

Abstract
Tumor protein p53 mutated/abnormal (p53abn) endometrial carcinomas account for over 50% of deaths but comprise only 15% of all endometrial carcinomas. Most patients show limited response to standard‐of‐care chemotherapy with or without radiotherapy, and only a minority of cases are amenable to targeted therapies like poly‐ADP ribose polymerase (PARP) inhibitors and HER2‐directed therapies. Recent immunotherapy clinical trials have demonstrated remarkable efficacy, not only in mismatch repair deficient (MMRd) tumors but also in a subset of mismatch repair‐proficient (MMRp) tumors. However, the immune microenvironment and its relationship to other therapeutic targets in MMRp endometrial carcinoma remains poorly understood. Here, we characterize the immune microenvironment of p53abn endometrial carcinoma, the most clinically aggressive subtype of MMRp endometrial carcinoma, and correlate antitumor immune signatures with other targetable alterations. We accrued 256 treatment‐naïve p53abn endometrial carcinomas and systemically profiled T‐cell, B‐cell, myeloid, and tumor‐cell populations with multiplex immunofluorescence to assess the tissue localization and functional status of immune cells. Shallow whole‐genome sequencing was performed on a subset of 126 cases. Patterns of immune infiltration were compared to survival outcomes and mutational signatures. Mixture modeling divided p53abn endometrial carcinoma into tumor‐infiltrating lymphocyte (TIL)‐rich and TIL‐poor subsets. Over 50% of tumors were TIL‐rich. TIL‐rich cases overexpressed targetable immune evasion molecules and were associated with longer overall and disease‐specific survival in multivariate analysis. This effect was particularly pronounced in advanced stage disease and in patients who did not receive adjuvant chemotherapy. TIL did not associate with homologous recombination deficient mutational signatures or HER2 amplification. Our findings demonstrate a biological rationale for immunotherapy in a substantial subset of patients with p53abn endometrial cancer and may help inform combination therapies with immune checkpoint inhibition, PARP inhibitors, and anti‐HER2 agents. © 2025 The Author(s). The Journal of Pathology published by John Wiley & Sons Ltd on behalf of The Pathological Society of Great Britain and Ireland.
Keywords: endometrial cancer, immunotherapy, tumor microenvironment, PARP inhibitors, HER2, tumor‐infiltrating lymphocytes, p53abn
Introduction
Endometrial cancer is the second most common type of gynecologic malignancy worldwide and the most common gynecologic malignancy in North America [1]. Molecular classification stratifies endometrial cancers into four prognostically distinct subtypes: DNA polymerase epsilon, catalytic subunit mutated (POLEmut), mismatch repair‐deficient (MMRd), tumor protein p53 mutated/abnormal (p53abn), and no specific molecular profile (NSMP) [2, 3, 4, 5]. p53abn cancers have the worst prognosis, comprising only 15% of all endometrial cancers but accounting for 50%–70% of mortalities [3, 6, 7, 8], with extrauterine involvement in over 50% of cases [5, 9]. Most p53abn cancers recur within 5 years of standard‐of‐care carboplatin‐paclitaxel chemotherapy with or without adjuvant radiotherapy, highlighting the need for alternative therapeutic options [6, 7, 10]. While several studies have investigated associations between immune infiltration and survival in endometrial carcinomas [11, 12, 13, 14], large‐scale studies investigating the immune microenvironment of p53abn tumors are lacking.
The interplay between different immune cell types within the tumor microenvironment determines the effectiveness of antitumor immunity. CD8+ cytotoxic T lymphocytes (CTL) are the main cells that recognize and kill tumor cells. Dendritic cells, macrophages and B cells are professional antigen‐presenting cells that activate CTL and CD4+ helper T cells (Th), which in turn secrete cytokines that potentiate the activity of CTL and antitumor macrophages. CD68+ macrophages can either help activate CTL, by presenting antigens and co‐stimulatory molecules, or inhibit CTL, by presenting antigens along with inhibitory ligands [15, 16]. CD4+FOXP3+ regulatory T cells (Tregs) inhibit antitumor immunity by secreting cytokines that block CTL maturation and induce macrophages to express immune‐inhibiting molecules [17, 18]. Finally, CD20+ B cells and CD79a+ plasma cells may potentiate antitumor immunity via multiple mechanisms [19, 20]. Key functional molecules that inhibit antitumor immunity include indoleamine 2,3‐dioxygenase 1 (IDO1) and programmed death‐ligand 1 (PD‐L1). Macrophages and tumor cells may express IDO1, which limits tryptophan availability, thereby inducing CTL death and proliferation of Tregs [21]. Tumors and macrophages may also express PD‐L1, blocking CTL‐mediated killing by ligating programmed cell death‐1 (PD‐1) on CTL and Th [22]. Cytokines such as IFN‐gamma, released by CTL upon tumor cell recognition, in turn cause upregulation of both IDO1 and PD‐L1 [22]. Therefore, PD‐L1 expression has been used as a marker of an active antitumor immune response across multiple different cancer types [23, 24, 25].
Immune checkpoint inhibitors disrupt the PD‐1/PD‐L1 pathway, reactivating exhausted T cells to attack tumor cells. These treatments are particularly effective in tumors with elevated numbers of mutations that generate neoantigens [26]. In endometrial cancer, POLEmut and MMRd tumors have over 10 times as many mutations as p53abn and NSMP tumors [2, 13] and, correspondingly, higher TIL densities [14]. While systemic therapy is often unnecessary in POLEmut cancers due to exceptionally favorable outcomes with hysterectomy alone, anti‐PD‐1 inhibitors have demonstrated remarkable efficacy in advanced, recurrent, and persistent MMRd endometrial cancers, even after multiple lines of therapy [27, 28, 29, 30]. More recently, Mirza et al [31] and Eskander et al [32] demonstrated the benefits of adding PD‐1 inhibitors to chemotherapy in both MMRd and in MMR‐proficient (MMRp) endometrial carcinomas. Subgroup analysis showed that the benefit in MMRp endometrial carcinomas was driven by p53abn cases [33]. As a result, pembrolizumab and dostarlimab have received FDA approval for the treatment of endometrial carcinoma, regardless of subtype. However, the factors underlying response to immune checkpoint inhibitors in p53abn endometrial cancer remain poorly understood.
Additional classes of targeted therapies under investigation in p53abn endometrial cancer include PARP inhibitors and HER2‐directed antibodies [34]. PARP inhibitors have become standard of care in multiple cancer types with BRCA1/BRCA2 mutations or homologous recombination‐deficiency (HRD) [35]. In high‐grade serous ovarian cancer (HGSOC), HRD tumors have higher immunogenicity than non‐HRD tumors. Additionally, markers of adaptive immunity are associated with longer overall survival in HRD but not non‐HRD tumors [36]. In p53abn endometrial cancer, approximately 25% of cases show evidence of HRD and fewer than 5% have BRCA1/2 mutations [37, 38]. Combination immune checkpoint and PARP inhibition has shown benefit in HRD endometrial cancers [39]. However, the relative immunogenicity of these cases has yet to be explored. p53abn endometrial cancers show a higher incidence of HER2 amplification than other molecular subtypes, with up to 25% of cases amplified [40]. Several phase I and II clinical trials assessing HER2‐targeted therapies are in progress [41, 42, 43, 44, 45], but to date, studies investigating the relative immunogenicity of HER2‐amplified endometrial carcinomas are lacking. Thus, a deeper understanding of the relationship between HRD, HER2 status, and the immune microenvironment in p53abn endometrial cancers may help inform combination therapies involving PARP inhibitor, HER2 blockade, and immunotherapy in clinical trials [39].
To understand the clinical relevance of the immune response to p53abn endometrial cancer, we systematically profiled the immune cell composition of n = 256 p53abn endometrial cancers with multiplex immunofluorescence to detect CTL, Th, Tregs, B cells, plasma cells, and macrophages. Furthermore, we evaluated the expression patterns of PD1, PD‐L1, and IDO1 – three pharmacologically actionable immunosuppressive molecules with translational relevance to current clinical trials in endometrial cancer. Finally, we investigated the relationship between immune composition, HRD, and HER2 expression/amplification in p53abn endometrial cancer.
Materials and methods
Ethics approval
Ethics approval was obtained from The University of British Columbia (UBC) Research Ethics Board (approval number H22‐03493) and the Institutional Review Boards from each center that supplied tissue.
Data acquisition
Sample acquisition and tissue microarray analysis construction
The cohort consisted of 256 treatment‐naive p53abn endometrial carcinomas retrospectively collected between 1993 and 2017 in Vancouver and in 10 tertiary and 19 community centers from across Canada, for which sufficient tissue for coring was available [3, 4, 46]. Clinicopathologic and outcome data were collected by chart review (end of follow‐up period: August 2023, median follow‐up: 3.52 years). All cancers were classified as p53abn according to the ProMisE algorithm [3], by immunohistochemistry (IHC) for TP53 and MMR proteins, and next‐generation sequencing for POLE hotspot mutations. Patients who received neoadjuvant therapy were excluded. Representative formalin‐fixed paraffin‐embedded tissue samples of p53abn endometrial carcinomas were cored at 0.6 mm in diameter in duplicate and arrayed as described previously [3]. All procedures involving human subjects were in accordance with the Declaration of Helsinki [47].
Multiplex immunofluorescence
In brief, tissue microarrays (TMAs) were cut at 4 μm for immunofluorescence. Slides were first deparaffinized with xylene and graded alcohol solutions (Thermo Fisher Scientific, Burnaby, BC, Canada), then incubated for 20 min in 10% neutral buffered formalin (Millipore Sigma Canada Ltd., Oakville, ON, Canada) followed by a wash in deionized water. Diva Decloaker heat retrieval solution (Biocare Medical, Pacheco, CA, USA) was used for antigen retrieval, and slides were stained in an Intellipath FLX Autostainer (Biocare Medical) in six rounds. Endogenous peroxidases were blocked with peroxidase‐1 (Biocare Medical), and nonspecific background staining was reduced using Background Sniper (Biocare Medical). Antibodies were diluted in Da Vinci Green Diluent (Biocare Medical) and incubated according to the times and concentrations specified in Supplementary materials and methods. Mach2 Rabbit‐HRP polymer or Mach2 Mouse‐HRP polymer (Biocare Medical) was applied, followed by the addition of the corresponding fluorophore for each round, as detailed in Supplementary materials and methods. After each round of staining, reagent proteins were denatured by microwaving in AR6 (Akoya Biosciences, Marlborough, MA, USA) in preparation for the next round. Nuclei were counterstained with Spectral DAPI (Akoya Biosciences), and a coverslip was mounted using Prolong Diamond coverslipping medium (Thermo Fisher Scientific). Both panels (B cell/T cell: CD8, CD3, FOXP3, CD20, CD79a, panCK; adaptive resistance: CD68, CD8, IDO1, PD‐L1, PD‐1, panCK) were imaged using the Vectra Polaris multispectral imaging system (Akoya Biosciences) and scanned using ‘motif’ settings. Detailed methods are described in Supplementary materials and methods.
Computational analysis
Cell and region counting
Computational analysis was performed using HALO (Version 3.6.4134.95, RRID:SCR_018350 Indica Labs, Albuquerque, NM, USA). In brief, three tissue segmentation and cell segmentation/phenotyping algorithms were trained using 10–30 representative images for each algorithm. Regions were classified as epithelial, stromal, glass, or other (including necrosis) for tissue segmentation. All areas of stroma within 0.6 mm of tumor epithelium (spot size) were considered tumor stroma. The mean and standard deviation of cell count and region area across all algorithms were calculated for each core and reviewed by a technologist. Any core with a standard deviation of >5 among the three algorithms, or with abnormal features as identified by the technologists, was flagged for review and manual annotation by a pathology resident and/or subspecialist gynecological pathologist. All personnel involved were blinded to patient identifiers during this process. Sarcomatoid areas in carcinosarcomas were considered epithelial, as all tumor cells within a carcinosarcoma originate from a common precursor epithelial cell. The intraepithelial area that was negative for CD68 and CD8 was designated as tumor cell area. All immune cells touching tumor cells were considered intraepithelial. TIL counts and areas for all duplicate cores from each sample were added together. Densities were computed as counts divided by areas, and log‐transformed values were computed using the formula log(TIL density + 1).
Mixture modeling of cell counts
TIL counts for epithelial and stromal areas were clustered according to a negative binomial mixture model, with the optimal number of clusters chosen by a Dirichlet process [48]. Further details of the methods are described in Supplementary materials and methods.
Shallow whole‐genome copy number analysis
Copy number (CN) signatures were generated as described in Jamieson et al [38] using the process outlined by Macintyre et al [49], as implemented by the Utanos R package (https://github.com/Huntsmanlab/utanos). In brief, CN features, such as DNA segment size, CN change points, segment CN, breakpoints per 10 Mb, length of segments with oscillating CN, and breakpoints per chromosome arm, were extracted using ExtractCopyNumberFeatures. The optimum number of signatures was determined by ChooseNumberSignatures; subsequently, GenerateSignatures was applied to the selected number of signatures. Finally, CN signatures for each sample were generated according to signature exposures. Five CN signatures were identified in our p53abn cohort, one of which (Vancouver Signature 5, VS5) was associated with BRCA1/2 CN loss [38]. ERBB2 (alias symbol, HER2) gain was defined as an absolute copy number (ACN) between 2.5 and 4.5, HER2 amplification as ACN between 4.5 and 8.5, and HER2 high‐level amplification as ACN > 8.5. The ACN was determined from relative CN profiles using the Rascal (Relative Absolute Copy Number) algorithm [50]. Immunohistochemical staining for HER2 was also performed using the VENTANA® anti‐HER2/neu (4B5) rabbit monoclonal primary antibody (Roche, Catalog No.: 790–2,991, RRID: AB_2335975, Laval, QC, Canada), at a dilution of 1:8 (ready to use).
Statistical analyses
The reporting criteria for Reporting Recommendations for Tumor Marker Prognostic Studies (REMARK) [51] were followed. All statistical analyses were performed in R version 4.3.2 [52], and p values <0.05 were considered statistically significant. Cases with missing data were excluded. Cox proportional hazards analysis was performed with the survival package (version 3.5‐7) [53, 54]. Overall, progression‐free and disease‐specific survival were evaluated as endpoints. No additional explanatory variables were considered for inclusion in multivariate models. Proportional hazards assumptions were evaluated with weighted Schoenfeld residuals [55]. Hazard ratios (HRs) and p values from the survival package were validated for consistency with a Bayesian implementation of the proportional hazards model. Sample size estimation was not performed. Hierarchical clustering was performed using Euclidean pairwise distance and Ward's linkage method [56].
Code availability
Code associated with this project is publicly available at https://github.com/Irrationone/tfri_halo.
Results
Cohort
We assembled a cohort of 256 treatment‐naive p53abn endometrial carcinomas diagnosed between 1993 and 2017 from institutional cohorts as well as a pan‐Canadian initiative (Figure 1) [14]. Histotypes of p53abn tumors included serous (n = 136), endometrioid (n = 52), carcinosarcoma (n = 31), clear cell (n = 15), mixed (n = 17), and other (n = 5). Patients received various treatment regimens in accordance with standard of care at the times of diagnoses, with most patients receiving adjuvant chemotherapy and a smaller proportion receiving adjuvant brachytherapy or radiotherapy (Figure 1). No patients received immunotherapy or neoadjuvant chemotherapy.
Figure 1.

Graphical overview of clinicopathologic parameters and data types for each sample, showing relationships between different parameters. Missing values are colored white. Grade 3 includes histotypes defined as high grade. Myo, depth of myometrial invasion; LVI, lymphovascular invasion.
Clustering based on immune composition
We performed multiplex immunofluorescence and automated image analysis to segment tumors into epithelial, stromal, and ‘other’ regions, followed by quantification of lymphocyte subsets, including CTL (CD3+CD8+), Th (CD3+CD8−FOXP3−), Treg (CD3+CD8−FOXP3+), B cells (CD20+CD79a+), and plasma cells (CD20−CD79a+) (Figure 2A). Cells within epithelial and stromal regions were counted separately. All TIL densities were highly correlated. Higher correlations between TIL types were seen within specific regions, and the highest correlations were seen between TIL types from the same cell lineage within regions (supplementary material, Figure S1).
Figure 2.
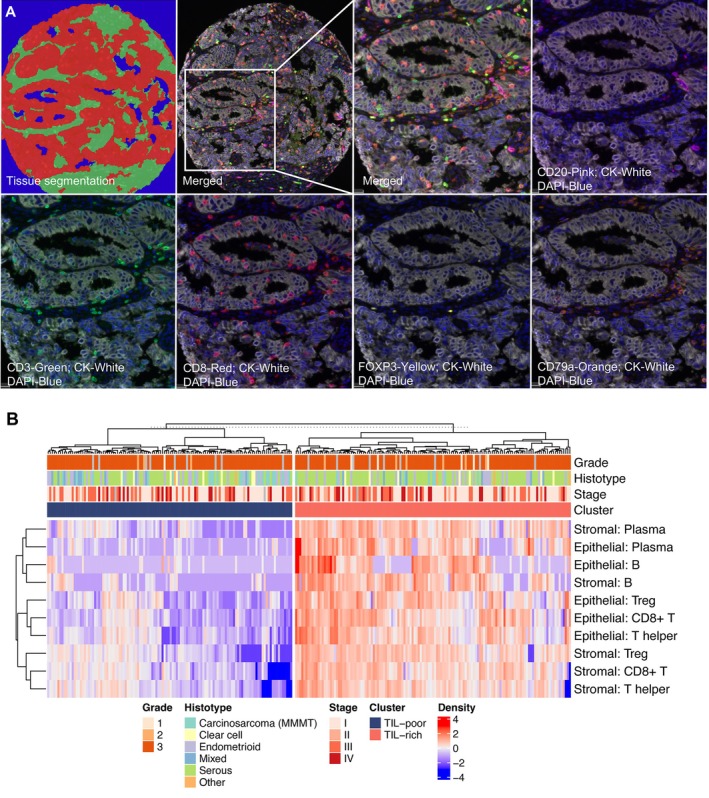
Derivation of TIL‐rich and TIL‐poor groups. (A) Representative multiplex immunofluorescence image of B/T cell panel. Representative segmentation of tumor (red), stromal (green), and glass/necrosis (blue) are shown in top left panel. Cytokeratin (white) and DAPI (blue) are shown in each image along with a single immune marker as annotated. (B) Heatmap of log‐transformed epithelial and stromal TIL densities for each sample. Results of hierarchical clustering by sample (top dendrogram) are split by TIL cluster.
Next, we clustered tumors based on epithelial and stromal counts normalized by region area with a negative binomial mixture model. As the number of immunologically distinct clusters was unknown a priori, the model was formulated to determine the optimal number of clusters using a Dirichlet process [48]. Two distinct clusters were found: TIL‐rich (135/255) and TIL‐poor (120/255). All T and B cell subsets infiltrated both tumor stroma and epithelium in TIL‐rich tumors (Figure 2B). We found no evidence of a subgroup with stroma‐restricted TIL, contrary to our findings of a distinct subgroup of tumors with stroma‐restricted TIL in high‐grade serous ovarian carcinoma [36]. TIL cluster was also not associated with the TP53 mutation type (Chi‐square p = 0.81, supplementary material, Table S2).
TIL‐rich tumors are associated with longer survival
We next assessed the relationships between TIL cluster, survival, and other clinicopathologic parameters in our cohort of p53abn endometrial carcinomas. Using multivariate Cox proportional hazards analysis, significant associations were identified between overall survival and adjuvant chemotherapy (HR 0.58, p = 0.041) and International Federation of Gynecology and Obstetrics (FIGO) stage [51] (HR 3.39 and 11.0 for stage III and IV compared to stage I, respectively, both p < 0.001), while older age (range: 35–94 years, median: 69 years) trended toward shorter overall survival (HR 1.02, p = 0.14). Importantly, TIL‐rich tumors were associated with prolonged overall (HR 0.63, p = 0.031) and disease‐specific survival (HR 0.58, p = 0.037) in multivariate Cox proportional‐hazards analysis accounting for age at diagnosis, FIGO stage [57], and adjuvant treatment (Table 1) and trended toward longer progression‐free survival (HR 0.74, p = 0.15). When TIL subtypes were assessed individually, each subtype was associated with mean HRs between 0.77 and 0.98, with statistical significance only in stromal CD8+ T cells, Th, and Tregs (supplementary material, Table S1). In contrast, univariate Kaplan–Meier analyses failed to identify a significant association between TIL‐rich cases or TIL densities and overall, progression‐free, or disease‐specific survival (all p > 0.175) (Figure 3A).
Table 1.
Hazard ratios, 95% confidence intervals, and significance values for TIL cluster and each clinicopathologic variable included in Cox proportional hazards multivariate survival analysis.
| Overall survival | Progression‐free survival | Disease‐specific survival | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Characteristic | HR | 95% CI | p | HR | 95% CI | p | HR | 95% CI | p |
| Age | 1.02 | 0.99, 1.04 | 0.14 | 1.00 | 0.98, 1.03 | 0.68 | 1.01 | 0.98, 1.03 | 0.57 |
| TIL | 0.63 | 0.42, 0.96 | 0.031 | 0.74 | 0.49, 1.12 | 0.15 | 0.58 | 0.35, 0.97 | 0.037 |
| Chemotherapy | 0.58 | 0.34, 0.98 | 0.041 | 0.98 | 0.57, 1.68 | 0.93 | 0.69 | 0.36, 1.34 | 0.28 |
| Radiotherapy | 0.82 | 0.48, 1.43 | 0.49 | 0.83 | 0.50, 1.38 | 0.47 | 0.95 | 0.49, 1.85 | 0.88 |
| Brachytherapy | 1.10 | 0.54, 2.23 | 0.80 | 1.08 | 0.61, 1.93 | 0.78 | 1.16 | 0.52, 2.58 | 0.72 |
| Stage | |||||||||
| I | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| II | 2.58 | 0.73, 9.06 | 0.14 | 1.05 | 0.24, 4.58 | 0.95 | 1.68 | 0.21, 13.6 | 0.63 |
| III | 3.39 | 1.94, 5.94 | <0.001 | 4.19 | 2.48, 7.08 | <0.001 | 5.27 | 2.54, 10.9 | <0.001 |
| IV | 11.0 | 5.84, 20.7 | <0.001 | 9.10 | 4.86, 17.1 | <0.001 | 16.6 | 7.54, 36.5 | <0.001 |
Abbreviations: CI, confidence interval; HR, hazard ratio.
Figure 3.
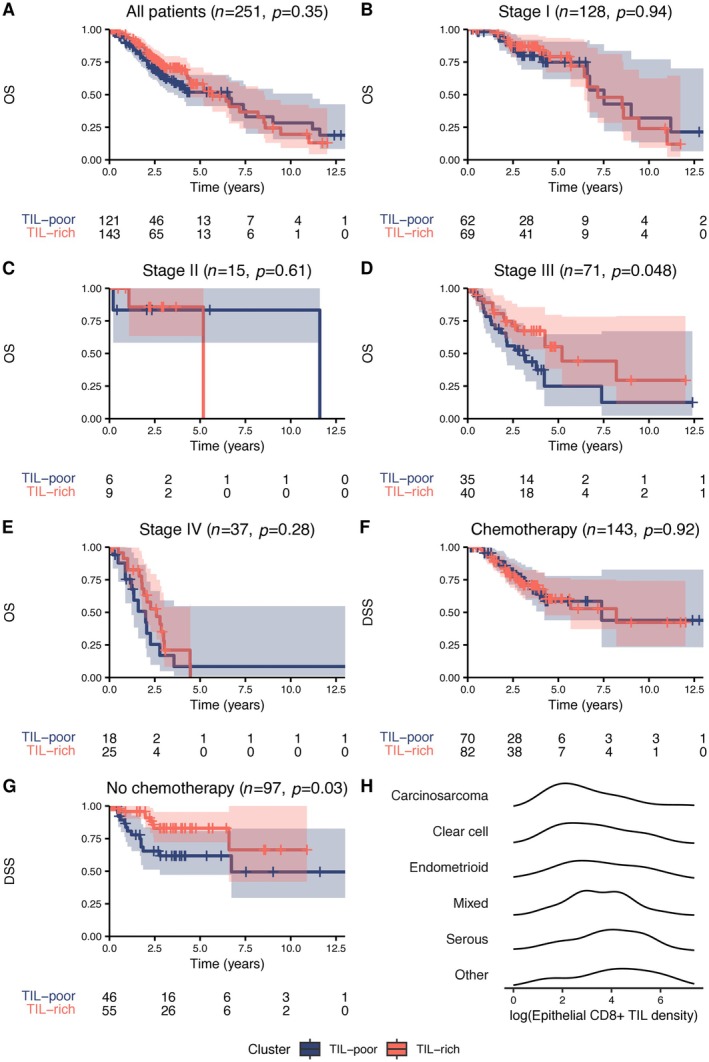
Univariate Kaplan–Meier survival curves and log‐rank p values of overall survival (OS) and TIL cluster. (A) All tumors in cohort with known stage and chemotherapy status. (B–E) Tumors grouped by stage. (F and G) Univariate Kaplan–Meier survival curves and log‐rank p values of disease‐specific survival (DSS) and TIL cluster grouped by adjuvant chemotherapy status. (H) Distribution of epithelial CD8+ TIL densities for each histotype.
To better understand the discordance in the effect of TIL between univariate and multivariate analyses, we assessed the TIL effect stratified by stage and adjuvant therapy. Univariate Kaplan–Meier analysis stratified by stage highlighted that the association between TIL cluster and survival was most pronounced in patients with stage III disease (Figure 3B–E). The median 5‐year overall survival in stage III disease for TIL‐poor cancers was 24.9% (95% CI: 9.6%–64.7%) versus 55.2% (95% CI: 38.3%–79.6%) for TIL‐rich stage III cancers (n = 71). In contrast, the median 5‐year overall survival in stage I disease for TIL‐poor cancers was 74.9% (95% CI: 61.9%–90.6%) versus 79.1% (95% CI: 67%–93.4%) for TIL‐rich stage I cancers (n = 118). Interpretability within stage II and IV tumors was limited by smaller sample sizes (Figure 3C,E). When we separated the cohort based on adjuvant chemotherapy use, we found that despite higher rates of chemotherapy use in stage III and IV disease in our cohort (p < 0.001), TIL‐rich cases were associated with significantly longer disease‐specific survival (p = 0.03) and trended toward longer progression‐free survival and overall survival (p = 0.067 and 0.09, respectively) only in patients who did not receive adjuvant chemotherapy (Figure 3F,G). Thus, TILs were associated with longer survival in p53abn endometrial carcinoma, particularly in patients with advanced‐stage disease and patients who did not receive adjuvant chemotherapy.
Differences in immune composition across histotypes of p53abn endometrial carcinoma
p53abn endometrial carcinomas comprise a mixture of histotypes including serous, carcinosarcoma, endometrioid, and clear cell. Histotype was significantly associated with TIL cluster (Fisher's exact test, p = 6.51 × 10−3), with carcinosarcomas the most TIL‐poor and serous carcinomas the most TIL‐rich (Figure 3H). Of the carcinosarcoma cases, 74% (23/31) were TIL‐poor, compared to only 43% (97/224) of all other histotypes (adjusted Fisher's p = 0.011) (Figure 2B). There were no significant pairwise differences in TIL cluster distribution between any of the other nonserous histotypes and serous carcinomas (the most common histotype). In multivariate survival analysis accounting for carcinosarcoma histotype and TIL cluster, carcinosarcomas trended towards shorter overall, progression‐free, and disease‐specific survival, consistent with prior findings [58, 59, 60], but this was not statistically significant (all p > 0.067). The association between the TIL‐rich cluster and overall survival remained significant when accounting for the carcinosarcoma histotype (HR 0.65, 95% CI: 0.426–0.989, p = 0.044), suggesting that TIL cluster is prognostic independently of histotype.
Immune composition and activity are altered in TIL‐rich samples
To further explore antitumor immunity and the tumor response to immunity, we assessed CD8+ T‐cell activation, immune subset composition, and macrophage and tumor expression of immune inhibitory molecules (supplementary material, Figure S2). Consistent with our previous findings [14], TIL‐rich tumors contained more CD8+PD1+ (activated CTL) and CD8+PD1− (naïve T cells), which was expected since CD8+ T cells helped define the TIL‐rich group (supplementary material, Figure S3). However, activated CTL made up a greater percentage of total CD8+ T cells in TIL‐rich tumors compared to TIL‐poor tumors (p = 0.011, Figure 4A). Furthermore, the CTL:Treg ratio (CTL:Treg) was significantly elevated within epithelium but not stroma of TIL‐rich tumors, suggestive of increased antitumor intraepithelial CTL activity (Figure 4B), whereas the CTL:Treg ratio was low in both the stroma and epithelium of TIL‐poor tumors.
Figure 4.
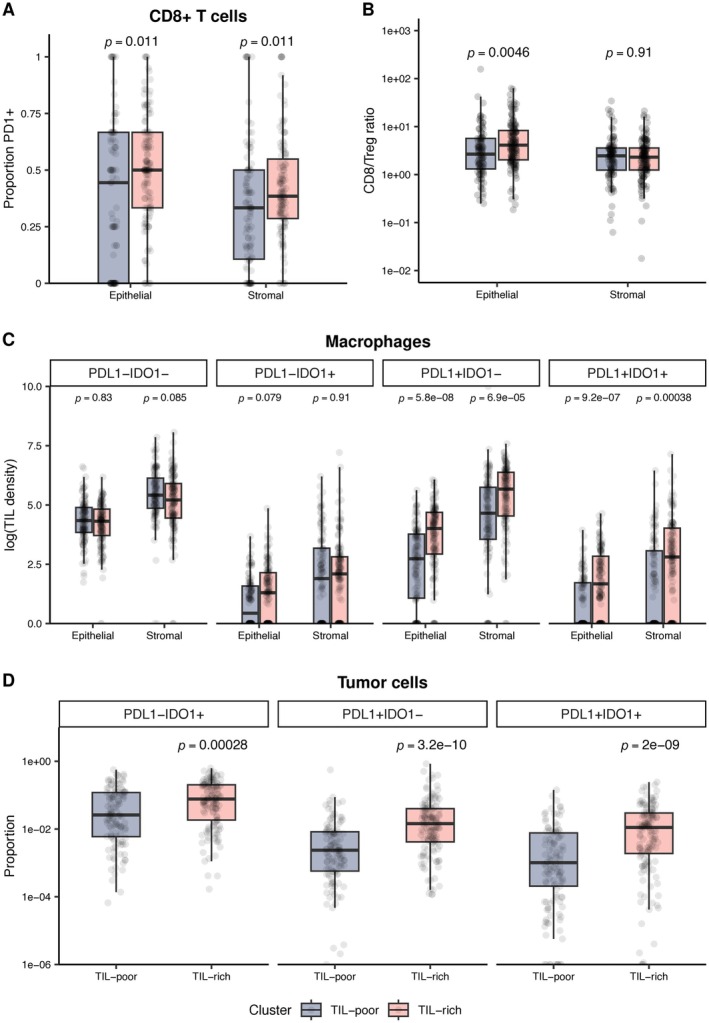
Expression of immune checkpoint molecules in TIL‐rich versus TIL‐poor p53abn endometrial cancer. (A) Relative proportions of CD8+ TIL that express PD‐1 for each TIL cluster. (B) Relative abundances of CD8+ T cell versus T regulatory cells (CD8/Treg) in TIL‐rich and TIL‐poor tumors. (C) Densities of CD68+ macrophages expressing PD‐L1, IDO1, both, or neither, in TIL‐rich versus TIL‐poor cases. (D) Proportions of tumor cells that express PD‐L1, IDO1, or both in TIL‐rich versus TIL‐poor cases. P values (Mann–Whitney U test) corrected for multiple comparisons within each heading are shown. Boxplot bars, boxes, and whiskers show median, first (Q1), and third (Q3) quartiles and Q1–1.5*IQR (interquartile range) and Q3 + 1.5*IQR, respectively. Mann–Whitney U test p values shown with multiple hypothesis testing correction by Holm method.
Tumor cells and macrophages upregulate PD‐L1 and IDO1 in response to CTL and Th expressed cytokines, thereby inhibiting antitumor immune attack [21, 22]. While there was no significant difference in the density of PD‐L1‐negative macrophages between TIL‐rich and TIL‐poor tumors, PD‐L1+IDO1− and PD‐L1+IDO1+ macrophages were significantly enriched in TIL‐rich tumors (both p < 0.001) (Figure 4C), as were PD‐L1+ and IDO1+ tumor cells (all p < 0.001) (Figure 4D). Together, these results suggest that the immune cells in TIL‐rich tumors actively participate in antitumor immunity that tumors attempt to resist.
Immune infiltration varies independently of targetable genomic alterations
Finally, we evaluated the relationship between TIL subgroups and therapeutically targetable genomic properties. We leveraged shallow whole‐genome sequencing‐derived CN signatures described in endometrial cancer [38] to identify HRD tumors (targetable with PARP inhibition) and HER2‐amplified tumors (targetable with anti‐HER2 antibodies) in a subset of 126 tumors from our cohort (Figure 5A, supplementary material, Figure S4A). HRD tumors (higher levels of the HRD signature than any other signature) comprised 21% of p53abn EC tumors, and 50% of tumors were HRD and/or HER2 positive/amplified by IHC or sequencing. Over half of the remaining cases were TIL‐rich, implying that 76% of all EC tested were potentially targetable with PARP inhibitors, HER2‐targeted therapies, and/or immunotherapy (Figure 5A). TIL cluster did not correlate with the HRD signature (Figure 5B), any other CN signature (supplementary material, Figure S4B), HER2 IHC status (p = 0.95), or HER2 amplification by shallow whole‐genome sequencing (p = 0.11) (Figure 5C,D). In addition, densities of individual TIL types in epithelial and stromal regions did not significantly correlate with any mutational signature. Thus, the TIL‐rich signature is distinct from HRD and HER2, possibly expanding the patient cohort amenable to targeted therapies.
Figure 5.
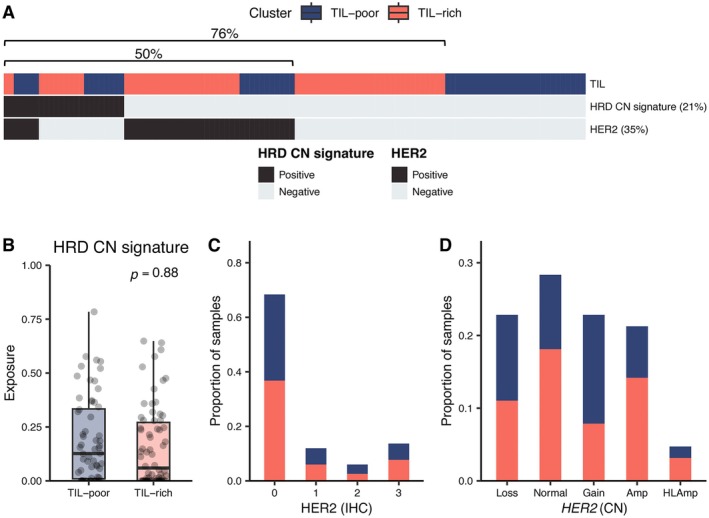
(A) Overview of relationship between TIL cluster, HRD‐associated CN signature (VS5), and HER2 status. Tumors where VS5 was the dominant CN signature are designated positive. HER2 positivity is defined by an IHC score of 3+ or amplification or high‐level amplification by whole‐genome sequencing. (B) Relative proportion (exposure) of HRD‐associated copy number signature in TIL‐poor and TIL‐rich samples. Boxplot bars, boxes, and whiskers show median, first (Q1), and third (Q3) quartiles and Q1–1.5*IQR (interquartile range) and Q3 + 1.5*IQR, respectively. (C and D) Number of TIL‐rich samples (orange) in comparison to TIL‐poor samples (blue) by (C) HER2 IHC score according to College of American Pathologist recommendations for endometrial carcinoma [74] and (D) HER2 CN status. HLAmp, high‐level amplification.
Discussion
Historically considered one of the less immunogenic subtypes of endometrial cancer, p53abn tumors had, until recently, received minimal attention in immunotherapy research. However, recent phase III clinical trials of immune checkpoint inhibitors in addition to standard‐of‐care chemotherapy demonstrated benefit in both MMRd and MMRp cancers [31, 32], with p53abn cases responding substantially better than NSMP cases [33]. These results motivated us to systematically profile the immune microenvironment in a large, multi‐institutional cohort of p53abn endometrial cancers. Analysis revealed two immunologically distinct subgroups defined by extensive and limited infiltration of T cells, B cells, and myeloid cells. Over half of cases were highly infiltrated by TIL, challenging the belief that p53abn cancers were immune depleted [61]. Moreover, these cases had increased markers of an active immune response, including higher CTL:Treg ratios, higher percentages of CTL expressing PD‐1, and increased PD‐L1 expression by tumor cells and macrophages, suggesting tumors were responding to active immune attack. In multivariate analysis, we found that TIL‐rich tumors were associated with longer overall and disease‐specific survival, in contrast to prior findings in smaller cohorts [13, 14]. Notably, the TIL‐rich group was strongly associated with survival in patients with advanced stage III disease and those who did not receive adjuvant chemotherapy. Finally, we found that TIL grouping was independent of HRD CN signature and HER2 status, thereby expanding the cohort of patients that may be treated with targeted therapies. Our findings may help to inform precision therapy for p53abn endometrial cancer by helping to identify patients most likely to respond to immunotherapy, PARP inhibition, and HER2‐directed therapies.
In analyzing the immune composition of p53abn endometrial cancer, we found multiple differences from HGSOC, which morphologically and genomically resembles p53abn endometrial cancer [2]. In HGSOC, we [36] and others [62, 63] identified a stroma‐restricted pattern of TIL that was associated with inferior prognosis compared to tumors with intraepithelial TIL. In the current study, the stroma‐restricted subtype was not identified in p53abn endometrial cancer (Figure 2) or in any histological subset of p53abn endometrial carcinoma (data not shown). Moreover, whereas immune infiltration correlated with HRD in HGSOC [36, 64, 65], immune infiltration failed to correlate with HRD and other mutational processes in p53abn endometrial cancer. The differences we observed in immune infiltration patterns between HGSOC and p53abn endometrial cancer have been reflected in immunotherapy clinical trial results: MMRp endometrial cancer shows a striking benefit from immunotherapy compared to HGSOC [31, 33, 66]. Thus, p53abn endometrial carcinomas and HGSOC have therapeutically relevant differences in immune microenvironment dynamics and antitumor immunity.
Our finding that TIL‐rich tumors were associated with improved survival in patients that did not receive adjuvant chemotherapy highlights several theoretical and practical considerations when choosing adjuvant therapies. Chemotherapy can induce immunogenic tumor cell death [67, 68, 69], releasing neoantigens and activating antitumor immunity. Conversely, chemotherapy can eliminate or prevent proliferation of antitumor immune cells, particularly when delivered at or near the maximum tolerable doses [68, 70]. Given that the association between TIL‐rich tumors and survival was strongest in patients who did not receive adjuvant chemotherapy, we hypothesized that the immunosuppressive effects of chemotherapy surpassed the immunogenic effects in our cohort. Conventional chemotherapy alone is strongly associated with survival in p53abn endometrial cancer and should remain a mainstay of treatment [6, 71]; however, careful titration and selection of chemotherapy agents to optimize immunogenic cell death may help to enable synergy with immunotherapy [68].
Our data examined for the first time the relationship between immune response and mutational processes in p53abn endometrial cancer. The presence of a TIL‐rich group of p53abn endometrial cancer associated with increased survival could help justify improved targeting of immunotherapies beyond molecular subtype‐ or histotype‐based strategies [10, 71]. While TIL‐rich tumors are more frequent in POLEmut and MMRd [13, 14] than other molecular subtypes, the variation within molecular subtype is greater than the variation between subtypes. Our data suggest immune profiling may help identify p53abn endometrial cancer patients with intrinsic antitumor immune responses that may be augmented by immunotherapy. Approximately 25% of cases in our cohort were TIL‐rich but did not show HRD or HER2 amplification/expression. The study was insufficiently powered to assess survival associations of TIL in tumors with and without HRD and HER2 amplification/expression. Thus, immunotherapies, anti‐HER2 therapies, and PARP inhibitors targeting HRD tumors may represent orthogonal approaches effective in different groups of p53abn endometrial cancer, with some tumors susceptible to multiple agents.
The limitations of our study include the use of TMA cores and the lack of immunotherapy‐treated patients in our cohort. We profiled TMAs instead of whole tissue sections, which may have underrepresented immune and tumor cell heterogeneity, as previously described [36, 72]. However, the large cohort size, use of duplicate cores, and systematic quantification of immune cell infiltrates in this study help to mitigate this sampling bias. Additionally, since none of our cohort received immunotherapy, clinical outcomes of TIL‐rich p53abn endometrial carcinoma patients treated with immunotherapy remain unknown. However, our results, combined with findings that (1) immune infiltration correlates with response to immunotherapy across other cancer types [25, 73] and that (2) a subset of MMRp endometrial cancers respond to immune checkpoint inhibitors [32, 33], support the hypothesis that TIL may predict robust responses to immunotherapy in p53abn endometrial cancer. Further studies in larger cohorts and with more extensive sampling of each tumor may help strengthen the relationship between TIL and outcomes in p53abn endometrial carcinoma.
As the use of immunotherapies extends to MMRp endometrial cancers, the immune microenvironment must be considered in addition to molecular subtype as a relevant factor. Our findings highlight the properties of the immune microenvironment that may indicate susceptibility to immune checkpoint inhibition in p53abn endometrial cancer and offer insights into the potential for rational personalized therapy for this aggressive disease.
Author contributions statement
JNM, LH, CBG, AJ, BHN, DGH and DRC conceived and designed the study; KM and KAM performed experiments; SDM, AWZ, ST, CC, JSdB, SL, JNM, LH, CBG, AJ, BHN, DGH and DRC interpreted the data; SDM and AWZ analyzed the data; AWZ performed statistical analysis; SDM, AWZ and JNM drafted the manuscript; and JNM obtained funding for the project. All authors read and approved the final manuscript.
Supporting information
Supplementary materials and methods
Figure S1. Pairwise correlations between epithelial and stromal TIL densities across the entire cohort
Figure S2. Representative multiplex immunofluorescence for the adaptive resistance panel
Figure S3. Epithelial and stromal TIL densities of PD‐1 positive and PD‐1 negative CD8+ T cells in TIL‐rich and TIL‐poor cases
Figure S4. Shallow whole‐genome sequencing‐derived copy number signature versus TIL cluster
Table S1. Hazard ratios, 95% confidence intervals and significance values for each TIL type in multivariate survival analysis, including all other clinicopathologic variables (age, chemotherapy, radiotherapy, brachytherapy, and stage)
Table S2. Contingency table showing relationship between TP53 mutation pattern by immunohistochemistry and TIL cluster for each case
Acknowledgements
The authors would like to acknowledge the expert opinion and guiding mentorship of Dr. Naveena Singh. Her enthusiasm for gynecological pathology and glowing personality will be sorely missed. Our hearts go out to her family and loved ones. We also thank the Cross Canada Consortium for data access and clinical data acquisition [46].
This work was funded by the Canadian Cancer Society Uterine Carcinosarcoma and Aggressive Uterine Cancer Research Grant (707034), Health Research BC, and the Terry Fox Research Institute (1116). This team has also been supported through funding from the Canada Research Chairs Program (JMc), the BC Cancer Foundation [Clinician Scientist Award (JMc)], the Chew Wei Chair in Gynecologic Oncology (JMc), the Vancouver Coastal Health Research Institute [Mentored Clinician Scientist Investigator Award (AJ)], and the Miller Mindell Fellowship (AJ) through the VGH & UBC Hospital Foundation.
No conflicts of interest were declared.
Data availability statement
Multiplex immunofluorescence data available via the MAPcore website https://mapcore.med.ubc.ca/martin-et-el-immune-infiltrates-predict-outcomes-in-p53abn-endometrial-cancer-citation-tbd/. Clinical data available on request from the authors due to privacy/ethical reasons. Clinical data are available upon reasonable request to the corresponding author.
References
- 1. Siegel RL, Giaquinto AN, Ahmedin J. Cancer statistics, 2024. CA Cancer J Clin 2024; 74: 12–49. [DOI] [PubMed] [Google Scholar]
- 2. Getz G, Gabriel SB, Cibulskis K, et al. Integrated genomic characterization of endometrial carcinoma. Nature 2013; 497: 67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Talhouk A, McConechy MK, Leung S, et al. A clinically applicable molecular‐based classification for endometrial cancers. Br J Cancer 2015; 113: 299–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Talhouk A, McConechy MK, Leung S, et al. Confirmation of ProMisE: a simple, genomics‐based clinical classifier for endometrial cancer. Cancer 2017; 123: 802–813. [DOI] [PubMed] [Google Scholar]
- 5. Kommoss S, McConechy MK, Kommoss F, et al. Final validation of the ProMisE molecular classifier for endometrial carcinoma in a large population‐based case series. Ann Oncol 2018; 29: 1180–1188. [DOI] [PubMed] [Google Scholar]
- 6. Lé On‐Castillo A, De Boer SM, Powell ME, et al. Molecular classification of the PORTEC‐3 trial for high‐risk endometrial cancer: impact on prognosis and benefit from adjuvant therapy. J Clin Oncol 2020; 38: 3388–3397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jamieson A, Thompson EF, Huvila J, et al. p53abn endometrial cancer: understanding the most aggressive endometrial cancers in the era of molecular classification. Int J Gynecol Cancer 2021; 31: 907–913. [DOI] [PubMed] [Google Scholar]
- 8. Siegenthaler F, Lindemann K, Epstein E, et al. Time to first recurrence, pattern of recurrence, and survival after recurrence in endometrial cancer according to the molecular classification. Gynecol Oncol 2022; 165: 230–238. [DOI] [PubMed] [Google Scholar]
- 9. Momeni‐Boroujeni A, Dahoud W, Vanderbilt CM, et al. Clinicopathologic and genomic analysis of TP53‐mutated endometrial carcinomas. Clin Cancer Res 2021; 27: 2613–2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ott PA, Bang YJ, Berton‐Rigaud D, et al. Safety and antitumor activity of pembrolizumab in advanced programmed death ligand 1–positive endometrial cancer: results from the KEYNOTE‐028 study. J Clin Oncol 2017; 35: 2535–2541. [DOI] [PubMed] [Google Scholar]
- 11. Asaka S, Yen T‐T, Wang T‐L, et al. T cell‐inflamed phenotype and increased Foxp3 expression in infiltrating T‐cells of mismatch‐repair deficient endometrial cancers. Mod Pathol 2019; 32: 576–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. López‐Janeiro Á, Villalba‐Esparza M, Brizzi ME, et al. The association between the tumor immune microenvironments and clinical outcome in low‐grade, early‐stage endometrial cancer patients. J Pathol 2022; 258: 426–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Eggink FA, Van Gool IC, Leary A, et al. Immunological profiling of molecularly classified high‐risk endometrial cancers identifies POLE‐mutant and microsatellite unstable carcinomas as candidates for checkpoint inhibition. Onco Targets Ther 2017; 6: e1264565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Talhouk A, Derocher H, Schmidt P, et al. Molecular subtype not immune response drives outcomes in endometrial carcinoma. Clin Cancer Res 2019; 25: 2537–2548. [DOI] [PubMed] [Google Scholar]
- 15. Mantovani A, Allavena P, Marchesi F, et al. Macrophages as tools and targets in cancer therapy. Nat Rev Drug Discov 2022; 21: 799–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chen Y, Song Y, Du W, et al. Tumor‐associated macrophages: an accomplice in solid tumor progression. J Biomed Sci 2019; 26: 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sakaguchi S, Mikami N, Wing JB, et al. Regulatory T cells and human disease. Annu Rev Immunol 2020; 38: 541–566. [DOI] [PubMed] [Google Scholar]
- 18. Wardell CM, MacDonald KN, Levings MK, et al. Cross talk between human regulatory T cells and antigen‐presenting cells: lessons for clinical applications. Eur J Immunol 2021; 51: 27–38. [DOI] [PubMed] [Google Scholar]
- 19. Laumont CM, Banville AC, Gilardi M, et al. Tumour‐infiltrating B cells: immunological mechanisms, clinical impact and therapeutic opportunities. Nat Rev Cancer 2022; 22: 414–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Laumont CM, Nelson BH. B cells in the tumor microenvironment: multi‐faceted organizers, regulators, and effectors of anti‐tumor immunity. Cancer Cell 2023; 41: 466–489. [DOI] [PubMed] [Google Scholar]
- 21. Feng X, Tang R, Zhang R, et al. A comprehensive analysis of IDO1 expression with tumour‐infiltrating immune cells and mutation burden in gynaecologic and breast cancers. J Cell Mol Med 2020; 24: 5238–5248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Topalian SL, Taube JM, Anders RA, et al. Mechanism‐driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat Rev Cancer 2016; 16: 275–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Santoro A, Angelico G, Inzani F, et al. The emerging and challenging role of PD‐L1 in patients with gynecological cancers: an updating review with clinico‐pathological considerations. Gynecol Oncol 2024; 184: 57–66. [DOI] [PubMed] [Google Scholar]
- 24. Holder AM, Dedeilia A, Sierra‐Davidson K, et al. Defining clinically useful biomarkers of immune checkpoint inhibitors in solid tumours. Nat Rev Cancer 2024; 24: 498–512. [DOI] [PubMed] [Google Scholar]
- 25. Martin SD, Bhuiyan I, Soleimani M, et al. Biomarkers for immune checkpoint inhibitors in renal cell carcinoma. J Clin Med 2023; 12: 4987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chan TA, Yarchoan M, Jaffee E, et al. Development of tumor mutation burden as an immunotherapy biomarker: utility for the oncology clinic. Ann Oncol 2019; 30: 44–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Colombo N, Biagioli E, Harano K, et al. Atezolizumab and chemotherapy for advanced or recurrent endometrial cancer (AtTEnd): a randomised, double‐blind, placebo‐controlled, phase 3 trial. Lancet Oncol 2024; 25: 1135–1146. [DOI] [PubMed] [Google Scholar]
- 28. Westin SN, Moore K, Chon HS, et al. Durvalumab plus carboplatin/paclitaxel followed by maintenance durvalumab with or without olaparib as first‐line treatment for advanced endometrial cancer: the phase III DUO‐E trial. J Clin Oncol 2024; 42: 283–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Makker V, Colombo N, Herráez AC, et al. Lenvatinib plus pembrolizumab for advanced endometrial cancer. N Engl J Med 2022; 386: 437–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. André T, Berton D, Curigliano G, et al. Antitumor activity and safety of dostarlimab monotherapy in patients with mismatch repair deficient solid tumors: a nonrandomized controlled trial. JAMA Netw Open 2023; 6: e2341165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mirza MR, Chase DM, Slomovitz BM, et al. Dostarlimab for primary advanced or recurrent endometrial cancer. N Engl J Med 2023; 388: 2145–2158. [DOI] [PubMed] [Google Scholar]
- 32. Eskander RN, Sill MW, Beffa L, et al. Pembrolizumab plus chemotherapy in advanced endometrial cancer. N Engl J Med 2023; 388: 2159–2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mirza MR, Sharma S, Herrstedt J, et al. 740MO dostarlimab + chemotherapy for the treatment of primary advanced or recurrent endometrial cancer (pA/rEC): analysis of progression free survival (PFS) and overall survival (OS) outcomes by molecular classification in the ENGOT‐EN6‐NSGO/GOG‐3031/RUBY trial. Ann Oncol 2023; 34: S507. [Google Scholar]
- 34. Bosse T, Creutzberg CL, Crosbie EJ, et al. Refining adjuvant treatment in endometrial cancer based on molecular features: the RAINBO clinical trial program. Int J Gynecol Cancer 2023; 33: 109–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Friedlander M, Lee YC, Tew WP. Managing adverse effects associated with poly (ADP‐ribose) polymerase inhibitors in ovarian cancer: a synthesis of clinical trial and real‐world data. Am Soc Clin Oncol Educ Book 2023; 43: e390876. [DOI] [PubMed] [Google Scholar]
- 36. Zhang AW, McPherson A, Milne K, et al. Interfaces of malignant and immunologic clonal dynamics in ovarian cancer. Cell 2018; 173: 1755–1769.e22. [DOI] [PubMed] [Google Scholar]
- 37. Wallbillich JJ, Morris RT, Ali‐Fehmi R. Comparing mutation frequencies for homologous recombination genes in uterine serous and high‐grade serous ovarian carcinomas: a case for homologous recombination deficiency testing in uterine serous carcinoma. Gynecol Oncol 2020; 159: 381–386. [DOI] [PubMed] [Google Scholar]
- 38. Jamieson A, de Sobral Barros J, Cochrane DR, et al. Targeted and shallow whole‐genome sequencing identifies therapeutic opportunities in p53abn endometrial cancers. Clin Cancer Res 2024; 30: 2461–2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Konstantinopoulos PA, Gockley AA, Xiong N, et al. Evaluation of treatment with talazoparib and avelumab in patients with recurrent mismatch repair proficient endometrial cancer. JAMA Oncol 2022; 8: 1317–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Vermij L, Léon‐Castillo A, Singh N, et al. p53 immunohistochemistry in endometrial cancer: clinical and molecular correlates in the PORTEC‐3 trial. Mod Pathol 2022; 35: 1475–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lumish M, Chui MH, Zhou Q, et al. A phase 2 trial of zanidatamab in HER2‐overexpressed advanced endometrial carcinoma and carcinosarcoma (ZW25‐IST‐2). Gynecol Oncol 2024; 182: 75–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Fader AN, Roque DM, Siegel E, et al. Randomized phase II trial of carboplatin‐paclitaxel compared with carboplatin‐paclitaxel‐trastuzumab in advanced (stage III‐IV) or recurrent uterine serous carcinomas that overexpress Her2/neu (NCT01367002): updated overall survival analysis. Clin Cancer Res 2020; 26: 3928–3935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Karpel HC, Slomovitz B, Coleman RL, et al. Treatment options for molecular subtypes of endometrial cancer in 2023. Curr Opin Obstet Gynecol 2023; 35: 270–278. [DOI] [PubMed] [Google Scholar]
- 44. Meric‐Bernstam F, Makker V, Oaknin A, et al. Efficacy and safety of trastuzumab deruxtecan in patients with HER2‐expressing solid tumors: primary results from the DESTINY‐PanTumor02 phase II trial. J Clin Oncol 2024; 42: 47–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. McNamara B, Greenman M, Pebley N, et al. Antibody–drug conjugates (ADC) in HER2/neu‐positive gynecologic tumors. Molecules 2023; 28: 7389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Jamieson A, Huvila J, Thompson EF, et al. Variation in practice in endometrial cancer and potential for improved care and equity through molecular classification. Gynecol Oncol 2022; 165: 201–214. [DOI] [PubMed] [Google Scholar]
- 47. World Medical Association . World medical association declaration of Helsinki: ethical principles for medical research involving human subjects. JAMA 2013; 310: 2191–2194. [DOI] [PubMed] [Google Scholar]
- 48. Neal RM. Markov chain sampling methods for Dirichlet process mixture models. J Comput Graph Stat 2000; 9: 249–265. [Google Scholar]
- 49. Macintyre G, Goranova TE, De Silva D, et al. Copy number signatures and mutational processes in ovarian carcinoma. Nat Genet 2018; 50: 1262–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sauer CM, Eldridge MD, Vias M, et al. Absolute copy number fitting from shallow whole genome sequencing data. bioRxiv 2021; p. 2021.07.19.452658[cited 2024 Jul 14]. Available from: https://www.biorxiv.org/content/10.1101/2021.07.19.452658v1. [Not peer reviewed].
- 51. McShane LM, Altman DG, Sauerbrei W, et al. REporting recommendations for tumour MARKer prognostic studies (REMARK). Br J Cancer 2005; 93: 387–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. R Core Team . R: A Language and Environment for Statistical Computing [Internet]. R Foundation for Statistical Computing: Vienna, Austria, 2023. Available from: https://www.R-project.org/. [Google Scholar]
- 53. Therneau TM, Grambsch PM. Modeling Survival Data: Extending the Cox Model. Springer New York: New York, NY, 2000. [cited 2017 Jul 20]. Available from: http://link.springer.com/10.1007/978-1-4757-3294-8. [Google Scholar]
- 54. Therneau TM. A Package for Survival Analysis in R [Internet]. R Package Version 3.5–7. 2023 [cited 2025 Mar 15], Available from: https://CRAN.R-project.org/package=survival.
- 55. Grambsch PM, Therneau TM. Proportional hazards tests and diagnostics based on weighted residuals. Biometrika 1994; 81: 515. [Google Scholar]
- 56. Ward JH Jr. Hierarchical grouping to optimize an objective function. J Am Stat Assoc 1963; 58: 236–244. [Google Scholar]
- 57. Creasman W. Revised FIGO staging for carcinoma of the endometrium. Int J Gynecol Obstet 2009; 105: 109. [DOI] [PubMed] [Google Scholar]
- 58. Zhang C, Hu W, Jia N, et al. Uterine carcinosarcoma and high‐risk endometrial carcinomas: a clinicopathological comparison. Int J Gynecol Cancer 2015; 25: 629–636. [DOI] [PubMed] [Google Scholar]
- 59. Bernardini MQ, Gien LT, Lau S, et al. Treatment related outcomes in high‐risk endometrial carcinoma: Canadian high risk endometrial cancer consortium (CHREC). Gynecol Oncol 2016; 141: 148–154. [DOI] [PubMed] [Google Scholar]
- 60. Raffone A, Travaglino A, Raimondo D, et al. Uterine carcinosarcoma vs endometrial serous and clear cell carcinoma: a systematic review and meta‐analysis of survival. Int J Gynaecol Obstet 2022; 158: 520–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Mendiola M, Pellinen T, Ramon‐Patino JL, et al. Prognostic implications of tumor‐infiltrating T cells in early‐stage endometrial cancer. Mod Pathol 2022; 35: 256–265. [DOI] [PubMed] [Google Scholar]
- 62. Sato E, Olson SH, Ahn J, et al. Intraepithelial CD8+ tumor‐infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc Natl Acad Sci U S A 2005; 102: 18538–18543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Komdeur FL, Wouters MCA, Workel HH, et al. CD103+ intraepithelial T cells in high‐grade serous ovarian cancer are phenotypically diverse TCRαβ+ CD8αβ+ T cells that can be targeted for cancer immunotherapy. Oncotarget 2016; 7: 75130–75144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Nelson BH, Greenberg PD, Schreiber H. New insights into tumor immunity revealed by the unique genetic and genomic aspects of ovarian cancer. Curr Opin Immunol 2015; 33: 93–100. [DOI] [PubMed] [Google Scholar]
- 65. Vázquez‐García I, Uhlitz F, Ceglia N, et al. Ovarian cancer mutational processes drive site‐specific immune evasion. Nature 2022; 612: 778–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Matulonis UA, Shapira‐Frommer R, Santin AD, et al. Antitumor activity and safety of pembrolizumab in patients with advanced recurrent ovarian cancer: results from the phase II KEYNOTE‐100 study. Ann Oncol 2019; 30: 1080–1087. [DOI] [PubMed] [Google Scholar]
- 67. Pfirschke C, Engblom C, Rickelt S, et al. Immunogenic chemotherapy sensitizes tumors to checkpoint blockade therapy. Immunity 2016; 44: 343–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Galluzzi L, Humeau J, Buqué A, et al. Immunostimulation with chemotherapy in the era of immune checkpoint inhibitors. Nat Rev Clin Oncol 2020; 17: 725–741. [DOI] [PubMed] [Google Scholar]
- 69. Casares N, Pequignot MO, Tesniere A, et al. Caspase‐dependent immunogenicity of doxorubicin‐induced tumor cell death. J Exp Med 2005; 202: 1691–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Yu W‐D, Sun G, Li J, et al. Mechanisms and therapeutic potentials of cancer immunotherapy in combination with radiotherapy and/or chemotherapy. Cancer Lett 2019; 452: 66–70. [DOI] [PubMed] [Google Scholar]
- 71. Jamieson A, Huvila J, Leung S, et al. Molecular subtype stratified outcomes according to adjuvant therapy in endometrial cancer. Gynecol Oncol 2023; 170: 282–289. [DOI] [PubMed] [Google Scholar]
- 72. Yuan Y. Spatial heterogeneity in the tumor microenvironment. Cold Spring Harb Perspect Med 2016; 6: a026583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Raskov H, Orhan A, Christensen JP, et al. Cytotoxic CD8+ T cells in cancer and cancer immunotherapy. Br J Cancer 2021; 124: 359–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Buza N. HER2 testing and reporting in endometrial serous carcinoma: practical recommendations for HER2 immunohistochemistry and fluorescent in situ hybridization: proceedings of the ISGyP companion society session at the 2020 USCAP annual meeting. Int J Gynecol Pathol 2021; 40: 17–23. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary materials and methods
Figure S1. Pairwise correlations between epithelial and stromal TIL densities across the entire cohort
Figure S2. Representative multiplex immunofluorescence for the adaptive resistance panel
Figure S3. Epithelial and stromal TIL densities of PD‐1 positive and PD‐1 negative CD8+ T cells in TIL‐rich and TIL‐poor cases
Figure S4. Shallow whole‐genome sequencing‐derived copy number signature versus TIL cluster
Table S1. Hazard ratios, 95% confidence intervals and significance values for each TIL type in multivariate survival analysis, including all other clinicopathologic variables (age, chemotherapy, radiotherapy, brachytherapy, and stage)
Table S2. Contingency table showing relationship between TP53 mutation pattern by immunohistochemistry and TIL cluster for each case
Data Availability Statement
Multiplex immunofluorescence data available via the MAPcore website https://mapcore.med.ubc.ca/martin-et-el-immune-infiltrates-predict-outcomes-in-p53abn-endometrial-cancer-citation-tbd/. Clinical data available on request from the authors due to privacy/ethical reasons. Clinical data are available upon reasonable request to the corresponding author.