

Abstract
The proteins encoded by the retinoblastoma gene family, pRB, p107, and p130, have been implicated in the regulation of cellular proliferation, differentiation, and transformation. Because interactions between p130 and E2F transcription factors have been proposed to play a role in the establishment and/or maintenance of quiescence in human peripheral T lymphocytes, we examined lymphoid differentiation and proliferation in p130-deficient mice. We show that p130−/− T cells proliferate normally in culture and exhibit normal cell-mediated immune function in vivo. However, p130−/− T lymphocytes expressed elevated levels of p107, and the characteristic p130-E2F DNA binding complex was replaced by a p107-E2F complex. Adoptive transfer of fetal liver lymphoid progenitors allowed us to circumvent the neonatal lethality associated with loss of p130 and p107 and to analyze the phenotype of p130−/−;p107−/− peripheral T lymphocytes. These cells achieved a quiescent state, exhibited derepression of a subset of E2F target genes, and were hypersensitive to concanavalin A stimulation. Interestingly, a significant portion of the E2F-4 in p130−/−;p107−/− T cells was detected in a complex with pRB and an as-yet-unidentified protein. These findings provide a biochemical basis for functional compensation between pRB family proteins.
The retinoblastoma tumor suppressor gene (RB) is the founding member of a family of genes that encode related cell cycle-regulatory proteins, including pRB, p107, and p130. Each pRB family protein can induce a G1-specific cell cycle arrest when overexpressed in tumor cells, and this effect is dependent upon a conserved bipartite protein binding domain present in pRB, p107, and p130 (15, 55, 77, 78). This domain mediates interactions with a number of proteins, including E2F transcription factors, and is also targeted by several viral oncoproteins that are known to displace these cellular binding proteins (51). Although the RB gene is mutated in different human tumors, similar disruptions of p107 or p130 have not been reported. This suggests that pRB family proteins regulate different cellular pathways, a concept supported by the observations that each protein can interact with distinct cellular proteins and that each inhibits cell cycle progression only in distinct subsets of tumor cell lines (reviewed in reference 70).
The analysis of mouse strains carrying inactivated alleles of the murine homologs of RB, p107, and p130 also indicates the existence of both shared and unique functions of this protein family. For example, Rb+/− mice develop pituitary and thyroid tumors, and Rb−/− embryos die at midgestation, exhibiting defective proliferation and differentiation in some lineages (13, 33, 43). In contrast, both p107−/− embryos and p130−/− embryos develop normally, and the mutant adults exhibit no obvious tumor predisposition (17, 44). However, embryos deficient for p130 and p107 exhibit excessive chondrocyte proliferation, bone defects, and rapid postnatal death, suggesting functional overlap or compensation between these two members of the gene family (17). Functional overlap was also observed upon interbreeding p107 and Rb mutant mouse strains. p107 deficiency accelerates the Rb−/− embryonic lethality by approximately 2 days, and Rb+/−;p107−/− mice are runted and exhibit bilateral retinal lesions (44). These results demonstrate that pRB, p107, and p130 play essential roles in the regulation of proliferation in vivo and also indicate complex genetic interactions within this gene family. However, the specific nature of these genetic interactions and their mechanism of action remains uncharacterized. In particular, it remains unclear if the absence of defects in p107−/− and p130−/− mice results from their redundant functions in most cell types or instead involves specific molecular compensation in particular lineages.
The E2F transcription factor family plays a central role in the regulation of cell cycle progression (58). Each of the five characterized E2F proteins dimerizes with a DP subunit, binds a consensus E2F site, and activates the expression of reporter genes that contain such sites (5, 24, 27, 29, 37, 45, 59; reviewed in reference 42). Overexpression of E2F-1 induces the expression of several endogenous cell cycle-regulatory genes, including those for B-myb, cdc2, cyclin A, and E2F-1 itself (21), and can drive quiescent cells to enter S phase (36, 47, 56). The regulation of E2F activity by pRB family proteins has been extensively investigated and suggests that the coordinated activity of these two protein families confines the expression of various genes to discrete phases of the cell cycle (16). pRB binds E2F-1, -2, and -3 with high affinity (45), while p107 and p130 interact specifically with E2F-4 and E2F-5 (5, 24, 29, 59); various experiments suggest that these complexes are formed in distinct phases of the cell cycle. In particular, p107-E2F complexes are first detected as cells near the G1/S transition and predominate in S-phase cells (18, 24, 60, 63), perhaps reflecting the increased synthesis of p107 as cells progress through the cell cycle (4). The p130-E2F interaction is most readily detected in quiescent or growth-arrested cells, including various serum-starved cells (18), differentiated muscle and neuronal cell cultures (19), and human peripheral T lymphocytes (11, 50, 69).
Numerous studies indicate that pRB family-E2F interactions can mediate promoter repression (35, 65, 68, 72, 73; reviewed in reference 16), and recent analysis of mouse embryo fibroblasts (MEFs) deficient for p107 and p130 indicates that these proteins are required for the repression of a subset of E2F-responsive genes (31). The increased expression of p130 in late-stage embryos (8) and p130’s proposed role in differentiated cell types suggest that analysis of fully differentiated adult tissues, rather than embryo-derived fibroblasts, may be particularly relevant for our understanding of these growth-regulatory molecules. However, the rapid postnatal death of p107−/−;p130−/− mice has precluded analysis of gene expression and, more importantly, of the proliferative properties of such mature tissues.
The analysis of human T lymphocytes in particular has suggested that p130 plays a key role in the regulation of E2F activity in the establishment of the resting state and/or as resting cells reenter the cell cycle (11, 64, 69). These lymphocytes constitute a uniform population of incompletely differentiated quiescent cells that, upon appropriate stimulation, undergo extensive proliferation followed by terminal differentiation (20, 79). Cultured lymphocytes can be induced to proliferate with various mitogens that mimic in vivo T-cell receptor activation, and this cell cycle entry is characterized by a lengthy G1 phase before cells initiate DNA synthesis (20, 40). Quiescent T cells contain a single E2F DNA binding complex that is composed of E2F-4 bound to p130 (69), and the expression of E2F target genes during cell cycle reentry is accompanied by gradual disappearance of the p130-E2F complex and progressive accumulation of free forms of E2F (11), the majority of which is E2F-4 (50). Thus, p130 function appears to be closely linked to the repression of cell cycle progression genes and the maintenence of cellular quiescence.
In this study we investigated directly the consequences of p130 loss of function on the control of quiescence and proliferation by utilizing peripheral T lymphocytes from p130−/− mice as a model system. Furthermore, we have explored the roles of other members of the pRB family in functional substitution for p130 in the establishment and maintenance of the quiescent state.
MATERIALS AND METHODS
Lymphoid cell collection and flow cytometry.
The spleen, thymus, and lymph nodes were dissected from 6- to 14-week old p130+/+ and p130−/− littermates or from recipient animals 6 to 12 weeks after adoptive transfer. Organs were lysed in phosphate-buffered saline (PBS) plus 2% calf serum by gentle pressure through 70-μm nylon mesh, and cells were washed briefly before flow cytometry, cell culture, or protein extraction for immunoblotting or electrophoretic mobility shift assay (EMSA). For analysis of T- and B-cell markers, 2.5 × 105 to 5 × 105 cells were incubated with appropriate fluorescein isothiocyanate- or phycoerythrin (PE)-conjugated monoclonal antibodies (0.1 to 0.4 μg in 100 μl) for 30 to 60 min on ice before being washed and stained with propidium iodide (PI) for analysis by Becton Dickinson FACScan. Analysis was restricted to viable lymphocytes or thymocytes by gating on appropriately sized, PI-negative populations. All flow cytometry antibodies were purchased from Pharmingen. For isolation of pure T cells, splenocytes were depleted of erythrocytes by hypotonic lysis, and 70 × 106 to 90 × 106 lymphocytes were incubated for 30 min on ice with PE-conjugated antibodies specific to B cells (B220; 2.5 μg) and macrophages (Mac1; 0.25 μg). Sorting of PE-negative cells from PE-positive cells (routinely 2 log units brighter) with a FACStar Plus or modified FACS II yielded T-cell-enriched populations that were consistently over 94% pure and generally provided 4 × 106 to 6 × 106 T cells for proliferation assays. T-cell fractions used for RNA and/or immunoblot analysis (75 to 90% pure) were generated by passing splenocytes or lymph node cells over T-cell enrichment columns (Biotex).
Contact hypersensitivity assay.
Assays were carried out as described previously (40). Briefly, four 10-week-old p130+/+ and p130−/− animals were sensitized by topical application of 2% oxazolone irritating agent (Sigma) in ethanol. Five days later, these animals and a control group that had not been sensitized were challenged by application of 1% oxazolone to the left ear (experimental) or 100% ethanol to the right ear (control). Ear thickness was measured on the day of challenge and 48 h later. ΔT is the change in ear thickness over this time period.
Proliferative assays.
Assays were carried out in triplicate, using 96-well round-bottom plates with 105 cells in 0.2 ml of RPMI supplemented with 10% fetal calf serum, 50 μM 2-mercaptoethanol, 2 mM l-glutamine, nonessential amino acids, 100 mg of penicillin per ml, 250 mg of streptomycin per ml, and 10 mM HEPES. Lymph node lymphocytes, splenocytes, or purified splenic T cells were washed, counted, diluted to 106/ml, and counted again before being cultured in the absence or presence of concanavalin A (0.1 to 3 μg/ml; Pharmacia), phorbol 12-myristate 13-acetate (PMA) (20 ng/ml; Sigma) plus ionomycin (100 ng/ml; Calbiochem), soluble CD3 antibody (1 to 5 μg/ml; Pharmingen), or plate-bound CD3. Antibody was bound to plates by incubation of 30 μl of CD3 antibody (10 μg/ml in PBS) per well for 90 min at 37°C, prior to three washes with 0.2 ml of PBS. Recombinant murine interleukin-2 (IL-2) (Boehringer-Mannheim) was used at 50 to 100 U/ml. Two hours prior to the indicated time points, [3H]thymidine (1 μCi/well; 6.7 Ci/mmol; Dupont/NEN) was added to each sample. At the indicated times, plates were frozen, and at the end of the 30-h time course all samples were collected on glass fiber filter mats with a Tomtec Harvester96. Incorporated radioactivity was quantified by liquid scintillation counting.
EMSA.
Whole-cell extracts were prepared from lymph node lymphocytes by lysis in 5× extraction buffer (100 mM HEPES [pH 7.4], 5 mM MgCl2, 2.5 mM EDTA, 0.5 M KCl, 20% glycerol, 0.5 mM phenylmethylsulfonyl fluoride, 20 μM sodium orthovanadate [pH 8.0]). The lysate concentration was determined with Bradford reagents and then adjusted to 1.5 mg/ml by dilution in 5× extraction buffer before use of 3 μl per reaction. E2F binding reactions were carried out as described previously (7), except for the use of reduced levels of total protein and salmon sperm nonspecific competitor DNA (0.5 μg). The saturating levels and specificities of antibodies to p130, p107, and pRB were determined by titration in wild-type and mutant whole-cell extracts (data not shown). The antibodies utilized are described below. Complexes were separated by electrophoresis on a 4% polyacrylamide (29.2:0.8)–0.25× Tris-borate-EDTA gel at 150 V for 3 h at 4°C. The sequence of the E2F consensus oligonucleotide was 5′-ATTTAAGTTTCGCGCCCTTTCTCAA-3′.
Adoptive transfer.
p130−/−;p107+/− animals either were intercrossed or were mated with p130+/−;p107−/− mice to obtain double-mutant and control embryos. The day of vaginal plug was taken as day zero. At embryonic day 13.5 (E13.5) or E14.5 fetal liver samples were collected into 550 μl of PBS and dissociated by passage through a 23-gauge needle. Fifty microliters of cell suspension was used for DNA purification and PCR genotyping of p107 (44) and p130 (17), while the remainder was used for intraocular injection of two recipient mice (4- to 8-week-old Rag2−/− or scid−/− mice) that had been sublethally irradiated (3 Gy from a 137C source) 2 h earlier. After 6 weeks, reconstitution was monitored by flow cytometry of T- and B-lymphocyte populations in peripheral blood and/or lymphoid organs. Identical results were observed with Rag2−/− or scid−/− recipients.
Western blotting.
The protein concentration in E2F extracts or Nonidet P-40 whole-cell lysates was determined with Bradford reagents, and 30 to 100 μg of protein per lane was loaded on sodium dodecyl sulfate-polyacrylamide (either 6.5% or 12%) (30:1) gels. Gels were electrophoresed at 150 V for 4 h or at 30 V overnight before transfer to nitrocellulose membrane (60 V for 4 h in 25 mM Tris–190 mM glycine–20% methanol). Blots were preblocked in PBS–0.1% Tween 20 plus 5% dried milk for 3 to 12 h before being probed with primary antibody diluted in block solution, usually overnight at 4°C. The blots were then washed with PBS–0.1% Tween 20 three times for 10 min each before incubation with peroxidase-linked secondary antibody (Amersham) (1:5,000 dilution; 1 h at room temperature) followed by another three washes. Blots were then developed with enhanced chemiluminescence reagents and exposed to Kodak X-OMAT 5 film for 1 to 20 min.
Antibodies.
The following antibodies were used for EMSA: Santa Cruz Biotechnology C-20 polyclonal (for p130), SD-15 monoclonal (for p107) (21a), 21C9 monoclonal (for pRB) (18), no. 207 polyclonal (a gift of M. Imperiale), Oncogene Science RB no. 2 polyclonal (for pRB), and LLF4-2 monoclonal (for E2F-4) (a gift of K. Moberg and J. Lees).
The following antibodies were used for immunoblot analysis: G245 monoclonal (for pRB) (from Pharmingen) and C-18 polyclonal (for p107), C-20 polyclonal (for p130), C-19G goat polyclonal (for p27), M2 polyclonal (for cdk2), C-21 polyclonal (for cdk6), M-20 polyclonal (for cyclin E), and 34B1-3 rat monoclonal (for cyclin D2) (all from Santa Cruz).
Northern analysis.
Lymph node cell suspensions or T-cell-enriched splenocyte fractions (of equivalent purity from each genotype) were collected, and total RNA was harvested by use of RNAzolB reagent. Five to 10 micrograms of RNA was electrophoretically separated on a 1% agarose–1× MOPS (morpholinepropanesulfonic acid)–8% formaldehyde gel. The RNAs were then transferred to nylon membranes in 20× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate), UV cross-linked to the membrane, and prehybridized, hybridized, and washed as described previously (59). All probes were labeled by random oligonucleotide labeling with a Quickprime kit (Stratagene).
RESULTS
Differentiation and proliferation of p130-deficient lymphocytes.
The abundance of specific p130-E2F complexes in various differentiated cell types, including cells of muscle, neuronal, and lymphoid lineages, suggests that this complex functions in the establishment or maintenance of the differentiated state. Although p130-deficient mice develop normally and exhibit no apparent tumor predisposition, we closely compared cells of the lymphoid lineages of wild-type and p130−/− littermates to determine if subtle abnormalities might result from p130 deficiency. Comparison of thymuses, lymph nodes, and spleens of wild-type and p130-deficient animals revealed no differences in cellularity or histological appearance (data not shown), and flow cytometric analysis of lymphoid organs indicated very similar expression of B-lymphocyte (B220+) and T-lymphocyte (T-cell receptor αβ+ [TCRαβ+]) surface markers (Fig. 1A). Similar populations of immunoglobulin M-expressing B cells and CD4- or CD8-positive T lymphocytes were also observed in suspensions of wild-type and p130−/− splenocytes (Fig. 1A). The appropriate expression of these and other markers of B cells (CD19 and immunoglobulin D) and T cells (CD62 and CD25) (data not shown) indicated that development of various lymphocyte populations proceeds normally in the absence of p130 function.
FIG. 1.
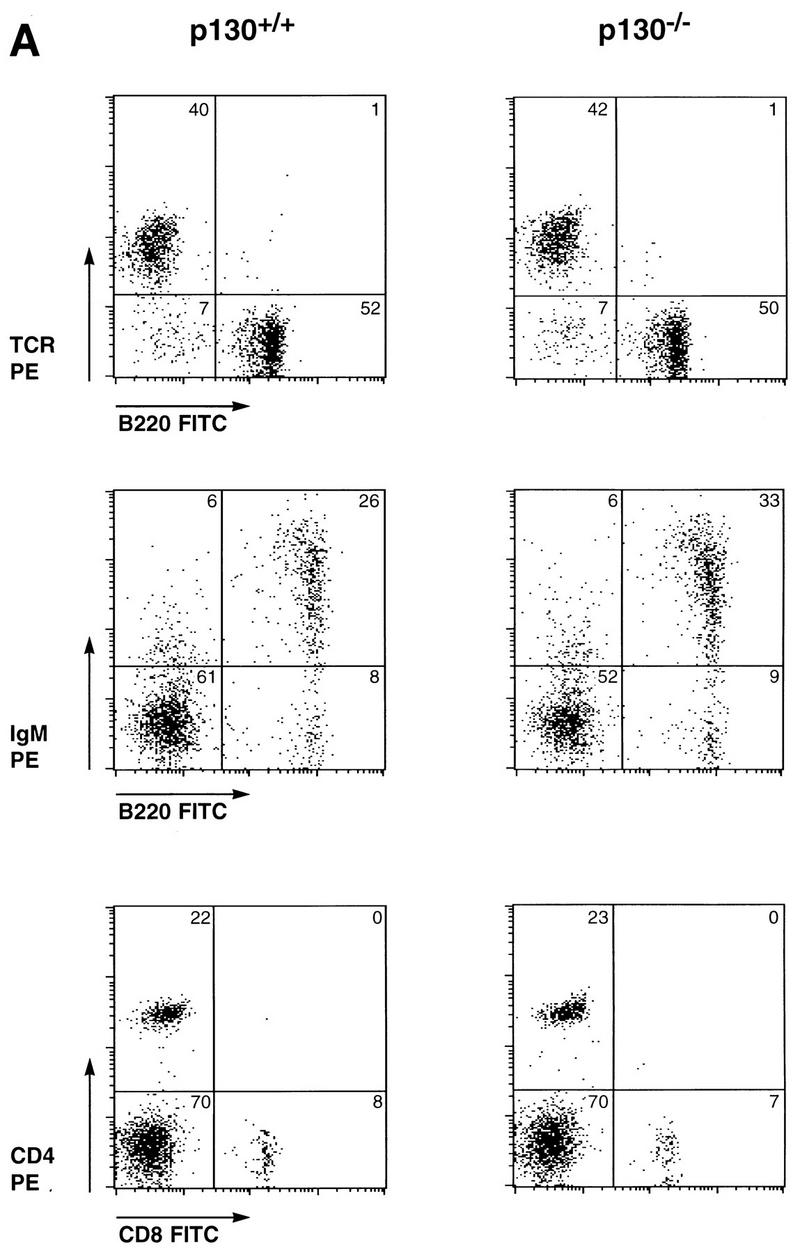

Normal differentiation and proliferation of p130−/− lymphocytes. (A) Wild-type and p130−/− splenocytes were stained for expression of the indicated cell surface markers and analyzed by flow cytometry. The percentages of cells in each quadrant are indicated. Each plot displays 2,000 events. FITC, fluorescein isothiocyanate; IgM, immunoglobulin M. (B, inset) Background incorporation of [3H]thymidine in cultures of unstimulated wild-type (open bar) and p130−/− (solid bar) lymph node lymphocytes cultured for 2 h in the presence of 1 μCi of [3H]thymidine. (B and C) Time course of [3H]thymidine incorporation, as a measure of DNA synthesis, upon PMA-plus-ionomycin (B) or concanavalin A (con A) (C) stimulation of lymph node lymphocytes (see Materials and Methods). Triplicate cultures were pulsed for 2 h at the times shown. Open squares, wild type; closed circles, p130−/−. Results are presented as mean counts per minute ± standard errors of the mean. Data are representative of three independent experiments. Similar results were obtained with wild-type and p130−/− purified splenic T cells. (D) In vivo T-cell responses as determined by contact hypersensitivity. The inflammatory responses of p130+/+ and p130−/− animals in reaction to topical application of exogenous hapten (2% oxazolone in ethanol) (solid bars) are presented as change in ear thickness 48 h after challenge. The control (open bars) represents the response to ethanol without hapten. Naive animals were not sensitized prior to challenge. Error bars indicate standard errors of the means.
Because p130 has been implicated in the maintenance of quiescence in G0 cell populations (50, 64, 69), we next wished to determine whether p130-deficient cells could achieve normal quiescence and whether they exhibited an abnormal response to mitogens. Lymphocytes isolated from the lymph nodes of wild-type and p130−/− littermates were cultured in the presence or absence of T-cell mitogens, and DNA synthesis was monitored by [3H]thymidine incorporation. Freshly isolated, unstimulated lymphocytes from animals of both genotypes exhibited background levels of thymidine incorporation after 2 h (Fig. 1B, inset) or 10 h (data not shown), demonstrating that lymphocytes can achieve quiescence in the absence of p130. Moreover, in both the wild-type and p130−/− cultures, proliferation induced by PMA plus ionomycin (Fig. 1B), the mitogenic lectin concanavalin A (Fig. 1C), or CD3 antibodies (data not shown) resulted in S-phase entry only after a 22- to 24-h period. Similar results were obtained with cultures of purified splenic T cells (data not shown). Thus, p130 is not required for the establishment of the quiescent state in peripheral T cells, nor does its absence promote premature S-phase entry following stimulation into the cell cycle.
We also assayed p130−/− T-lymphocyte activation and function in vivo. Contact hypersensitivity provides a measure of cell-mediated immune function in response to epidermal exposure to an exogenous hapten and is characterized by localized inflammation (40). The normal responses of p130−/− mice in this assay when compared to p130+/+ controls (Fig. 1D) indicated appropriate regulation of antigen presentation, CD4 T-cell activation, and lymphokine secretion in the absence of p130.
Effects of p130 loss on p107 function.
To investigate the consequence of p130 loss of function for regulation of E2F transcription factors, we performed EMSA with extracts prepared from wild-type and p130-deficient lymphocytes. Initial characterization of wild-type murine peripheral T lymphocytes indicated that these cells possess a single E2F DNA binding activity that is composed of p130 and the E2F-4 member of the E2F transcription factor family (Fig. 2A), much like the complex described for human T cells (69). Figure 2A demonstrates that addition of a p130 polyclonal antibody (lane 2) or an E2F-4-specific monoclonal antibody (lane 3) supershifted this complex. Inclusion of both antibodies produced a single complex with even slower mobility (lane 4). Thus, in murine T cells, as had been shown previously for human cells, the predominant E2F-regulatory complex contains p130.
FIG. 2.
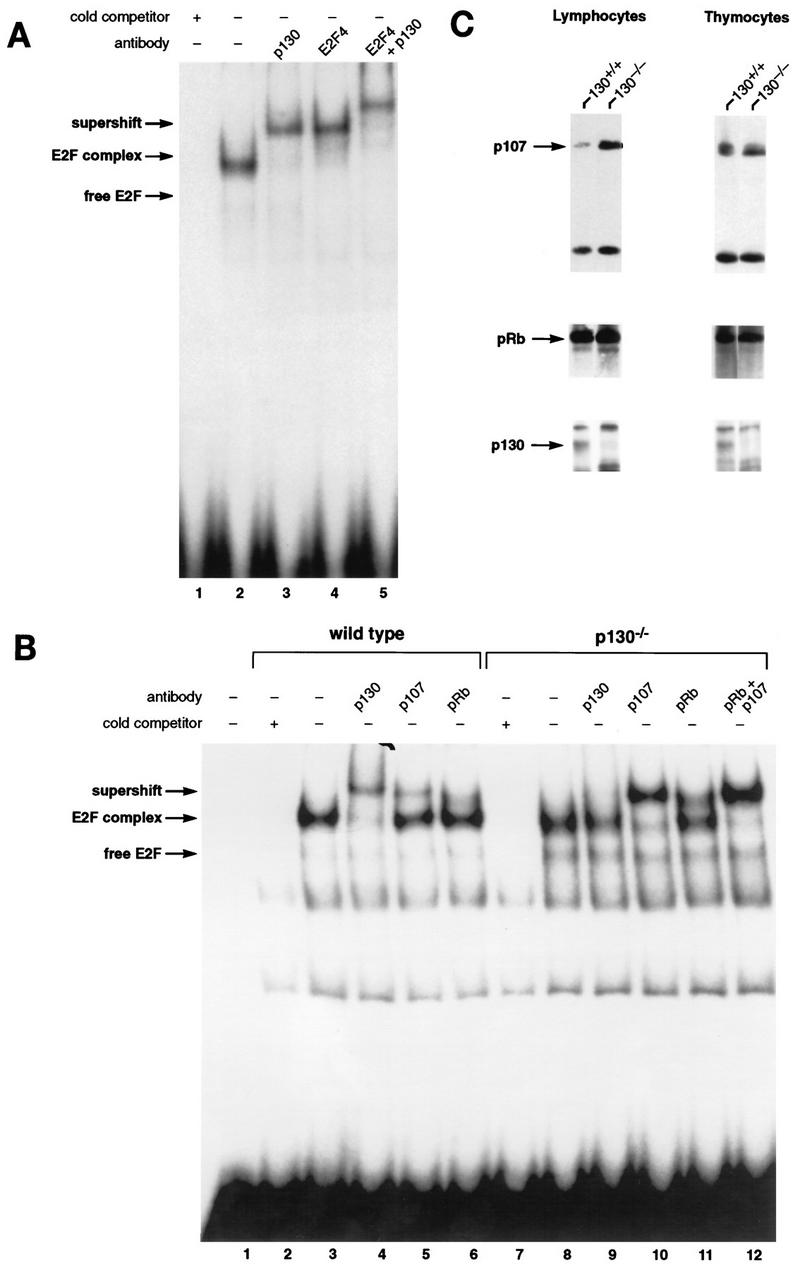
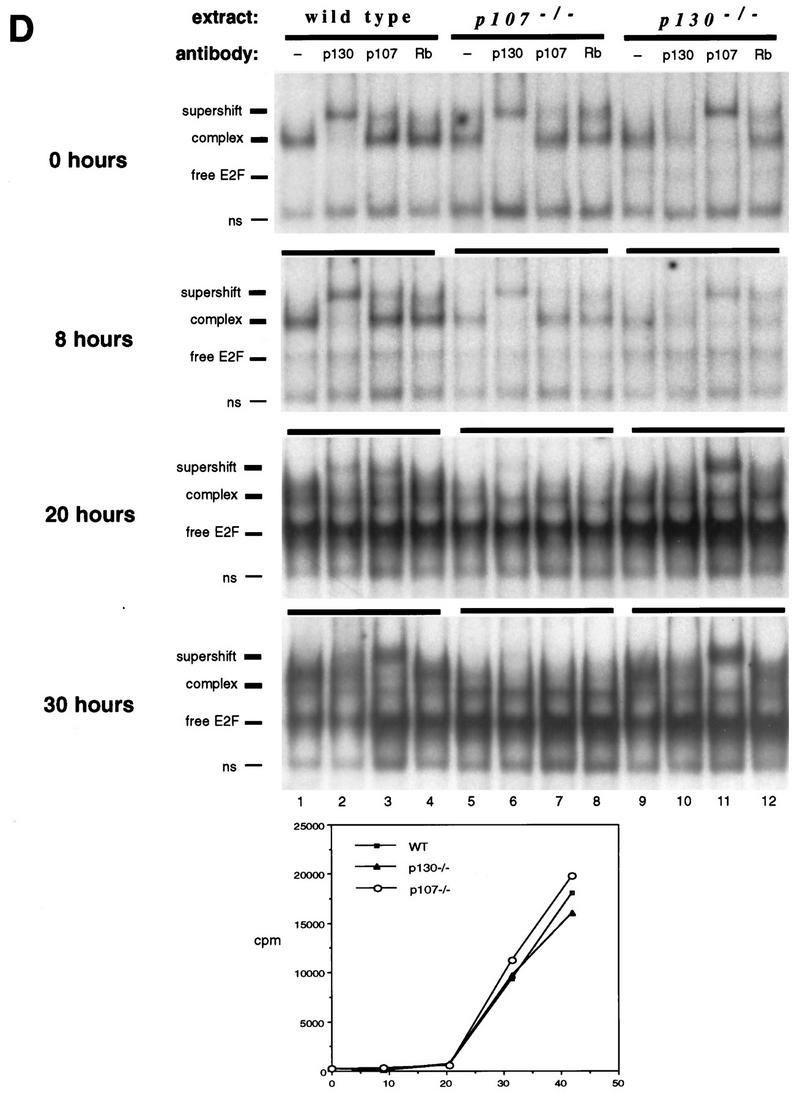
Characterization of E2F complexes in wild-type and mutant peripheral T lymphocytes. (A) Whole-cell extracts of wild-type lymph node lymphocytes were analyzed by EMSA with a radiolabeled E2F probe. The single complex (lane 2) was supershifted by antibody to p130 (lane 3) and antibody to E2F-4 (lane 4). Addition of both antibodies induced a second supershift of the entire complex (lane 5). Specificity of the complex was demonstrated by competition with a 100-fold excess of cold E2F oligonucleotide (lane 1). (B) Characterization of E2F complexes present in wild-type and p130−/− T-lymphocyte extracts. The complex present in p130−/− cells (lane 8) appears to be similar in mobility to that in wild-type cells (lane 3). These complexes were analyzed with antibodies to p130 (lanes 4 and 9), p107 (lanes 5 and 10), or pRB (lanes 6 and 11). The positions of free E2F and higher-order E2F complexes are indicated. The specificity of these complexes was shown by competition with a 100-fold excess of cold E2F oligonucleotide (lanes 2 and 7). Analyses of purified T-cell populations or lymphocyte nuclear extracts yielded similar results (data not shown). (C) Elevated expression of p107 in peripheral T lymphocytes of p130−/− mice. Whole-cell extracts (30 μg) from wild-type and p130−/− thymocytes and lymph node T lymphocytes were subjected to immunoblot analysis with p107, pRB, or p130 antibodies. The lower band in the p107 panel is a cross-reacting protein that serves as a loading control. Similar results were obtained upon analyses of nuclear extracts. Quantitation indicates that p107 levels increased 25% in thymocytes and sevenfold in peripheral lymphocytes. (D) E2F complex analysis as wild-type and mutant T cells reenter the cell cycle. Lymph node lymphocytes were isolated from wild-type (lanes 1 to 4), p107−/− (lanes 5 to 8), and p130−/− (lanes 9 to 12) animals and stimulated with PMA-ionomycin. Cell extracts were prepared at the indicated time points, and E2F complexes were identified by using antibodies to p130, p107, and pRB. In addition, a fraction of each culture was labeled with [3H]thymidine to determine S-phase entry (bottom panel). The positions of the nonspecific DNA binding complex (ns), free E2F, E2F complex, and antibody-induced supershift are indicated at the left.
We next compared E2F complexes in extracts of wild-type and p130−/− T cells (Fig. 2B). Strikingly, the E2F DNA binding activity in p130-deficient cells (lane 8) was very similar to that in wild-type cells (lane 3). Although some free E2F was detectable in the p130-deficient extracts, the vast majority (85 to 90%) was present in a slower-mobility complex that migrates at a position similar to that of the p130-E2F complex. This complex was supershifted upon addition of a p107-specific antibody (Fig. 2B, lane 10), indicating that p107 can replace p130 as the major component of the E2F complex in p130-deficient cells. The appearance of this novel p107-E2F complex in p130−/− T lymphocytes suggests that p107 contributes to E2F regulation in these resting cells, perhaps as part of a transcriptional repressor complex (65). We also utilized the E2F-4-specific antibody to confirm that all DNA-bound E2F in p130−/− extracts is E2F-4 (data not shown).
Interestingly, p107 expression is normally highest in cycling cells, particularly during S phase of the cell cycle (4), and p107 levels are usually inversely correlated with expression of p130 in proliferating and growth-arrested cells (64). We therefore analyzed the expression of p107 in the lymphoid organs of wild-type and p130−/− mice. Although the thymocytes of both animals expressed similar, relatively high levels of p107 and pRB, mature T cells isolated from the lymph nodes of p130 mutant animals expressed significantly higher levels of p107 than did wild-type cells (Fig. 2C). Thus, a specific overexpression of p107 in response to p130 deficiency occurs only in late stages of lymphoid development, coincident with the differentiation of quiescent peripheral T cells. This observation, coupled with the detection of novel p107-E2F complexes in peripheral T cells, suggested that E2F regulation or another conserved function of p130 or p107 may be crucial for lymphoid maturation and/or cell cycle regulation.
We also analyzed the nature of E2F complexes as wild-type, p130−/−, and p107−/− T cells reentered the cell cycle upon mitogen stimulation (Fig. 2D). In extracts of wild-type T lymphocytes, elevated levels of free E2F correlated with disappearance of the p130-E2F complex in G1, and S-phase extracts contained both free E2F and a prominent p107-E2F complex (Fig. 2D, lanes 1 to 4) (11, 50). In p130-deficient T-cell extracts (Fig. 2D, lanes 9 to 12), the p107-E2F complex detected in resting cells was maintained during the G1 phase of the cell cycle (8 and 20 h poststimulation), and the levels of free E2F detected in late-G1-phase (20-h) and S-phase (30-h) extracts were similar to wild type. S-phase extracts of p107-deficient cells lacked the prominent p107-E2F complex (Fig. 2D, lanes 9 to 12, 30 h poststimulation), and there was no apparent substitution by p130 or pRB, despite the relatively high levels of pRB in S-phase lymphocytes (references 11 and 50 and data not shown). This suggests that the pRB family-E2F interactions detected in these experiments are specific and do not result merely from two interacting proteins spuriously associating within a cellular extract.
Lymphopoiesis in the absence of p107 and p130.
In order to examine the requirement for p107 in the context of a p130 mutation in regard to both T-cell development and function, we intercrossed animals carrying mutant alleles in both genes. As we have shown previously, embryos lacking both p107 and p130 exhibit excessive epiphyseal chondrocyte proliferation and defective long-bone development and die shortly after birth (17). Although this phenotype dramatically demonstrates that some tissues require p130 and/or p107 function for proper regulation of proliferation, it also precludes the analysis of adult tissues that lack both proteins. In the mouse, mature T lymphocytes expressing T-cell receptor (TCR) and either CD4 or CD8 begin to be exported to the periphery at approximately E18, but this differentiation process peaks only after birth (79). As a first test of the effects of combined elimination of p130 and p107, we examined thymic T-cell development in double-mutant embryos and their control littermates by dissecting the fetal thymus and performing flow cytometry analysis of CD4, CD8, TCRαβ, and CD25 cell surface markers. For each of these markers, fetal thymocytes from E18 p130−/−;p107−/− embryos exhibited staining patterns very similar to those of control littermates. Figure 3A demonstrates that both control and double-mutant fetal thymuses contained 92% CD4- and CD8-expressing (double-positive) thymocytes and very low levels (2 to 4%) of mature single-positive cells.
FIG. 3.
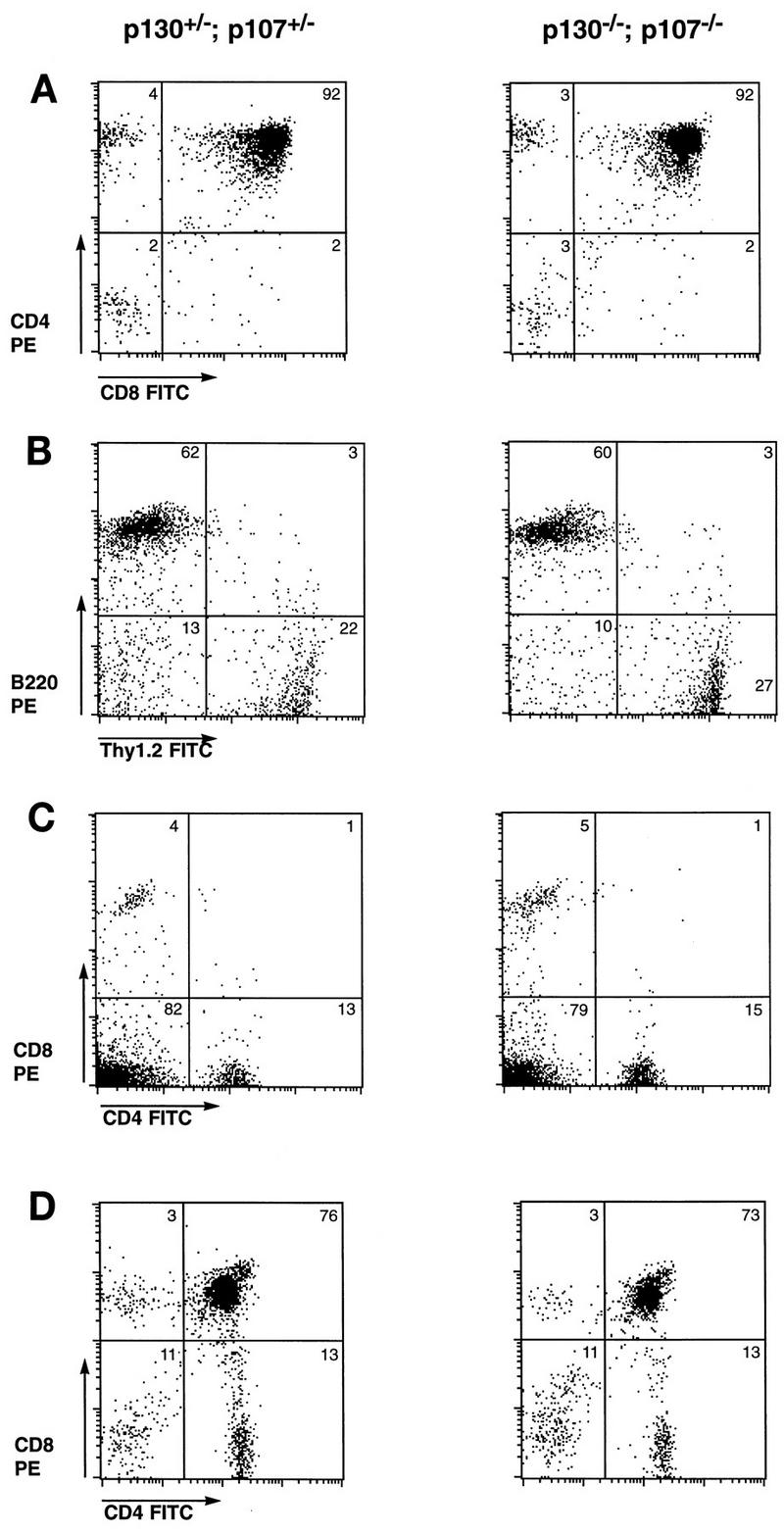
Mature lymphocytes develop in the absence of p130 and p107. (A) Fetal thymocytes of p130−/−;p107−/− and control embryos at day 18 of gestation were subjected to flow cytometric analysis for expression of CD4 and CD8. Results are representative of at least two embryos of each genotype. FITC, fluorescein isothiocyanate. (B and C) The lymphoid compartments of Rag2−/− animals were reconstituted via adoptive transfer of fetal liver from p130−/−;p107−/− and control embryos. After 6 weeks, the presence of mature B and T lymphocytes was assessed by flow cytometric analysis of B220 and Thy1.2 expression, respectively (B), and CD4 and CD8 expression (C) on splenocytes of reconstituted animals. Data are representative of three independent analyses, and the ratio of T to B lymphocytes in each was influenced by the time elapsed since reconstitution, as the B-cell lineage matures more rapidly than the T-cell lineage. Splenocytes of Rag2−/− controls were devoid of mature lymphocytes, as expected (data not shown). (D) Thymocytes of animals reconstituted with p130−/−;p107−/− or control fetal liver exhibit similar levels of CD4+ CD8+ (double-positive) and CD4+ or CD8+ (single-positive) T cells.
In order to circumvent the limitations on the analysis of lymphocytes from germline double homozygotes imposed by the attendant neonatal lethality, we turned to adoptive transfer of fetal liver lymphoid progenitor cells of various genotypes. This enabled us to analyze later stages of T-cell development and the resulting phenotypes of peripheral T cells. p130+/−;p107−/− mice were mated with p130−/−;p107+/− animals, and E14.5 to E15.5 fetal liver samples were collected, genotyped, and injected into lymphoid-deficient (Rag2−/− or scid−/−) recipient animals as described in Materials and Methods. Six to 12 weeks after injection, the lymphoid compartments of recipient animals were analyzed both histologically and for expression of specific differentiation markers of the B-cell and T-cell lineages. The sizes, cellularities, and histological appearances of the thymuses, lymph nodes, and spleens were generally similar in animals reconstituted with p130+/−;p107+/−, p130−/−;p107+/−, or p130−/−;p107−/− cells; occasional differences in cellularity were observed, which likely reflect the efficiency of injection (data not shown). Flow cytometric analysis of splenocytes from these reconstituted animals consistently revealed similar levels of Thy1.2-expressing T cells (20 to 30%) and B220-positive B cells (60 to 70%), regardless of donor cell genotype (Fig. 3B). Comparison of p130+/−;p107+/− and p130−/−;p107−/− peripheral T cells or thymocytes also revealed closely matched percentages of mature (CD4 or CD8 single-positive) or immature (CD4 and CD8 double-positive) T-lymphoid cells, respectively (Fig. 3C and D). These data indicated that despite their high-level expression in developing lymphoid organs (8, 39) and putative role in lymphocyte cell cycle transitions (11, 50, 69), p130 and p107 are dispensable for many aspects of lymphoid maturation and differentiation.
In addition to conducting cell cycle analysis of p130/p107 double-mutant lymphocytes (see below), we wished to characterize the status of various cell cycle regulatory molecules, including pRB family proteins, cyclin-dependent kinases, and the p27 and p21 cyclin-dependent kinase inhibitors. To do so, we performed immunoblot analyses to determine if these cell cycle components were altered in lymphocytes lacking p130 or lacking p130 and p107. Cell extracts were made from the lymph nodes of animals reconstituted with p130+/−;p107+/−, p130−/−;p107+/−, or p130−/−;p107−/− lymphocytes. Figure 4A shows immunoblot analysis with antibodies specific for p130, p107, pRB, cdk6, cyclin D2, cdk2, and cyclin E, as well as p27. As expected, p107 levels were elevated in p130-deficient lymphocytes (lane 2), and lysates of p130−/−;p107−/− cells lacked both proteins (lane 3). The absence of both p107 and p130 did not induce a change in the levels or phosphorylation status of pRB. Moreover, when compared with that in control cells, expression of cdk2, the cdk4 homolog cdk6, and cyclin D2 (Fig. 4A) and cyclin D1 (data not shown) in lymphocytes lacking p130 (lane 2) or p130 and p107 (lane 3) was unchanged. A slight reduction in levels of cyclin E was observed in double-mutant cells (lane 3). Although a minor effect, this was observed in extracts prepared by either hypotonic or Nonidet P-40 lysis. Analysis of expression of cyclin-dependent kinase inhibitor during lymphoid proliferation indicates that resting lymphocytes express low levels of p21 but that p27 levels are elevated and play an important role in rapamycin-mediated inhibition of cell cycle progression (53). The lymphocytes from reconstituted animals expressed low levels of p21 and high levels of p27; both were unaffected by the status of p130 and p107 (Fig. 4A and data not shown).
FIG. 4.
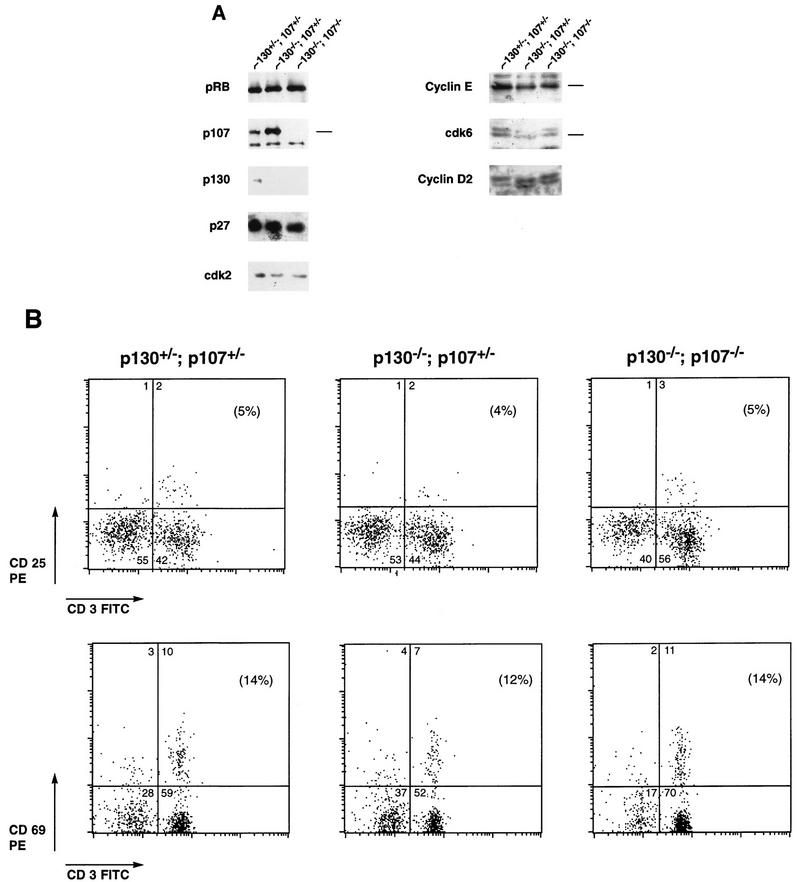

p130−/−;p107−/− peripheral T lymphocytes achieve quiescence. (A) Expression of cell cycle-regulatory molecules in peripheral T cells lacking p130 or both p130 and p107. Whole-cell extracts (60 μg) of p130+/−;p107+/−, p130−/−;p107+/−, and p130−/−;p107−/− lymph node lymphocytes were resolved by sodium dodecyl sulfate–7% polyacrylamide gel electrophoresis, transferred to nitrocellulose, and subjected to immunoblot analysis. This blot was sequentially probed with antibodies to pRB, p107, p130, p27, and cdk2. A separate blot from a 12% gel was probed with antibodies to cyclin E, cdk6, and cyclin D2. The antibodies are described in Materials and Methods. (B) Comparison of expression of T-lymphocyte activation markers on reconstituted splenocytes. The expression of CD25/IL-2 receptor and the CD69 early activation marker on splenic T lymphocytes was evaluated in Rag2−/− animals 6.5 weeks postreconstitution with p130+/−;p107+/−, p130−/−;p107+/−, or p130−/−;p107−/− fetal liver. The percentage of CD3-positive T cells expressing each marker is indicated in parentheses. CD69 antibody detects transiently activated cells, while CD25 is expressed on proliferating T-cell populations (67). FITC, fluorescein isothiocyanate. (C) Comparison of CD45RB and CD62 expression on reconstituted splenocytes. The percentage of Thy1.2-positive T cells expressing each marker is indicated in parentheses. CD45RB antibody (clone 16A) reacts with an exon B-dependent epitope of CD45 that is highly expressed on naive lymphocytes (22). The CD62/MEL14 antibody detects lymphocyte L-selectin expression, which is rapidly down-regulated upon activation. In each case these markers of lymphocyte activation are expressed similarly on p130+/−;p107+/−, p130−/−;p107+/−, and p130−/−;p107−/− splenic T cells.
To characterize further the state of differentiation and cell cycle activity of lymphocytes lacking either p130 or both p130 and p107, these cells were subjected to flow cytometric analysis of various lymphoid surface proteins that are specifically expressed either on resting (CD62, CD45RB, and B7.2) or activated (CD25 and CD69) lymphocytes; cell cycle activity was determined by assessing DNA content. The CD25/IL-2 receptor and CD69 early activation marker were each present on only a small percentage of p130+/−;p107+/−, p130−/−;p107+/−, and p130−/−;p107−/− CD3-positive T cells (Fig. 4B). Consistent with this, a large proportion (81 to 88%) of both the mutant and control T lymphocytes expressed the CD45RB naive T-cell marker (22) and the CD62/L-selectin adhesion molecule that is expressed on resting lymphocytes (67) (Fig. 4C). In addition, analysis of DNA content by PI staining indicated that more than 97% of both p130+/−;p107+/− and double-mutant cells contained a 2N DNA content, and less than 1.5% of cells were in S phase (data not shown). Taken together, the analysis of cell cycle-regulatory proteins, expression of surface markers, and DNA content indicates that lymphocytes deficient in p130 and p107 do not proliferate inappropriately in vivo and instead achieve a quiescent state similar to that of control lymphocytes. Considering the documented association of p130 with E2F in resting T cells (11, 69) and the manner in which p107 substitutes in the absence of p130, the ability of double-mutant cells to achieve and maintain quiescence is noteworthy.
E2F DNA binding activity in p130−/−;p107−/− T cells.
To assess the regulation of E2F activity in p130−/−;p107−/− T cells, we performed EMSA with extracts of double-mutant and control peripheral T lymphocytes. Figure 5A compares the E2F complexes present in extracts of p130−/−;p107+/+ and p130−/−;p107−/− lymphocytes. Double-mutant extracts exhibited markedly increased levels of free E2F (lane 6) compared to the p130−/− single mutant (lane 2). However, double-mutant cells also contained a slower-mobility E2F complex that constituted 50 to 60% of the total E2F DNA binding activity. While the E2F complex in p130-deficient cells contained predominantly p107 (lane 3), the majority of the complex in double-mutant cells was supershifted by a pRB-specific antibody (lane 8). We further analyzed the pRB-containing complex in double-mutant cells by addition of E2F-4 antibody (Fig. 5B). This antibody shifted all of the free and complexed E2F (lane 3), demonstrating that this G0 E2F complex contained, in part, pRB bound to E2F-4.
FIG. 5.
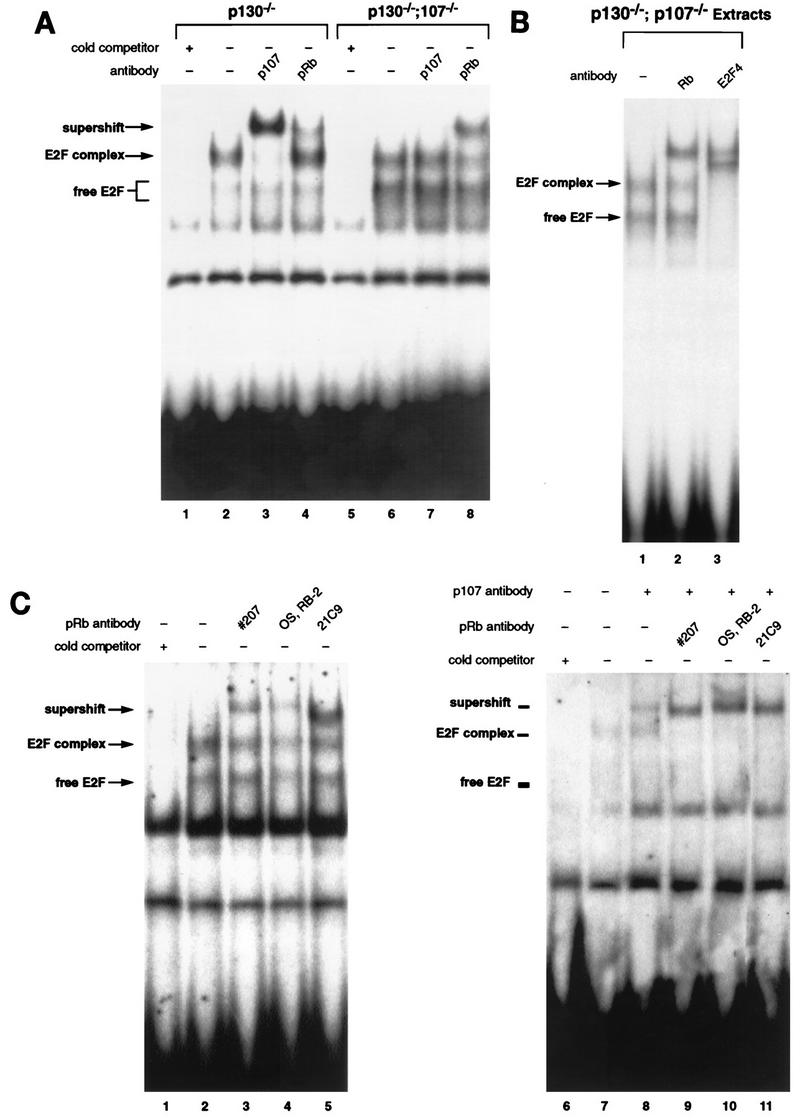
Characterization of E2F complexes in p130−/−;p107−/− peripheral lymphocytes. (A) E2F EMSA with extracts of p130−/−;p107−/− double-mutant lymph node lymphocytes indicated significantly elevated levels of free E2F (lane 6) compared to p130−/− lymphocytes (lane 2). However, both extracts contain E2F complexes of similar mobility. These complexes were characterized by addition of antibodies to p107 (lanes 3 and 7) and pRB (lanes 4 and 8). The specificity of these complexes was shown by competition with a 100-fold excess of cold E2F oligonucleotide (lanes 1 and 5). (B) The E2F complex in p130−/−;p107−/− extracts contains E2F-4 and pRB. Addition of pRB monoclonal antibody 21C9 supershifted a portion of the E2F complex (lane 2), and the E2F-4 antibody supershifted all of the free and complexed forms of E2F (lane 3). (C) A portion of the E2F complex in p130−/−;p107−/− lymphocyte extracts remains unaffected by several different pRB antibodies. Polyclonal serum raised against an 800-amino-acid fragment of murine pRB (no. 207) (lane 3) or an anti-C-terminal peptide antibody (Oncogene Science) (lane 4), as well as the 21C9 monoclonal antibody (lane 5), supershifted only a portion of the E2F complex detected in double-mutant lymphocytes. Extracts of asynchronously cycling MEFs were used (right panel) to confirm that each antibody can supershift murine pRB (lanes 8 to 10). p107-specific antibody is included in lanes 7 to 10 so that the pRB-E2F complex can be discerned.
We also noted that a portion of the E2F complex in double-mutant cells was unaffected by the pRB antibody (Fig. 5A, lane 8, and Fig. 5B, lane 2), despite the addition of a twofold-greater amount of antibody than necessary for the observed supershift (data not shown). Therefore, we investigated the nature of this complex with additional antibodies, including an anti-pRB C-terminal peptide polyclonal antibody, polyclonal antiserum raised against a murine pRB antigen (Fig. 5C), and the E2F-4-specific antibody (Fig. 5B). While all three antibodies directed against the retinoblastoma protein failed to completely supershift the E2F complex (Fig. 5C, lanes 3, 4, and 5), the E2F-4 antibody effectively supershifted all E2F complexes (Fig. 5B, lane 3). The ability of each antibody to supershift murine pRB was verified by using extracts of asynchronously cycling MEFs (Fig. 5C, right panel). These observations suggest that p130- and p107-deficient cells possess a novel E2F-4 binding activity that is distinct from the known pRB-family proteins. In contrast, the vast majority of E2F DNA binding activity in wild-type or p130−/− T-cell extracts can be identified with pRB family antibodies (Fig. 2A, lane 3, and Fig. 5A, lane 3), suggesting that this novel activity is present at significant levels only in p130−/−;p107−/− cells.
Regulation of E2F-responsive genes in p130−/−;p107−/− T cells.
Previous studies have shown that transcriptional repression of putative E2F target genes correlates with the formation of pRB family-containing protein complexes on upstream E2F DNA binding sites (41, 68). With regard to G0 cell populations, such as wild-type peripheral T lymphocytes, p130-E2F complexes have been proposed to repress E2F target gene expression to prevent cell cycle entry (64, 69). The detection of significant free E2F DNA binding activity in double-mutant lymphocytes suggested that cellular E2F transcription factors were not fully regulated by pRB family protein binding. Recent analysis of serum-starved MEFs that lack p130 and p107 detected derepression of certain putative E2F target genes, including those for B-myb, cdc2, E2F-1, and, to a lesser degree, cyclin A2, thymidine synthase, and ribonucleotide reductase M2 (RRM2) (31). To characterize the effects of p130 and p107 loss on E2F target gene expression in mature peripheral lymphocytes, we harvested RNAs from various mutant T lymphocytes derived via adoptive transfer experiments and performed Northern blot analysis. Unpurified p130−/−;p107−/− lymphocytes isolated from lymph nodes expressed elevated levels of the mRNAs encoding B-myb, cdc2, E2F1, cyclin A2, and RRM2 compared with p130+/−;p107+/− and p130−/−;p107+/− controls (data not shown). In contrast to analyses of serum-starved cell cultures, these lymphocytes were uniformly quiescent in vivo, thereby ensuring that the elevated expression of these genes did not reflect a subset of cells that failed to undergo growth arrest. In order to verify that T lymphocytes exhibit this defect, we depleted B cells from splenocyte suspensions and purified RNAs from equivalent populations of double-mutant and control T lymphocytes (see Materials and Methods). As shown in Fig. 6, the expression of cdc2, cyclin A2, B-myb, and RRM2 was notably elevated in T cells deficient in p130 and p107. However, the expression of various other genes that are presumed targets of E2F regulation was unaffected, including the genes for cyclin E, E2F-1, TS1, Rb, and E2F-4 (Fig. 6) and dihydrofolate reductase, c-myc, and thymidine kinase (data not shown). This data indicated that in the absence of p130 and p107, a specific subset of E2F target genes is derepressed. The appropriate regulation of these genes in p130−/−;p107+/− T cells (Fig. 6) indicated that the loss of both proteins was essential for this defect to become manifest. It remains unclear if the subset of E2F target genes that are unaffected by loss of p130 and p107 are regulated by non-E2F-related mechanisms in these cells or if this reflects E2F repression mediated by pRB and/or the uncharacterized E2F binding protein.
FIG. 6.
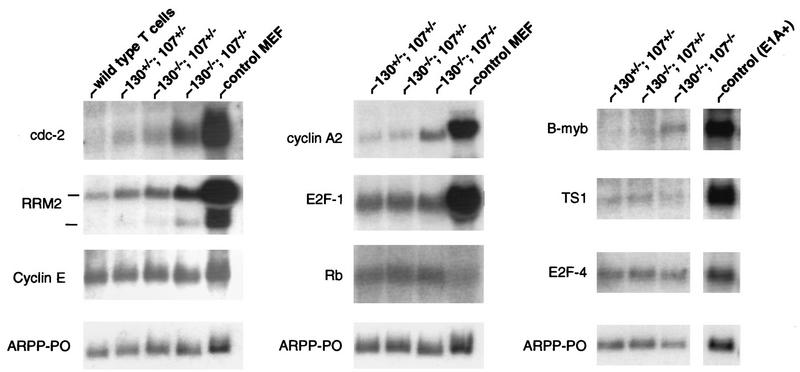
A subset of E2F target genes is derepressed specifically in p130−/−;p107−/− peripheral T lymphocytes. Total RNA was purified from T-cell-enriched (85 to 90% pure) splenocytes of reconstituted animals and analyzed by Northern blotting (10 μg/lane). (Left and middle panels) Blots with T-lymphocyte RNA from chimeric and control wild-type animals, as well as cycling MEFs, were sequentially hybridized with the indicated probes. (Right panel) RNAs from the indicated lymphocytes and E1A-expressing MEFs were resolved and hybridized with the indicated probes. Acidic ribosomal phosphoprotein PO (ARPP-PO) was used as a loading control.
Proliferation of p130−/−;p107−/− lymphocytes.
The increased free E2F and associated partial deregulation of E2F target genes observed specifically in T lymphocytes lacking both p130 and p107 raised the possibility that mitogen treatment of these cells might reveal aberrant proliferative properties that were not observed in cells lacking only p130 (Fig. 1). To determine if double-mutant T cells progress to S phase more rapidly than normal T lymphocytes, purified splenic T cells were cultured in the presence of specific mitogens, and S-phase entry was monitored over the next 30 h (Fig. 7). In both the p130+/−;p107+/− and p130−/−;p107−/− T-cell cultures, concanavalin A treatment induced S-phase entry at 20 to 22 h after stimulation, and the kinetics of cell cycle entry were not significantly altered by the addition of exogenous murine IL-2 (Fig. 7A). Similar results were obtained upon treatment of double-mutant and control T lymphocytes with PMA plus ionomycin (data not shown).
FIG. 7.
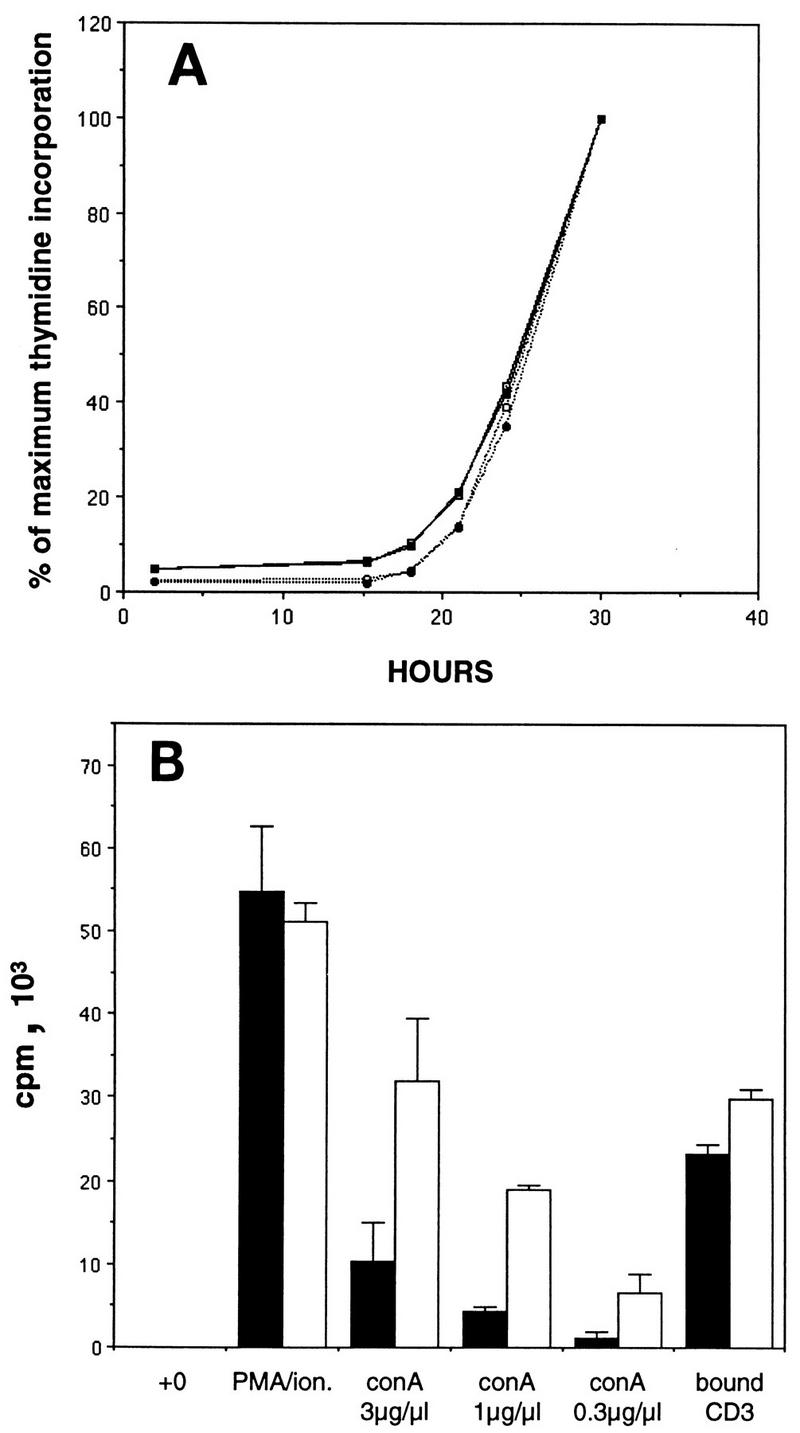
Cell cycle reentry and mitogen sensitivity of p130−/−;p107−/− T lymphocytes. (A) p130+/−;p107+/− (solid lines) and p130−/−;p107−/− (dotted lines) purified splenic T cells were stimulated with concanavalin A (3 μg/ml) and incubated in the presence (closed symbols) or absence (open symbols) of murine IL-2 (100 U/ml). S-phase entry was monitored as described in the legend to Fig. 1, and results are presented as the percentage of maximum thymidine incorporation. Similar results were obtained upon stimulation with PMA plus ionomycin (data not shown). Data are representative of two independent experiments. (B) p130+/−;p107+/− (solid bars) and p130−/−;p107−/− (open bars) purified splenic T lymphocytes were either untreated or stimulated with PMA plus ionomycin (ion.) various concentrations of concanavalin A (conA), or plate-bound CD3 antibody. Twenty-eight hours after stimulation (middle of the first S phase), the cultures were pulsed with [3H]thymidine (1 μCi) for 2 h. Results are presented as means ± standard errors of the means. Data are representative of two independent experiments.
In contrast to the similar rates at which double-mutant and control lymphocytes entered the cell cycle, analysis of total thymidine incorporation in these cultures revealed a striking elevation in the response of the double-mutant cells to concanavalin A (Fig. 7B). The 2.5- to 3-fold elevation in thymidine incorporation by cells lacking both p130 and p107 indicated a distinct difference in the concanavalin A sensitivities of these lymphocytes. Mitogenic lectins such as phytohemaglutinin and concanavalin A efficiently cross-link many cell surface proteins, and their proliferative effects on lymphocytes require the TCR (40). The comparable responses of p130+/−;p107+/− and p130−/−;p107−/− lymphocytes to PMA-ionomycin treatment (Fig. 7B) indicated that the double-mutant cells are not inherently hypersensitive, while the relatively minor increase in the sensitivity to CD3 antibodies (Fig. 7B) may suggest the involvement of non-TCR signaling pathways in this defect. Thus, the removal of p130 and p107 function in quiescent lymphocytes increased the sensitivity of these cells to surface protein-mediated mitogenic signals, suggesting a role for these proteins in a pathway that restricts lymphocyte activation.
DISCUSSION
The retinoblastoma gene family encodes three closely related proteins that have been implicated in the regulation of cellular proliferation, differentiation, and transformation. We have analyzed murine T-cell development and proliferation in the absence of two members of this family, p130 and p107, in an effort to address the importance of these genes in the establishment and maintenance of the quiescent G0 state as well as to probe the nature of functional overlap within the Rb gene family. The demonstration of a novel phenotype in T cells lacking p130 and p107 indicates a form of genetic interaction, which may be explained in three general ways. First, the protein products of the two genes regulate distinct pathways which, when disrupted, produce additive effects that are deleterious. Alternatively, the proteins may contribute equally to the regulation of a specific cellular function, a phenomenon generally described as functional redundancy. In a third scenario, most appropriately termed functional overlap or functional compensation, one protein is capable of a function usually performed by the other but in normal circumstances does not exercise such a function. Although compensation may be associated with increased protein levels, this is not strictly necessary.
Our analysis of lymphocyte differentiation and proliferation strongly supports the concept that p107 and p130 are capable of specific functional compensation. First, p107 protein levels were significantly elevated in p130-deficient peripheral T cells. This modulation of p107 expression occurred during the last stages of lymphocyte development, as these cells became quiescent (79). Second, p130-deficient lymphocytes contained a new p107-E2F complex that effectively replaced the prominent p130-E2F complex characteristic of G0 lymphocytes. The p130-E2F complex is a common feature of differentiated cells (19, 62, 69) and correlates with the repression of E2F target genes (35, 68) and quiescence (64, 69). The possibility that p107 functionally substituted for p130 in this setting is consistent with the ability of p107 to efficiently bind and inhibit E2F-4 activity (5, 24) and with the ability of p107 to mediate repression when tethered to an E2F DNA binding domain (65). Finally, the functional importance of p107 activity in p130-deficient cells was demonstrated by the defective regulation of both E2F-mediated gene expression and mitogen responsiveness in lymphocytes lacking p130 and p107 but not in cells deficient in p130 alone.
Recent studies of the MyoD and myf-5 myogenic proteins demonstrate that a complete understanding of apparently redundant regulatory pathways requires detailed biochemical analyses. The hypothesis that these myogenic factors function redundantly during muscle cell commitment is based on the lack of muscle tissue in myoD−/−;myf-5−/− animals, while mice lacking MyoD or myf-5 alone exhibit normal muscle; also, MyoD-deficient muscle contains elevated levels of myf-5 protein (57). However, it has recently been shown that myf-5 and MyoD proteins are expressed in distinct cell types and determine different muscle lineages (6), suggesting that the redundancy in this MyoD/myf-5 system exists not at the molecular level but rather through regulation of separate muscle cell lineages (6).
In contrast, our analysis of p130-deficient T lymphocytes not only demonstrates compensatory overexpression of p107 but also defines a compensatory biochemical activity of p107, namely, the binding and regulation of E2F transcription factors. The normal expression of various lymphoid markers on these mutant cells makes it unlikely that any alteration of T-cell lineage contributes to these effects. Therefore, this provides the first direct evidence that loss of function within the pRB family leads to specific compensation at the biochemical and molecular level.
These analyses of E2F regulation by p130 and p107 are particularly relevant to our understanding of the G0 status of mature tissues, which may differ substantially from the growth-arrested state associated with serum-starved fibroblasts in culture. Interestingly, despite increased p107 levels in p130-deficient MEFs, the levels of p107-E2F complexes remain unchanged in these cells (31), suggesting a mechanistic difference between the regulation of E2F activity in resting lymphocytes and fibroblasts.
The appearance of defects in gene expression and proliferation through the progressive disruption of p130 and p107 function highlights the importance of E2F regulation and the E2F complex observed in resting lymphocytes. Whereas p130-deficient lymphocytes exhibit little free E2F and are not measurably defective in gene expression or cell cycle regulation, the observed increase in free E2F in double-mutant cells correlates with specific defects. The fact that these defects are more limited than those observed upon overexpression of E2Fs via transfection (36, 56) or viral transduction (21) likely reflects both the physiological E2F levels in primary lymphocytes and the fact that only a portion of the E2F is in a free form (see below).
The complex process of T-cell activation requires integration of various positive and negative signaling pathways (34). The concanavalin A hypersensitivity of p130−/−;p107−/− T cells suggests a role for these proteins in a pathway constraining activation and might therefore indicate that these cells would respond inappropriately to stimuli in vivo. However, such experiments are exceedingly difficult with chimeric animals like the p130−/−;p107−/− mice, primarily due to the limited effectiveness of antigen presentation, which is essential for cell-mediated immune responses. Future studies will address the molecular basis of mitogen hypersensitivity of double-mutant lymphocytes.
Various studies have suggested a link between pRB family proteins and the repression of E2F-regulated genes. pRB and p107 are each capable of silencing adjacent promoter elements when bound to an E2F site containing promoter (65, 72), and recent in vivo analyses of endogenous E2F sites in the B-myb (80), cdc2 (68), and cyclin A (30) promoters revealed site occupancy exclusively during the G0 and G1 phases of the cell cycle, coincident with promoter repression. The derepression of gene expression observed in p130−/−;p107−/− quiescent lymphocytes demonstrates that p130 and/or p107 is an essential component of the repressive complex for a specific subset of E2F target genes. Although this subset of genes is similar to that derepressed in serum-starved p130−/−;p107−/− MEFs (31), derepression of E2F-1 and thymidine synthase was undetectable in resting T cells, while the degree of RRM2 derepression (particularly of the 1.6-kb mRNA) was greater than that observed in MEFs. Interestingly, these genes are distinct from those affected by loss of pRB (2, 28, 31). These differences may reflect the differential affinities of pRB family proteins for the various E2Fs as well as the complexity of interactions within each specific promoter, each of which may also vary between cell types.
The ability of p107 to effectively compensate for p130 loss has important implications for pRB family regulation of proliferation and tumor suppression. First, it suggests that mutational inactivation of p130 and p107 function during tumorigenesis would require disruption of four distinct genetic loci (two alleles of each gene), which would be expected to be rare. In addition, our data indicates an even more extensive degree of compensation between pRB family proteins. Although slightly more distantly related, pRB has considerable homology with p130 and p107, particularly within the protein binding domain termed the pocket, which mediates interactions with E2Fs as well as a majority of the other pRB family binding proteins (23, 25, 46). The detection of pRB–E2F-4 complexes in p130−/−;p107−/− lymphocytes suggests biochemical compensation between the more distantly related proteins of this family. Although pRB–E2F-4 interactions have been detected in G1- or S-phase populations of cells (32, 50), this is the first demonstration of this interaction in quiescent cells, and it may provide insight regarding mechanisms of gene repression in naturally resting cells. The failure of pRB to bind all of the E2F-4 in p130−/−;p107−/− cells may reflect pRB’s lower affinity for this E2F family member or may be due simply to insufficient pRB concentrations. Strikingly, a significant portion of the E2F-4 in extracts of p130−/−;p107−/− lymphocytes interacts with a protein immunologically distinct from pRB. A similar non-pRB-E2F complex has been observed in extracts of p130−/−;p107−/− MEFs (31). Although the identity and function of this activity remain unknown, the observation suggests the existence of a novel protein functionally and possibly structurally related to the pRB family.
The pRB–E2F-4 interaction detected in p130−/−;p107−/− quiescent lymphocytes suggests that, at least in some cell types, pRB may compensate for the loss of these functions. pRB compensation for p130 and p107 could permit repression of certain E2F target genes or provide other growth-regulatory functions that contribute to cell cycle arrest and maintenance of quiescence. Similarly, specific compensation by pRB might provide some explanation for both the relatively limited developmental phenotype of p130−/−;p107−/− embryos (17) and the lack of tumor development in p130−/−;p107+/− or p130+/−;p107−/− mice. Sporadic loss of the remaining allele of p107 or p130 in p130−/−;p107+/− or p130+/−;p107−/− animals would be expected to reveal any tumor suppressor function unique to these two proteins, but to date they have not exhibited obvious predisposition for tumors (unpublished observations).
It is also possible that the effects of the absence of pRB in some cell types are ameliorated by p130 and p107. A genetic interaction between p107 and Rb has been demonstrated, such that Rb−/−;p107−/− embryos die more rapidly than Rb−/− embryos and Rb+/−;p107−/− mice develop bilateral retinal lesions (44). Although the mechanism of this genetic interaction remains to be fully characterized, we do detect elevated p107 protein levels in some tissues of Rb−/− embryos (unpublished observation). The suggestion that p107 and p130 might mitigate some defects associated with pRB deficiency may also help to explain the normal development and proliferation of Rb−/− lymphoid cells (9) and the striking ability of Rb−/− ES cells to contribute extensively to all tissues of adult chimeric mice (48, 74). There are, however, clear limitations on the extent of functional compensation within the Rb family, as illustrated by the distinct embryonic and cellular defects associated with loss of Rb or p130/p107. These distinct phenotypes may be related to the differential expression of pRB family proteins or to specific differences in their ability to interact with other cellular proteins.
Functional compensation within the pRB family of cell cycle-regulatory proteins should enhance the ability of this family to inhibit inappropriate proliferation and suggests that the proteins function as a concerted barrier to cellular transformation. Interestingly, pRB, p107, and p130 are each specifically bound by viral oncoproteins, including simian virus 40 large T antigen, adenovirus E1A, and human papillomavirus E7 (52). Mutational analyses of T-antigen demonstrate that the pRB family binding domain is necessary for transformation (10, 12, 76), and it has recently been shown that a T-antigen domain that specifically inactivates p130 and p107 is required for T-antigen-induced growth in low serum concentrations and growth to high cell density (66). Our data strongly supports the notion that the broadly conserved ability of viral oncoproteins to inactivate each member of the pRB family is related to the ability of these proteins to functionally compensate for the regulation of E2F or other cellular functions.
Accordingly, compensation within this gene family may also influence the mechanisms by which tumors subvert normal growth control. A cell cycle-regulatory pathway involving pRB, cyclin D/cdk4, and its specific inhibitor p16 is frequently mutated in human cancer (61, 71). Disruption of this pathway is believed to bypass pRB function, either by direct mutation or through pRB phosphorylation mediated by excessive activity of cyclin D/cdk4. However, experiments suggest that cyclin D/cdk4 also phosphorylates p107 (4) and p130 (49), in addition to its well documented role in phosphorylation of pRB (61). Moreover, cell cycle arrest mediated by p107 or p130 is reversed by cotransfection of cyclin D (4, 14). Therefore, the mutational deregulation of this CDK pathway may inactivate functions of p130 and p107 as well as pRB and thus provide some cell types with a growth advantage not achieved via inactivation of RB alone. Indeed, mutation of p16 is detected in a wider spectrum of human tumors than that of RB and also appears to occur more frequently in several tumor types (1, 3, 26, 38, 54, 75).
Although functional compensation within the pRB family may help to explain the relatively limited tumor spectrum associated with the RB heterozygous state, it remains unclear why certain tissues, such as human retina and murine pituitary gland, are uniquely sensitive to loss of pRB. This sensitivity could reflect failed compensation in these tissues, due perhaps to insufficient expression of pRB-related proteins or an inability of these proteins to fulfill a specific biochemical activity of pRB. At present, the mechanism and extent of compensation in vivo remain uncharacterized. However, the most direct test of functional compensation within this gene family involves analysis of cells deficient for p130, p107, and pRB. Appropriate interbreeding of mice carrying combined mutations of these genes would yield embryos that lack all three proteins. Alternatively, triple mutant cells could be derived via repeated gene targeting in embryonic stem cells. Analysis of chimeric mice generated with such embryonic stem cells would help to dissect the role of this gene family in both differentiation and tumor suppression.
ACKNOWLEDGMENTS
We thank Bart Williams, Kay MacLeod, Elias Theodorou, and all members of the Jacks lab for helpful discussions and Bart Williams, Bob Weinberg, Mariana Nacht, Laura Attardi, Jenny-Sue Lanni, Sunil Hingorhani, and Karen Chikowski for critical reading of the manuscript. We also thank David Gerber, Glenn Paradis, and Arlene Sharpe for assistance in lymphocyte proliferative assays and cell sorting and advice on in vivo immune analysis, respectively. Thanks also go to M. Imperiale and J. Lees for gifts of pRB and E2F-4 antibodies and to Rob Hurford and Nick Dyson for communicating results prior to publication and providing cDNA probes for acidic ribosomal phosphoprotein PO, thymidine kinase, E2F-1, and E2F-4. Thanks also go to Phil Steiner and Lee Johnson for cyclin E and thymidine synthase probes, respectively.
T.J. is an Associate Investigator of the Howard Hughes Medical Institute.
REFERENCES
- 1.Aagaard L, Lukas J, Bartkova J, Kjerulff A A, Strauss M, Bartek J. Aberrations of p16Ink4 and retinoblastoma tumour-suppressor genes occur in distinct subsets of human cancer cell lines. Int J Cancer. 1995;61:115–120. doi: 10.1002/ijc.2910610120. [DOI] [PubMed] [Google Scholar]
- 2.Almasan A, Kelly R, Lee E, Bradley A, Li W, Bertino J, Wahl G. Deficiency of the retinoblastoma protein leads to inappropriate S-phase entry, activation of E2F responsive genes, and apoptosis. Proc Natl Acad Sci USA. 1995;92:5436–5440. doi: 10.1073/pnas.92.12.5436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bartkova J, Lukas J, Guldberg P, Alsner J, Kirkin A, Zeuthen J, Bartek J. The p16-cyclinD/cdk4-pRb pathway as a functional unit frequently altered in melanoma pathogenesis. Cancer Res. 1996;56:5475–5483. [PubMed] [Google Scholar]
- 4.Beijersbergen R L, Carlee L, Kerkhoven R M, Bernards R. Regulation of the retinoblastoma protein-related p107 by G1 cyclin complexes. Genes Dev. 1995;9:1340–1353. doi: 10.1101/gad.9.11.1340. [DOI] [PubMed] [Google Scholar]
- 5.Beijersbergen R L, Kerkhoven R M, Zhu L, Carlee L, Voorhoeve P M, Bernards R. E2F-4, a new member of the E2F family, has oncogenic activity and associates with p107 in vivo. Genes Dev. 1994;8:2680–2690. doi: 10.1101/gad.8.22.2680. [DOI] [PubMed] [Google Scholar]
- 6.Braun T, Arnold H. myf-5 and myoD genes are activated in distinct mesenchymal stem cells and determine different skeletal muscle cell lineages. EMBO J. 1996;15:310–318. [PMC free article] [PubMed] [Google Scholar]
- 7.Cao L, Faha B, Dembski M, Tsai L H, Harlow E, Dyson N. Independent binding of the retinoblastoma protein and p107 to the transcription factor E2F. Nature. 1992;355:176–179. doi: 10.1038/355176a0. [DOI] [PubMed] [Google Scholar]
- 8.Chen G, Guy C T, Chen H W, Hu N, Lee E Y, Lee W H. Molecular cloning and developmental expression of mouse p130, a member of the retinoblastoma gene family. J Biol Chem. 1996;271:9567–9572. [PubMed] [Google Scholar]
- 9.Chen J, Gorman J, Stewart V, Williams B, Jacks T, Alt F W. Generation of normal lymphocyte populations of Rb-deficient ES cells: analysis of gene function by RAG-2-deficient blastocyst complementation. Curr Biol. 1993;3:405–413. doi: 10.1016/0960-9822(93)90347-q. [DOI] [PubMed] [Google Scholar]
- 10.Chen S, Paucha E. Identification of a region of simian virus 40 large T antigen required for cell transformation. J Virol. 1990;64:3350–3357. doi: 10.1128/jvi.64.7.3350-3357.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chittenden T, Livingston D M, DeCaprio J A. Cell cycle analysis of E2F in primary human T cells reveals novel E2F complexes and biochemically distinct forms of free E2F. Mol Cell Biol. 1993;13:3975–3983. doi: 10.1128/mcb.13.7.3975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Christensen J, Imperiale M. Inactivation of the retinoblastoma susceptibility protein is not sufficient for the transforming function of the conserved region 2-like domain of simian virus 40 large T antigen. J Virol. 1995;69:3945–3948. doi: 10.1128/jvi.69.6.3945-3948.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Clarke A R, Maandag E R, van Roon M, van der Lugt N M T, van der Valk M, Hooper M L, Berns A, te Riele H. Requirement for a functional Rb-1 gene in murine development. Nature. 1992;359:328–330. doi: 10.1038/359328a0. [DOI] [PubMed] [Google Scholar]
- 14.Claudio P P, De Luca A, Howard C M, Baldi A, Firpo E J, Koff A, Paggi M G, Giordano A. Functional analysis of pRb2/p130 interaction with cyclins. Cancer Res. 1996;56:2003–2008. [PubMed] [Google Scholar]
- 15.Claudio P P, Howard C M, Baldi A, De Luca A, Fu Y, Condorelli G, Sun Y, Colburn N, Calabretta B, Giordano A. p130/pRb2 has growth suppressive properties similar to yet distinctive from those of retinoblastoma family members pRb and p107. Cancer Res. 1994;54:5556–5560. [PubMed] [Google Scholar]
- 16.Cobrinik D. Regulatory interactions among E2Fs and cell cycle control proteins. Curr Top Microbiol Immunol. 1996;208:31–61. doi: 10.1007/978-3-642-79910-5_2. [DOI] [PubMed] [Google Scholar]
- 17.Cobrinik D, Lee M, Hannon G, Mulligan G, Bronson R, Dyson N, Harlow E, Beach D, Weinberg R, Jacks T. Shared role of the pRB-related p130 and p107 proteins in limb development. Genes Dev. 1996;10:1633–1644. doi: 10.1101/gad.10.13.1633. [DOI] [PubMed] [Google Scholar]
- 18.Cobrinik D, Whyte P, Peeper D S, Jacks T, Weinberg R A. Cell cycle-specific association of E2F with the p130 E1A-binding protein. Genes Dev. 1993;7:2392–2404. doi: 10.1101/gad.7.12a.2392. [DOI] [PubMed] [Google Scholar]
- 19.Corbeil H B, Whyte P, Branton P E. Characterization of transcription factor E2F complexes during muscle and neuronal differentiation. Oncogene. 1995;11:909–920. [PubMed] [Google Scholar]
- 20.Crabtree G R. Contingent genetic regulatory events in T lymphocyte activation. Science. 1989;243:355–361. doi: 10.1126/science.2783497. [DOI] [PubMed] [Google Scholar]
- 21.DeGregori J, Kowalik T, Nevins J. Cellular targets for activation by the E2F1 transcription factor include DNA synthesis- and G1/S-regulatory genes. Mol Cell Biol. 1995;15:4215–4224. doi: 10.1128/mcb.15.8.4215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21a.Dyson N, Dembski M, Fattaey A, Ngwu C, Ewen M, Helin K. Analysis of p107-associated proteins: p107 associates with a form of E2F that differs from pRB-associated E2F-1. J Virol. 1993;66:7641–7647. doi: 10.1128/jvi.67.12.7641-7647.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ernst D N, Weigle D, Noonan D J, McQuitty D N, Hobbs M V. The age-associated increase in IFN synthesis by mouse CD8+ T cells correlates with shifts in the frequencies of cell subsets defined by membrane CD44, CD45RB, 3G11 and MEL-14 expression. J Immunol. 1993;151:575–581. [PubMed] [Google Scholar]
- 23.Ewen M E, Xing Y, Lawrence J B, Livingston D M. Molecular cloning, chromosomal mapping and expression of the cDNA for p107, a retinoblastoma gene product-related protein. Cell. 1991;66:1155–1164. doi: 10.1016/0092-8674(91)90038-z. [DOI] [PubMed] [Google Scholar]
- 24.Ginsberg D, Vairo G, Chittenden T, Xiao Z, Xu G, Wydner K L, DeCaprio J A, Lawrence J B, Livingston D M. E2F-4, a new member of the E2F transcription factor family, interacts with p107. Genes Dev. 1994;8:2665–2679. doi: 10.1101/gad.8.22.2665. [DOI] [PubMed] [Google Scholar]
- 25.Hannon G J, Demetrick D, Beach D. Isolation of the Rb-related p130 through its interaction with CDK2 and cyclins. Genes Dev. 1993;7:2378–2391. doi: 10.1101/gad.7.12a.2378. [DOI] [PubMed] [Google Scholar]
- 26.He J, Olson J, James C. Lack of p16INK4 or retinoblastoma protein (pRb), or amplification-associated overexpression of cdk4 is observed in distinct subsets of malignant glial tumors and cell lines. Cancer Res. 1995;55:4833–4836. [PubMed] [Google Scholar]
- 27.Helin K, Lees J A, Vidal M, Dyson N, Harlow E, Fattaey A. A cDNA encoding a pRB-binding protein with properties of the transcription factor E2F. Cell. 1992;70:337–350. doi: 10.1016/0092-8674(92)90107-n. [DOI] [PubMed] [Google Scholar]
- 28.Herrera R E, Sah V P, Williams B O, Weinberg R A, Jacks T. Altered cell cycle kinetics, gene expression, and G1 restriction point regulation in Rb-deficient fibroblasts. Mol Cell Biol. 1996;16:2402–2407. doi: 10.1128/mcb.16.5.2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hijmans E M, Voorhoeve P M, Beijersbergen R L, van’t Veer L J, Bernards R. E2F-5, a new E2F family member that interacts with p130 in vivo. Mol Cell Biol. 1995;15:3082–3089. doi: 10.1128/mcb.15.6.3082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huet X, Rech A, Plet A, Blanchard J. Cyclin A expression is under negative transcriptional control during the cell cycle. Mol Cell Biol. 1996;16:3789–3798. doi: 10.1128/mcb.16.7.3789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hurford R K, Cobrinik D, Lee M, Dyson N. pRB and p107/p130 are required for the regulated expression of different sets of E2F responsive genes. Genes Dev. 1997;11:1447–1463. doi: 10.1101/gad.11.11.1447. [DOI] [PubMed] [Google Scholar]
- 32.Ikeda M, Jakoi L, Nevins J. A unique role for the Rb protein in controlling E2F accumulation during cell growth and differentiation. Proc Natl Acad Sci USA. 1996;93:3215–3220. doi: 10.1073/pnas.93.8.3215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jacks T, Fazeli A, Schmidt E, Bronson R, Goodell M, Weinberg R. Effects of an Rb mutation in the mouse. Nature. 1992;359:295–300. doi: 10.1038/359295a0. [DOI] [PubMed] [Google Scholar]
- 34.Janeway C, Bottomly K. Signals and signs for lymphocyte responses. Cell. 1994;76:275–285. doi: 10.1016/0092-8674(94)90335-2. [DOI] [PubMed] [Google Scholar]
- 35.Johnson D. Regulation of E2F-1 gene expression by p130 (Rb2) and D-type cyclin kinase activity. Oncogene. 1995;11:1685–1692. [PubMed] [Google Scholar]
- 36.Johnson D G, Schwarz J K, Cress W D, Nevins J R. Expression of transcription factor E2F1 induces quiescent cells to enter S phase. Nature. 1993;365:349–352. doi: 10.1038/365349a0. [DOI] [PubMed] [Google Scholar]
- 37.Kaelin W G, Krek W, Sellers W R, DeCaprio J A, Ajchenbaum F, Fuchs C S, Chittenden T, Li Y, Blanar M A, et al. Expression cloning of a cDNA encoding a retinoblastoma binding protein with E2F-like properties. Cell. 1992;70:351–364. doi: 10.1016/0092-8674(92)90108-o. [DOI] [PubMed] [Google Scholar]
- 38.Kamb A, Gruis N A, Weaver-Feldhaus J, Liu Q, Harshman K, Tavtigian S V, Stockert E, Day R S, Johnson B E, Skolnick M H. A cell cycle regulator potentially involved in genesis of many tumor types. Science. 1994;264:436–440. doi: 10.1126/science.8153634. [DOI] [PubMed] [Google Scholar]
- 39.Kim K K, Soonpaa M H, Wang H, Field L J. Developmental expression of p107 mRNA and evidence for alternative splicing of the p107 (RBL1) gene product. Genomics. 1995;28:520–529. doi: 10.1006/geno.1995.1184. [DOI] [PubMed] [Google Scholar]
- 40.Kruisbeek A. Units 4.2.1 and 3.12.1. In: Coico R, editor. Current protocols in immunology. New York, N.Y: John Wiley & Sons; 1991. [Google Scholar]
- 41.Lam E, Morris J D, Davies R, Crook T, Watson R, Vousden K H. HPV16 E7 oncoprotein deregulates B-myb expression: correlation with targetting of p107/E2F complexes. EMBO J. 1994;13:871–878. doi: 10.1002/j.1460-2075.1994.tb06330.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.La Thangue N. DP and E2F proteins: components of a heterodimeric transcription factor implicated in cell cycle control. Curr Opin Cell Biol. 1994;6:443–450. doi: 10.1016/0955-0674(94)90038-8. [DOI] [PubMed] [Google Scholar]
- 43.Lee E Y-H P, Chang C-Y, Hu N, Wang Y-C J, Lai C-C, Herrup K, Lee W-H. Mice deficient for Rb are nonviable and show defects in neurogenesis and haematopoiesis. Nature. 1992;359:288–295. doi: 10.1038/359288a0. [DOI] [PubMed] [Google Scholar]
- 44.Lee M, Williams B, Mulligan G, Mukai S, Bronson R, Dyson N, Harlow E, Jacks T. Targeted disruption of p107: functional overlap between p107 and Rb. Genes Dev. 1996;10:1621–1632. doi: 10.1101/gad.10.13.1621. [DOI] [PubMed] [Google Scholar]
- 45.Lees J A, Saito M, Vidal M, Valentino M, Look T, Harlow E, Dyson N, Helin K. The retinoblastoma protein binds to a family of E2F transcription factors. Mol Cell Biol. 1993;13:7813–7825. doi: 10.1128/mcb.13.12.7813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li Y, Graham C, Lacy S, Duncan A M V, Whyte P. The adenovirus E1A-associated 130-kD protein is encoded by a member of the retinoblastoma gene family and physically interacts with cyclins A and E. Genes Dev. 1993;7:2366–2377. doi: 10.1101/gad.7.12a.2366. [DOI] [PubMed] [Google Scholar]
- 47.Lukas J, Petersen B O, Holm K, Bartek J, Helin K. Deregulated expression of E2F family members induces S-phase entry and overcomes p16INK4A-mediated growth suppression. Mol Cell Biol. 1996;16:1047–1057. doi: 10.1128/mcb.16.3.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Maandag E C R, van der Valk M, Vlaar M, Feltkamp C, O’Brien J, van Roon M, van der Lugt N, Berns A, te Riele H. Developmental rescue of an embryonic-lethal mutation in the retinoblastoma gene in chimeric mice. EMBO J. 1994;13:4260–4268. doi: 10.1002/j.1460-2075.1994.tb06746.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mayol X, Garriga J, Grana X. Cell cycle-dependent phosphorylation of the retinoblastoma-related protein p130. Oncogene. 1995;11:801–808. [PubMed] [Google Scholar]
- 50.Moberg K, Starz M A, Lees J A. E2F-4 switches from p130 to p107 and pRB in response to cell cycle reentry. Mol Cell Biol. 1996;16:1436–1449. doi: 10.1128/mcb.16.4.1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nevins J. E2F: a link between the Rb tumor suppressor protein and viral oncoproteins. Science. 1992;258:424–429. doi: 10.1126/science.1411535. [DOI] [PubMed] [Google Scholar]
- 52.Nevins J R. Cell cycle targets of the DNA tumor viruses. Curr Opin Genet Dev. 1994;4:130–134. doi: 10.1016/0959-437x(94)90101-5. [DOI] [PubMed] [Google Scholar]
- 53.Nourse J, Firpo E, Flanagan W M, Coats S, Polyak K, Lee M, Massague J, Crabtree G, Roberts J M. Interleukin-2-mediated elimination of the p27 kip1 cyclin-dependent kinase inhibitor prevented by rapamycin. Nature. 1994;372:570–573. doi: 10.1038/372570a0. [DOI] [PubMed] [Google Scholar]
- 54.Okamoto A, Demetrick D, Spillare A, Hagiwara K, Hussain S, Bennet W, Forrester K, Gerwin B, Serrano M, Beach D, et al. Mutations and altered expression of p16INK4A in human cancer. Proc Natl Acad Sci USA. 1994;91:11045–11049. doi: 10.1073/pnas.91.23.11045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Qin X Q, Chittenden T, Livingston D M, Kaelin W., Jr Identification of a growth suppression domain within the retinoblastoma gene product. Genes Dev. 1992;6:953–964. doi: 10.1101/gad.6.6.953. [DOI] [PubMed] [Google Scholar]
- 56.Qin X Q, Livingston D M, Kaelin W, Jr, Adams P D. Deregulated transcription factor E2F-1 expression leads to S-phase entry and p53-mediated apoptosis. Proc Natl Acad Sci USA. 1994;91:10918–10922. doi: 10.1073/pnas.91.23.10918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rudnicki M A, Schnegelsberg P, Stead R, Braun T, Arnold H, Nevins J R. MyoD or Myf-5 is required for the formation of skeletal muscle. Cell. 1993;75:1351–1359. doi: 10.1016/0092-8674(93)90621-v. [DOI] [PubMed] [Google Scholar]
- 58.Sanchez A, Dynlacht B. Transcriptional control of the cell cycle. Curr Opin Cell Biol. 1996;8:318–324. doi: 10.1016/s0955-0674(96)80004-4. [DOI] [PubMed] [Google Scholar]
- 59.Sardet C, Vidal M, Cobrinik D, Geng Y, Onufryk C, Chen A, Weinberg R A. E2F-4 and E2F-5, two members of the E2F family, are expressed in the early phases of the cell cycle. Proc Natl Acad Sci USA. 1995;92:2403–2407. doi: 10.1073/pnas.92.6.2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schwarz J K, Devoto S H, Smith E J, Chellapan S P, Jakoi L, Nevins J R. Interactions of the p107 and Rb proteins with E2F during the cell proliferation response. EMBO J. 1993;12:1013–1020. doi: 10.1002/j.1460-2075.1993.tb05742.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sherr C J. Cancer cell cycles. Science. 1996;274:1672–1677. doi: 10.1126/science.274.5293.1672. [DOI] [PubMed] [Google Scholar]
- 62.Shin E K, Shin A, Pauldin C, Schaffhausen B, Yee A. Multiple changes in E2F function and regulation occur upon muscle differentiation. Mol Cell Biol. 1995;15:2252–2262. doi: 10.1128/mcb.15.4.2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shirodkar S E, M, DeCaprio J A, Morgan J, Livingston D, Chittenden T. The transcription factor E2F interacts with the retinoblastoma product and a p107-cyclin A complex in a cell cycle-regulated manner. Cell. 1992;68:157–166. doi: 10.1016/0092-8674(92)90214-w. [DOI] [PubMed] [Google Scholar]
- 64.Smith E J, Leone G, Degregori J, Nevins J. The accumulation of an E2F-p130 transcriptional repressor distinguishes a G0 cell state from a G1 cell state. Mol Cell Biol. 1996;16:6965–6976. doi: 10.1128/mcb.16.12.6965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Starostik P, Chow K N B, Dean D C. Transcriptional repression and growth suppression by the p107 pocket protein. Mol Cell Biol. 1996;16:3606–3614. doi: 10.1128/mcb.16.7.3606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Studbal H, Zalvide J, Campbell K S, Schweitzer C, Roberts T, DeCaprio J. Inactivation of the RB-related proteins p107 and p130 mediated by the J domain of simian virus 40 large T antigen. Mol Cell Biol. 1997;17:4979–4989. doi: 10.1128/mcb.17.9.4979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tivol E, Borriello F, Schweitzer A N, Lynch W P, Bluestone W, Sharpe A H. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity. 1995;3:541–547. doi: 10.1016/1074-7613(95)90125-6. [DOI] [PubMed] [Google Scholar]
- 68.Tommasi S, Pfeifer G. In vivo structure of the human cdc2 promoter: release of a p130-E2F4 complex from sequences immediately upstream of the transcription initiation site coincides with induction of cdc2 expression. Mol Cell Biol. 1995;15:6901–6913. doi: 10.1128/mcb.15.12.6901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Vairo G, Livingston D, Ginsberg D. Functional interaction between E2F-4 and p130: evidence for distinct mechanisms underlying growth suppression by different retinoblastoma protein family members. Genes Dev. 1995;9:869–881. doi: 10.1101/gad.9.7.869. [DOI] [PubMed] [Google Scholar]
- 70.Wang J Y, Knudsen E S, Welch P J. The retinoblastoma tumor suppressor protein. Adv Cancer Res. 1994;64:25–85. doi: 10.1016/s0065-230x(08)60834-9. [DOI] [PubMed] [Google Scholar]
- 71.Weinberg R A. The retinoblastoma protein and cell cycle control. Cell. 1995;81:323–330. doi: 10.1016/0092-8674(95)90385-2. [DOI] [PubMed] [Google Scholar]
- 72.Weintraub S J, Chow K N, Luo R X, Zhang S H, He S, Dean D C. Mechanism of active transcriptional repression by the retinoblastoma protein. Nature. 1995;375:812–815. doi: 10.1038/375812a0. [DOI] [PubMed] [Google Scholar]
- 73.Weintraub S J, Prater C A, Dean D C. Retinoblastoma protein switches the E2F site from positive to negative element. Nature. 1992;358:259–261. doi: 10.1038/358259a0. [DOI] [PubMed] [Google Scholar]
- 74.Williams, B. O., E. M. Schmitt, L. Remington, R. T. Bronson, D. M. Albert, R. A. Weinberg, and T. Jacks. Extensive contribution of Rb-deficient cells to adult chimeric mice with limited histopathological consequences. EMBO J., in press. [DOI] [PMC free article] [PubMed]
- 75.Yeager T, Stadler W, Belair C, Puthenveettil J, Olopade O, Reznikoff C. Increased p16 levels correlate with pRb alterations in human urothelial cells. Cancer Res. 1995;55:493–497. [PubMed] [Google Scholar]
- 76.Zalvide J, DeCaprio J A. Role of pRb-related proteins in simian virus 40 large-T-antigen-mediated transformation. Mol Cell Biol. 1995;15:5800–5810. doi: 10.1128/mcb.15.10.5800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhu L, Enders G, Lees J A, Beijersbergen R L, Bernards R, Harlow E. The pRb-related protein p107 contains two growth suppression domains: independent interactions with E2F and cyclin/cdk complexes. EMBO J. 1995;14:1904–1913. doi: 10.1002/j.1460-2075.1995.tb07182.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhu L, van den Heuvel S, Helin K, Fattaey A, Ewen M, Livingston D, Dyson N, Harlow E. Inhibition of cell proliferation by p107, a relative of the retinoblastoma protein. Genes Dev. 1993;7:1111–1125. doi: 10.1101/gad.7.7a.1111. [DOI] [PubMed] [Google Scholar]
- 79.Zuniga-Pflucker J C, Lenardo M. Regulation of thymocyte development from immature progenitors. Curr Opin Immunol. 1996;8:215–224. doi: 10.1016/s0952-7915(96)80060-4. [DOI] [PubMed] [Google Scholar]
- 80.Zwicker J, Liu N, Engeland K, Lucibello F C, Muller R. Cell cycle regulation of E2F site occupancy in vivo. Science. 1996;271:1595–1597. doi: 10.1126/science.271.5255.1595. [DOI] [PubMed] [Google Scholar]