). Unsupervised annotation identified cluster 12 as a population of enteroendocrine cells (EECs), while cluster 4 marked a population of goblet cells (Fig. 2B). To better characterize these populations, we performed differential expression on all Il18r1− versus Il18r1+ IECs and determined that Il18r1+ cells were enriched for markers of serotonin biosynthesis (Tph1) and enterochromaffin cell transcription factors (Neurod1, Lmx1a) (Fig. 2G). Moreover, GO term analysis revealed upregulation of processes involved with neurotransmitter secretion (SI Appendix, Fig. S2A). In contrast, we found only scattered expression of Il18r1, without obvious cellular specificity in our analysis of a colonic scRNA-seq dataset (SI Appendix, Fig. S2 D–F). As such, we focused the subsequent analysis on the small intestine.
Fig. 2.
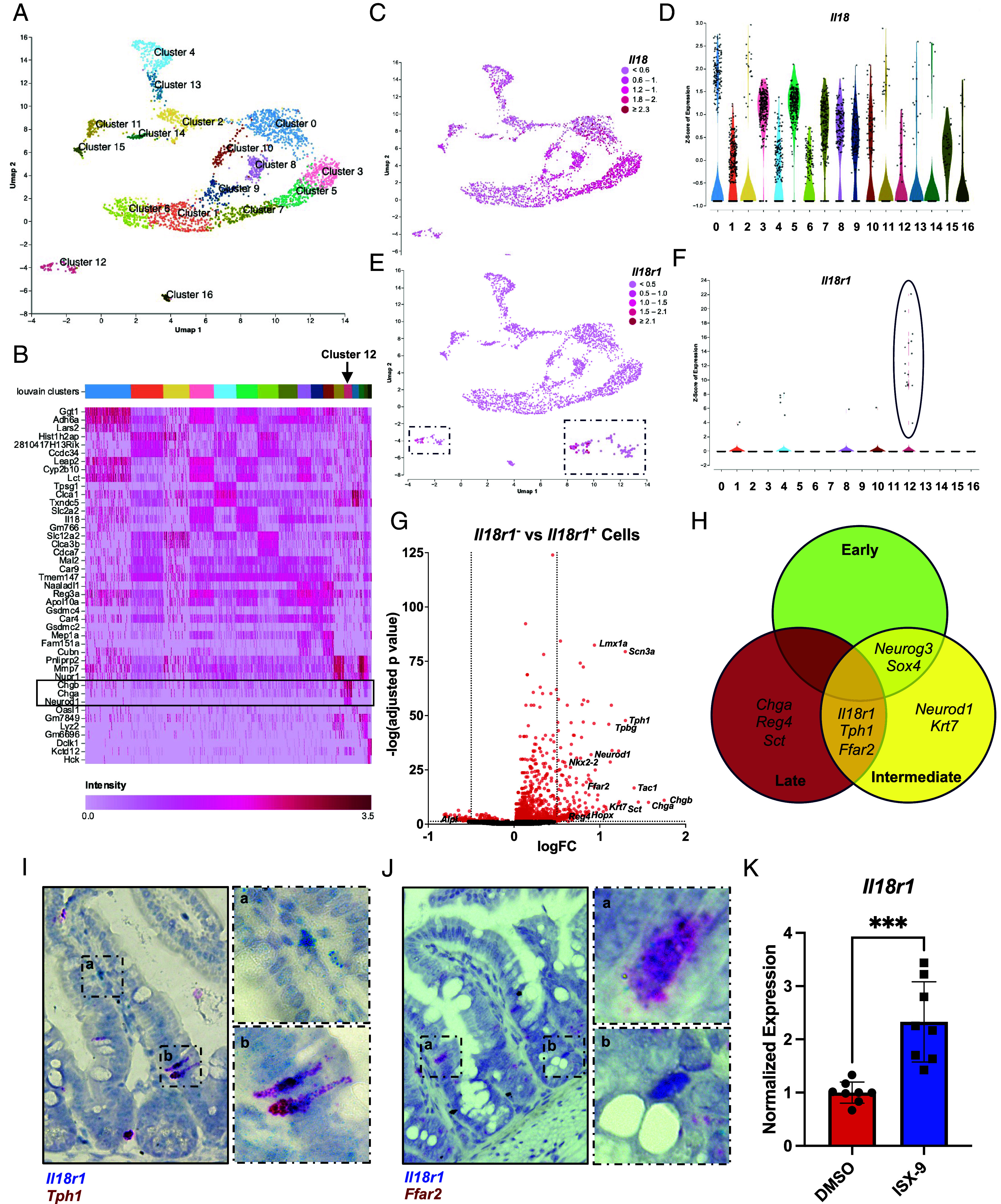
Enterochromaffin cells (ECC) are the predominant Il18r1+ population in the murine small intestine. (A) UMAP of murine small-intestinal IECs with Louvain clusters labeled. Accessed from NCBI GEO: GSE123516. (B) Heatmap of Louvain clusters with top three genes identified per cluster. Cluster 12 is identified with a box and arrow. (C) Expression of Il18 overlaid on UMAP plot. (D) Violin plot of expression of Il18 in each Louvain cluster. (E) Expression of Il18r1 overlaid on UMAP plot. (F) Violin plot of expression of Il18r1 in each Louvain cluster, with cluster 12 is highlighted. (G) Volcano plot of differential expression of Il18r1+ vs Il18r1− cells (P < 0.05, log2FC > 0.5). (H) Venn diagram of selected population-specific genes (P < 0.01, log2FC > 1.5) from bulk RNA-seq on small-intestinal IECs of FACS-sorted Neurog3Chrono mice. Accessed from NCBI GEO: GSE113561. (I) Duplex RNA in situ hybridization in ileal tissue showing colocalization of Il18r1 and Tph1 transcripts in ileal ECC (n > 2). Enlarged areas indicated by hashed. boxes, displaying staining in Il18r1+ immune cells (a) and double-positive ECC (b). n > 2 per probe. (J) Duplex RNA in situ hybridization in ileal tissue showing colocalization of Il18r1 and Ffar2 transcripts in ileal ECC (n > 2). Enlarged areas indicated by hashed boxes, displaying double (a) and single positive IECs (b). n > 2 per probe. (K) RT-qPCR for Il18r1 from wild-type ileal organoids treated with ISX-9 for 48 h. Data were analyzed with Student’s t test. Mean ± SD is shown with ***P ≤ 0.001. n = 8 from 4 biologic repeats.
ECC express an array of chemo- and mechanoreceptors which responds to microbial cues, including free fatty acid receptors (FFARs), such as FFAR2, which are required to regulate the production of serotonin (5-hydroxytryptamine; 5-HT) (33). Therefore, we asked whether microbial colonization altered expression of Il18r1 and found no difference in expression between germ-free (GF) and Specific Pathogen Free (SPF) conventionalized Tph1+ ECC by scRNA-seq (30; SI Appendix, Fig. S2 G–K). To confirm expression of Il18r1+ on ECC, we made use of previously published Neurog3Chrono reporter mouse bulk RNA-seq dataset, which labels enteroendocrine cells (34). Neurogenin-3 (Neurog3) is transiently expressed in the common progenitor of EECs (34, 35). In Neurog3Chrono animals, Neurog3+ cells shift from yellow to red fluorescence as a function of time, thereby allowing for analysis of early versus late EEC populations. Importantly, fluorescence-activated cell sorting (FACS)-sorted Neurog3+ cells were found to express Il18r1 transcripts (Fig. 2H). Moreover, Il18r1 was coexpressed in the intermediate to late EEC population along with Tph1 and Ffar2, two markers identified from our earlier single-cell analysis (Fig. 2G), which we further validated via duplex RNA in situ hybridization to identify Tph1+Il18r1+ and Ffar2+Il18r1+ cells in the ileum (Fig. 2 I and J). Finally, we asked whether we could modulate expression of Il18r1 by inducing EECs. Accordingly, we cultured murine ileal organoids in the presence of isoxazole-9 (ISX-9), a small-molecule agonist of the EEC transcription factor Neurod1 (36), and found that ISX-9 induced profound upregulation of Il18r1 after 48 h (Fig. 2K). Importantly, this response was specific to EECs, as while Neurod1 was induced (SI Appendix, Fig. S2B), Atoh1—the master regulator of intestinal secretory cells—remained unchanged (SI Appendix, Fig. S2C).
IL-18 does not Induce 5-HT Release from ECC.
Intestinal ECC produce the majority of serotonin outside of the central nervous system, which is required for intestinal homeostasis and for proper regulation of gut peristalsis and inflammation during enteric infection (37–39). Notably, germ-free mice display low levels of circulating 5-HT (40–42) suggesting that microbial stimulus is required for proper function. Given that IL-18 is matured in response to microbial colonization (Fig. 3G), we wondered whether IL-18 regulated 5-HT levels. Surprisingly, we observed that germ-free mice had higher levels of small-intestinal 5-HT+ cells (Fig. 3 A and B), despite lower circulating serotonin (Fig. 3E). Likewise, SPF mice displayed greater inflammasome activity and more mature IL-18 relative to germ-free animals (Fig. 3G). However, IL-18 deficiency did not affect serotonin levels (Fig. 3E), number of ECC (Fig. 3B), or spatial distribution of ECC along the crypt–villus axis (Fig. 3 C and D) relative to SPF wild-type littermates, suggesting that IL-18 does not regulate 5-HT levels in the homeostatic intestine. Given that IL-18 is constitutively released, we wondered whether ECC were unresponsive to homeostatic IL-18 levels and that increased concentrations would be required to modulate serotonin. Accordingly, we intraperitoneally injected mice with recombinant IL-18 (rIL-18) and assessed circulating serotonin from two to 24 h postinjection and observed no change in circulating 5-HT at any assessed timepoint (Fig. 3F). As Il18r1 marks only a subset of ECC, we wondered whether measuring total 5-HT in circulation could mask a local IL-18-dependent effect. TPH1 is the rate-limiting enzyme for serotonin synthesis. In ECC, 5-HT is dynamically regulated and released as a result of ion flux (38). In support, treatment with thapsigargin, which raises intracellular Ca2+ levels, robustly induced Tph1 (Fig. 3H), which was independent of changes to overall EECs, as read out by Chromogranin A (Chga) transcripts (SI Appendix, Fig. S3A), a marker for EECs (43). However, treatment with rIL-18 did not alter Tph1 transcription (Fig. 3H).
Fig. 3.

IL-18 does not induce 5-HT release from ECC. (A) Representative image of 5-HT+ ECC in the ileum of wild-type GF, SPF colonized Il18+/+ and SPF colonized Il18−/− mice. (B) Quantification of 5-HT+ ECC in the ileum of wild-type GF, SPF colonized Il18+/+ and SPF colonized Il18−/− mice. One point represents one mouse. One-way ANOVA with Tukey’s multiple comparisons test. Mean ± SEM with ns (nonsignificant), *P ≤ 0.05 and **P ≤ 0.01 displayed. (C and D) Frequency of 5-HT+ ECC in the crypt (C) or villi (D) of wild-type GF, SPF colonized Il18+/+ and SPF colonized Il18−/− mice. One point represents one mouse. One-way ANOVA with Tukey’s multiple comparisons test. Mean ± SEM with ns (nonsignificant) and *P ≤ 0.05 displayed. (E) ELISA of circulating 5-HT levels from whole blood of wild-type GF, SPF colonized Il18+/+ and SPF colonized Il18−/− mice, normalized to SPF Il18+/+ mice. One point represents one mouse. One-way ANOVA with Tukey’s multiple comparisons test. Mean ± SD with ns (nonsignificant), **P ≤ 0.01 and ***P ≤ 0.001 displayed. (F) ELISA of circulating 5-HT levels from whole blood of wild-type mice intraperitoneally injected with rIL-18 for stated timepoints. Normalized to control (sham) mice. One point represents one mouse. One-way ANOVA with Dunnett’s multiple comparisons test. Mean ± SD with ns (nonsignificant) displayed. (G) Whole tissue immunoblot of wild-type GF, SPF colonized Il18+/+ and SPF colonized Il18−/− ileum for stated inflammasome components. Each lane represents one mouse. (H) RT-qPCR of Tph1 transcripts from wild-type ileal organoids treated with rIL-18 or thapsigargin (thapsi) for 6 h. One-way ANOVA with Dunnett’s multiple comparisons test. Mean ± SD with ns (nonsignificant) and ****P ≤ 0.0001 displayed. n = 12 from 4 biological repeats. (I) RT-qPCR of Tph1 transcripts from Il18+/+ and Il18−/− ileal organoids treated with FlaTox for 6 h. Two-way ANOVA with Tukey’s multiple comparisons test. Mean ± SD is shown with ns (nonsignificant) displayed. n = 6 from 3 biological repeats.
Physiologically, elevated IL-18 levels occur alongside the release of other alarmins during pyroptosis (44, 45). Therefore, we asked whether pyroptosis was required for IL-18 signaling on ECC. FlaTox is a potent inducer of the NLRC4 inflammasome (6) and rapidly induced pyroptosis and IL-18 maturation and release in ileal organoids (SI Appendix, Fig. S3B). Six-hour FlaTox treatment resulted in a mild induction of Tph1 (Fig. 3I) which was independent of changes to overall EEC transcripts (Chga, SI Appendix, Fig. S3C) or IL-18. Collectively, these findings suggest that although Il18r1 is expressed on intestinal ECC, IL-18 signaling is dispensable for serotonin biosynthesis or release.
Il18r1 Appears on a Subset of revSCs Following Irradiation-Induced Injury.
Given that IL-18 did not appear to regulate serotonin levels in the intestine, we asked what other features were unique to EECs. Notably, EECs, including ECC, are known to be radioresistant and are capable of dedifferentiating into alternative stem cell populations, which replace homeostatic Lgr5+ crypt-base intestinal stem cells (ISCs) that are ablated upon injury (46, 47). Accordingly, we observed enrichment of Hopx, a marker of alternate, reserve stem cells (46), and Nkx2-2, an EEC transcription factor that marks a population of ECC with expanded lineage potential (48) at baseline in Il18r1+ cells (Fig. 2G). Therefore, we asked whether crypt injury would induce IL-18-dependent effects in Il18r1+ cells. We began by analyzing a scRNA-seq dataset (32) generated from the small intestine of mice 72 h postexposure to total body irradiation (IR). As a synchronized model of DNA damage, irradiation induces cell death in rapidly proliferating crypt-base Lgr5+ ISCs, thereby facilitating the study of ISC dynamics following crypt injury. Regardless of treatment, unsupervised clustering revealed strong expression of Il18r1+ in ECC (cluster 15, Fig. 4 A–C). Importantly, Il18r1 was also found to be induced in an IR-dependent cluster (cluster 8, Fig. 4 A–C), which has previously been annotated as a population of Clu+ injury-induced revSC [Ayyaz et al. (32) annotation replicated in Fig. 4D, subclustering in SI Appendix, Fig. S4 A–D], that emerge as a consequence of YAP/TAZ signaling in the irradiated crypt (32, 49). In support, differential expression of irradiated Il18r1− versus irradiated Il18r1+ IECs revealed enrichment of a mixture of enterochromaffin cell as well as some YAP/TAZ markers, including Clu and Ly6a (Fig. 4E) and duplex RNA in situ hybridization confirmed the presence of both Il18r1+Tph1 +and Il18r1+Clu+ double-positive cells in the crypt (Fig. 4 F and G). Notably, both Il18r1+Tph1 +and Il18r1+Clu+ double-positive cells appeared in similar locations in the irradiated crypt and were consistently found adjacent to granular crypt-base cells, which are likely Paneth cells (PC), suggesting that Il18r1 marks a single population which express a mixture of EEC and revSC markers. Importantly, this position is normally occupied by Wnt-receiving crypt-base stem cell populations (50, 51). Accordingly, differential expression revealed that in addition to revSC and EEC markers, Il18r1+ cells were enriched for transcripts associated with Wnt signaling (Fig. 4E) including the canonical effector Tcf4 (52), noncanonical/planar cell polarity (PCP) component Cfap126 (also known as Flattop) (53), and trophoblast glycoprotein (Tpbg), the latter of which functions as a molecular switch to regulate canonical and noncanonical/PCP Wnt signaling (54).
Fig. 4.
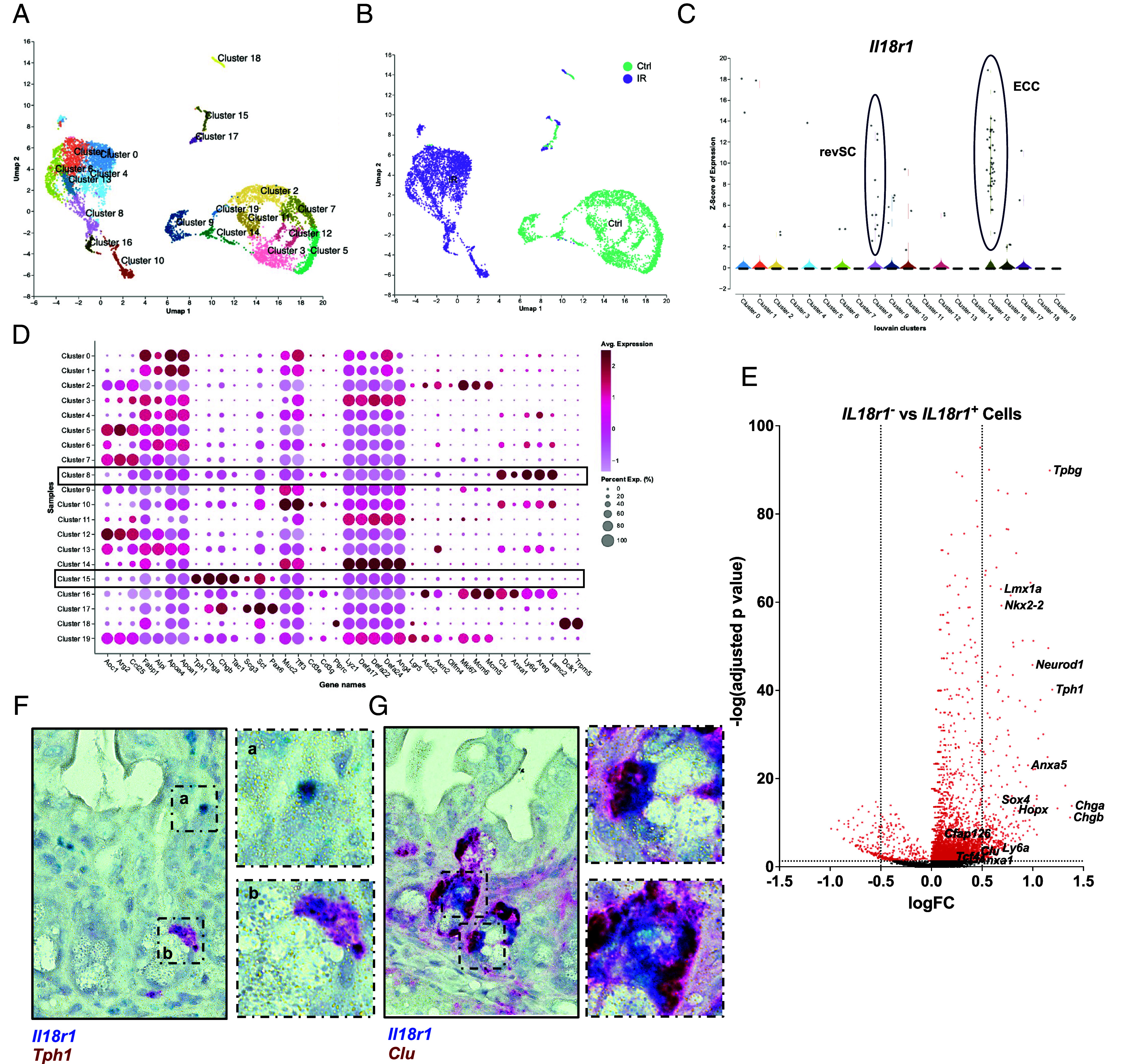
Il18r1 appears on a subset of revSCs following irradiation induced injury. (A) UMAP of murine small-intestinal IECs from control and irradiated animals with Louvain clusters labeled. Accessed from NCBI GEO: GSE123516. (B) Treatment groups overlaid on UMAP plot. (C) Violin plot of expression of Il18r1 in each Louvain cluster, with revSCs (Cluster 8) and ECC (Cluster 15) highlighted. (D) Dot plot of marker genes for indicated IEC clusters with revSC (Cluster 8) and ECC (Cluster 15) highlighted. (E) Volcano plot of differential expression of Il18r1− vs Il18r1+ cells (P < 0.05, log2FC > 0.5) from the irradiated treatment group. (F) Duplex RNA in situ hybridization in ileal tissue showing colocalization of Il18r1 and Tph1 transcripts in crypt residing cells, 72 h post-IR (n > 2). Enlarged areas indicated by hashed boxes, displaying staining in Il18r1+ immune cells (a) and double-positive ECC (b). n > 2 per probe. (G) Duplex RNA in situ hybridization in ileal tissue showing colocalization of Il18r1 and Clu transcripts in crypt cells, 72 h post-IR (n > 2). Enlarged areas indicated by hashed boxes.
Taken together, these data suggest that following irradiation Il18r1 is expressed in a population of ECC associated with expanded-lineage potential, as well as a Wnt-receiving revSC population.
IL-18 Regulates Proliferation in the Regenerating crypt.
We next sought to characterize the functional consequence of IL-18 signaling during IR injury. EECs are known to possess homeostatic and IR injury-induced stem cell activity (47), and upon PC depletion, form an alternative niche to support Lgr5+ ISCs (55). Therefore, we asked whether IL-18 promotes the dedifferentiation of ECC to become an immature, recovery population. While specific lineage tracing models are required to fully explore this hypothesis, we reasoned that the frequency of II18r1+ cells in the recovering crypt could serve as a proxy marker. At baseline, Il18r1+ cells were rare, but were induced post-IR, where they were found residing in the crypt, ranging from the crypt base to the +4/+10 position (Fig. 5A). However, we observed no difference in Il18r1+ levels in Il18+/+ versus Il18−/− tissue (Fig. 5B), suggesting that IL-18 is dispensable for the induction of revSCs. Notably, the pattern of Il18r1 staining differed from Clu. While Il18r1+ appeared as a discrete cell (Fig. 5A), Clu marked multiple cells and resulted in patchy staining through the regenerating crypt (Fig. 5C). Indeed, the pattern of Clu staining closely mirrored Ki67 immunofluorescence (Fig. 5E), which marks all proliferating cells. Thus, we concluded that Clu in situ hybridization stained both revSCs and direct progeny, while Il18r1 and Tph1 marked an initial population of EECs/revSCs in the regenerating crypt. Accordingly, we asked whether IL-18 was required for proliferation following irradiation and observed reduced Clu (Fig. 5 C and D) and Ki67 (Fig. 5 E and F) staining in Il18−/− animals relative to wild-type controls. Importantly, IL-18 was dispensable for the regulation of baseline proliferation (SI Appendix, Fig. S5 J and K), suggesting that injury, and ensuing appearance of Il18r1+ cells in the crypt is required to mediate IL-18-dependent proliferation of revSC progeny.
Fig. 5.
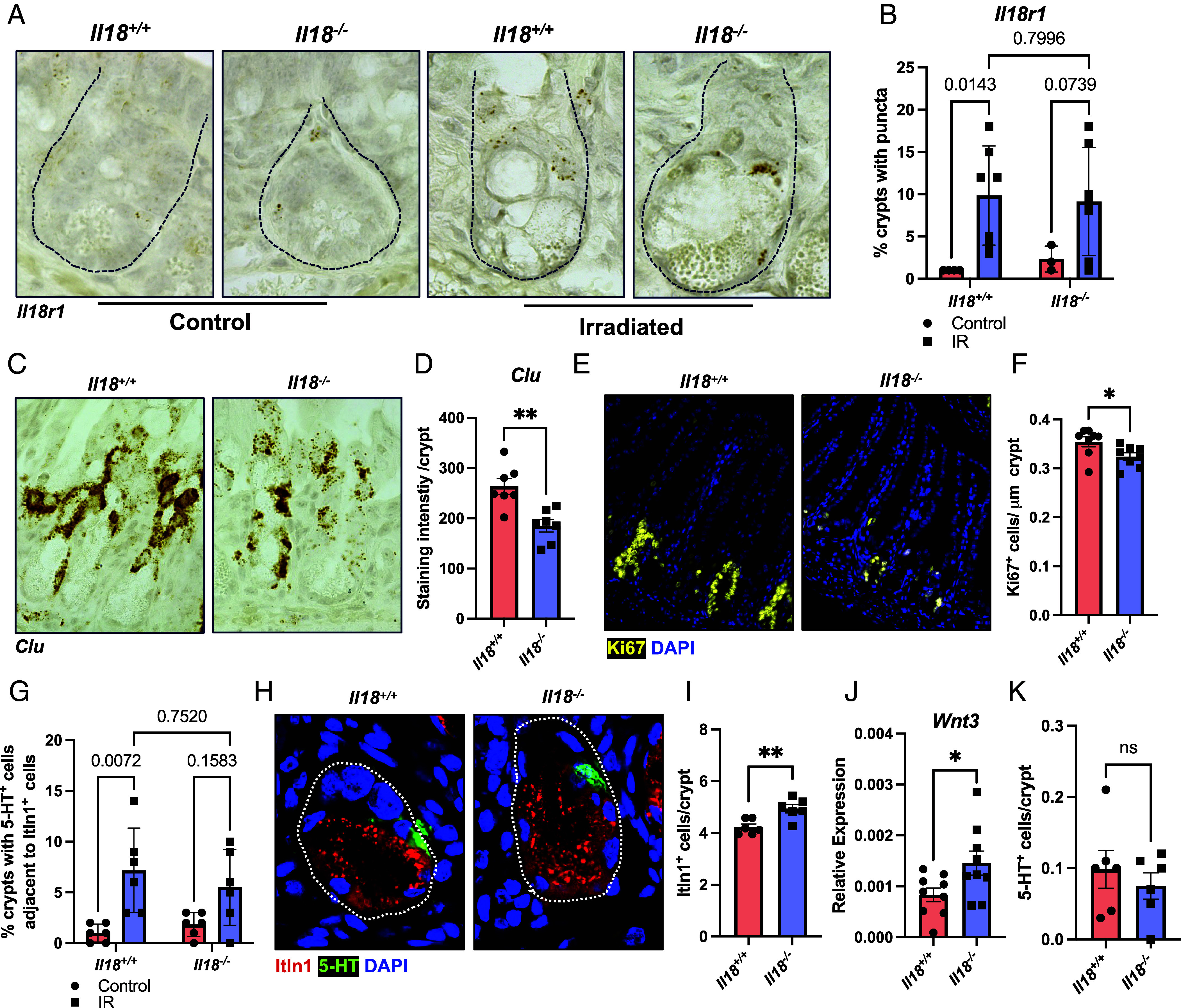
IL-18 regulates proliferation in the regenerating crypt. (A) Representative image of RNA in situ hybridization for Il18r1 transcripts in ileal crypt-resident cells from Il18+/+ and Il18−/− animals at baseline and 72 h post-IR. Crypts indicated by hashed lines. (B) Quantification of the frequency of crypts with Il18r1 staining relative to total crypts from Il18+/+ and Il18−/− animals at baseline and 72 h post-IR. One point represents one mouse. Two-way ANOVA with Tukey’s multiple comparisons test. Mean ± SEM is shown with indicated P values. (C) Representative image of RNA in situ hybridization for Clu transcripts in the crypts of Il18+/+ and Il18−/− animals 72 h post-IR. (D) Quantification of Clu staining intensity in the crypts of Il18+/+ and Il18−/− animals 72 h post-IR. One point represents one mouse. Data were analyzed with Student’s t test. Mean ± SEM is shown with **P ≤ 0.01 displayed. (E) Representative image of Ki67+ cells of Il18+/+ and Il18−/− mice 72 h post-IR. (F) Quantification Ki67+ cells of Il18+/+ and Il18−/− mice 72 h post-IR, standardized to crypt length. One point represents one mouse. Data were analyzed with Student’s t test. Mean ± SEM is shown with *P ≤ 0.05 displayed. (G) Quantification of the frequency of crypts with 5-HT+ cells adjacent to Itln1+ cells, relative to total crypts, from Il18+/+ and Il18−/− animals at baseline and 72 h post-IR. One point represents one mouse. Two-way ANOVA with Tukey’s multiple comparisons test. Mean ± SEM is shown with indicated P values. (H) Representative image of Itln1+ Paneth and 5-HT+ ECC in the crypt of Il18+/+ and Il18−/− mice 72 h post-IR. Crypts indicated with hashed line. (I) Quantification of Itln1+ Paneth cells (PC) in the crypt of Il18+/+ and Il18−/− animals 72 h post-IR. One point represents one mouse. Data were analyzed with Student’s t test. Mean ± SEM is shown with *P ≤ 0.05 displayed. (J) RT-qPCR for Wnt3 transcripts from ileal tissue of Il18+/+ and Il18−/− animals 72 h post-IR. One point represents one mouse. Data were analyzed with Student’s t test. Mean ± SD is shown with *P ≤ 0.05 displayed. (K) Quantification of 5-HT+ cells in the crypt of Il18+/+ and Il18−/− animals 72 h post-IR. One point represents one mouse. Data were analyzed with Student’s t test. Mean ± SEM is shown with nonsignificance (ns) displayed.
Secretory Populations are Dysregulated in Regenerating Il18−/− crypts.
It has been suggested that PC express the IL-18 receptor, where IL-18 signaling at baseline and during adherent-invasive Escherichia coli infection is reportedly required for cellular function, such that Il18−/− animals have fewer functional PC (23). While our analysis of multiple scRNA-seq datasets and our RNA in situ hybridization data do not support the notion that Il18r1 is expressed on PC, given the appearance of Il18r1+ cells in the irradiated crypt, we wondered whether Il18r1+ cells support PC during IR-induced injury. In accordance with our Il18r1 staining (Fig. 5 A and B), we observed an IL-18-independent increase in the frequency of crypts with 5-HT+ cells adjacent to Itln1+ PC post-IR, which is a site normally occupied by ISCs (Fig. 5 G and H). Quantification of Itln1+ PC revealed an increase in irradiated Il18−/− animals (Fig. 5 H and I), which was accompanied by elevated effector transcripts, including Wnt3 (Fig. 5J) and trending increases in Lyz1 (SI Appendix, Fig. S5A) and Egf (SI Appendix, Fig. S5B) from qPCR of ileal tissue. We queried whether crypt-injury was required and found no IL-18-dependent differences in PC numbers at baseline (SI Appendix, Fig. S5 L and M). Next, we asked whether the increase of PC post-IR was a result of IL-18-dependent regulation of cell proliferation or cell death. However, we observed no Ki67+Itln1+ or cleaved caspase-3+ (Cl.Casp3+) PC in the recovering crypt (SI Appendix, Fig. S5 C and D), and no IL-18-dependent difference in crypt cell death following irradiation (SI Appendix, Fig. S5E). Notably, our analysis was performed 72 h post-IR, which marks a time of peak revSC-mediated regeneration (32, 56), but is likely too late a timepoint to observe rapid apoptotic or early proliferative events. Collectively, these results suggest that IL-18 signaling negatively regulates PC abundance following crypt-injury, through mechanisms which remain unclear.
Next, we asked whether other secretory populations were similarly affected by IL-18 deficiency. Like PC, we observed no IL-18-dependent change in goblet (Muc2, SI Appendix, Fig. S5 N and O) or tuft (Dclk1, SI Appendix, Fig. S5 P and Q) cell populations at baseline. Similarly, upon irradiation, crypt residing ECC (Fig. 5K), and villus goblet cell numbers were equivalent between wild-type and Il18−/− mice (SI Appendix, Fig. S5 F and G). However, tuft cells were significantly increased in irradiated Il18−/− animals (SI Appendix, Fig. S5 H and I). Importantly, our profiling of secretory populations suggests that the reduced proliferation observed in irradiated Il18−/− mice is likely a result of changes to the absorptive enterocyte lineage, as we found assessed secretory populations to be unchanged, or in the case of tuft and PC populations, exhibit a concomitant increase in Il18−/− animals. Given that neither Paneth nor tuft cells appear to express the IL-18 receptor, it is currently unclear whether changes to these secretory populations are a result of shared IL-18-dependent regulation of an Il18r1+ progenitor or indirectly induced as a result of dysregulated proliferation in IL-18-deficient mice.
Epithelial Intrinsic IL-18 Signaling Regulates Absorptive Cell Differentiation.
In an effort to deconvolute our in vivo data, we utilized ileal organoids to assess which IL-18 phenotypes were a result of epithelial-intrinsic signaling. Organoid passaging requires physical disruption of the mature organoid into crypt fragments, which induces regenerative programs in ISC populations (57). Given that IL-18 may play a role in regulating the regenerative capacity of crypt-specific IECs, we began by assessing growth in Il18−/− organoids following passaging. Surprisingly, at 24 h postpassage, Il18−/− cystic organoids were larger (Fig. 6 A and B), possessed more Itln1+ PC (Fig. 6 C and D), and expressed increased levels of Lyz1, Lgr5, and Clu transcripts when compared to Il18+/+ counterparts (Fig. 6E). Notably, the elevated PC observed postirradiation in our in vivo model persisted in vitro in Il18−/− organoids, suggesting an epithelial-intrinsic mechanism. In contrast, Il18−/− organoids exhibited reduced expression of Dclk1 (Fig. 6E), suggesting the involvement of additional epithelial-extrinsic signaling in tuft cell differentiation, or alternatively, that it was a specific feature of organoid culture. Indeed, when cultured in the absence of Wnt3a—which is the experimental condition we use—murine ileal organoid growth is dependent on the function of PC (50), such that the increased PC in Il18−/− organoids would mask revSC defects.
Fig. 6.

Epithelial intrinsic IL-18 signaling regulates absorptive cell differentiation. (A) Representative brightfield image of Il18+/+ and Il18−/− organoids, 24 h postpassage. (B) Violin plot of the area of cystic Il18+/+ and Il18−/− organoids, 24 h postpassage. Data were analyzed with Student’s t test. Mean ± SD is shown with ****P ≤ 0.0001. Data pooled from 3 biologic repeats. (C) Representative image of Itln1+ PC in Il18+/+ and Il18−/− organoids, 24 h postpassage. (D) Quantification of Itln1+ PC in Il18+/+ and Il18−/− organoids, 24 h postpassage. Data were analyzed with Student’s t test. Mean ± SEM is shown with *P ≤ 0.05 displayed. n = 5. (E) RT-qPCR for stated transcripts from Il18+/+ and Il18−/− organoids 24 h postpassage. Data were analyzed with Student’s t test. Mean ± SD is shown with *P ≤ 0.05, **P ≤ 0.01 and ****P ≤ 0.0001 displayed. n = 8 from 4 biological repeats. (F) Frequency of cystic (<1 crypt-bud) or budding (2 + crypt-buds) organoids from Il18+/+ and Il18−/− mice, 72 h postpassage. Data were analyzed with Student’s t test. Mean ± SD is shown with *P ≤ 0.05 displayed, n = 4. (G) Violin plot of number of crypt-buds in budding organoids from Il18+/+ and Il18−/− mice, 72 h postpassage. Data were analyzed with Student’s t test. Mean ± SD is shown with nonsignificance (ns) displayed. Data pooled from 4 biologic repeats. (H) Representative brightfield image of Il18+/+ and Il18−/− organoids, 72 h postpassage. (I) Representative image of Itln1+ PC and Ki67+ cells in crypt-buds of Il18+/+ and Il18−/− organoids, 72 h postpassage. (J and K) Quantification of Ki67+ (J) and Itln1+ Paneth (K) cells as a frequency of DAPI+ cells per crypt-bud of Il18+/+ and Il18−/− organoids, 72 h postpassage. Data were analyzed with Student’s t test. Mean ± SD is shown with nonsignificance (ns) displayed. n = 5. (L) RT-qPCR for stated transcripts from Il18+/+ and Il18−/− organoids 72 h postpassage. Data were analyzed with Student’s t test. Mean ± SD is shown with **P ≤ 0.0and ***P ≤ 0.001 displayed. n = 8 from 4 biological repeats. (M) Representative immunoblot for IL-18 maturation in response to treatment with stated TLR ligands from Tlr5+/+ and Tlr5−/− ileal organoids for 24 h, n = 3. (N) RT-qPCR for stated transcripts from mature (72 h postpassage) Il18+/+ and Il18−/− ileal organoids treated with flagellin for 48 h. Two-way ANOVA with Tukey’s multiple comparisons test. Mean ± SD is shown with *P ≤ 0.05, **P ≤ 0.01, and nonsignificance (ns) displayed. n = 6 from 3 biological repeats. (O) Model. Abbreviations are as follows: ECC, PC, revival stem cell (revSC).
In order to explore whether epithelial-intrinsic IL-18 signaling was sufficient to regulate cell fate, we assessed growth and cell differentiation in developing Il18−/− organoids. After 72 h, Il18−/− organoids were composed of a greater percent of cysts (Fig. 6F). However, in budding organoids, there was no change in the number of crypt-buds (Fig. 6 G and H), despite an increase in PC at 24 h, which are thought to be required for crypt budding (58, 59). Likewise, despite elevated Lgr5 and Clu transcripts (Fig. 6L), we observed no change in Ki67 staining (Fig. 6 I and J) in Il18−/− organoids. Indeed, at 72 h postpassage the IL-18-dependent change in PC number had largely resolved, with no difference in Lyz1 (Fig. 6L) and only a trending increase in Itln1+ cells observed in Il18−/− organoids relative to wild-type controls (Fig. 6K). To confirm that the transient increase in PC postpassage was not a hard-wired property of IL-18-deficient tissues, we made use of the gamma-secretase inhibitor DAPT [N-(N-(3,5-difluorophenacetyl)-l-alanyl)-s-phenylglycinet-butyl ester] to induce Atoh1-dependent secretory lineages in mature organoids. As expected, 48-hour DAPT treatment resulted in a strong induction of Atoh1 and Spdef, which is a marker of goblet and PC progenitors (60, 61), as well as Lyz1 (SI Appendix, Fig. S6A). However, we observed no IL-18-dependent changes in any assessed transcripts, suggesting that IL-18-dependent regulation of PC is transient and occurs in response to crypt injury.
Multiple pathways contribute to the balance between stemness and differentiation in the crypt. While Wnt signaling promotes the maintenance of ISCs, and the differentiation of PC (62), Notch signaling is required for lineage commitment, where Hes1, a Notch target gene induces differentiation of absorptive enterocytes through inhibition of Atoh1 (63). Profiling of Atoh1 and Hes1 transcripts revealed an IL-18-dependent decrease in Hes1, and an accumulation of Il18r1 in Il18−/− organoids 72 h postpassage (Fig. 6L). These data are reminiscent of our in vivo proliferation results and suggest that IL-18 may be required for proper absorptive differentiation from an Il18r1+Clu+ progenitor pool.
Finally, we wondered whether IL-18 regulated IEC differentiation during conditions of elevated inflammasome activation. Salmonella infection induces robust inflammasome activation and increases IL-18 levels in the gut (64, 65), which is accompanied by a shift toward absorptive enterocytes (66), suggesting a potential role for the cytokine in regulating IEC differentiation. Likewise, reanalysis of a published ileal bulk RNA-seq dataset (SI Appendix, Fig. S6B) revealed that microbial colonization of germ-free animals, which induces intestinal IL-18 production (Fig. 3G), also results in a reduction of stem (Lgr5, Ascl2) and TA transcripts (Olfm4) and a shift toward absorptive enterocytes (Alpi, Fabp1), which was accompanied by increased Il18 transcripts, the latter of which we confirmed by RNA in situ hybridization (SI Appendix, Fig. S6 C and D).
Organoids produce low levels of IL-18 at baseline, which can be increased when inflammasomes are activated, such as with the NLRC4 ligand, flagellin (Fig. 6M). Importantly, murine ileal IECs also express a limited array of TLRs, including TLR2, -4, and -5 (67), where TLR5 also detects flagellin. To determine whether inflammasome activation was dependent on TLR signaling, we assessed IL-18 maturation in Tlr5−/− organoids in response to PAM3CSK4 (TLR2), LPS (TLR4), and flagellin (TLR5) treatment and found that only flagellin was capable of induce IL-18 processing, which was itself TLR5-independent (Fig. 6M). These results support the notion that flagellin treatment is a viable method to activate the NLRC4 inflammasome. Moreover, while FlaTox rapidly induces widespread pyroptosis (6), we found flagellin to be a comparatively mild stimulus, allowing for analysis at longer time points. In agreement with our previous data, 48-hour flagellin treatment of mature organoids (72 h postpassage) revealed reduced enterocyte differentiation in Il18−/− organoids, as marked by Alpi and a concomitant increase in the PC transcription factor Spdef (Fig. 6N), which was independent of TLR5 (SI Appendix, Fig. S6E).
Collectively, these data suggest that IL-18, matured downstream of epithelial inflammasomes, signals through a plastic Il18r1+ enterochromaffin/revSC population to direct proliferation and absorptive enterocyte lineage differentiation of revSC progeny. In the absence of IL-18, the number of PC and associated growth factors increase, such that IL-18 deficiency is sufficient to alter cell fate in the regenerating crypt (model, Fig. 6O).
Discussion
In this study, we characterized the expression of Il18r1 in the murine intestine and report that Il18r1 is expressed on small-intestinal Tph1+ ECC. IL-33, a related type-1 family cytokine, was recently shown to induced serotonin release from ECC (39). Surprisingly, despite the presence of Il18r1 in ECC, IL-18 signaling did not regulate serotonin levels. Instead, we found that epithelial-intrinsic IL-18 signaling was required for epithelial lineage commitment following crypt injury. In the intestine, IR-induced damage abolishes rapidly cycling Lgr5+ ISCs, ultimately inducing populations of radioresistant revival stem cells (68), which are characterized by high levels of YAP/TAZ signaling, including expression of Clu, Anxa1, and Ly6a. Critically, we demonstrate that Il18r1 marks a population of revSCs. YAP/TAZ signaling transduces key mechanosensory cues from the extracellular environment and is correspondingly repressed upon cell–cell contact and induced as a result of actin-remodeling (69). Notably, inflammasome activation in the intestinal barrier results in actin remodeling and cell extrusion (6, 64, 70), suggesting a link between pyroptosis, IL-18, and epithelial restitution.
Many cell populations have been shown to dedifferentiate and become stem-like during injury. For example, subpopulations of tuft and PC possess stemness potential and can drive injury-induced recovery programs, as well as cancer (71–75). Likewise, Tph1+ ECC located at the +4 position are radioresistant and can dedifferentiate to a recovery stem cell state after radiation-induced injury (46). At baseline, Il18r1+ ECC were enriched for markers of plasticity, and upon IR injury, Tph1+ Il18r1+ cells appeared in the recovering crypt and occupied a site adjacent to PC which is typical of rapidly cycling ISC populations. While lineage trace models are required to determine whether Il18r1+ revSCs originate from ECC, we observed Tph1+Il18r1+, Clu+Il18r1+, and 5-HT+ cells in similar locations adjacent to PC in the recovering crypt, suggesting that these cells belong to a single plastic population.
Notably, IL-18 did not regulate the abundance of Il18r1+ cells in the recovering crypt, suggesting that IL-18 is dispensable for enterochromaffin cell dedifferentiation or de novo induction of revSCs. Rather, IL-18 deficiency resulted in blunted proliferation and reduced Clu staining in the recovering crypt, suggesting that IL-18 is required for proliferation of the revSC progeny. This observation is commensurate with the previously proposed role of IL-18 in inducing stem cells via Akt-Tcf4 signaling (23), and fits well within the paradigm of IL-1 family cytokine-dependent regulation of ISC dynamics. For example, IL-1 signaling via IL-1R1+ GREM1+ mesenchymal cells is known to upregulate RSPO3, a Wnt agonist, to promote ISC maintenance (76), while IL-38 functions directly as a Wnt3a agonist and a growth factor in organoid cultures (77).
While we focused on the functional consequence of IL-18 deficiency in the epithelium, we have not provided a mechanism by which IL-18 signaling on Il18r1+ IECs enacts these changes. In this regard, the finding that Il18r1+ cells are Wnt-receiving cells presents a promising hypothesis—that IL-18 may tune PC-derived proliferative signals in revSCs. It is plausible that IL-18 directly regulates Wnt signals through the induction of Wnt inhibitors in IL18R1+ recipient cells. Alternatively, IL-18 may modulate the switch between canonical and noncanonical/ PCP Wnt signaling in these cells, thereby influencing the lineage fate of daughter cells (53, 62). However, further work using IL18R1 lineage trace models is required.
Irradiation induces activation of the AIM2 inflammasome in the intestinal epithelium (78), which is accompanied by an increase in circulating IL-18 that is thought to exacerbate systemic radiation-induced damage (79, 80). In the small intestine, we found that IL-18 deficiency resulted in reduced proliferation, and increased PC following irradiation. In vitro, Il18−/− organoids displayed altered cell differentiation, and upon inflammasome activation, IL-18-deficient organoids exhibited transcriptional responses characteristic of blunted absorptive enterocyte differentiation. In support, absorptive enterocyte populations are known to be induced following Salmonella infection (66). Given that inflammasome transcripts are enriched in enterocytes of the mid-villus region (30, 31), are reduced in the crypt base, and are effectors of pyroptosis, these findings suggest a model wherein IL-18 is released by pyroptotic enterocytes, and signals back to IL18R1+ revSCs to direct enterocyte differentiation in order to replace lost cells.
Importantly, lineage differentiation was dysregulated in flagellin-treated IL-18-deficient organoids, and accompanied by an increase in Spdef, a marker of PC differentiation. These results suggest potential crosstalk between abrogated revSC differentiation and induction of PC in the recovering crypt. Moreover, despite elevated PC immediately postpassage, mature Il18−/− organoids did not display greater numbers of crypt-buds or proliferation. Indeed, at later timepoints, an increased proportion of Il18−/− organoids failed to bud and remained cystic. Collectively, these results suggest that Il18−/− crypts may exist in a compensatory state, where blunted revSC proliferation induces a secondary response in PC populations, which is not sufficient to restore normal homeostasis. As PC do not appear to express the IL-18 receptor, signaling from IL18R1+ revSCs is likely required to direct neighboring PC function. In support, IL-18 has previously been implicated in regulating antimicrobial peptides (AMPs), including PC-specific AMPs such as lysozyme (23), while EECs are known to support an alternative stem-cell niche upon PC depletion (55). However, further work is required to uncover the precise signaling network. Promisingly, recent studies have begun to demonstrate that along with IL-1 family cytokines, pyroptotic pores release a variety of damage-associated molecular patterns (DAMPs) which modulate tissue repair pathways (6, 45). It is currently unclear to what extent these DAMPs may serve as a signal to PC in the context of IL-18 deficiency. Finally, the model of ileal organoid culture used in this study, while widespread in the field, is highly dependent on Paneth-cell-derived Wnt3a. Accordingly, IL-18-dependent changes in IEC populations are likely affected by initial imbalances in PC numbers in vitro, such that additional phenotypes are obscured. This is a limitation of our study, and future work utilizing organoids derived from single revSCs, grown with recombinant Wnt3a, is required to better characterize the epithelial-intrinsic role of IL-18 in the regenerating crypt.
In sum, our findings highlight an underappreciated role for epithelial-intrinsic IL-18 signaling in the gut and position a plastic population of IL18R1+ ECC as a key mediator of this program in the recovering crypt.
Materials and Methods
Animal studies were conducted under protocols approved by University of Toronto Committee on Use and Care of Animals. Detailed information relating to all animal experiments, associated tissue collection protocols (81), and analysis can be found in the SI Appendix. For revSC (32) and spatial-transcriptomic (31) cluster identification, cell types were annotated using previously defined cell markers. Generation of murine ileal organoids (82), as well as organoid immunofluorescence staining was performed according to previously described protocols (83). For generation of FlaTox, the original LFn-Fla sequence (84) was obtained from Addgene (RRID:Addgene_84871) and Protective Antigen (PA) was purified as previously described (85). Detailed information relating to organoid experiments, associated protocols, and analysis can be found in the SI Appendix.
Supplementary Material
Appendix 01 (PDF)
Acknowledgments
We thank Bruce Vallance for their gift of Il18−/− mice. NJW was supported by both an Ontario Graduate Scholarship and Canadian Graduate Scholarship, and this project was funded by Grants from the Canadian Institutes for Health Research (CIHR) and Crohn’s Colitis Canada (CCC) to S.E.G. and D.J.P. Portions of this manuscript were developed from the thesis of N.J.W.
Author contributions
N.J.W., D.K.T., A.R., O.S., S.G., D.J.P., and S.E.G. designed research; N.J.W., D.K.T., A.R., O.S., and S.G. performed research; N.J.W., D.K.T., A.R., and S.G. contributed new reagents/analytic tools; N.J.W., D.K.T., A.R., O.S., and S.G. analyzed data; D.J.P. and S.E.G. supervised Research; and N.J.W., D.K.T., and S.G. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
This article is a PNAS Direct Submission. I.R. is a guest editor invited by the Editorial Board.
Data, Materials, and Software Availability
All study data are included in the article and/or supporting information.
Supporting Information
References
- 1.Broz P., Dixit V. M., Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 16, 407–420 (2016). [DOI] [PubMed] [Google Scholar]
- 2.Kayagaki N., et al. , Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 526, 666–671 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Lamkanfi M., Dixit V. M., Mechanisms and functions of inflammasomes. Cell 157, 1013–1022 (2014). [DOI] [PubMed] [Google Scholar]
- 4.Shi J., et al. , Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526, 660–665 (2015). [DOI] [PubMed] [Google Scholar]
- 5.Crowley S. M., et al. , Intestinal restriction of Salmonella Typhimurium requires caspase-1 and caspase-11 epithelial intrinsic inflammasomes. PLoS Pathog. 16, e1008498 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rauch I., et al. , NAIP-NLRC4 inflammasomes coordinate intestinal epithelial cell expulsion with Eicosanoid and IL-18 release via activation of caspase-1 and -8. Immunity 46, 649–659 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mitchell P. S., et al. , NAIP–NLRC4-deficient mice are susceptible to shigellosis. eLife 9, e59022 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Harrison O. J., et al. , Epithelial-derived IL-18 regulates Th17 cell differentiation and Foxp3(+) Treg cell function in the intestine. Mucosal Immunol. 8, 1226–1236 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Weiss E. S., et al. , Interleukin-18 diagnostically distinguishes and pathogenically promotes human and murine macrophage activation syndrome. Blood 131, 1442–1455 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chudnovskiy A., et al. , Host-protozoan interactions protect from mucosal infections through activation of the inflammasome. Cell 167, 444–456 e14 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Escalante N. K., et al. , The common mouse protozoa Tritrichomonas muris alters mucosal T cell homeostasis and colitis susceptibility. J. Exp. Med. 213, 2841–2850 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lemire P., et al. , The NLR protein NLRP6 does not impact gut microbiota composition. Cell Rep. 21, 3653–3661 (2017). [DOI] [PubMed] [Google Scholar]
- 13.Hoshino K., et al. , Cutting edge: Generation of IL-18 receptor-deficient mice: Evidence for IL-1 receptor-related protein as an essential IL-18 binding receptor. J. Immunol. 162, 5041–5044 (1999). [PubMed] [Google Scholar]
- 14.Adachi O., et al. , Targeted disruption of the MyD88 gene results in loss of IL-1- and IL- 18-mediated function. Immunity 9, 143–150 (1998). [DOI] [PubMed] [Google Scholar]
- 15.Born T. L., Thomassen E., Bird T. A., Sims J. E., Cloning of a novel receptor subunit, AcPL, required for interleukin-18 signaling. J. Biol. Chem. 273, 29445–29450 (1998). [DOI] [PubMed] [Google Scholar]
- 16.Nowarski R., et al. , Epithelial IL-18 equilibrium controls barrier function in colitis. Cell 163, 1444–1456 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Okamura H., et al. , A novel costimulatory factor for gamma interferon induction found in the livers of mice causes endotoxic shock. Infect. Immun. 63, 3966–3972 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Robinson D., et al. , IGIF does not drive Th1 development but synergizes with IL-12 for interferon-gamma production and activates IRAK and NFkappaB. Immunity 7, 571–581 (1997). [DOI] [PubMed] [Google Scholar]
- 19.Smeltz R. B., Chen J., Hu-Li J., Shevach E. M., Regulation of interleukin (IL)-18 receptor alpha chain expression on CD4(+) T cells during T helper (Th)1/Th2 differentiation. Critical downregulatory role of IL-4. J. Exp. Med. 194, 143–153 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sellin M. E., Maslowski K. M., Maloy K. J., Hardt W. D., Inflammasomes of the intestinal epithelium. Trends Immunol. 36, 442–450 (2015). [DOI] [PubMed] [Google Scholar]
- 21.Huber S., et al. , IL-22BP is regulated by the inflammasome and modulates tumorigenesis in the intestine. Nature 491, 259–263 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang Z., et al. , IL-22–induced cell extrusion and IL-18–induced cell death prevent and cure rotavirus infection. Sci. Immunol. 5, eabd2876 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chiang H.-Y., et al. , IL-22 initiates an IL-18-dependent epithelial response circuit to enforce intestinal host defence. Nat. Commun. 13, 874 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang B., et al. , Prevention and cure of rotavirus infection via TLR5/NLRC4–mediated production of IL-22 and IL-18. Science 1979, 861–865 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Novick D., et al. , Interleukin-18 binding protein: A novel modulator of the Th1 cytokine response. Immunity 10, 127–136 (1999). [DOI] [PubMed] [Google Scholar]
- 26.Ludwiczek O., et al. , Elevated systemic levels of free interleukin-18 (IL-18) in patients with Crohn’s disease. Eur. Cytokine Netw. 16, 27–33 (2005). [PubMed] [Google Scholar]
- 27.Rivas M. A., et al. , Deep resequencing of GWAS loci identifies independent rare variants associated with inflammatory bowel disease. Nat. Genet. 43, 1066–73 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Festen E. A. M., et al. , A meta-analysis of genome-wide association scans identifies IL18RAP, PTPN2, TAGAP, and PUS10 as shared risk loci for Crohn’s disease and celiac disease. PLoS Genet. 7, e1001283 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhernakova A., et al. , Genetic analysis of innate immunity in Crohn’s disease and ulcerative colitis identifies two susceptibility loci harboring CARD9 and IL18RAP. Am. J. Hum. Genet. 82, 1202–10 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tsang D. K. L., et al. , A single-cell survey of the microbial impacts on the mouse small intestinal epithelium. Gut. Microbes. 14, 2108281 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moor A. E., et al. , Spatial reconstruction of single enterocytes uncovers broad zonation along the intestinal villus axis. Cell 175, 1156–1167.e15 (2018). [DOI] [PubMed] [Google Scholar]
- 32.Ayyaz A., et al. , Single-cell transcriptomes of the regenerating intestine reveal a revival stem cell. Nature 569, 121–125 (2019). [DOI] [PubMed] [Google Scholar]
- 33.Akiba Y., et al. , FFA2 activation combined with ulcerogenic COX inhibition induces duodenal mucosal injury via the 5-HT pathway in rats. Am. J. Physiol. Gastrointest. Liver Physiol. 313, G117–G128 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gehart H., et al. , Identification of enteroendocrine regulators by real-time single-cell differentiation mapping. Cell 176, 1158–1173.e16 (2019). [DOI] [PubMed] [Google Scholar]
- 35.Mellitzer G., et al. , Loss of enteroendocrine cells in mice alters lipid absorption and glucose homeostasis and impairs postnatal survival. J. Clin. Invest. 120, 1708–1721 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tsakmaki A., Fonseca Pedro P., Pavlidis P., Hayee B. H., Bewick G. A., ISX-9 manipulates endocrine progenitor fate revealing conserved intestinal lineages in mouse and human organoids. Mol. Metab. 34, 157–173 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Koopman N., et al. , The multifaceted role of serotonin in intestinal homeostasis. Int. J. Mol. Sci. 22, 9487 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bellono N. W., et al. , Enterochromaffin cells are gut chemosensors that couple to sensory neural pathways. Cell 170, 185–198.e16 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen Z., et al. , Interleukin-33 promotes serotonin release from enterochromaffin cells for intestinal homeostasis. Immunity 54, 151–163.e6 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yano J. M., et al. , Indigenous bacteria from the gut microbiota regulate host serotonin biosynthesis. Cell 161, 264–276 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wikoff W. R., et al. , Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc. Natl. Acad. Sci. U.S.A. 106, 3698–3703 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sjögren K., et al. , The gut microbiota regulates bone mass in mice. J. Bone Miner. Res. 27, 1357–1367 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Engelstoft M. S., et al. , Research resource: A Chromogranin A reporter for serotonin and histamine secreting enteroendocrine cells. Mol. Endocrinol. 29, 1658–1671 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Volchuk A., Ye A., Chi L., Steinberg B. E., Goldenberg N. M., Indirect regulation of HMGB1 release by gasdermin D. Nature Commun. 11, 1–11 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mehrotra P., et al. , Oxylipins and metabolites from pyroptotic cells act as promoters of tissue repair. Nature 631, 8019631 (2024). [DOI] [PubMed] [Google Scholar]
- 46.Sei Y., et al. , Mature enteroendocrine cells contribute to basal and pathological stem cell dynamics in the small intestine. Am. J. Physiol. Gastrointest. Liver Physiol. 315, G495–G510 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yan K. S., et al. , Intestinal enteroendocrine lineage cells possess homeostatic and injury-inducible stem cell activity. Cell Stem. Cell 21, 78–90.e6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gross S., et al. , Nkx2.2 is expressed in a subset of enteroendocrine cells with expanded lineage potential. Am. J. Physiol. Gastrointest. Liver Physiol. 309, G975–G987 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gregorieff A., Liu Y., Inanlou M. R., Khomchuk Y., Wrana J. L., Yap-dependent reprogramming of Lgr5+ stem cells drives intestinal regeneration and cancer. Nature 526, 7575526 (2015). [DOI] [PubMed] [Google Scholar]
- 50.Sato T., et al. , Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts. Nature 469, 415–418 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fevr T., Robine S., Louvard D., Huelsken J., Wnt/β-catenin is essential for intestinal homeostasis and maintenance of intestinal stem cells. Mol. Cell Biol. 27, 7551–7559 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.van Es J. H., et al. , A critical role for the Wnt effector Tcf4 in adult intestinal homeostatic self-renewal. Mol. Cell. Biol. 32, 1918–1927 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Böttcher A., et al. , Non-canonical Wnt/PCP signalling regulates intestinal stem cell lineage priming towards enteroendocrine and Paneth cell fates. Nature Cell Biol. 23, 123 (2021). [DOI] [PubMed] [Google Scholar]
- 54.Kagermeier-Schenk B., et al. , Waif1/5T4 inhibits Wnt/β-catenin signaling and activates noncanonical Wnt pathways by modifying LRP6 subcellular localization. Dev. Cell 21, 1129–1143 (2011). [DOI] [PubMed] [Google Scholar]
- 55.Van Es J. H., et al. , Enteroendocrine and tuft cells support Lgr5 stem cells on Paneth cell depletion. Proc. Natl. Acad. Sci. U.S.A. 116, 26599–26605 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen L., et al. , TGFβ1 induces fetal reprogramming and enhances intestinal regeneration. Cell Stem. Cell 30, 1520–1537.e8 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sato T., et al. , Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 459, 262–265 (2009). [DOI] [PubMed] [Google Scholar]
- 58.Serra D., et al. , Self-organization and symmetry breaking in intestinal organoid development. Nature 569, 66–72 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Farin H. F., et al. , Visualization of a short-range Wnt gradient in the intestinal stem-cell niche. Nature 530, 340–343 (2016). [DOI] [PubMed] [Google Scholar]
- 60.Noah T. K., Kazanjian A., Whitsett J., Shroyer N. F., SAM pointed domain ETS factor (SPDEF) regulates terminal differentiation and maturation of intestinal goblet cells. Exp. Cell Res. 316, 452–465 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gregorieff A., et al. , The Ets-domain transcription factor Spdef promotes maturation of goblet and Paneth cells in the intestinal epithelium. Gastroenterology 137, 1333–1345.e3 (2009). [DOI] [PubMed] [Google Scholar]
- 62.Colozza G., et al. , Intestinal Paneth cell differentiation relies on asymmetric regulation of Wnt signaling by Daam1/2. Sci. Adv. 9, eadh9673 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Noah T. K., Shroyer N. F., Notch in the intestine: Regulation of homeostasis and pathogenesis. Annu. Rev. Physiol. 75, 263–288 (2013). [DOI] [PubMed] [Google Scholar]
- 64.Sellin M. E., et al. , Epithelium-intrinsic NAIP/NLRC4 inflammasome drives infected enterocyte expulsion to restrict Salmonella replication in the intestinal mucosa. Cell Host. Microbe 16, 237–248 (2014). [DOI] [PubMed] [Google Scholar]
- 65.Fattinger S. A., et al. , Epithelium-autonomous NAIP/NLRC4 prevents TNF-driven inflammatory destruction of the gut epithelial barrier in Salmonella-infected mice. Mucosal Immunol. 14, 615–629 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Haber A. L., et al. , A single-cell survey of the small intestinal epithelium. Nature 551, 333–339 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Price A. E., et al. , A map of Toll-like receptor expression in the intestinal epithelium reveals distinct spatial, cell type-specific, and temporal patterns. Immunity 49, 560–575.e6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kim C.-K., Yang V. W., Bialkowska A. B., The role of intestinal stem cells in epithelial regeneration following radiation-induced gut injury. Curr. Stem. Cell Rep. 3, 320–332 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Deng F., Wu Z., Zou F., Wang S., Wang X., The Hippo–YAP/TAZ signaling pathway in intestinal self-renewal and regeneration after injury. Front. Cell Dev. Biol. 10, 894737 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Samperio Ventayol P., et al. , Bacterial detection by NAIP/NLRC4 elicits prompt contractions of intestinal epithelial cell layers. Proc. Nat. Acad. Sci. 118, e2013963118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yu S., et al. , Paneth cell multipotency induced by notch activation following injury. Cell Stem. Cell 23, 46–59.e5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schmitt M., et al. , Paneth cells respond to inflammation and contribute to tissue regeneration by acquiring stem-like features through SCF/c-kit signaling. Cell Rep. 24, 2312–2328.e7 (2018). [DOI] [PubMed] [Google Scholar]
- 73.Verhagen M. P., et al. , Non-stem cell lineages as an alternative origin of intestinal tumorigenesis in the context of inflammation. Nature Genet. 56, 1456–1467 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Middelhoff M., et al. , Prox1-positive cells monitor and sustain the murine intestinal epithelial cholinergic niche. Nat. Commun. 11, 111 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Westphalen C. B., et al. , Long-lived intestinal tuft cells serve as colon cancer–initiating cells. J. Clin. Invest. 124, 1283–1295 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cox C. B., et al. , IL-1R1-dependent signaling coordinates epithelial regeneration in response to intestinal damage. Sci. Immunol. 6, 8856 (2021). [DOI] [PubMed] [Google Scholar]
- 77.Dinarello A., et al. , IL-38 regulates intestinal stem cell homeostasis by inducing WNT signaling and beneficial IL-1β secretion. Proc. Natl. Acad. Sci. U.S.A. 120, e2306476120 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hu B., et al. , The DNA-sensing AIM2 inflammasome controls radiation-induced cell death and tissue injury. Science 1979, 765–768 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ha C. T., et al. , Circulating interleukin-18 as a biomarker of total-body radiation exposure in mice, minipigs, and nonhuman primates (NHP). PLoS One 9, e109249 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Li X., et al. , IL-18 binding protein (IL-18BP) as a novel radiation countermeasure after radiation exposure in mice. Sci. Rep. 10, 18674 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lavoie B., et al. , Gut-derived serotonin contributes to bone deficits in colitis. Pharmacol. Res. 140, 75–84 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mazzone R. J., et al. , NLRP1B expressed in intestinal epithelial cells is refractory to activation with Val-boro-Pro. Microbes Infect. 26, 105398 (2024), 10.1016/J.MICINF.2024.105398. [DOI] [PubMed] [Google Scholar]
- 83.Foerster E. G., et al. , ATG16L1 protects from interferon-γ-induced cell death in the small intestinal crypt. Mucosal Immunol. 16, 135–152 (2023). [DOI] [PubMed] [Google Scholar]
- 84.Rauch I., et al. , NAIP proteins are required for cytosolic detection of specific bacterial ligands in vivo. J. Exp. Med. 213, 657–665 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Batty S., Chow E. M. C., Kassam A., Der S. D., Mogridge J., Inhibition of mitogen-activated protein kinase signalling by Bacillus anthracis lethal toxin causes destabilization of interleukin-8 mRNA. Cell Microbiol. 8, 130–138 (2006). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Data Availability Statement
All study data are included in the article and/or supporting information.