

Abstract
Previous studies have shown that a mitogen activated protein (MAP) kinase (MEK)-independent signaling pathway is required by activated Raf or fibroblast-derived growth factor (FGF) for the differentiation of rat hippocampal neuronal H19-7 cells. We now demonstrate that both Raf and FGF similarly induce prolonged transcription and translation of the immediate early gene pip92 in the absence of activation of the MAP kinases (MAPKs) ERK1 and ERK2. To determine the mechanism by which this occurs and to identify novel Raf-activated signaling pathways, we investigated the induction of the pip92 promoter by both FGF and an estradiol-activated Raf-1–estrogen receptor fusion protein (ΔRaf-1:ER) in H19-7 cells. Deletion analysis of the pip92 promoter indicated that activation by the MAPK-independent pathway occurs primarily within the region containing a serum response element (SRE). Further analysis of the SRE by using a heterologous thymidine kinase promoter showed that both an Ets and CArG-like site are required. Elk1, which binds to the Ets site, was phosphorylated both in vitro and in vivo by the MAPK-independent pathway, and phosphorylation of an Elk1-GAL4 fusion protein by this pathway was sufficient for transactivation. Finally, at least two Elk1 kinases were fractionated by gel filtration, and analysis by an in-gel kinase assay revealed at least three novel Raf-activated Elk1 kinases. These results indicate that both FGF and Raf activate MAPK-independent kinases that can stimulate Elk1 phosphorylation and immediate early gene transcription.
Signal transduction is the process of integrating a variety of stimuli from the external environment, resulting in the activation of signaling cascades that converge on a defined target such as the phosphorylation of a transcription factor. There are numerous examples of molecules such as Ras that have been shown to activate multiple effectors, eliciting a variety of outcomes. Despite increasing evidence that most signaling molecules can activate more than one effector, many of these downstream signaling molecules have not been identified.
The best-characterized signaling cascade involves the successive activation of Ras, Raf, mitogen-activated protein (MAP) kinases 1 and 2 (MEK), and MAP kinases (MAPKs) ERK1 and ERK2 (26, 30). Although MEK is the only known kinase directly downstream of Raf, there are several lines of evidence suggesting that Raf can also activate other effectors. Thus, PC12 cells transfected with the activated raf-1 oncogene were reported to differentiate without MAPK activation (38, 39). Activation of MAPK by the oncogenes ras and raf was not detected in Rat 1a cells (13). Similarly, expression of the ΔRaf-1:ER protein in Rat 1a cells led to activation of MEK by estradiol in the absence of detectable MAPK stimulation (29). Raf-1 has been shown to activate the promoters for atrial natriuretic factor and myosin light chain-2 in cardiac muscle cells, whereas activated MEK was inhibitory (33). Finally, we have demonstrated that MEK is necessary but insufficient for the neuronal differentiation of hippocampal cells by Raf (18). These data suggest that the effects of Raf may be mediated by MEK- and MAPK-independent signal transduction pathways. To identify these novel signaling pathways, it is important to have a biological target.
Using differential display, we have identified pip92 as an immediate early gene induced during differentiation of a conditionally immortalized rat hippocampal cell line, H19-7 (10). pip92 (also known as chx1 or ETR101) was cloned from serum-stimulated BALB/c 3T3 fibroblasts (3) and from activated T lymphocytes treated with cycloheximide (6). Human pip92 cDNA was cloned from the myeloid leukemia cell line HL-60 (31). pip92 is rapidly and transiently induced by stimulation with serum growth factors and the tumor promoter 12-O-tetradecanoylphorbol-13-acetate (TPA) in fibroblasts and by treatment with nerve growth factor in PC12 cells (3). pip92 encodes a short-lived, proline-rich protein with no significant sequence similarity with any known protein. The function of its encoded protein is unknown, and the downstream signal transduction cascades leading to the induction of pip92 in response to external stimuli are not well understood.
The pip92 promoter has been cloned and shown to respond to induction by serum in mouse 3T3 fibroblasts via a serum response element (SRE) (20). The SRE, studied most extensively in the c-fos promoter, consists of a CArG box which has a consensus sequence CC(A/T)6GG that binds to the serum response factor (SRF) (34). When SRF is bound to the c-fos SRE, it recruits a ternary complex factor (TCF) to an upstream Ets-like binding site (8, 16). In the pip92 promoter, the SRE consists of at least one Ets protein binding site and a CArG site that binds SRF (20). Gel shift analyses demonstrated that the pip92 Ets sites bind to Elk1/TCF, a ternary complex factor that is a member of the Ets family of transcription factors (reviewed in reference 23). Elk1 is active as a transcription factor upon phosphorylation of primarily two sites, serines 383 and 389 (S383/S389) (17, 24). Three members of the MAPK family, the classic MAPKs (ERK1 and ERK2), JNKs (Jun kinases), and p38, are all able to phosphorylate Elk1 (14, 17, 24, 37).
In this study, we have focused on the role of the MEK/MAPK pathway in the induction of pip92 gene expression by either fibroblast growth factor (bFGF) in rat hippocampal H19-7 cells or activated Raf in H19-7 cells stably transfected with an oncogenic human raf-1–estrogen receptor fusion gene (encoding ΔRaf-1:ER) (29). The results indicate the presence of at least two pathways, one MAPK dependent and the other MAPK independent, for induction of pip92 in response to bFGF or to activated Raf in differentiating rat neuronal H19-7 cells. To identify a discrete target of the MAPK-independent pathway activated by Raf and FGF, we analyzed the pip92 promoter. Studies using pip92 promoter deletion constructs and promoter fragments linked to the heterologous thymidine kinase (tk) promoter showed that the SRE is required and sufficient for activation. Furthermore, Elk1 was phosphorylated by both Raf and FGF via a MAPK-independent kinase at S383/S389, which was sufficient to induce transactivation of an Elk1-Gal4 fusion protein. Finally, in-gel kinase assays revealed at least three novel Raf-activated kinases that specifically phosphorylate Elk1 at these sites.
MATERIALS AND METHODS
Materials.
Protein A-Sepharose and glutathione-Sepharose 4B were purchased from Pharmacia. Fetal bovine serum, Dulbecco modified Eagle medium, Bluo-gal, and Geneticin were purchased from Life Technologies Inc. Hygromycin and estradiol were purchased from Sigma (St. Louis, Mo.). Epidermal growth factor (EGF; receptor grade) was purchased from Biomedical Technologies (Stoughton, Mass.). Human bFGF (receptor grade) was purchased from Bachem (Torrance, Calif.). Myelin basic protein (MBP) was from Sigma. Monoclonal antibody against the hemagglutinin (HA) epitope was purchased from BabCo (Emeryville, Calif.). The MKP-1 expression vector was generated as previously described (4). The pip92 promoter-chloramphenicol acetyltransferase (CAT) reporter fusion constructs (−1281pip92/CAT, −1111pip92/CAT, −287pip92/CAT, −159pip92/CAT, and −89pip92/CAT) were made as described previously (20). The heterologous pip92 promoter-tk promoter-CAT reporter fusion constructs −1270/−970pip92, −1231/−1158pip92, −1231/1158**pip92 with a mutated SRE site, −1111/−970pip92, P (a pip92 fragment containing the proximal Ets site and a mutated distal Ets site), D (a pip92 fragment containing the distal Ets site and a mutated proximal Ets site), and C (a pip92 fragment containing three copies of the SRE site) were described previously (20). The GAL4 reporter plasmid Gal2-TATA-luciferase and constructs expressing amino acids 307 to 428 of wild-type Elk1 or a Ser→Ala Elk1 mutant fused to the GAL4 binding domain [GAL-ElkC and GAL-ElkC(A383/A389)] were kindly provided by R. Treisman (24). Plasmid GST-ElkC, expressing glutathione S-transferase (GST) fused to the C-terminal peptide (residues 307 to 428) of wild-type Elk1, was obtained from R. Treisman (24). Plasmid GST-ElkC(A383/A389), expressing GST fused to the C-terminal peptide (residues 307 to 428) of the A383/A389 Elk1 mutant, was generated by ligating a BglII/SpeI fragment expressing the C-terminal mutant peptide into the GEX30X vector. The cytomegalovirus–β-galactosidase expression vector was a gift from V. Sukhatme. Plasmid DNAs were prepared by CsCl-ethidium bromide gradient centrifugation or by purification through columns as specified by the manufacturer (Qiagen). PD098059 was a generous gift from A. Saltiel. AmpliTaq DNA polymerase was purchased from Perkin-Elmer Corp., and [α-35S]dATP (1,200 Ci/mmol) was obtained from Dupont, NEN Research Products.
Cell culture.
The rat neuronal hippocampal progenital cell line H19-7 was generated by transduction with the retroviral vectors containing the temperature-sensitive simian virus 40 large T antigen that is functionally active at 33°C and inactive at 39°C as previously described (10). The Raf-1:ER cells were made by stable transfection of H19-7 cells with estradiol-regulated Raf-1 generated by fusing a constitutively active, oncogenic portion of human Raf-1 to the hormone binding domain of human estrogen receptor as previously described (18, 29). The undifferentiated and proliferating cells were cultured at 33°C in medium containing 10% fetal bovine serum and 200 mg of G418 per ml to maintain selection pressure on the transduced immortalization vector. ΔRaf-1:ER cells were also maintained in hygromycin. Prior to differentiation, cells were shifted to 39°C in N2 medium (5 mg of insulin per ml, 100 mg of transferrin per ml, 20 nM progesterone, 20 nM selenium sodium salt, 60 mM putrescine, 0.11 mg of sodium pyruvate per ml, 2 mM glutamine) for 2 days (H19-7 cells) or 1 day (ΔRaf-1:ER cells). H19-7 cells were differentiated with 10 ng of bFGF per ml and ΔRaf-1:ER cells were differentiated with either 1 μM or 10 nM estradiol. When specified, cells were pretreated with the synthetic MEK inhibitor PD098059 (9) 10 min prior to bFGF or estradiol stimulation.
Isolation and reamplification of pip92 cDNA probe.
mRNA differential display was performed essentially as previously described (22), using an RNAmap kit (GenHunter), except that routinely 0.2 μg of total RNA or 0.1 μg of poly(A)+ RNA was used for reverse transcription. The amplified cDNAs were then separated on a 6% DNA sequencing gel. The DNA sequencing gel was blotted on to a piece of Whatmann 3MM paper and dried without methanol-acetic acid fixing. The autoradiogram and dried gel were oriented with either radioactive ink or needle punches. After development of the film, cDNA bands of interest were located by either marking with a clean pencil or cutting through the film. The gel slice along with the 3MM paper was incubated in 100 ml of distilled H2O for 10 min. After rehydration of the polyacrylamide gel, the cDNA was diffused out by boiling the gel slice for 15 min in a tightly capped microcentrifuge tube. cDNA was recovered by ethanol precipitation in the presence of 0.3 M sodium acetate and 5 ml of glycogen (10 mg/ml) as a carrier and redissolved in distilled H2O. Some of the eluted cDNA probe was reamplified by using the same primer set and PCR conditions as used in the mRNA differential display except that the deoxynucleoside triphosphate concentration was in each case 20 mM instead of 2 to 4 mM. The reamplified cDNA probes were subcloned into a PCRII vector by using the TA cloning system (Invitrogen) for DNA sequencing. DNA sequencing was carried out by the dideoxynucleotide chain terminator method, using Sequenase (United States Biochemical) and a T7 or SP6 primer (GIBCO-BRL).
DNA transfection and CAT assay.
Transient transfections were performed by the calcium phosphate precipitation method (28). Plasmid pCMV-GAL, which contains the Escherichia coli β-galactosidase gene driven by the cytomegalovirus promoter, was used as an internal control to determine transfection efficiency. β-Galactosidase was assayed as described elsewhere (28). The CAT assay was done with an enzyme-linked immunosorbent assay CAT assay kit (5′ Æ3′).
RNA preparation and Northern blot hybridization.
Total cellular RNA from H19-7 or ΔRaf-1:ER cells was isolated by the guanidium isothiocyanate procedure described elsewhere (5). Poly(A)+ RNA was selected by oligo(dT)-cellulose column chromatography. For Northern blot analysis, total RNA (10 μg for each sample) was denatured by glyoxal, separated by electrophoresis, stained with acridine orange (15 μg/ml) in 10 mM sodium phosphate, transferred to a nitrocellulose membrane (Schleicher & Schuell), and hybridized to a pip92 probe. The 32P-labeled 125-bp pip92 cDNA probe was made by using a random primer labeling kit (Boehringer Mannheim).
Cell labeling and immunoprecipitation of Pip92.
Quiescent H19-7 or ΔRaf-1:ER cells were incubated for 1 h in medium without methionine prior to labeling. Cells were then stimulated with 10 mM bFGF or 1 or 10 nM estradiol for the indicated times. The cells were metabolically labeled with [35S]methionine (1,269 Ci/mmol, 100 mCi/ml of medium; ICN) during the last 30 min of stimulation. Cells were washed in cold phosphate-buffered saline (PBS) and lysed in radioimmunoprecipitation assay (RIPA) buffer (150 mM NaCl, 1% Nonidet P-40, 0.5% deoxycholate, 0.1% sodium dodecyl sulfate [SDS], 50 mM Tris [pH 8.0], 1 mM phenylmethylsulfonyl fluoride [PMSF]). Lysates were cleared by measuring trichloroacetic acid-precipitable counts). For immunoprecipitation, the cell lysates were immunoprecipitated as described elsewhere (15) with anti-Pip92 rabbit antiserum (3). Protein A-beads were washed by RIPA buffer once, incubated with anti-Pip92 antiserum for 2 h, and then washed with RIPA buffer three times. The beads were then incubated with cell lysates overnight. Sample was washed once in RIPA buffer, three times in buffer I (150 mM NaCl, 10 mM Tris [pH 7.5], 2 mM EDTA, 1% Nonidet P-40), once in buffer II (500 mM NaCl, 10 mM Tris [pH 7.5], 2 mM EDTA), and once in distilled water. Samples were analyzed on SDS–12.5% polyacrylamide gels. The gel was treated with En3Hance (Dupont Research Products) in order to improve detection of radiolabeled protein and then subjected to fluorography. The image of autoradiographic film or gel bands was analyzed by using AMBIS software.
Immunoprecipitation and assay of HA-tagged ERK2.
ERK2 kinase activity was measured by immunoprecipitation of the epitope-tagged ERK2, followed by an in vitro phosphorylation assay (18, 25). Transfected cells were stimulated and lysed with 1% TLB solution, consisting of 20 mM Tris (pH 7.9), 137 mM NaCl, 5 mM Na2EDTA, 10% glycerol, 1% Triton X-100, 0.2 mM PMSF, 1 μg of aprotinin per ml, 20 μM leupeptin, 1 mM sodium orthovanadate (pH 10.0), 1 mM EGTA, 10 mM NaF, 1 mM tetrasodium pyrophosphate, 1 mM β-glycerophosphate (pH 7.4), and 0.1 g of p-nitrophenylphosphate per ml. The cell lysates were then incubated with protein A-Sepharose precoupled with the antibody 12CA5, which binds to the HA epitope, for 24 h at 4°C. The immune complexes were washed with lysis buffer and with kinase reaction buffer containing 20 mM HEPES (pH 7.4), 10 mM MgCl2, 1 mM dithiothreitol, 200 mM orthovanadate, and 10 mM p-nitrophenylphosphate. The MBP phosphorylation assay with immunoprecipitated HA-tagged ERK2 was done using MBP as a substrate as described previously (18).
Elk1 transactivation assay with the GAL4-luciferase system.
Transient transfections with GAL-ElkC, GAL-ElkC(A383/A389), and the GAL4 reporter plasmid Gal2-TATA-luciferase were performed by calcium phosphate precipitation (28). Luciferase activity was measured by using a luciferase assay kit (Promega) and luminometer.
In vitro Elk1 phosphorylation using glutathione-Sepharose 4B beads.
The Sepharose 4B beads prebound to bacterially expressed wild-type or mutant GST-ElkC (or just GST as a negative control) were prepared by using Bulk GST purification modules (Pharmacia catalog no. XY-045-00-08). The cell extracts or fractions (200 μl, or 20 to 30 μg of total protein) were added to 50 μl of resuspended beads in 1× kinase buffer (see below), 3 μM ATP, and 30 μCi of [32P]ATP. The bead and protein mixtures were incubated at room temperature for 2 h with extensive shaking and washed with PBS three times and PBS containing 0.1 M NaCl two times. Phosphorylated GST-Elk1 products were eluted by incubating the beads with 30 to 40 μl of Pharmacia elution buffer. Eluates were resolved by polyacrylamide gel electrophoresis (PAGE) on a 12.5% polyacrylamide gel, and the 32P-labeled protein bands were identified by autoradiography.
In-gel kinase renaturation assay.
A 12.5% gel for SDS-PAGE was prepared by using 50 μg of bacterially expressed GST-ElkC, mutant GST-ElkC(A383/389), or GST per ml as the substrate for phosphorylation. H19-7 or Raf-1:ER cell extracts stimulated with FGF or estradiol in the absence or presence of MEK inhibitor (30 μg/ml) were applied to the gel. All gel renaturation and phosphorylation protocols were performed as previously described (2).
Fractionation of Elk1 kinases.
ΔRaf-1:ER cells were grown for 3 days on 150-mm-diameter dishes at 33°C, shifted to 39°C in N2 medium for 24 h, and then treated with 10 nM estradiol for 1 h. The cells were trypsinized, washed in PBS, and then lysed in buffer A (20 mM Tris [pH 7.5], 150 mM NaCl, 10 mM MgCl2, 2 mM EGTA) plus 0.2 mM PMSF, 1 μg of aprotinin per ml, and 50 mM NaF. The cell extract was passed through a 26.5-gauge needle and then centrifuged at 10,000 rpm for 10 min at 4°C; 200 μl (2.6 μg/μl) of the cell lysate was applied to a Superose 6 HR column (Pharmacia) that was equilibrated in buffer A and had previously been calibrated with molecular weight standards (2 mg each of immunoglobulin G and chicken ovalbumin per ml); 0.5-ml fractions were collected and assayed for Elk1 kinase activity or Western blot analysis with anti-active MAPK antibody.
Western blot analysis with anti-active MAPK antibody.
Fractions from the Superose 6 column (40 μl) were resolved by SDS-PAGE (10% gel) and transferred to a membrane for immunoblotting with anti-active MAPK antibody (Promega) according to the Promega protocol.
RESULTS
mRNA expression of pip92 in H19-7 and Raf-1:ER cells is induced by FGF or activated Raf.
H19-7 cells differentiate in response to FGF but not EGF at 39°C, the temperature at which the simian virus 40 large T antigen is not active (10). Using RNA display, we identified a number of immediate early genes that were selectively induced by FGF in H19-7 cells, including cyr61, egr-2, c-fos, and pip92 (data not shown). We chose pip92 as a target and investigated the signaling pathways leading to its induction. Northern blot analysis using total RNA from H19-7 cells stimulated with FGF showed that pip92 mRNA was induced by 30 min, sustained until 2 h, and decreased thereafter (Fig. 1A). Treatment of H19-7 cells with EGF also induced pip92 mRNA, but the level of induction was substantially lower than that induced by FGF (Fig. 1B).
FIG. 1.
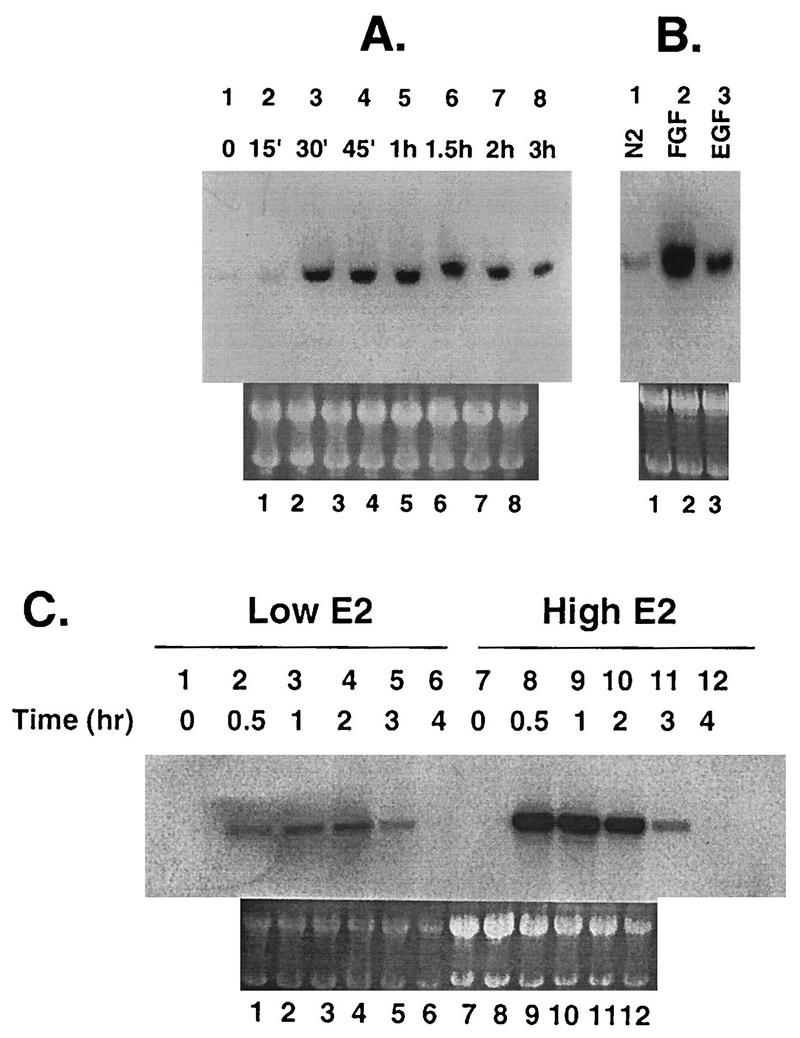
Northern blot analysis of pip92 mRNA from H19-7 and ΔRaf-1:ER cells. A 10-μg aliquot of total RNA was extracted from cells, applied to each lane, and hybridized to a 125-bp 32P-labeled pip92 cDNA fragment as described in Materials and Methods. (A) Time-dependent expression of pip92 mRNA in H19-7 cells treated with bFGF (10 ng/ml) for the indicated times. (B) Expression of pip92 mRNA after H19-7 cells were stimulated for 1 h with either N2 alone, 10 μM EGF, or 10 ng of bFGF per ml. (C) Time-dependent expression of pip92 mRNA from ΔRaf-1:ER cells treated with either 10 nM or 1 μM estradiol for the indicated times. As a control for RNA loading, total RNA was visualized under UV light by acridine orange staining as described in Materials and Methods (lower panels).
In ΔRaf-1:ER cells expressing an estradiol-regulated Raf-1 kinase, Raf activation in response to 10 nM to 1 μM estradiol treatment leads to neuronal differentiation (18). The induction of pip92 mRNA by activated Raf was kinetically similar to that observed for pip92 in H19-7 cells, reaching a maximum by 30 min at both low and high concentrations of estradiol and decreasing after 2 h of estradiol stimulation (Fig. 1C). The amount of expressed pip92 mRNA was much higher in response to 1 μM estradiol than in response to the lower estradiol dose. This difference was due to differential Raf activation, since no induction of pip92 mRNA by estradiol was observed in the parent cell line (H19-7) lacking the ΔRaf-1:ER fusion protein (data not shown).
An MEK inhibitor suppresses MAPK activation and partially suppresses pip92 mRNA induction.
To investigate the role of the MEK/MAPK pathway in the induction of pip92 expression, cells were pretreated for 10 min with the synthetic MEK inhibitor PD098059, which is highly specific for the MEK family (9, 27). We have previously used this approach to show that activation of MAPK is not necessary for differentiation by FGF in H19-7 cells (19). To determine the conditions in ΔRaf-1:ER cells for suppression of MAPK activity by the MEK inhibitor, cells were initially transfected with an HA epitope-tagged ERK2 expressing plasmid. Cells were then stimulated with 1 μM estradiol following pretreatment in the presence or absence of 0 to 50 μM PD098059, and the MAPK activity was determined by immunoprecipitation of ERK2 with anti-HA antibody and phosphorylation of MBP. The results indicate that 10 μM PD098059 suppressed MAPK activity below the level in unstimulated cells, and 30 μM completely inhibited MAPK in ΔRaf-1:ER cells (Fig. 2). The apparent activity in the immunoprecipitates from the mock-transfected cells (no DNA lanes) is due to a nonspecific kinase. A similar dose-response curve was obtained when H19-7 cells were stimulated with FGF in the presence of PD098059 (19). In the latter experiments, both transfected ERK2 and endogenous ERK1 and ERK2 were assayed, and the results were the same.
FIG. 2.

Inhibition of MAPK by the MEK inhibitor (MI) PD098059. ΔRaf-1:ER cells were either mock transfected (no DNA) or transiently transfected with 10 μg of HA-tagged ERK2 plasmid as described in Materials and Methods. After being transferred to 39°C in N2 medium for 24 h, the cells were stimulated with 1 μM estradiol for 1 h following a 10-min pretreatment in the absence or presence of 0 to 50 μM PD098059 as indicated. Cell lysates were immunoprecipitated with anti-HA antibody, and the HA-tagged ERK2 was assayed for kinase activity by using MBP as described in Materials and Methods. The reaction mixtures were resolved by SDS-PAGE, and the gel was dried and exposed to X-ray film for autoradiography. (A) Autoradiograph depicting dose response for inhibition of ERK2 by PD098059. (B) Plot of dose response for inhibition of ERK2 by PD098059. The autoradiograph in (A) was scanned and quantitated with an AMBIS radioanalytic scanner.
Transcriptional activation of the pip92 gene occurs by MEK-dependent and -independent pathways.
Transcriptional activation of the pip92 gene was examined by using a CAT reporter plasmid linked to a 1,281-bp pip92 promoter fragment (−1281pip92/CAT) (20) transiently expressed in H19-7 or ΔRaf-1:ER cells. Treatment of H19-7 cells with bFGF (10 ng/ml) rapidly stimulated pip92 transcription, which reached a plateau by 4 h as monitored by CAT activity (Fig. 3A). A similar pattern for induction of pip92 transcription was observed in ΔRaf-1:ER cells upon stimulation with 1 μM estradiol (Fig. 3B).
FIG. 3.

Inhibition of transcriptional activation of pip92 by pretreatment with the MEK inhibitor. (A and B) Time course of transcriptional activation of pip92 by bFGF in H19-7 cells or by activated Raf in ΔRaf-1:ER cells. The −1281pip92/CAT fusion plasmid (10 μg) was transiently transfected into H19-7 or ΔRaf-1:ER cells. Where indicated, samples were pretreated with 10 μM MEK inhibitor (MI) for 10 min prior to stimulation. The cells were stimulated with 10 ng of bFGF per ml (•) or 10 ng of bFGF per ml plus 10 μM MEK inhibitor (○) for H19-7 cells (A) or with 10 nM estradiol (□), 1 μM estradiol (•), or 1 μM estradiol plus 10 μM MEK inhibitor (○) for ΔRaf-1:ER cells (B). (C and D) Dose response for inhibition of pip92 transcription by the MEK inhibitor. Cells were stimulated with either 10 ng of FGF per ml (H19-7 cells) (C) or 1 μM estradiol (ΔRaf-1:ER cells) plus 10 to 50 μM PD098059 (D). The activity of the expressed CAT enzyme in 40 to 60 μg of cell lysate was measured at the indicated times as described in Materials and Methods. Extracts prepared from cells without any transfected DNA were used for a negative control. Data are plotted as the mean plus the range of samples from two independent experiments.
Following pretreatment of H19-7 cells for 10 min with 10 μM PD098095, activation of the pip92 promoter by bFGF was decreased by approximately 50% (Fig. 3A). Analysis of the dose response for inhibition (Fig. 3C) indicated that no further inhibition of pip92 transcription could be detected up to 50 μM PD098059, a dose at which MAPK activity is completely suppressed (19). Pretreatment of ΔRaf-1:ER cells with the MEK inhibitor prior to 1 μM estradiol stimulation also decreased pip92 transcription by about 50%, comparable to the level observed following treatment with 10 nM estradiol alone (Fig. 3B). This result is consistent with the observation that treatment of the ΔRaf-1:ER cells with 10 nM estradiol causes little stimulation of ERK2 above background (Fig. 1), as previously observed (18). Again, analysis of the dose response for PD098059 showed that almost 50% of the Raf-stimulated pip92 transcriptional activity was maintained at PD098095 concentrations up to 50 μM (Fig. 3D). Similar results were obtained when pip92 mRNA was quantitated by Northern analysis (data not shown). These data indicate that pip92 gene transcription can be induced in the absence of MEK/MAPK activity, suggesting that the 1,281-bp pip92 promoter fragment is responsive to at least two signaling pathways for induction of gene expression by bFGF or activated Raf.
Kinetics of Pip92 protein synthesis is biphasic.
The kinetics of Pip92 synthesis were examined in both H19-7 and ΔRaf-1:ER cells. Both cells were metabolically labeled with [35S]methionine for the last 30 min before harvest, and cell lysates were immunoprecipitated with anti-Pip92 rabbit antibodies. In H19-7 cells, the rate of Pip92 synthesis appeared to be biphasic, reaching a peak by 1 h after bFGF stimulation and declining rapidly to a low level that was sustained up to 6 h. Pretreatment of H19-7 cells for 10 min with 10 μM MEK inhibitor prior to bFGF stimulation inhibited induction of the early transient peak, yielding only the sustained peak of Pip92 expression which was maintained for at least 5 to 6 h poststimulation (Fig. 4A and B). This inhibition appeared to be specific since the MEK inhibitor alone had no effect on total [35S]methionine-labeled protein in trichloroacetic acid precipitates (data not shown).
FIG. 4.

Time course of synthesis of Pip92. Cells were pulse-labeled with [35S]Met during the last 30 min of stimulation, and Pip92 was immunoprecipitated and resolved by SDS-PAGE (12.5% gel) as described in Materials and Methods. (A) Autoradiograph showing synthesis of Pip92 by bFGF (10 ng/ml) in H19-7 cells. Samples were untreated or pretreated with 10 μM MEK inhibitor (MI) for 10 min prior to stimulation for the indicated times. (B) Plot of the time course of FGF-stimulated Pip92 protein synthesis, quantitated with an AMBIS radioanalytic scanner. Samples were treated with 10 ng of bFGF per ml alone (•) or 10 ng of bFGF per ml plus 10 μM MEK inhibitor (○). Data are plotted as the percent of maximum Pip92 synthesis (6,250 cpm) and represent the mean plus the range of samples from two independent experiments. (C) Autoradiograph showing synthesis of Pip92 by activated Raf in ΔRaf-1:ER cells. Cells were stimulated for the indicated times with 10 nM or 1 μM estradiol or pretreated for 10 min with 10 μM MEK inhibitor prior to stimulation with 1 μM estradiol. (D) Plot of the time course of estradiol-stimulated Pip92 protein synthesis quantitated with an AMBIS radioanalytic scanner. Samples were treated with 1 μM estradiol (•), 10 ng of estradiol per ml (□), or 1 μM estradiol plus 10 μM MEK inhibitor (○). Data are plotted as the percent of maximum Pip92 synthesis (3,100 cpm) and represent the mean plus the range of samples from two independent experiments.
A similar pattern of Pip92 protein synthesis was observed in ΔRaf-1:ER cells following stimulation by 1 μM estradiol, with maximum levels of newly synthesized Pip92 protein observed at 1 to 2 h poststimulation (Fig. 4C and D). Pretreatment of ΔRaf-1:ER cells with 10 μM MEK inhibitor prior to 1 μM estradiol stimulation again resulted in loss of the early peak, leaving only the sustained peak of newly synthesized Pip92 protein. When ΔRaf-1:ER cells were stimulated with the lower (10 nM) dose of estradiol, only the sustained peak of Pip92 expression was observed. These results demonstrate the presence of at least two kinetically distinct pathways for induction of Pip92 protein synthesis in response to bFGF or Raf; the first mechanism involves a MEK- and MAPK-sensitive pathway, and the second, more sustained signal does not require activation of MAPK.
Deletion analysis of pip92 promoter constructs.
The studies described above indicate that the pip92 promoter contains a domain responsive to a MAPK-independent signaling pathway activated by FGF or Raf. To identify these domains, the pip92 promoter was subjected to deletion analysis (Fig. 5A). The pip92 promoter (−1271pip92/CAT) has two Ets and two SRF binding domains (CArG boxes) between −1111 and −1271. Deletion of these two SREs (−1111pip92CAT) resulted in complete loss of Raf-activated transcription and loss of most (≥70%) of the FGF-stimulated transcription (Fig. 5B and C). Further deletion of the promoter up to −89 (−287CAT, −159CAT, and −89pip92CAT) had no effect. Similar results were observed in cells pretreated with 30 μM PD098059, which reduced overall transcription by about 50%. These results indicate that the two pathways for pip92 induction, MAPK independent and MAPK dependent, activate transcriptional regulatory elements between −1271 and −1111, a domain that contains SREs.
FIG. 5.

CAT assay of intact and deleted pip92/CAT promoter constructs. A 10-μg aliquot of DNA of deletion pip92 promoter/CAT constructs was transiently transfected into H19-7 or ΔRaf-1:ER cells. (A) Diagram depicting deletions of pip92 promoter. Transcriptional regulatory sites include a proximal (Ets-P) and distal (Ets-D) Ets domain for Elk1 binding, a SRE (CAArG) site for binding SRF, an Sp1 binding site, and a CRE site for binding CREB. (B) H19-7 cells were either untreated or pretreated with 30 μM MEK inhibitor (MI) for 10 min and then stimulated for 1 h with 10 ng of bFGF per ml. CAT activity was assayed as described in Materials and Methods. Data are plotted as the mean plus the range of samples from two independent experiments. (C) ΔRaf-1:ER cells were either untreated or pretreated with 30 μM MEK inhibitor (MI) for 10 min and then stimulated for 1 h with either 1 μM estradiol or 10 ng of estradiol per ml. CAT activity was assayed as described in Materials and Methods.
The MAPK-independent signaling pathway activates an SRE within the pip92 promoter.
The SRE enhancer region (−1270 to −970) of the pip92 promoter was further analyzed by linking it or its binding domains to a heterologous tk promoter (Fig. 6A). The results indicate that a domain containing a CArG-like box and Ets elements (1231 to −1158) is sufficient for full pip92 transcriptional activation by FGF or activated Raf-1 (Fig. 6B and C). Elimination of both the Ets and CArG-like domains or the CArG-like box alone resulted in complete loss of transcriptional activation. Furthermore, neither the CArG-like nor the Ets binding domain alone was sufficient to mediate transcription. Similar data were obtained for cells that had been pretreated with 30 μM PD098059 to suppress the MEK/MAPK pathway. These results indicate that transcriptional activation of pip92 (−1270 to −970) by the FGF- or Raf-activated MAPK-independent pathway is mediated by an SRE containing both an Ets binding and CArG-like element.
FIG. 6.

CAT assay of heterologous pip92-tk fusion promoter construct. Ten-microgram aliquots of the designated heterologous pip92-tk fusion promoter constructs were transiently transfected into H19-7 or ΔRaf-1:ER cells. (A) Diagram of heterologous pip92-tk promoter-CAT constructs. The transcriptional regulatory sites are described in the legend to Fig. 5A. (B) H19-7 cells were either untreated or pretreated for 10 min with 30 μM MEK inhibitor (MI) and then stimulated for 1 h with 10 ng of bFGF per ml. (C) ΔRaf-1:ER cells were either untreated or pretreated with 30 μM MEK inhibitor (MI) for 10 min and then stimulated for 1 h with either 1 μM estradiol or 10 ng of estradiol per ml. CAT activity was measured as described in Materials and Methods. Data are plotted as the mean plus the range of samples from two independent experiments.
Raf and FGF stimulate MAPK-independent kinases that phosphorylate Elk1 in vitro.
Previous studies have shown that the pip92 SRE can interact with recombinant SRF and Elk1 proteins at the CArG and Ets binding sites, respectively, forming a ternary complex (20). While SRF is not a target for MAPKs, Elk1 can be phosphorylated by MAPKs and is critical for transcriptional activation of target genes such as c-fos (17, 24). Therefore, we determined whether Elk1 can be similarly phosphorylated by the Raf-activated MAPK-independent pathway. Cell lysates were prepared from H19-7 cells stimulated with 10 ng of bFGF per ml or ΔRaf-1:ER cells stimulated with 1 μM estradiol to activate Raf. The lysates were then incubated in the presence of radioactively labeled ATP with a GST-Elk1 C-terminus fusion protein (GST-ElkC) prebound to Sepharose 4B beads. As expected, GST-ElkC was phosphorylated by extracts from cells stimulated with either FGF in H19-7 cells (Fig. 7A) or activated Raf-1 in ΔRaf-1:ER cells (Fig. 7B). Pretreatment of cells with 10 to 30 μM PD098059 for 10 min prior to stimulation resulted in a 50% decrease in GST-ElkC phosphorylation by cell lysates (Fig. 7).
FIG. 7.

Effect of MEK inhibition on in vitro Elk1 phosphorylation. H19-7 (A) or ΔRaf-1:ER (B) cells in N2 medium were untreated or pretreated for 10 min with the indicated amount of MEK inhibitor (MI) and then untreated or stimulated with 10 ng of bFGF per ml (H19-7) or 1 μM estradiol (ΔRaf-1:ER) for 1 h. With cell extracts containing 15 to 20 μg of protein and 50 μl of Sepharose 4B beads resuspended with Tris-buffered saline (1:1 ratio), the in vitro Elk1 phosphorylation was performed as described in Materials and Methods. Sepharose 4B beads prebound to bacterially expressed fusion proteins of GST, GST-ElkC (wild-type [wt] Elk1), or the GST-ElkC(A383/A389) mutant (mt) were prepared as described in Materials and Methods and used for substrates as indicated. The final eluates from the beads were resolved by SDS-PAGE (12.5% gel) and visualized by autoradiography.
Growth factor-regulated transcriptional activation of Elk1 is dependent on phosphorylation of S383/S389 in the C terminus (17, 24). When extracts from stimulated cells were incubated with a mutant GST-ElkC fusion protein in which S383/S389 of Elk1 had been mutated to A383/A389, no phosphorylation of the mutated Elk was detected (Fig. 7). These results indicate that MAPK-independent as well as MAPK-dependent kinases mediate Raf- and FGF-induced phosphorylation of Elk1 at S383/S389.
Raf and FGF stimulate MAPK-independent kinases that transcriptionally activate Elk1.
To determine whether Elk1 could become a transcriptional activator in response to phosphorylation by the MAPK-independent kinases in vivo, cells were transfected prior to treatment with a plasmid encoding ElkC fused to the GAL4 DNA binding domain (GAL-ElkC) along with a reporter plasmid containing a GAL4 binding site linked to the luciferase gene. As shown in Fig. 8A and B, both FGF (in H19-7 cells) and estradiol-activated Raf (in ΔRaf-1:ER cells) stimulated Elk1-mediated GAL4 transcription as monitored by luciferase activity. Pretreatment of cells with up to 30 μM PD0980959 inhibited only about 50% of the Elk1-mediated transcription (Fig. 8C and D), similar to results described above for transcription of pip92. Substitution of a plasmid encoding mutant GAL-ElkC with the S383/S389 phosphorylation sites mutated to A383/A389 resulted in complete loss of Gal4 transcription (Fig. 8A and B). These results indicate that Elk1 can become a transcriptional activator in response to phosphorylation by Raf- and FGF-stimulated MAPK-independent kinases, and the stimulation is specific for the phosphorylation of S383/S389 in Elk1. Thus, it is possible that phosphorylation of Elk1 accounts for the transcriptional activation of pip92 by this pathway.
FIG. 8.

Effect of MEK inhibition on Elk1 phosphorylation and transcriptional activation of GAL4. Ten micrograms of plasmids expressing either wild-type (wt Elk1) or mutant (mt Elk1) GAL4-ElkC(A383/A389) fusion proteins and 10 μg of the GAL4-luciferase reporter plasmid were transiently cotransfected into cells. (A and B) Selective activation of wild-type but not mutant GAL4-ElkC fusion protein. H19-7 cells (A) or ΔRaf-1:ER cells (B) in N2 medium were untreated or pretreated for 10 min with 30 μM MEK inhibitor (MI) and then untreated or stimulated with 10 ng of bFGF per ml (H19-7) or 1 μM estradiol (ΔRaf-1:ER) for 1 h. Luciferase activity was measured as described in Materials and Methods. Data are plotted as the percent of maximum luciferase activity and represent the mean plus the range of samples from two independent experiments. (C and D) Dose response of inhibition of GAL4 transactivation by the MEK inhibitor. H19-7 (C) or ΔRaf-1:ER (D) cells in N2 medium were untreated or pretreated for 10 min with the indicated amount of MEK inhibitor and then untreated or stimulated with 10 ng of bFGF per ml (H19-7) or 1 μM estradiol (ΔRaf-1:ER) for 1 h. Samples were assayed as described in Materials and Methods. These results are representative of two independent experiments.
Identification of novel Elk1 kinases that are activated by Raf.
To identify the MAPK-independent Elk1 kinases, in-gel kinase assays were performed with either wild-type or mutant (A383/A389) GST-ElkC fusion protein as the substrates (Fig. 9). Extracts containing equal protein from ΔRaf-1:ER cells that had been stimulated with 10 nM or 1 μM estradiol were resolved by SDS-PAGE, renatured, and assayed for Elk1 phosphorylation in the gel. The results showed a 44-kDa band, a constitutively active kinase of ca. 80 kDa, and three previously unreported Elk1 kinases of 220, 135, and 69 kDa (Fig. 9A). No significant kinase activity was detected when the mutant GST-ElkC(A383/A389) was used as a substrate (Fig. 9B). As observed for pip92 transcription, 10 nM estradiol was sufficient to induce maximal kinase activity, and Raf activation of the novel kinases was sustained for at least 3 h. Some induction of the constitutively activated 80-kDa band was also observed. The induction of these kinases was due to Raf activation rather than estradiol stimulation through the endogenous estrogen receptor, since no significant induction by estradiol was observed in the parent H19-7 cells (35a). Pretreatment of the cells with 30 μM PD098059 suppressed the 1-h peak of 44-kDa kinase activity, which presumably corresponds to MAPK, but did not affect the activities of the other kinases.
FIG. 9.

Identification of novel Raf-activated Elk1 kinases. ΔRaf-1:ER cells were untreated or pretreated with 30 μM MEK inhibitor (MI) and then stimulated for the indicated times with either 1 μM estradiol or 10 ng of estradiol per ml as indicated. Cell extracts containing 20 to 30 μg of proteins were resolved by SDS-PAGE on a 10% gel containing 50 μg of bacterially expressed wild-type GST-ElkC (wt Elk1) (A) or mutant GST-ElkC(A383/A389) (mt Elk1) (B) per ml as a substrate. The in-gel kinase renaturation assay was performed as described in Materials and Methods. Elk1 kinases that are activated by Raf and are distinct from MAPK are indicated by arrows.
Interestingly, analysis of samples from FGF-treated H19-7 cells showed only slight induction of these kinases at best (data not shown). Since FGF is upstream of Raf and activates a more complex set of signaling pathways, this result is not surprising. The signal from activated Raf appeared to be simpler and more robust; therefore, we focused on the Raf-stimulated kinases.
To ensure that the kinases that we identified are not artifacts of the renaturation process, we have verified that there are other Elk1 kinase activities distinct from MAPK by column chromatography. ΔRaf-1:ER cells that had been stimulated with 10 nM estradiol were lysed and fractionated by fast protein liquid chromatography gel filtration using a Superose 6 column. Fractions were then assayed for phosphorylation of Elk1 by the in vitro GST-Elk1 kinase assay and for activated MAPK by immunoblotting with anti-phospho-MAPK antibodies. As shown in Fig. 10, there are at least two peaks of Elk1 kinase activity that are comparable in size to kinases resolved by the gel renaturation assay. The peak of the activated MAPK activity does not correspond to that of the second Elk1 peak, suggesting that there may be more than one kinase in this molecular weight range. Unfortunately the activities of the other kinases could not be accurately quantitated and are probably underestimated due to their instability over time, whereas the MAPK activities were relatively stable (35b). Taken together, these results illustrate that there are Raf-activated kinases other than MAPK that can phosphorylate Elk1.
FIG. 10.

Fractionation of Raf-activated Elk1 kinases by gel filtration. ΔRaf-1:ER cells were shifted to 39°C in N2 medium for 24 h and then treated with 10 nM estradiol for 1 h to activate Raf. The cells were lysed, and 200 μl of the extract was applied to a Superose 6 HR column as described in Materials and Methods. Fractions of 0.5 ml were collected and assayed for protein (A) or Elk1 kinase (B) activity, using GST-Elk1 as a substrate, and for MAPK activity by immunoblotting with anti-active MAPK antibody as described in Materials and Methods. Prior to fractionation of the cell lysates, the column was calibrated by using molecular weight standards, and the arrows in panel A point to the peak fractions of immunoglobulin G (150 kDa) and chicken ovalbumin (46 kDa).
DISCUSSION
In this report, we have identified pip92 as an immediate early gene that is selectively induced by differentiating factors (bFGF or activated Raf-1) in rat hippocampal neuronal cells. These studies reveal the presence of at least two pathways for pip92 gene induction in response to bFGF or Raf which lead to differences in the amplitude and kinetics of Pip92 protein synthesis. The first mechanism for pip92 gene induction, which involves a MEK- and MAPK-dependent pathway, apparently results in the rapid activation and then deactivation of Pip92 protein synthesis. The second mechanism, which is MAPK independent, rapidly reaches a steady state for Pip92 protein synthesis that is maintained for hours. It is possible that this second pathway plays a key role in neuronal differentiation, since recent studies from our laboratory have shown that a MEK/MAPK-independent signaling pathway is required for the differentiation of H19-7 cells (18, 19).
Our results indicate that FGF and Raf can activate a common SRE in the promoter of the immediate early gene pip92 via MAPK-dependent and -independent signaling pathways. Both Raf-activated kinases are able to phosphorylate the SRE binding protein Elk1/TCF on S383/S389, enabling transactivation of target genes. The MAPK-independent pathway accounts for approximately 50% of the pip92 transcriptional activity, which was localized to the SRE, as well as 50% of the Elk1 transactivation of GAL4, a correlation consistent with Elk1 as the target of the MAPK-independent kinase. Analysis of the rate of Pip92 protein synthesis showed that the MEK/MAPK-dependent activation was transient (within an hour), whereas the activation by the MAPK-independent pathway was prolonged. Similarly, analysis of Raf-activated Elk1 kinases showed transient activation of a kinase corresponding to MAPK, whereas at least three other kinases that remained activated to a similar extent for several hours were identified. These results reveal a mechanism whereby the same signaling molecule, Raf, activates two distinct kinase pathways that can act on similar targets but with different kinetics and therefore different outcomes.
The role of MAPKs in neuronal differentiation is still under investigation. Previous studies based on transient expression in PC12 cells have suggested that MEK is both necessary and sufficient for neuronal differentiation (7, 11). However, studies based on mutational analysis of platelet-derived growth factor or Trk receptors have indicated that activation of the Ras/Raf/MAPK signaling pathway by differentiating factors is insufficient to induce PC12 cell differentiation (32, 35). Recent studies from our laboratory using H19-7 cells have shown that activated MEK is not sufficient to stimulate neuronal differentiation (18). Furthermore, activation of MEK is not even required for differentiation in response to FGF, a physiological activator (19). This result is consistent with the observation that MAPK stimulates only transient synthesis of Pip92 in H19-7 cells even though MAPK activation by FGF is sustained for hours at a low level (18). In hippocampal H19-7 cells, it is likely that the MAPK-independent signaling pathway, which is able to maintain synthesis of Pip92 and presumably other SRE-responsive proteins for hours, plays an important role in neuronal differentiation.
The induction of Pip92 expression under all known differentiating conditions in H19-7 cells raises the possibility that it is a key component in neuronal differentiation. To date, little is known about the function of the proline-rich Pip92. In PC12 pheochromocytoma cells, Pip92 is a cytoplasmic protein with a very short half-life that is slightly induced by factors that cause proliferation or membrane depolarization and is highly induced by factors that cause neuronal differentiation (3). Pip92 is also induced in fibroblasts by serum (3), and its human homolog is induced by TPA during HL-60 cell differentiation (31). The observation that Pip92 is expressed in the presence of the MEK inhibitor at concentrations that block Raf-induced differentiation suggests that Pip92 is not sufficient to mediate differentiation, consistent with its induction by a variety of biological stimuli.
Several lines of evidence suggest that Raf may phosphorylate other proteins besides MEK. For example, a kinase activity distinct from MAPK that is activated by Raf-1 and phosphorylates c-fos has been described (1a). Although controversial, Raf-1 has also been reported to phosphorylate and activate Cdc25, a cell cycle-regulated phosphatase (12). In addition, Raf has been reported to phosphorylate and inactivate BAD, resulting in enhanced protection by Bcl-2 against apoptosis (36, 40). Finally, a recent report of a study using a similar approach based on MKP-1 inactivation suggests that Raf can stimulate p70 S6 kinase through a MAPK-independent mechanism (21). Whether it is the same Raf-activated kinase that activates pip92 remains to be determined. Although some of these observations remain to be verified, taken together these data suggest that the effects of Raf may be mediated by MEK- and/or MAPK-independent signal transduction pathways.
Although Elk1 can be phosphorylated and activated by other members of the MAPK family as well some downstream kinases, the Elk1 kinases that we have identified do not appear to correspond to these reported kinases. Previously, ERK1 and ERK2 as well as JNKs (stress-activated kinases) and p38 (HOG) have all been reported to phosphorylate Elk1 (14, 17, 24, 37). However, the molecular weights of the novel kinases do not correspond to the reported sizes of JNK or p38. Furthermore, neither JNKs nor p38 is significantly activated by Raf within the first few hours of stimulation in H19-7 cells (1). Thus, the Elk1 kinases that we have identified appear to be distinct from these previously reported enzymes.
While signaling through Raf occurs exclusively through the SRE, FGF can also activate a region of the pip92 promoter that contains a CREB protein binding site near the site of transcription initiation. This result indicates that FGF is able to activate a target that is biochemically distinct from the SRE through a MAPK-independent mechanism. Consistent with this observation, phosphorylation of CREB at its transcriptional activation site was detected in response to FGF but not Raf stimulation (5a). Recent results from our laboratory suggest that FGF requires two discrete signaling pathways to mediate neuronal differentiation: the Ras/Raf pathway and the Src family kinase pathway (19). Since Raf stimulates pip92 transcription via the SRE in the H19-7 cells, it is possible that the Src pathway is the mediator of FGF-induced CREB phosphorylation. In either case, however, CREB phosphorylation does not appear to be required for neuronal differentiation since Raf is not able to activate this factor but can promote differentiation.
The results implicating the SRE in stimulation of pip92 by Raf and FGF in H19-7 cells are similar to those obtained previously for serum induction of pip92 in mouse 3T3 cells (20). While Elk1 was shown to bind to the Ets-like site in vitro, we cannot rule out the possibility that other TCFs mediate pip92 induction in vivo. However, the dose response for PD098059 inhibition of pip92 induction, the temporal pattern of pip92 induction, and the amplitude of pip92 induction by Raf and FGF parallel those for Elk1 phosphorylation, consistent with a common phosphorylation event responsible for both effects.
The detailed nature of the MAPK-independent signaling pathway downstream of Raf remains to be determined. If this pathway parallels the MEK/MAPK pathway, then it is possible that there is another kinase downstream of Raf that activates the Elk1 kinases that we have identified. Since different kinases renature to different extents, the relative levels of activation by the in-gel kinase assay may not be a true reflection of their in vivo activity. Whether all of the Elk1 kinases that we have identified are responsible for the in vivo transcriptional activity, or whether one is the primary kinase, cannot be determined at present. The activity that we measured (consistently about half of the total pip92-CAT transcriptional activation, GAL4-ElkC transactivation, or GST-ElkC phosphorylation) may reflect the action of a single kinase or result from multiple enzymes being activated. Since the Raf-activated, MAPK-independent Elk1 kinases appear to mediate prolonged Elk1 phosphorylation, it is possible that one or more can also play a role in differentiation.
ACKNOWLEDGMENTS
We thank L. Hill for assistance with the manuscript, G. Bowie for photographic assistance with the figures, and A. Salteil, R. Treisman, and V. Sukhatme for generously providing reagents.
This work was supported by National Institute of Health grants NS33858 (to M.R.R.) and CA52220 (to L.F.L.) and a gift from the Cornelius Crane Trust (to M.R.R.).
REFERENCES
- 1.Abe, M., and M. R. Rosner. Unpublished data.
- 1a.Agarwal S, Corbley M J, Roberts T M. Reconstitution of signal transduction from the membrane to the nucleus in a baculovirus expression system: activation of Raf-1 leads to hypermodification of c-jun and c-fos via multiple pathways. Oncogene. 1995;11:427–438. [PubMed] [Google Scholar]
- 2.Chao T-S O, Abe M, Hershenson M B, Gomes I, Rosner M R. Src tyrosine kinase mediates stimulation of Raf-1 and mitogen-activated protein kinase by the tumor promoter thapsigargin. Cancer Res. 1997;57:3168–3173. [PubMed] [Google Scholar]
- 3.Charles C H, Simske J F, O’Brien T P, Lau L F. Pip92: a short lived, growth factor-inducible protein in BALB/c 3T3 and PC12 cells. Mol Cell Biol. 1990;10:6769–6774. doi: 10.1128/mcb.10.12.6769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Charles C H, Sun H, Lau L F, Tonks N K. The growth factor inducible immediate early gene 3CH134 encodes a protein-tyrosine-phosphatase. Proc Natl Acad Sci USA. 1993;90:5292–5296. doi: 10.1073/pnas.90.11.5292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 5a.Chung, K.-C., and M. R. Rosner. Unpublished data.
- 6.Coleclough C, Kuhn L, Lefkovits I. Regulation of mRNA abundance in activated T lymphocytes: identification of mRNA species affected by the inhibition of protein synthesis. Proc Natl Acad Sci USA. 1990;87:1753–1757. doi: 10.1073/pnas.87.5.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cowley S, Paterson H, Kemp P, Marshall C J. Activation of MAP kinase kinase is necessary and sufficient for PC12 differentiation and for transformation of NIH 3T3 cells. Cell. 1994;77:841–852. doi: 10.1016/0092-8674(94)90133-3. [DOI] [PubMed] [Google Scholar]
- 8.Dalton S, Treisman R. Characterization of SAP-1, a protein recruited by serum response factor to the c-fos serum response element. Cell. 1992;68:597–612. doi: 10.1016/0092-8674(92)90194-h. [DOI] [PubMed] [Google Scholar]
- 9.Dudley D T, Pang L, Decker S J, Bridges A J, Saltiel A R. A synthetic inhibitor of the mitogen-activated protein kinase cascade. Proc Natl Acad Sci USA. 1995;92:7686–7689. doi: 10.1073/pnas.92.17.7686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eves E M, Tucker M S, Roback J D, Downen M, Rosner M R, Wainer B H. Immortal rat hippocampal cell lines exhibit neuronal and glial lineages and neurotrophin gene expression. Proc Natl Acad Sci USA. 1992;89:4373–4377. doi: 10.1073/pnas.89.10.4373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fukuda M, Gotoh Y, Tachibana T, Dell K, Hattori S, Yoneda Y, Nishida E. Induction of neurite outgrowth by MAP kinase in PC12 cells. Oncogene. 1995;11:239–244. [PubMed] [Google Scholar]
- 12.Galaktionov K, Jessus C, Beach D. Raf1 interaction with Cdc25 phosphatase ties mitogenic signal transduction to cell cycle activation. Genes Dev. 1995;9:1046–1058. doi: 10.1101/gad.9.9.1046. [DOI] [PubMed] [Google Scholar]
- 13.Gallego C, Gupta S K, Heasley L E, Oian N-X, Johnson G I. Mitogen-activated protein kinase activation resulting from selective oncogene expression in NIH 3T3 and Rat 1a cells. Proc Natl Acad Sci USA. 1992;89:7355–7359. doi: 10.1073/pnas.89.16.7355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gille H, Strahl T, Shaw P E. Activation of ternary complex factor Elk-1 by stress-activated protein kinases. Curr Biol. 1995;5:1191–1200. doi: 10.1016/s0960-9822(95)00235-1. [DOI] [PubMed] [Google Scholar]
- 15.Harlow E, Lane P. Antibodies: a laboratory manual. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory; 1988. [Google Scholar]
- 16.Hipskind R A, Rao V N, Mueller C G F, Reddy E S P, Nordheim A. Ets-related protein Elk-1 is homologous to the c-fos regulatory factor p62TCF. Nature. 1991;354:531–534. doi: 10.1038/354531a0. [DOI] [PubMed] [Google Scholar]
- 17.Janknecht R, Ernst W H, Pingoud V, Nordheim A. Activation of ternary complex factor Elk-1 by MAP kinases. EMBO J. 1993;12:5097–5104. doi: 10.1002/j.1460-2075.1993.tb06204.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kuo W-L, Abe M, Rhee J, Eves E M, McCarthy S A, Yan M, Templeton D J, McMahon M, Rosner M R. Raf, but not MEK or ERK, is sufficient for differentiation of hippocampal neuronal cells. Mol Cell Biol. 1996;16:1458–1470. doi: 10.1128/mcb.16.4.1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kuo W-L, Chung K-C, Rosner M R. Differentiation of central nervous system neuronal cells by fibroblast-derived growth factor requires at least two signaling pathways: roles for Ras and Src. Mol Cell Biol. 1997;17:4633–4643. doi: 10.1128/mcb.17.8.4633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Latinkic B V, Lau L F. Transcriptional activation of immediate early gene pip92 by serum growth factors requires both Ets and CArG-like elements. J Biol Chem. 1994;269:23163–23170. [PubMed] [Google Scholar]
- 21.Lenormand P, McMahon M, Pouyssegur J. Oncogenic Raf-1 activates p70 S6 kinase via a mitogen-activated protein kinase-independent pathway. J Biol Chem. 1996;271:15762–15768. doi: 10.1074/jbc.271.26.15762. [DOI] [PubMed] [Google Scholar]
- 22.Liang P, Pardee A B. Differential display of eukaryotic messenger RNA by means of the polymerase chain reaction. Science. 1992;257:967–971. doi: 10.1126/science.1354393. [DOI] [PubMed] [Google Scholar]
- 23.Macleod K, Leprince D, Stehelin D. The ets gene family. Trends Biochem. 1992;17:251–256. doi: 10.1016/0968-0004(92)90404-w. [DOI] [PubMed] [Google Scholar]
- 24.Marais R, Wynne J, Treisman R. The SRF accessory protein Elk-1 contains a growth factor-regulated transcriptional activation domain. Cell. 1993;73:381–393. doi: 10.1016/0092-8674(93)90237-k. [DOI] [PubMed] [Google Scholar]
- 25.Meloche S, Pages G, Pouyssegur J. Functional expression and growth factor activation of an epitope-tagged p44 mitogen-activated protein kinase, p44mapk. Mol Biol Cell. 1992;3:63–71. doi: 10.1091/mbc.3.1.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nishida E, Gotoh Y. The MAP kinase cascade is essential for diverse signal transduction pathways. Trends Biochem Sci. 1993;18:128–131. doi: 10.1016/0968-0004(93)90019-j. [DOI] [PubMed] [Google Scholar]
- 27.Pang L, Sawada T, Decker S, Saltiel A. Inhibition of MAP kinase kinase blocks the differentiation of PC-12 cells induced by nerve growth factor. J Biol Chem. 1995;270:13585–13588. doi: 10.1074/jbc.270.23.13585. [DOI] [PubMed] [Google Scholar]
- 28.Sambrook J, Fritsch E F, Maniatis T. Molecular cloning: a laboratory manual. 2nd ed. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 29.Samuels M L, Weber M J, Bishop J M, McMahon M. Conditional transformation of cells and rapid activation of the mitogen-activated protein kinase cascade by an estradiol-dependent human Raf-1 protein kinase. Mol Cell Biol. 1993;13:6241–6252. doi: 10.1128/mcb.13.10.6241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Seger R, Krebs E G. The MAPK signaling cascade. FASEB J. 1995;9:726–735. [PubMed] [Google Scholar]
- 31.Shimizu N, Ohta M, Fujiwara C, Sagara J, Mochizuki N, Oda T, Utiyama H. Expression of a novel immediate early gene during 12-o-tetradecanoylphorbol-13-acetate-induced macrophagic differentiation of HL-60 cells. J Biol Chem. 1991;266:12157–12161. [PubMed] [Google Scholar]
- 32.Stephens R M, Loeb D M, Copeland T D, Pawson T, Greene L A, Kaplan D R. Trk receptors use redundant signal transduction pathways involving SHC and PLC-gamma1 to mediate NGF response. Neuron. 1994;12:691–705. doi: 10.1016/0896-6273(94)90223-2. [DOI] [PubMed] [Google Scholar]
- 33.Thorburn J, Xu S, Thorburn A. MAP kinase- and Rho-dependent signals interact to regulate gene expression but not actin morphology in cardiac muscle cells. EMBO J. 1997;16:1888–1900. doi: 10.1093/emboj/16.8.1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Treisman R. Identification and purification of a polypeptide that binds to the c-fos serum response element. EMBO J. 1987;6:2711–2717. doi: 10.1002/j.1460-2075.1987.tb02564.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vaillancourt R R, Heasley L E, Zamarripa J, Storey B, Valius M, Kazlauskas A, Johnson G L. Mitogen-activated protein kinase activation is insufficient for growth factor receptor-mediated PC12 cell differentiation. Mol Cell Biol. 1995;15:3644–3653. doi: 10.1128/mcb.15.7.3644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35a.Wang, D., and M. R. Rosner. Unpublished data.
- 35b.Wang, D., K.-C. Chung, and M. R. Rosner. Unpublished data.
- 36.Wang H-G, Rapp U R, Reed J C. Bcl-2 targets the protein kinase Raf-1 to mitochondria. Cell. 1996;87:629–638. doi: 10.1016/s0092-8674(00)81383-5. [DOI] [PubMed] [Google Scholar]
- 37.Whitmarsh A J, Shore P, Sharrocks A D, Davis R J. Integration of MAP kinase signal transduction pathways at the serum response element. Science. 1995;269:403–407. doi: 10.1126/science.7618106. [DOI] [PubMed] [Google Scholar]
- 38.Wood K W, Qi H, D’Arcangelo G, Armstrong R C, Roberts T M, Halegoua S. The cytoplasmic raf oncogene induces a neuronal phenotype in PC12 cells: a potential role for cellular raf kinases in neuronal growth factor signal transduction. Proc Natl Acad Sci USA. 1993;90:5016–5020. doi: 10.1073/pnas.90.11.5016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wood K W, Sarnecki C, Roberts T M, Blenis J. ras mediates nerve growth factor receptor modulation of three signal-transducing protein kinases: MAP kinase, Raf-1, and RSK. Cell. 1992;68:1041–1050. doi: 10.1016/0092-8674(92)90076-o. [DOI] [PubMed] [Google Scholar]
- 40.Zha J, Harada H, Yang E, Jockel J, Korsmeyer S J. Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14-3-3 not BCL-XL. Cell. 1996;87:619–628. doi: 10.1016/s0092-8674(00)81382-3. [DOI] [PubMed] [Google Scholar]