

Abstract
The human double-stranded RNA-dependent protein kinase (PKR) is an important component of the interferon response to virus infection. The activation of PKR is accompanied by autophosphorylation at multiple sites, including one in the N-terminal regulatory region (Thr-258) that is required for full kinase activity. Several protein kinases are activated by phosphorylation in the region between kinase subdomains VII and VIII, referred to as the activation loop. We show that Thr-446 and Thr-451 in the PKR activation loop are required in vivo and in vitro for high-level kinase activity. Mutation of either residue to Ala impaired translational control by PKR in yeast cells and COS1 cells and led to tumor formation in mice. These mutations also impaired autophosphorylation and eukaryotic initiation factor 2 subunit α (eIF2α) phosphorylation by PKR in vitro. Whereas the Ala-446 substitution substantially reduced PKR function, the mutant kinase containing Ala-451 was completely inactive. PKR specifically phosphorylated Thr-446 and Thr-451 in synthetic peptides in vitro, and mass spectrometry analysis of PKR phosphopeptides confirmed that Thr-446 is an autophosphorylation site in vivo. Substitution of Glu-490 in subdomain X of PKR partially restored kinase activity when combined with the Ala-451 mutation. This finding suggests that the interaction between subdomain X and the activation loop, described previously for MAP kinase, is a regulatory feature conserved in PKR. We found that the yeast eIF2α kinase GCN2 autophosphorylates at Thr-882 and Thr-887, located in the activation loop at exactly the same positions as Thr-446 and Thr-451 in PKR. Thr-887 was more critically required than was Thr-882 for GCN2 kinase activity, paralleling the relative importance of Thr-446 and Thr-451 in PKR. These results indicate striking similarities between GCN2 and PKR in the importance of autophosphorylation and the conserved Thr residues in the activation loop.
PKR, the double-stranded RNA (dsRNA)-activated protein kinase (also known as DAI), is transcriptionally induced by interferon and activated in virus-infected mammalian cells, where it plays an important role in cellular antiviral defense mechanisms. PKR interferes with virus replication by phosphorylating eukaryotic initiation factor 2 (eIF2) subunit α (eIF2α), converting eIF2 from a substrate to an inhibitor of its guanine nucleotide exchange factor, eIF2B. This reduction in the recycling of eIF2 by eIF2B leads to a general inhibition of translation that limits viral protein synthesis (30). PKR may also have an important role in controlling cellular proliferation, as the expression of catalytically defective PKR alleles transforms mammalian cells in culture and leads to tumor formation in mice (25, 31).
The yeast Saccharomyces cerevisiae also contains an eIF2α kinase, known as GCN2, that is activated by uncharged tRNA when cells are starved for one or more amino acids. Limited phosphorylation of eIF2 under these conditions leads to increased translation of GCN4 mRNA, encoding a transcriptional activator of amino acid biosynthetic genes. This induction of GCN4 translation in response to diminished eIF2 recycling occurs because ribosomes are prevented from initiating at short upstream open reading frames in the GCN4 mRNA leader, enabling them to initiate at the GCN4 start codon instead. The newly synthesized GCN4 protein leads to increased expression of amino acid biosynthetic genes, reversing the amino acid limitation which triggered the activation of GCN2 (reviewed in reference 20). Low-level expression of PKR in yeast mutants lacking GCN2 leads to eIF2α phosphorylation at a level sufficient to induce GCN4 expression without inhibiting general translation initiation (13). When expressed at higher levels, PKR phosphorylates eIF2α to an extent that inhibits general protein synthesis and prevents yeast cell growth (9, 13).
PKR kinase activity is stimulated in vitro by dsRNA, and the N-terminal 171 amino acids of the protein contain two copies of a dsRNA-binding motif (dsRBM) found in numerous other dsRNA-binding proteins (reviewed in reference 30). Binding of dsRNA stimulates the autokinase activity of PKR, and autophosphorylation appears to lock the enzyme into an active conformation which can bind and phosphorylate eIF2α in the absence of dsRNA (16, 17, 26). Sequence analysis of phosphopeptides derived from PKR following autophosphorylation in vitro led to the identification of a cluster of autophosphorylation sites located between the dsRBMs and the kinase domain of the protein. Mutation of one site, Thr-258, reduced the efficiency of autophosphorylation and substrate phosphorylation by PKR in vitro and partially impaired kinase function in yeast and mammalian cells. Mutations at two neighboring autophosphorylation sites (Ser-242 and Thr-255) had relatively little effect on kinase function alone but exacerbated the defects associated with the Ala-258 substitution (42). Because the PKR S242A,T255A,T258A triple mutant retains significant levels of autophosphorylation and substrate phosphorylation activities, these cannot be the only autophosphorylation sites in the protein. Additional sites have been detected in the linker region between the two dsRBMs; however, it is unknown whether these sites are important for PKR function (42a).
Several protein kinases, but not all, are activated by phosphorylation of residues within the kinase domain itself, in a segment located between kinase subdomains VII and VIII called the activation loop (reviewed in references 19 and 21). As shown in Fig. 1, such activation sites are located between 5 and 10 residues N terminal to the conserved Ala-Pro-Glu (APE) motif in kinase subdomain VIII. In the crystal structures of MAP kinase Erk2 (50), cyclin-dependent kinase Cdk2 (12), and cyclic AMP-dependent protein kinase (PKA) (23), this loop faces the cleft between the N-terminal and C-terminal lobes, where the active site resides. In the case of MAP kinase and Cdk2, it has been proposed that phosphorylation of these residues alters the conformation of the activation loop to permit binding of the peptide substrate. In the case of PKA, kinetic analysis of mutant proteins has suggested that phosphorylation of the activation loop stimulates the binding of ATP and the rate of phosphoryl transfer, rather than increasing the binding of the peptide substrate (1). It has also been suggested that in all three enzymes, the phosphorylated residue in the activation loop contributes to the proper alignment of catalytic residues, particularly the conserved Asp in subdomain VI-B, or promotes the correct relative orientation of the N-terminal and C-terminal lobes of the kinase domain by interacting with basic residues in the vicinity of the catalytic center (reviewed in reference 21). In any case, phosphorylation of residues in the activation loop is a key event in stimulating substrate phosphorylation by each of these kinases. In phosphorylase kinase, it appears that the role of the phosphorylated residue is carried out by a glutamate residue in the activation loop (21).
FIG. 1.

Established phosphorylation sites in the activation loops of several protein kinases. Sequence alignments of the segments between kinase subdomains VII and VIII are shown for the protein kinases listed on the left (19, 40). Highly conserved residues common to all of the kinases are shown in bold type without underlining. Phosphorylated residues required for activation of the kinases are shown in bold type and underlined. The kinases were Erk2 (extracellular signal-regulated kinase), Cdk2 (cyclin-dependent kinase), PKA (cyclic AMP-dependent protein kinase), RSK2 (ribosomal protein S6 kinase), GSK3α (glycogen synthase kinase), dGCN2 (Drosophila GCN2 homolog), and HRI (heme-regulated inhibitor). Potential autophosphorylation sites examined in the PKR and GCN2 activation loops are indicated (positions 446, 448, and 451 and positions 882 and 887, respectively). The identification of Thr-446 in PKR and Thr-882 and Thr-887 in GCN2 as autophosphorylation sites is the subject of this report.
PKR contains two Thr residues and one Ser residue within 10 amino acids of the APE motif in subdomain VIII (SPE in PKR) and, interestingly, all of the known eIF2α kinases contain two Thr residues at exactly the same positions relative to subdomain VIII (Fig. 1). Therefore, we wished to determine whether these conserved Thr residues are additional autophosphorylation sites required for kinase catalytic function. To address this possibility, we altered each of the residues to nonphosphorylatable residues and examined the effects on kinase function in yeast and mammalian cells. The results of this analysis indicated that Thr-446 and Thr-451 in the PKR activation loop are both required for high-level kinase function in vivo and in vitro. Mass spectrometry (MS) analysis of phosphopeptides in PKR purified from yeast cells confirmed that Thr-446 is a site of autophosphorylation in vivo. Interestingly, we also found that yeast eIF2α kinase GCN2 autophosphorylates at the two conserved Thr residues in its activation loop, and both sites were found to be important for GCN2 kinase function. Thus, autophosphorylation in the activation loop is required for high-level kinase function by two different members of the eIF2α kinase family.
MATERIALS AND METHODS
Plasmids and yeast strains.
Construction of the PKR alleles containing the point mutations K296R (38) and T258A (42) and the triple mutation S242A,T255A,T258A (42) in the yeast expression vector pEMBLyex4 has been previously described. A PCR fusion technique (49) was used to create the following single and multiple point mutations in the PKR kinase domain. Thr-446 was mutated to Ser, Asp, or Ala by changing the Thr-446 ACA codon to TCA, GAC, or GCA, respectively. Ser-448 was mutated to Ala by changing the AGT codon to GCT. Thr-451 was changed to Ser, Asp, or Ala by changing the Thr-451 ACT codon to TCT, GAT, or GCT, respectively. Glu-490 was mutated to Gln by changing the Glu-490 GAA codon to CAA.
AccI-BamHI fragments containing the desired mutations were isolated from the PCR fusion products and subcloned into AccI-BamHI-digested p1543, which contains the wild-type PKR coding sequence as a HindIII-BamHI fragment in pUC18 (38). The full-length PKR coding sequences containing the desired point mutations were isolated from the pUC18 clones as HindIII-BamHI fragments and ligated with a modified version of pEMBLyex4 (18) digested with HindIII and BamHI. This method was used to generate the following plasmids carrying PKR proteins with point mutations (indicated in parentheses): p2156(T446S), p2157(T446D), p1689(T446A), p2110(S448A), p2131(T451S), p2119(T451D), p2111(T451A), p2117(T446S,T451S), p2118(T446D,T451D), p2109(T446A,T451A), and p2163(E490Q). To make p1688(M365V), the Met-365 AUG codon was changed to GUG and a BglII fragment containing the desired mutation was isolated from the PCR fusion product and ligated with BglII-digested p1469, which contains the wildtype PKR coding sequence in pEMBLyex4 (38). To create p2161(T258A,T446A) and p2158(T258A,T451A), a BglII fragment containing the T258A point mutation (42) was ligated with BglII-digested p1689 and p2111, respectively. To create p2587(S242A,T255A,T258A,T446A), a HindIII-BglII fragment containing the S242A,T255A,T258A triple point mutation (42) was ligated with p1689 digested with HindIII-BglII. To create pR117(M365V,E490Q), a BglII fragment containing the M365V point mutation was isolated from p1688 and ligated with BglII-digested p2163. To create pR113(T451A,E490Q), an AflII-BamHI fragment containing the point mutations was isolated from the PCR fusion product and ligated with AflII-BamHI-digested p1543, creating pR108. A HindIII-BamHI fragment containing the point mutations was isolated from pR108 and ligated with HindIII-BamHI digested p1469. For the expression of PKR alleles in mammalian cells under the control of a cytomegalovirus (CMV) promoter, the PKR coding sequences were isolated as HindIII-BamHI fragments from the appropriate pEMBLyex4 derivatives and ligated with vector pcDNA3 (Invitrogen) digested with HindIII and BamHI.
A low-copy-number plasmid containing the wild-type PKR cDNA with the coding sequence for amino acids MRGSHHHHHH inserted at the 5′ end was kindly provided by Makiko Kobayashi. This six-His-tagged PKR coding sequence was subcloned as an ApaI-HindIII fragment into the ApaI and HindIII sites of pEMBLyex4, creating plasmid pR156. Plasmid pR181, carrying 6×His-PKR-K296R, was created by isolating from p1470 (38) a 500-bp BglII fragment containing the K296R point mutation and subcloning this fragment into BglII-digested pR156.
Construction of GCN2 plasmids p587, p585, p630, p631, and p722 has been described elsewhere (37). Single and double point mutations in the GCN2 kinase domain were generated by PCR fusion (49). Thr-882 and Thr-887 were mutated to Ser and Ala by changing the ACA Thr codons to TCT and GCC, respectively. KpnI-XhoI fragments encoding the Ala substitution mutations were isolated from the PCR fusion products and subcloned into KpnI-XhoI-digested pB2. This plasmid contains the BamHI fragment of p585 containing the N-terminal half of GCN2 and upstream sequences in a derivative of pBSII KS− (Stratagene), modified to leave only the SmaI, BamHI, and SpeI sites in its polylinker. The resulting BamHI fragments containing the mutated sequences were isolated from these intermediate plasmids and used to replace the corresponding fragment in p585, a single-copy plasmid bearing wild-type GCN2 derived from YCp50 (32). Insertion of the BamHI fragment in the correct orientation was verified by DNA sequence analysis of sequences flanking the BamHI site. This method was used to generate the following plasmids carrying GCN2 proteins with point mutations indicated in parentheses: pB9(T882A), pB10(T887A), and pB11(T882A,T887A). KpnI-XhoI fragments containing the desired Ser substitutions were isolated from the PCR fusion products and subcloned into KpnI-XhoI-digested p722, a low-copy-number GCN2 plasmid, generating pB17(T813S), pB87(T887S), and pB15(T882S,T887S). The KpnI-XhoI fragment from p631 containing the K628R mutation was subcloned in both p630 and p722 to generate pB34 and pB31, respectively. High-copy-number GCN2 plasmids carrying Ala or Ser substitutions at positions 882 and 887 were generated by subcloning the corresponding KpnI-XhoI fragments from the low-copy-number plasmids described above into p630 digested with the same enzymes to produce plasmids pB65(T882A), pB60(T887A), pB61(T882A,T887A), pB66(T882S), pB63(T887S), and pB25-(T882S,T887S).
Construction of the yeast strains H1894 (a gcn2Δ ura3-52 leu2-3 leu2-112 trp1-Δ63) and H1816 (a ura3-52 leu2-3 leu2-112 gcn2Δ sui2Δ trp1-Δ63 p1108 [GCN4-lacZ TRP1] at trp1-Δ63 p1097 [SUI2 LEU2]) has been described elsewhere (13). Strain H1817 is isogenic to strain H1816 except that p1097 (SUI2 LEU2) was replaced with p1098 (SUI2-S51A LEU2). Yeast strain GP3299 (a ura3-52 leu2-3 leu2-112 trp1-Δ63 gcn2Δ gcd2Δ::hisG pAV1033 [GCD2-K627T TRP1]) is a derivative of strain H1515 (14, 35) and was kindly provided by Graham Pavitt.
GCN2 antisera.
Specific antibodies recognizing the N terminus of GCN2 were raised against a TrpE-GCN2 fusion protein bearing the GCN2 coding sequences of the EcoRI fragment of GCN2 inserted into pATH11 (ATCC 37699). A second TrpE-GCN2 fusion protein containing the C-terminal region of GCN2 was generated by subcloning the BamHI-NheI fragment of GCN2 into the pATH3 (ATCC 37697) vector, kindly provided by Matthew Marton. The fusion proteins were expressed in Escherichia coli HB101-9 and purified for use as antigens as described previously (24). Antibodies were generated by Covance Laboratories Inc.
Immunoblot analysis of protein expression in yeast cells.
Transformants of the H1816 and H1817 strains containing various PKR alleles were grown in synthetic minimal dextrose (SD) medium at 30°C for ∼30 h and then were shifted to inducing conditions, synthetic minimal medium containing 10% galactose and 2% raffinose (SGAL medium), for ∼12 h. Whole-cell extracts were prepared by breaking the cells with glass beads in lysis buffer (100 mM Tris-HCl [pH 8.0], 20% glycerol, 1 mM β-mercaptoethanol) containing complete protease inhibitor (CPI) cocktail (Boehringer Mannheim Biochemicals). For analysis of phosphatase-treated proteins, 1 μl (∼400 U) of λ protein phosphatase (New England BioLabs) was added to samples of whole-cell extracts (prepared as just described) containing 30 μg of total protein and 1× λ protein phosphatase buffer (50 mM Tris-HCl [pH 7.8], 5 mM dithiothreitol, 2 mM MnCl2, 100 μg of bovine serum albumin per ml); the mixtures were incubated at 30°C for 1 h. Extracts were separated by electrophoresis in sodium dodecyl sulfate (SDS)–8 to 16% polyacrylamide gradient gels and transferred to nitrocellulose. Blots were treated with blocking solution (20 mM Tris-HCl [pH 7.6], 150 mM NaCl, 0.1% Tween 20) containing 5% nonfat dry milk. Immunodetection of PKR was done with PKR-specific monoclonal antibodies (lot 71/10; RiboGene) at a dilution of 1:1,000 in the blocking solution just described. Immune complexes were visualized with the enhanced chemiluminescence (ECL) detection system (Amersham) following the vendor’s instructions.
Yeast extracts from transformants of strain H1894 containing different GCN2 alleles and grown in SD medium were prepared as previously described (45). Proteins were separated by electrophoresis in SDS–4 to 16% polyacrylamide gradient gels and transferred to polyvinylidene difluoride (PVDF) membranes. Immunodetection was performed as described for PKR, except that specific antibodies against the N-terminal portion of GCN2 or GCD6 (6) were used at a dilution of 1:2,000 or 1:1,000, respectively.
In vitro protein kinase assays.
Transformants of strain H1817 containing various plasmid-borne PKR alleles were grown for ∼16 h in SD medium, diluted 1:50 in SGAL medium, and grown for another ∼16 h to induce PKR expression. Whole-cell extracts were prepared, PKR was immunoprecipitated from the extracts with anti-PKR polyclonal antibodies bound to protein A-Sepharose beads (Pharmacia), and the immune complexes were assayed for kinase activity, all as previously described (38) but with the following modification. After the proteins were resolved by electrophoresis in SDS–8 to 16% polyacrylamide gradient gels, they were transferred to nitrocellulose filters, which were subjected to autoradiography with Kodak XAR film to visualize the radiolabeled proteins. Following autoradiography, the blots were treated with blocking solution and immunodetection of PKR was carried out as described above. The recombinant yeast eIF2α used in these assays was expressed in bacteria and purified as described previously (52).
For kinase assays with peptide substrates, 4 μg of the appropriate synthetic peptides was added to the kinase reaction mixtures with 10 μCi of [γ-32P]ATP and incubated for 15 min at 30°C. After the addition of Laemmli sample buffer (27), the samples were boiled for 10 min and analyzed by SDS-polyacrylamide gel electrophoresis (PAGE). Gels were stained with Coomassie blue, dried under vacuum, and subjected to autoradiography with Kodak XAR film. The synthetic peptides were prepared by Peptide Technologies Corp. (Gaithersburg, Md.) and were purified and analyzed by high-pressure liquid chromatography (HPLC) to verify both synthesis and purity.
In vitro kinase activities of GCN2 were assayed as described previously (45), except that after SDS-PAGE with 4 to 16% gels, the proteins were transferred to PVDF membranes. The labeled proteins bound to the membrane were visualized by autoradiography, after which the membranes were subjected to immunoblot analysis with antibodies against GCN2 as described above.
In vivo labeling of PKR in yeast cells.
Transformants of yeast strain H1817 expressing wild-type and mutant PKR alleles were grown for ∼16 h at 30°C in synthetic low-phosphate medium containing dextrose [20 mM KCl, 2 mM MgSO4, 2 mM CaCl2, 15 mM (NH4)2SO4, 8.5 mM NaCl, 0.1% yeast extract, 2% glucose] and then were shifted to low-phosphate medium containing 10% galactose and 2% raffinose instead of dextrose for ∼12 h to induce PKR expression. About 4 units of cells at an optical density of 600 nm (OD600) was resuspended in the same galactose-raffinose medium containing 1.5 mCi of [32P]orthophosphate and incubated at 30°C for 4 to 6 h. Cells were broken in kinase reaction breaking buffer (KRBB; 20 mM Tris-HCL [pH 8.0], 50 mM KCl, 400 mM NaCl, 20% glycerol, 1% Triton X-100, 0.5 mM EDTA) containing phosphatase inhibitors (50 mM NaF, 20 mM β-glycerol phosphate, 5 mM sodium pyrophosphate, 125 μM sodium orthovanadate) and CPI cocktail. For immunoprecipitation of PKR, 30 μl of protein A-Sepharose beads was resuspended in 200 μl of KRBB and incubated with 1 μl of PKR-specific polyclonal antiserum for 30 min at room temperature. The beads were pelleted and washed with 400 μl of KRBB. Total protein extract from 4 OD600 units of cells in 200 μl of KRBB was added to the washed beads and incubated for 2 h at 4°C. Immune complexes were collected as described above and resuspended in Laemmli sample buffer (27). Samples were boiled for 10 min and resolved by SDS-PAGE with 10% polyacrylamide gels. Gels were dried under vacuum and subjected to autoradiography with Kodak XAR film.
Analysis of PKR expression in mammalian cells.
COS1 cells were grown in 10-cm tissue culture plates to 50 to 60% confluence in Dulbecco modified Eagle medium with 10% fetal bovine serum. Plasmids bearing wild-type or mutant PKR alleles under the control of a CMV promoter (pcDNA3) were introduced into COS1 cells by the calcium phosphate precipitation procedure (39). DNA (20 μg) was dissolved in 0.25 M CaCl2, and an equal volume of 2× HEPES-buffered saline (280 mM NaCl, 50 mM HEPES, 1.5 mM Na2HPO4 [pH 7.1]) was added to form a precipitate. After 30 min at room temperature, the precipitate was added to the monolayer in a dropwise manner. The medium was changed at 18 h posttransfection. At 48 h posttransfection, the cells were washed twice with cold phosphate-buffered saline and lysed with 10 mM phosphate buffer (10 mM phosphate [pH 7.4], 100 mM NaCl, 0.5% Nonidet P-40, 10% glycerol, 50 μg each of aprotinin and leupeptin per ml, 2 mM phenylmethylsulfonyl fluoride, 0.1% NaN3, 1 mM Na3VO4) or by a freeze-thaw-vortex procedure with 0.25M Tris-HCl (pH 8.0)–0.5% Nonidet P-40. After removal of debris, proteins were resolved in a 10% polyacrylamide–SDS gel and transferred to a 0.2 μm-pore-size nitrocellulose filter. After incubation in 5% nonfat dry milk dissolved in 20 mM Tris-HCl (pH 8.0)–150 mM NaCl–0.1% Tween 20, the blot was probed with mouse monoclonal anti-PKR antibodies (RiboGene) at a 1:300 dilution and developed with a horseradish peroxidase-conjugated anti-mouse secondary antibody by use of an ECL procedure (Amersham).
For metabolic labeling of transfected COS1 cells, cultures were incubated in methionine-free Dulbecco modified Eagle medium with 10% fetal bovine serum for 1 h at 45 h posttransfection and then labeled for 1 h with [35S]methionine (NEN Life Science; 0.1 mCi/ml; 5 ml per dish). For immunoprecipitation of PKR, 500 μl of extract containing 500 μg of protein in 10 mM phosphate buffer was incubated with 30 μl of protein A-Sepharose beads for 1 h at 4°C to preclear the beads. After centrifugation to remove the beads, the supernatant was incubated with 2 μl of polyclonal anti-PKR antiserum (18) and then with 30 μl of protein A-Sepharose beads for 3 h at 4°C. The immune complexes were collected and washed twice with 400 μl of 10 mM phosphate buffer. Proteins were solubilized in Laemmli sample buffer. After separation of samples in 10% polyacrylamide gels, the gels were fixed with 40% methanol–10% acetic acid, dried under vacuum, and then visualized by autoradiography with Kodak XAR film.
Tumorigenesis assays.
Two to four nude mice (BALB/c nude; Taconic Farms, N.Y.) were injected in the left or right flank with transfected cells containing different PKR constructs at 5 × 106 cells per injection per mouse (25). Tumor growth was monitored every 3 days, and tumors larger than 3 mm were regarded as positive.
Phosphoamino acid analysis of peptide substrates.
In vitro kinase reaction mixtures with synthetic peptides as substrates were fractionated by electrophoresis in Tricine–SDS–10 to 20% polyacrylamide gradient gels and transferred to PVDF membranes. The membranes were exposed to film (Kodak XAR), and the labeled PKR and peptide bands were visualized by autoradiography. The strip of PVDF membrane containing the radiolabeled peptide was subjected to acid hydrolysis as described previously (22). The supernatant containing the hydrolyzed peptide was removed to a separate tube and lyophilized. Deionized water (100 μl) was added, and the sample was again lyophilized to dryness. Radiolabeled phosphoamino acids were separated by electrophoresis on thin-layer chromatography plates (EM 5716-7; Merck) at pH 3.5 (4). Phosphoserine, phosphothreonine, and phosphotyrosine standards (Sigma) were analyzed in parallel and visualized by ninhydrin staining. Radiolabeled phosphoamino acids were visualized by autoradiography.
In vitro GCN2 autokinase reaction mixtures were fractionated by SDS-PAGE with 6% gels and transferred to PVDF membranes as described above. The radiolabeled GCN2 bands were visualized by autoradiography, and the strip of PVDF membrane containing the radiolabeled protein was subjected to phosphoamino acid analysis following the protocol described above.
Isolation of autophosphorylated PKR from yeast cells for phosphopeptide analysis.
Plasmids pR156 and pR181 carrying six-His-tagged PKR and 6×His-PKR-K296R were introduced into yeast strain GP3299. To provide a negative control, an untagged version of wild-type PKR expressed from plasmid p1469 (38) was also transformed into yeast strain GP3299. Single colonies of each yeast strain were inoculated into 5.0 ml of SD medium and cultured overnight at 30°C. This 5.0-ml starter culture was inoculated into 100 ml of SGAL medium and grown overnight at 30°C. One liter of SGAL medium was inoculated with the 100-ml overnight SGAL culture, and the culture was grown an additional 48 h at 30°C. Cells were harvested by centrifugation at 4,500 × g for 10 min at 4°C. The pellet was resuspended in 50 ml of H2O containing CPI cocktail and phosphatase inhibitors (50 mM NaF, 20 mM β-glycerolphosphate, 125 μM Na3VO4). Cells were pelleted by centrifugation in a swinging-bucket rotor at 4,000 rpm for 5 min at 4°C. The pellet was resuspended in 4.0 ml of nickel column binding buffer (20 mM sodium phosphate, 500 mM NaCl, 10 mM imidazole, 0.1% Triton X-100) plus CPI cocktail and the phosphatase inhibitors just described, and the cells were broken with glass beads. An aliquot of the resulting extract containing 3.0 mg of total cell protein was incubated with 100 μl of Ni-NTA Silica resin (Qiagen) by rotation in a cold room for 1 h. The resin was pelleted and washed twice in nickel column binding buffer containing 40 mM imidazole. Proteins bound to the resin were eluted with Ni column elution buffer (20 mM sodium phosphate, 500 mM NaCl, 400 mM imidazole) containing protease and phosphatase inhibitors. Proteins eluted from the Ni-NTA Silica resin were incubated with poly(I) · poly(C)-agarose (Pharmacia) for 1 h at 4°C. The poly(I) · poly(C) agarose was pelleted and washed three times with buffer A (150 mM KCl, 20 mM HEPES, 10% glycerol, 5 mM magnesium acetate) plus CPI cocktail and phosphatase inhibitors. The pellet was resuspended in 100 μl of buffer A, and 20 μl of 6× Laemmli sample buffer was added. The samples were boiled for 5 min and separated by electrophoresis in SDS–8 to 16% polyacrylamide gradient gels. The PKR protein bands were visualized by Cu staining (Bio-Rad).
MS.
In-gel digestions of the Cu-stained PKR proteins obtained from the SDS-PAGE separations just described were carried out with ∼200 ng of protein and sequencing-grade modified trypsin or sequencing-grade Asp-N or Lys-C (Boehringer) at a protein/enzyme ratio of 1:1 in 50 mM NH4HCO3 for 2 h at 37°C, and the resulting peptides were extracted as described previously (36). The extracted peptides were redissolved in 10 μl of acetonitrile-H2O (1:1) for MS analysis. For phosphatase treatment of the extracted peptides, 2 μl of the peptides was mixed with 2 μl (0.5 U) of calf intestinal phosphatase (CIP) (New England BioLabs) in 50 mM NH4HCO3 and incubated at 37°C for 30 min.
A matrix-assisted laser desorption-ionization time-of-flight mass spectrometer (MALDI/TOF) with delayed extraction and equipped with a 2-GHz digital oscilloscope (Voyager-DE; Perseptive Biosystems, Framingham, Mass.) was used to locate the phosphopeptides by peptide mapping, whereby the masses of peptides resulting from in-gel enzymatic digestions of PKR protein were measured and compared with the calculated theoretical masses based on the specificity of the protease. A twofold dilution of saturated 2,5-dihydroxybenzoic acid in acetonitrile-H2O (1:1) solution was used as the working matrix solution for MALDI/TOF MS analysis. Peptide mixture (0.5 μl) was mixed with 0.5 μl of the working matrix solution on a sample plate, air dried, and inserted into the mass spectrometer for MS measurements.
An electrospray ion-trap mass spectrometer (LCQ; Finnigan, San Jose, Calif.) coupled on-line with a microbore HPLC apparatus (Magic 2002; Michrom BioResources, Auburn, Calif.) was used for MS sequencing of selected peptides. A monitor C18 column (5-mm particle diameter; 150-Å pore size; 0.5 by 150 mm) with mobile phases A (acetonitrile–n-propanol–water–acetic acid–trifluoroacetic acid, 1:1:98:0.1:0.02) and B (acetonitrile–n-propanol–water–acetic acid–trifluoroacetic acid, 70:20:10:0.09:0.02) was developed with a gradient of 2 to 98% mobile phase B over a period of 5 min for on-line liquid chromatography (LC)-MS-MS analysis. The rest of the extracted peptide mixture after MALDI/TOF MS measurements was dried, redissolved in 5 μl of mobile phase A and loaded on the HPLC apparatus. LC-MS-MS was carried out in the constant MS-MS mode, in which the doubly charged phosphopeptide ions, as determined from MALDI/TOF MS measurements, were subjected to collision-induced dissociation (CID) as they eluted from the HPLC column (50a).
To sequence the tryptic phosphopeptide containing residues 430 to 447 by LC-MS-MS, the doubly charged phosphopeptide ion produced by electrospray ionization was isolated in the ion-trap mass spectrometer and resonantly excited to cause fragmentation of the peptide backbone through CID. The masses of the resulting fragments were measured; from these, the sequence of the peptide could be identified from the peptide ladder that was generated. The position of the phosphoamino acid was determined from the fact that the masses of the fragment ions containing the phosphoamino acid were 80 Da higher than the calculated masses of the corresponding fragments, whereas the masses of other fragment ions not containing the phosphoamino acid matched the calculated masses without phosphorylation. In this manner, we unambiguously identified Thr-446 as the only site of phosphorylation in the tryptic phosphopeptide containing residues 430 to 447. In a similar fashion, we confirmed that the Lys-C peptide of 2,060.2 Da corresponded to the unphosphorylated form of the peptide containing residues 450 to 467.
RESULTS
Substitutions at Thr-446 and Thr-451 impair general and GCN4-specific translational control by PKR in yeast cells.
We altered the potential autophosphorylation sites Thr-446 and Thr-451 in the PKR activation loop to phosphorylatable Ser residues or nonphosphorylatable Ala or Asp residues and examined the effects on PKR function in yeast cells. cDNAs encoding the mutant proteins were expressed from a galactose-inducible GAL-CYC1 promoter in yeast strains lacking GCN2. High-level expression of PKR from this promoter on galactose medium is lethal due to extensive phosphorylation of eIF2α at Ser-51. This growth inhibition is completely eliminated by a mutation that inactivates kinase subdomain II in PKR (substitution of Lys-296 with Arg; K296R) (Fig. 2A). The degree of growth inhibition on galactose medium in strains expressing PKR from the GAL1-CYC1 promoter provides a sensitive indicator of PKR function in vivo (9, 13). Low-level expression of wild-type PKR on medium containing dextrose as a carbon source does not inhibit cell growth but is sufficient to restore the translation of GCN4 mRNA in strains lacking GCN2. gcn2Δ mutants are unable to grow on medium containing 3-aminotriazole (3-AT), a competitive inhibitor of histidine biosynthesis, because they cannot derepress GCN4 and its target genes in the histidine biosynthetic pathway. Consequently, the ability to complement the 3-AT sensitivity of a gcn2Δ mutant provides a second indicator of PKR function in yeast cells (13).
FIG. 2.
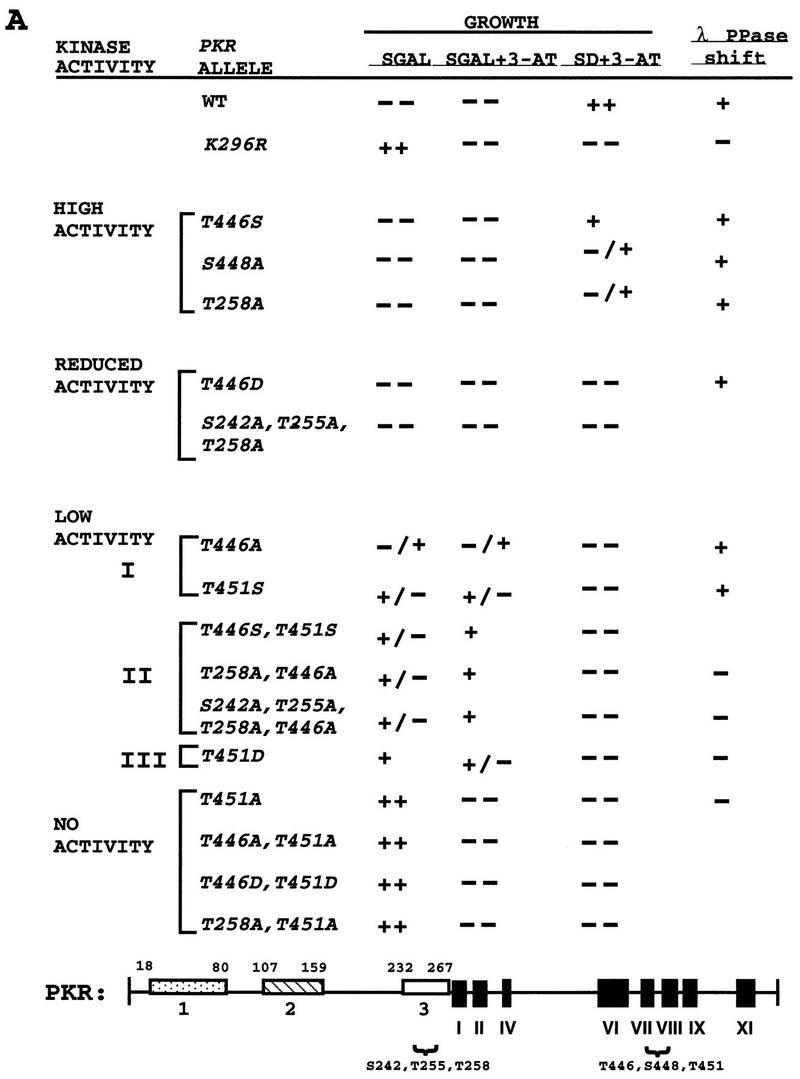

In vivo analysis of mutant PKR alleles by growth tests in yeast. (A) The schematic at the bottom represents the full-length wild-type PKR protein sequence. The boxes in the amino-terminal half represent the three regions rich in basic residues. Boxes 1 and 2 contain DRBM-1 and -2, respectively. Box 3 is a region rich in basic amino acids of unknown function. The amino acid residues spanning these regions are indicated. The black boxes in the C-terminal half (I to XI) represent conserved kinase subdomains found in the catalytic regions of all protein kinases. The locations of known (S242, T255, and T258) or suspected (T446, S448, and T451) autophosphorylation sites are indicated. Plasmids carrying the indicated PKR alleles were introduced into the gcn2Δ yeast strain H1816 expressing wild-type eIF2α. Patches of transformants were grown to confluence in SD medium and replica plated to SD medium plus 30 mM 3-AT, SGAL medium, and SGAL medium plus 30 mM 3-AT. Plates were incubated for 3 to 4 days at 30°C, and growth was scored relative to that of the wild-type (WT) PKR transformants. Relative growth from strongest to weakest was scored as ++, +, +/−, −/+, and −−. Whole-cell lysates from the indicated strains that were either treated or not treated with λ protein phosphatase (λ PPase) were subjected to SDS-PAGE fractionation and immunoblot analysis as described in the legends to Fig. 3 and 4. PKR proteins whose mobilities increased when treated with phosphatase are indicated by +; proteins that showed no discernible change in mobility when treated with phosphatase are indicated by −. (B) Selected transformants of H1816 bearing the indicated PKR constructs were streaked on SGAL medium and incubated for 6 days at 30°C.
The mutant PKR alleles shown in Fig. 2A were introduced into a gcn2Δ strain containing wild-type eIF2α (H1816), and the resulting transformants were tested for growth on medium containing galactose (SGAL medium) or dextrose (SD medium) as a carbon source in the presence or absence of 3-AT. Based on the results of the growth tests, the mutant alleles could be assigned to one of six groups displaying different reductions in kinase activity relative to that of wild-type PKR (Fig. 2A). Substitution of Ser-448 with Ala (S448A) caused a modest reduction in PKR function, manifested by lethality on SGAL medium and diminished growth relative to that conferred by wild-type PKR in SD medium containing 3-AT. The phenotype of the S448A allele was the same as that observed previously for an Ala substitution in the Thr-258 phosphorylation site (T258A) (42) (Fig. 2A). An Ala substitution at Thr-446 or Thr-451 led to a substantially greater reduction in PKR function. The T451A allele was indistinguishable from catalytically inactive K296R allele, whereas the T446A allele had low-level activity, allowing weak growth on SGAL medium but not conferring 3-AT resistance on SD medium. These results suggested that T446 is required only for full PKR activity, whereas T451 is required for any measurable activity.
If T446 and T451 functioned as autophosphorylation sites, then substitutions with phosphorylatable Ser residues might have relatively little effect on PKR function. Moreover, substitutions with Asp, which structurally resembles phosphoserine, might impair kinase function less than Ala substitutions would. These expectations were borne out for position 446 in that the T446S allele had high-level activity, the T446D allele had reduced activity, and the T446A allele had low-level activity (Fig. 2A). Similarly, the T451S allele was slightly more functional than the T451D allele, and both were more functional than the T451A allele (Fig. 2A). (The T451D allele was judged to be less functional than the T451S allele because it conferred less toxicity on SGAL medium.) The results of these experiments are consistent with the idea that T446 is an important autophosphorylation site. The results for T451 are less conclusive because the T451S and T451D alleles had low-level function compared to wild-type PKR (Fig. 2A).
The PKR alleles containing substitutions at two of the three residues analyzed in the activation loop had lower-level activity in yeast cells than did alleles containing the corresponding single-residue substitutions (Fig. 2A). We also observed an additive effect of combining the T446A substitution with the Ala substitutions in the previously established autophosphorylation sites at Ser-242, Thr-255, and Thr-258 (42) (Fig. 2A and B). It was not surprising to find that all of the double mutants containing the T451A substitution were completely inactive because T451A alone was sufficient to abolish PKR function in yeast cells.
Substitutions at Thr-446 and Thr-451 impair autoregulation of PKR expression in yeast cells.
It has been shown that the expression of PKR is negatively autoregulated in yeast cells, such that the wild-type protein is expressed at very low levels, whereas the catalytically inactive K296R protein is expressed at about 20-fold-higher levels (13, 38). Accordingly, an independent assessment of PKR function can be achieved by comparing the in vivo steady-state levels of mutant and wild-type PKR proteins by immunoblot analysis. In addition, by measuring PKR expression in isogenic strains containing either wild-type eIF2α or eIF2α containing an S51A mutation (eIF2α-S51A), we can distinguish between effects on PKR expression attributable to autoregulation (those occurring with wild-type eIF2α) from differences in protein stability or immunological cross-reactivity (seen in the presence of eIF2α-S51A). Figure 3 shows the effects of selected substitutions at positions 446, 451, and 258 on PKR protein levels in a strain containing wild-type eIF2α, where autoregulation of PKR expression is intact. Ala substitutions at positions 446 and 451 increased PKR expression in proportion to their deleterious effects on PKR function, as judged by the growth assays described above. Thus, the inactive T451A protein was expressed at higher levels than was the T446A protein, which retained low-level activity (Fig. 3, compare lanes 5 and 8). In most experiments, the T446D and T451D proteins were expressed at slightly lower levels than were the T446A and T451A proteins, respectively (Fig. 3, lanes 12 and 13 and lanes 14 and 15) (data not shown), consistent with the idea that Asp is more functional than Ala at these two positions. The T258A mutation led to a smaller increase in PKR expression than did the T446A mutation, in agreement with the conclusion that T446 is more critical than T258 for PKR activity (Fig. 3, lanes 2 and 5). In addition, the T258A,T446A double-mutant protein was expressed at higher levels than was either the T258A or the T446A single-mutant protein, confirming that these mutations have additive effects on PKR function.
FIG. 3.

Immunoblot analysis of PKR protein levels in gcn2Δ yeast cells expressing wild-type eIF2α. Transformants of yeast strain H1816 bearing the indicated wild-type (WT) or mutant PKR alleles were grown in SD medium at 30°C for 30 h and then shifted to SGAL medium for 12 h to induce PKR expression. Thirty micrograms of total cell protein from each strain was fractionated by SDS-PAGE with 10% polyacrylamide gels and subjected to immunoblot analysis with monoclonal antibodies against PKR (71/10) and monoclonal antibodies against PAB1 (PAB), the yeast poly(A)-binding protein. The latter were used to confirm that equal amounts of cell protein were analyzed for each strain. ECL was used to visualize immune complexes. Lanes 10 and 11, 12 and 13, and 14 and 15 contain extracts isolated from two independent transformants bearing the indicated PKR alleles.
In a strain containing eIF2α-S51A, where autoregulation of PKR expression is disrupted (13, 38), nearly equivalent levels of the mutant proteins containing various substitutions at positions 446, 448, and 451 were observed (Fig. 4A and B). This result showed that the elevated protein expression observed with the Ala substitutions at positions 446 and 451, compared to that observed with wild-type PKR in the strain with wild-type eIF2α (Fig. 3), resulted from diminished autoregulation rather than from altered protein stabilities.
FIG. 4.

Immunoblot analysis of protein levels and relative mobilities of PKR proteins expressed in yeast cells containing nonphosphorylatable eIF2α-S51A. (A and B) Thirty micrograms of total cell protein from yeast strain H1817 expressing wild-type (WT) PKR protein or the indicated PKR mutant proteins was subjected to SDS-PAGE with 10% (A) or 7.5% (B) polyacrylamide gels and immunoblot analysis with PKR-specific monoclonal antiserum and ECL to detect immune complexes. (C) Comparison of the relative mobilities of PKR proteins with and without phosphatase treatment. Thirty micrograms of total cell protein from transformants of the H1817 strain were treated with λ protein phosphatase (+ λ PPase). Phosphatase-treated and untreated samples were subjected to SDS-PAGE (7.5% polyacrylamide gel) and immunoblot analysis. The broken lines in panels B and C are drawn midway through the K296R PKR bands. The displacements of the midpoints of the untreated wild-type and T451S PKR bands from these lines were 2.7 and 2.0 mm, respectively, whereas the protein bands of the phosphatase-treated samples were displaced from these lines by 1.2 and 0.5 mm, respectively.
The mobilities of the PKR proteins observed in the immunoblots in Fig. 3 and 4A and B also correlated with their functional assignments based on the growth tests (Fig. 2) and the ability to autoregulate protein expression (Fig. 3). This correlation can be explained by assuming that higher levels of PKR autophosphorylation lead to lower electrophoretic mobilities. In Fig. 3, lanes 1 to 9, where the mutant proteins were arranged in order of decreasing deduced kinase activities from left to right, the electrophoretic mobilities also increased from left to right. The mobility differences between the mutant proteins were even more pronounced in Fig. 4B, where it is evident that the T451S, T451D, and T451A mutants exhibited a progressive increase in mobility, consistent with the idea that Ser is more functional than Asp and that Asp is more functional than Ala at position 451. Treatment of the samples with λ phosphatase prior to gel electrophoresis diminished the differences in electrophoretic mobilities between mutant and wild-type PKR proteins, as shown in Fig. 4C for the T451A and T451S proteins. (We attribute the slight differences in mobilities between the wild-type and T451S proteins versus the nonfunctional T451A and K296R proteins after phosphatase treatment to incomplete dephosphorylation of the former two proteins.) As summarized in the last column of Fig. 2A, we discerned an increase in electrophoretic mobility following λ phosphatase treatment for all of the PKR proteins with high-level and reduced activities and for subgroup I proteins with low-level activity but not for other PKR proteins with even lower activity. Presumably, the latter proteins are autophosphorylated at too few sites to produce a significant increase in electrophoretic mobility upon phosphatase treatment.
Finally, we compared the relative levels of autophosphorylation by the T446A and T451A PKR proteins in vivo by metabolically labeling cells with [32P]orthophosphate and immunoprecipitating the PKR proteins. The mutant and wild-type PKR proteins were expressed at similar levels because the strain contained eIF2α-S51A. As shown in Fig. 5, the T451A and T446A,T451A mutant proteins were labeled at very low levels under these conditions, comparable to that seen for the K296R protein, whereas the T446A mutant protein incorporated label at a level intermediate between that of the wild-type and K296R PKR proteins. These results support the idea that the T451A protein is extremely defective for autophosphorylation, whereas the T446A protein has an intermediate level of autokinase activity in vivo.
FIG. 5.

In vivo autophosphorylation of wild-type and mutant PKR proteins in yeast cells containing eIF2α-S51A. Transformants of yeast strain H1817 (containing nonphosphorylatable eIF2α-S51A) bearing the indicated wild-type (WT) or mutant PKR alleles were grown in SGAL medium to induce PKR expression and metabolically labeled with [32P]orthophosphate for 4 to 6 h (see Materials and Methods). PKR proteins were immunoprecipitated from aliquots of labeled whole-cell extracts with polyclonal antibodies against PKR, and the immune complexes were analyzed by SDS-PAGE (10% polyacrylamide) followed by autoradiography.
Substitutions at Thr-446 and Thr-451 impair PKR kinase activity in vitro.
We conducted in vitro kinase assays on a subset of the mutant PKR proteins containing substitutions at position 446, 448, and 451. The proteins were immunoprecipitated from transformants of eIF2α-S51A strain H1817 (in which the wild-type and mutant PKR proteins were expressed at similar levels) and incubated in the presence of [γ-32P]ATP and purified eIF2. Following SDS-PAGE separation, the proteins were transferred to nitrocellulose and subjected to autoradiography to visualize the radioactivity incorporated into PKR and eIF2α. The filters then were probed with PKR antibodies to measure the amounts of PKR recovered in the immune complexes. In accordance with our in vivo estimates of PKR function, the T446D, T446A, T451S, and T451D proteins showed greatly reduced autophosphorylation and eIF2α kinase activities, whereas the T451A protein was completely inactive (Fig. 6, lanes 1 to 7). Note that the Asp-451 protein had higher autokinase activity than did the Ala-451 protein and that the Asp-446 protein had higher eIF2α kinase activity than did the Ala-446 protein. In addition, Ala substitutions at the previously established phosphorylation sites Ser-242, Thr-255, and Thr-258 were less deleterious than was the Ala-446 substitution (Fig. 6, lanes 10 to 12).
FIG. 6.

In vitro kinase activities of immunopurified PKR proteins isolated from yeast strains containing eIF2α-S51A. (A) Transformants of strain H1817 bearing the indicated wild-type (WT) or mutant PKR alleles were grown in SGAL medium to induce PKR expression, and the PKR proteins were immunoprecipitated from whole-cell extracts containing 150 μg of total protein with polyclonal antibodies against PKR. Immune complexes were incubated in kinase reaction buffer (KRB) (38) in the presence of [γ-32P]ATP and 1 μg of purified recombinant eIF2α for 15 min at 30°C. Radiolabeled samples were separated by SDS-PAGE (8 to 16% polyacrylamide gradient gel) and transferred to a nitrocellulose filter. The radiolabeled PKR and eIF2α proteins were visualized by autoradiography. It was shown previously that PKR immunopurified from yeast extracts was activated without the addition of dsRNA activators and that the addition of poly(I) · poly(C) produced no significant increase in activity for either mutant or wild-type enzymes (38); therefore, these assays were conducted without the addition of exogenous dsRNA. (B) The same nitrocellulose filter was subjected to immunoblot analysis with monoclonal antibodies against PKR as described in the legends to Fig. 3 and 4 to visualize the amounts of PKR immunoprecipitated for each sample.
Substitution of a conserved residue between kinase subdomains IX and X partially suppresses the catalytic defect imposed by the Ala-451 mutation.
The crystal structure of the unphosphorylated (inactive) form of the MAP kinase ERK2 showed that the activation loop interacts with residues between kinase subdomains IX and X, located within and near the G helix. In particular, Tyr-231 of ERK2 makes two hydrogen bonds with the activation loop. Phosphorylation of Tyr-185 and Thr-183 in the ERK2 activation loop causes it to refold, resulting in a loss of contacts between the loop and neighboring segments as the kinase assumes an active conformation (7). From the yeast MAP kinase FUS3 a dominant mutation (D227N) that partially compensated for reduced phosphorylation of the activation loop of FUS3 in mutant strains containing a defective form of the MAP kinase kinase STE7 was isolated (5). Asp-227 in FUS3 is located at a position closely corresponding to that of Tyr-231 in ERK2 (10); therefore, mutation of Asp-227 to Asn in FUS3 could alter interactions between the G helix and activation loop to increase kinase activity in the hypophosphorylated form of FUS3, mimicking the effect of phosphorylation by STE7.
We noted that the three known eIF2α kinases, PKR, GCN2, and HRI, all contain a Glu residue at the position corresponding to Asp-227 in FUS3 and that this Glu residue is 1 of only 11 residues that are conserved among eIF2α kinases but absent in the majority of other protein kinases (37). We reasoned that if autophosphorylation in the activation loop is a critical step in the activation of PKR, it might be possible to reactivate the PKR Ala-451 mutant protein by changing the conserved Glu residue (at position 490) to Gln, as this would be the equivalent of changing Asp-227 to Asn-227 in FUS3. In accordance with this prediction, the E490Q mutation partially restored the growth inhibition phenotype characteristic of wild-type PKR in SGAL medium when introduced into the Ala-451 mutant protein. Based on its growth phenotype (Fig. 7A), the T451A,E490Q double-mutant protein fell into low-level activity group I or II as defined in Fig. 2A. The phenotype of the E490Q single-mutant protein in SD medium containing 3-AT suggested that it had higher-than-wild-type levels of kinase activity in vivo (Fig. 7A). In contrast, the Gln-490 substitution had no effect on the phenotype of the less severe defect conferred by the M365V substitution in the N-terminal lobe of PKR (Fig. 7A), which would be expected to impair ATP binding. The results of in vitro autokinase assays confirmed that the E490Q substitution restored low-level kinase activity to the inactive Ala-451 mutant (Fig. 7B, lanes 3 and 4) but had no stimulatory effect on the less impaired Val-465 mutant kinase activity (lanes 8 and 9 versus lanes 10 and 11). These findings suggest that a specific interaction occurs between the G-helix region and the autophosphorylation loop of PKR, as described for the MAP kinases ERK2 and FUS3.
FIG. 7.

Substitution of Glu-490 partially suppresses the impairment of kinase activity imposed by the T451A mutation in the PKR activation loop in vivo and in vitro. (A) In vivo analysis. Plasmids carrying the indicated PKR alleles were introduced into the gcn2Δ yeast strain H1816 expressing wild-type eIF2α. Patches of transformants were grown to confluence in SD medium and replica plated to the indicated media. Growth was scored relative to that of the strain containing wild-type (WT) PKR as described in the legend to Fig. 2. (B) In vitro analysis. Transformants of strain H1817 (expressing eIF22-S51A) bearing the indicated wild-type (WT) or mutant PKR alleles were grown in SGAL medium to induce PKR expression, and the PKR proteins were immunoprecipitated from whole-cell extracts containing 150 μg of total protein with polyclonal antibodies against PKR. In vitro kinase assays were carried out as described in Materials and Methods. Radiolabeled samples were separated by SDS-PAGE (10% polyacrylamide gel) and transferred to a nitrocellulose filter. The radiolabeled PKR proteins were visualized by autoradiography (upper panel). The same nitrocellulose filter was subjected to immunoblot analysis with monoclonal antibodies against PKR to visualize the amounts of PKR immunoprecipitated for each sample (lower panel).
Effects of Thr-446 and Thr-451 substitutions on PKR activity in mammalian cells.
We wished to determine whether mutations at positions 446 and 451 would impair PKR activity in mammalian cells. It has been shown that PKR negatively autoregulates its expression in mammalian cells in a manner similar to that seen in yeast cells (2, 43). Therefore, we transfected a subset of the mutant PKR alleles into COS1 cells and measured rates of synthesis and steady-state levels of the encoded proteins as a measure of their ability to autoregulate PKR expression. The PKR cDNAs were expressed under the control of the CMV immediate-early promoter. At 48 h after transfection, the cells were metabolically labeled with [35S]methionine for 1.5 h and cytoplasmic extracts were prepared and immunoprecipitated with anti-PKR antibodies (Fig. 8A). In addition, cytoplasmic extracts prepared from the transfected cells were subjected to immunoblot analysis with antibodies against PKR (Fig. 8B). The results in Fig. 8 indicate that the T446A,T451A and T446A,T451A proteins were synthesized at levels comparable to that of the K296R protein, indicating profound defects in kinase function for all three mutant proteins in COS1 cells. The T446S,T451S protein was produced at lower levels than the corresponding Ala doublemutant protein, supporting the conclusion that Ser is a functional replacement for Thr at these positions. The T258A and S242A,T255A,T258A mutant proteins were produced at levels only slightly higher than that of the wild-type protein, consistent with relatively small reductions in kinase function. These findings indicate that Thr-446 and Thr-451 are critically required for kinase function in COS1 cells and that each makes a stronger contribution to PKR activity than do Ser-242, Thr-255, and Thr-258.
FIG. 8.
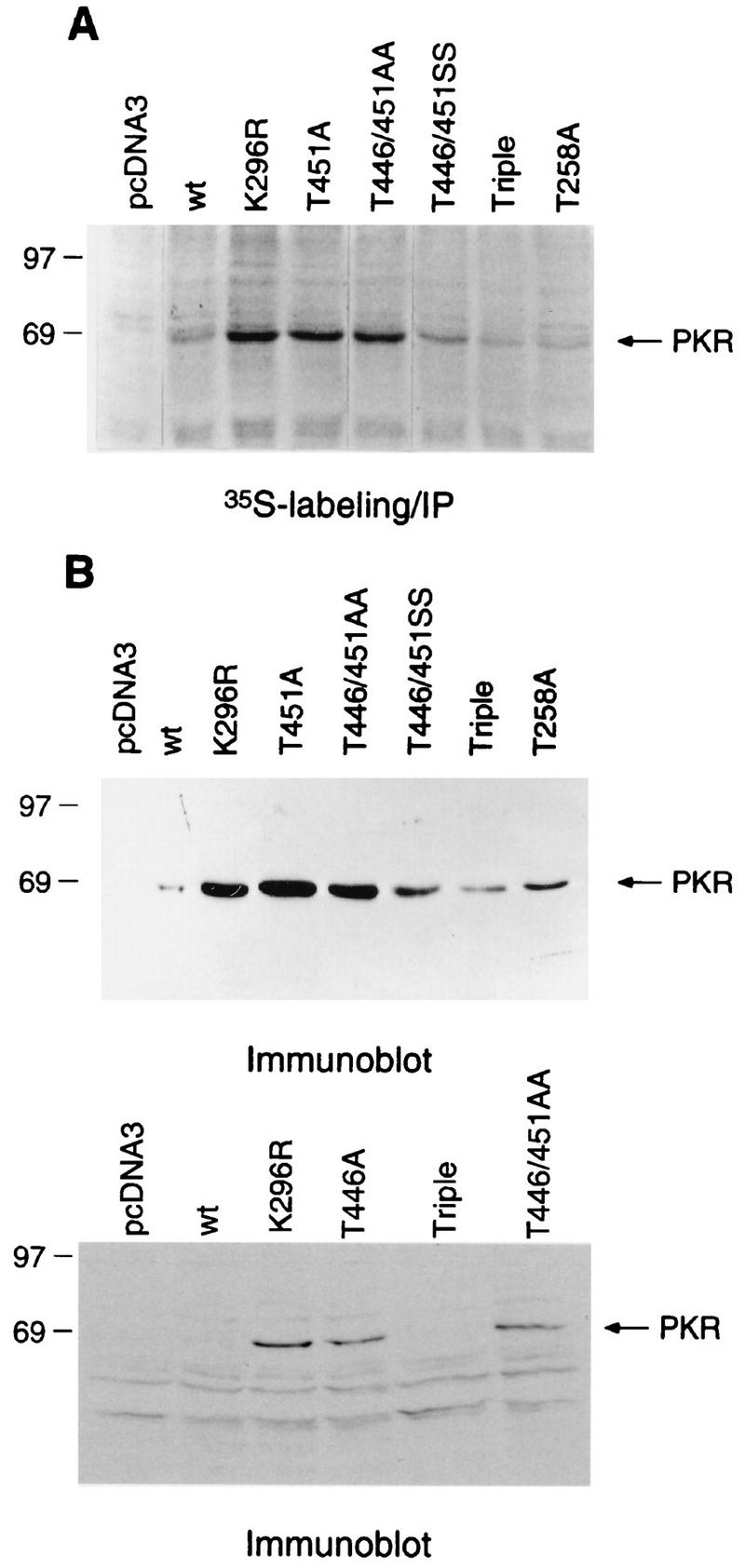
Analysis of autoregulation of PKR expression in mammalian cells. The indicated PKR alleles under the control of a CMV promoter were transfected into COS1 cells (see Materials and Methods), and the cells were harvested 48 h postinfection. (A) Transfected cells were labeled with [35S]methionine, and the PKR proteins were immunoprecipitated (IP) from 500 μg of total cytoplasmic protein with anti-PKR polyclonal antibodies. Radiolabeled samples were resolved by SDS-PAGE followed by autoradiography. wt, wild type. (B) Immunoblot analysis of PKR expression levels in COS1 cells. Total cytoplasmic protein (75 μg) was subjected to SDS-PAGE followed by immunoblot analysis with monoclonal antibodies against PKR, and immune complexes were visualized by ECL.
Substitutions in PKR autophosphorylation sites lead to tumor formation in mice.
As a second means of assessing the effects of mutations at positions 446 and 451 on PKR function in mammalian cells, we examined the ability of selected mutant proteins to elicit tumors in mice. Stable transfectants of NIH 3T3 cells bearing cDNAs encoding the mutant proteins were injected into nude mice, and the mice were monitored for the appearance of tumors. In accordance with previous results, the K296R and Δ6 alleles elicited tumor formation (Table 1). This phenomenon has been attributed to dominant interference with endogenous wild-type PKR by the catalytically inactive mutant proteins through the formation of defective heterodimers (25, 31). The T451A and S242A,T255A,T258A alleles were also effective in eliciting tumors (Table 1), indicating that their products were functionally impaired and capable of interfering with endogenous PKR.
TABLE 1.
Tumorigenicity of NIH 3T3 cells expressing PKR variants
| Clone | No. of tumorsa | Latency (days) |
|---|---|---|
| pcDNA3 (vector) | 0 | >21b |
| Wild-type PKR | 0 | >21c |
| K296R | 4 | 7–14 |
| Δ6 | 4 | 7–10 |
| T451A | 4 | 10–11 |
| T446A,T451A | 4 | 12–14 |
| S242A,T255A,T258A | 4 | 12–14 |
Four animals were used for each clone.
A tumor appeared in 1 mouse after 96 days.
Tumors appeared in 3 mice at 59 to 80 days.
PKR specifically phosphorylates Thr-446 and Thr-451 in synthetic peptides in vitro.
To obtain biochemical evidence that Thr-446 and Thr-451 are autophosphorylation sites, we first conducted in vitro kinase assays with immunopurified PKR using synthetic peptides corresponding to residues 439 to 458 as exogenous substrates. The wild-type peptide and two mutant peptides containing single Ala substitutions at residues 446 and 451 were phosphorylated in vitro at levels higher than that seen for the Ala-446,Ala-451 double-mutant peptide (Fig. 9A). We confirmed that phosphorylation of the wild-type peptide was completely dependent on PKR by showing that no phosphorylation occurred with the K296R catalytically inactive kinase (data not shown). Analysis of the phosphoamino acids produced by PKR in the wild-type peptide revealed that the majority of phosphorylation occurred at threonine residues (Fig. 9B, left panel). Taken together, these results indicated that PKR phosphorylated both Thr-446 and Thr-451 in the synthetic peptides and did so at much higher efficiencies than it phosphorylated the two Ser residues present in the peptides at positions 448 and 456. To provide additional support for this conclusion, we analyzed the phosphoamino acids produced in a peptide containing Ser residues in place of Thr-446 and Thr-451. This mutant peptide was phosphorylated more efficiently than was the Ala-446,Ala-451 peptide (Fig. 9A), and phosphoamino acid analysis confirmed that only phosphoserine was produced in the in vitro reaction (Fig. 9B, right panel). (A larger amount of radiolabeled product was subjected to phosphoamino acid analysis for the Ser-446,Ser-451 peptide than for the Thr-446,Thr-451 peptide.) In addition, phosphoamino acid analysis of peptides containing single Ala substitutions at positions 446 and 451 showed that the bulk of the phosphorylation occurred at the sole Thr residue remaining in these peptides (data not shown). Thus, when provided with the putative activation loop peptide as an exogenous substrate, PKR showed a preference for phosphorylating threonine residues at positions 446 and 451.
FIG. 9.

In vitro phosphorylation and phosphoamino acid analysis of synthetic peptide substrates. (A) Transformants of strain H1817 (containing nonphosphorylatable eIF2α-S51A) bearing the wild-type PKR allele were grown in SGAL medium, and PKR was immunoprecipitated from whole-cell extracts as described in the legend to Fig. 6. Immune complexes were incubated in kinase reaction buffer in the presence of [γ-32P]ATP and 4 μg of a synthetic 20-mer peptide (lanes 2 to 6) or no added peptide (lane 1) for 15 min at 30°C. Radiolabeled samples were analyzed by Tricine-SDS-PAGE (10 to 20% polyacrylamide gradient gel). The gels were stained with Coomassie blue, dried under vacuum, and subjected to autoradiography. The wild-type peptide (whose sequence is shown on the left in panel B) was present in the reaction in lane 2. The mutant peptides analyzed in lanes 3 to 6 contained the indicated substitutions at positions 446 and 451. (B) Phosphoamino acid analysis of in vitro-labeled peptide substrates. In vitro kinase reactions were carried out as described above, and proteins were transferred to PVDF membranes and subjected to autoradiography. The strip of membrane containing the radiolabeled peptide was hydrolyzed with HCl, and soluble amino acids were resolved by thin-layer electrophoresis (see Materials and Methods), after which the phosphoamino acids were visualized by autoradiography. Unlabeled phosphoamino acid standards (phosphoserine [S], phosphothreonine [T], and phosphotyrosine [Y]) were separated along with each sample and visualized by ninhydrin staining. The positions of the standards are indicated to the right of each autoradiogram.
MS evidence for autophosphorylation of Thr-446 in PKR in vivo.
To determine whether Thr-446 and Thr-451 are autophosphorylated by PKR in vivo, we used MS to evaluate the phosphorylation status of these residues in PKR expressed in yeast cells. A polyhistidine-tagged form of PKR was purified by nickel affinity and poly(I) · poly(C) affinity chromatography and SDS-PAGE (see Materials and Methods). The His-tagged PKR allele conferred on strain H1816 the same growth phenotypes in SGAL medium and SD medium containing 3-AT that we observed with wild-type PKR expressed from the same vector (data not shown), indicating that the tagged protein had wild-type eIF2α kinase activity in vivo.
Purified PKR was subjected to trypsin digestion and analyzed by MALDI/TOF MS to determine the molecular masses of the resulting tryptic peptides (see Materials and Methods). When these masses were compared with those of all predicted tryptic peptides produced by partial or complete trypsin digestion of PKR, we could identify predicted peptides covering 79% of the PKR sequence. From these comparisons, we identified a peptide with a measured mass of 2,057.2 Da, 80 Da higher than the calculated mass of a predicted PKR peptide spanning residues 430 to 447 and containing Thr-437, Ser-438, and Thr-446 (1,977.3 Da) (Table 2, row 2). These findings suggested that the 2,057.2-Da peptide corresponds to the peptide spanning residues 430 to 447 and bearing a single phosphate group. Supporting this deduction, CIP treatment of the trypsin-digested sample led to the appearance of a peptide 80 Da smaller than the putative phosphopeptide spanning residues 430 to 447. Only the 2,057.2-Da phosphorylated form of the peptide spanning residues 430 to 447 was detected in the sample prior to CIP treatment (Table 2, row 1). The same MS measurements detected a peptide of 1,717.9 Da, very close to the predicted mass (1,720.0 Da) of the unphosphorylated tryptic peptide spanning residues 430 to 445 (Table 2, row 3), and a phosphorylated form of this peptide was not detected. These findings indicated that Thr-437 and Ser-438 were not phosphorylated at a detectable level in the PKR preparation, from which we deduced that Thr-446 is the phosphorylated residue in the 2,057.2-Da phosphopeptide (Table 2, row 2). To confirm this conclusion, we sequenced the phosphopeptide spanning residues 430 to 447 by LC-MS-MS analysis, in which the doubly charged phosphopeptide ions are subjected to CID as they elute from an HPLC column (see Materials and Methods). Using this technique, we unambiguously identified Thr-446 as the only site of phosphorylation in this peptide (50a).
TABLE 2.
MS assignments of measured peptides containing Thr-446 and Thr-451a
| PKR allele | Measured mass (Da) | Calculated mass (Da) | Sequence (residues) | Comments |
|---|---|---|---|---|
| Wild type | Not observed | 1,977.3 | 430–447 | Tryptic peptide; unphosphorylated peptide of residues 430 to 447 was not detected |
| Wild type | 2,057.2b,c,d | 2,057.3b | 430–447 + PO3 | Tryptic peptide; Thr-437, Ser-438, or Thr-446 could be phosphorylated; MS-MS sequencing of this peptide unambiguously identified Thr-446 as the site of phosphorylation |
| Wild type | 1,717.9b | 1,720.0b | 430–445 | Tryptic peptide; Thr-437 and Ser-438 were not phosphorylated |
| Wild type | 2,060.2b,d | 2,060.3b | 450–467 | Lys-C peptide; Thr-451 was not phosphorylated; verified by MS-MS sequencing |
| K296R | 2,525.9b | 2,525.8b | 442–463 | Asp-N peptide; Thr 446 and Thr 451 were not phosphorylated |
We were able to identify predicted peptides covering 79% of the entire PKR sequence.
Averaged mass.
This peptide loses 80 Da upon CIP treatment of the total trypsin digest.
Peptides sequenced by MS-MS.
In the same analysis, we failed to detect any tryptic peptides that contained Thr-451, presumably because they were too small to be measured by MS. Consequently, we carried out a second in-gel digestion of PKR using Lys-C instead of trypsin and detected a peptide with a predicted molecular weight corresponding to that of an unphosphorylated Lys-C peptide spanning residues 450 to 467 and containing Thr-451 (Table 2, row 4). We confirmed that the Lys-C peptide of 2,060.2 Da corresponded to the unphosphorylated form of the peptide spanning residues 450 to 467 by LC-MS-MS sequencing (data not shown). We did not detect the phosphorylated form of this peptide, suggesting either that Thr-451 was not phosphorylated in our purified PKR or that the phosphorylated form of the Lys-C peptide containing Thr-451 was below the MS detection limit.
It is conceivable that Thr-446 is not a site of autophosphorylation but is phosphorylated by another protein kinase present in yeast. To eliminate this possibility, we purified from yeast a His-tagged form of the catalytically inactive K296R mutant kinase by the same procedure as that described for the wild-type kinase and subjected it to MS analysis. We observed only the unphosphorylated form of a tryptic peptide containing Thr-446 (Table 2, last row). We conclude that Thr-446 is a site for autophosphorylation by PKR in vivo.
Autophosphorylation sites in the activation loop of the yeast eIF2α kinase GCN2 are required for kinase function in vitro and in vivo.
We noted that GCN2 contains two Thr residues at exactly the same positions relative to kinase subdomain VIII as those occupied by Thr-446 and Thr-451 in PKR (Fig. 1) and set out to determine whether these Thr residues are autophosphorylation sites required for GCN2 kinase function. Mutations made in a plasmid-borne GCN2 allele that substituted Thr-882 or Thr-887 with alanine partially or completely impaired complementation of the 3-AT-sensitive phenotype of a gcn2Δ strain (Fig. 10A), respectively, without reducing the steady-state level of the GCN2 protein, as measured by immunoblot analysis (Fig. 10B). Substitution of Thr-882 with Ser had no detectable effect on GCN2 function, whereas the Ser substitution at residue 887, individually or in combination with Ser-882, led to a partial reduction in complementation activity (Fig. 10A and B). Glu or Asp substitutions at both positions resulted in the same phenotypes as the corresponding Ala substitutions; however, the extent to which Asp or Glu will substitute for a phosphorylated residue in the activation loop is known to vary considerably, from no activity to partial activity for the kinases with substitutions (21). The results of immune complex assays of GCN2 autophosphorylation activity for the Ala- and Ser-substituted mutant GCN2 proteins paralleled the complementation data in showing reduced activities for the Ser-887 and Ser-882,Ser-887 mutant proteins (Fig. 10C, lanes 6 to 9) and little or no activity for proteins with single or double Ala substitutions at positions 882 and 887 (lanes 1 to 4 and 11 to 14). After normalizing the amounts of labeled GCN2 produced in these assays (Fig. 10C, top panel) for the amounts of GCN2 protein recovered in the immune complexes (bottom panel), we concluded that the Ala-882 mutant protein showed higher levels of autokinase activity than did the Ala-887 and Ala-882,Ala-887 mutant proteins (Fig. 10C, lanes 2 to 5 and 12 to 14), in agreement with the complementation data.
FIG. 10.
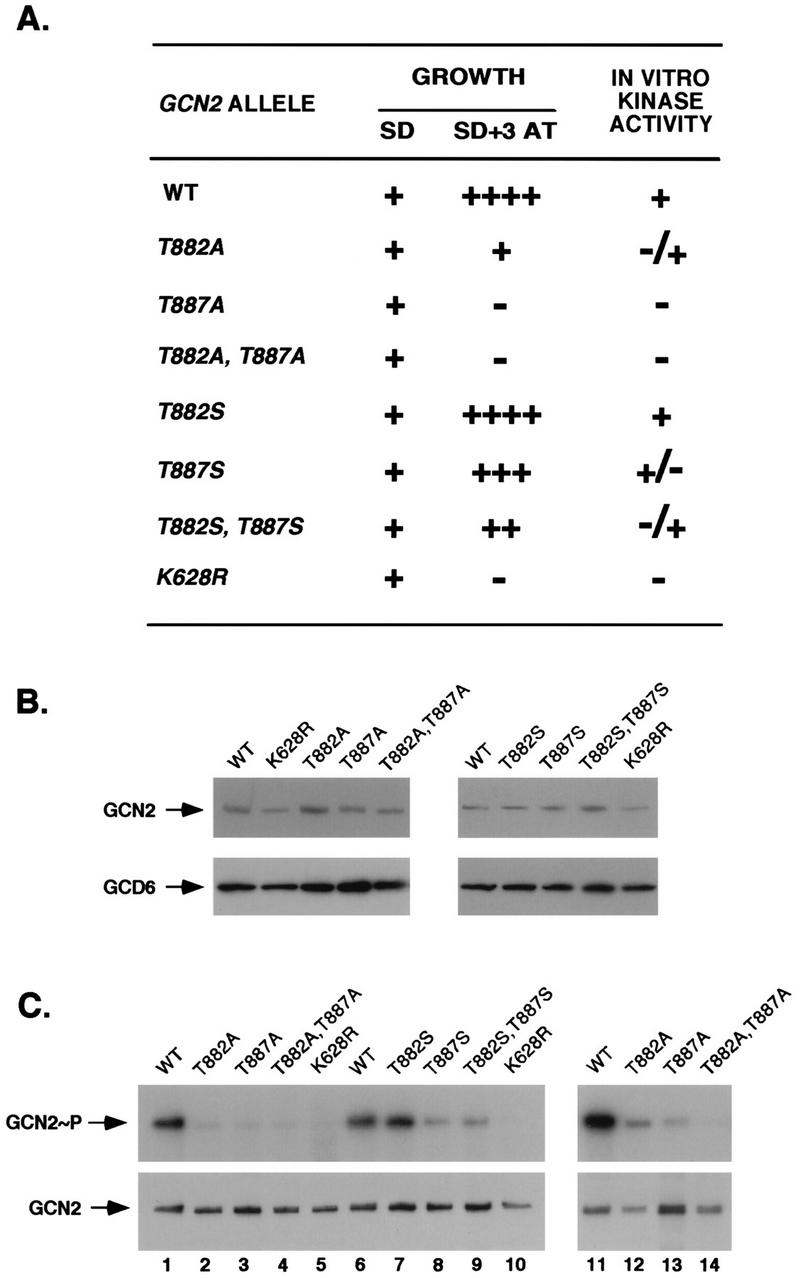
Analysis of substitutions at Thr-882 and Thr-887 on GCN2 function in vivo and autokinase activity in vitro. (A) In vivo analysis of GCN2 function. Patches of transformants of strain H1894 containing the indicated GCN2 alleles on low-copy-number plasmids were grown to confluence in SD medium and replica plated to SD medium or SD medium supplemented with increasing concentrations of 3-AT ranging from 7.5 to 150 mM. Growth relative to that of the wild-type (WT) control was scored as follows: ++++, +++, ++, and +, the growth of the mutant was indistinguishable from that of the wild type on 3-AT concentrations of 150, 100, 75, and 10 mM, respectively; −, no growth at any of these 3-AT concentrations. The levels of in vitro kinase activities were ranked as follows: +, +/−, −/+, and −, activities that were identical to that of wild type, somewhat reduced, or greatly reduced relative to wild type, or undetectable. (B) Immunoblot analysis of GCN2 expression. Fifteen micrograms of whole-cell extracts prepared from transformants of strain H1894 bearing the indicated GCN2 alleles on low-copy-number plasmids were separated by SDS-PAGE with 6% polyacrylamide gels and transferred to PVDF membranes. Immunoblot analysis was performed with antibodies against GCN2 and GCD6 and ECL to detect the immune complexes. (C) In vitro autokinase activities of GCN2 proteins. Transformants of strain H1894 bearing the indicated GCN2 alleles on low-copy-number (lanes 1 to 10) or high-copy-number (lanes 11 to 14) plasmids were grown in liquid SD medium to an OD600 of 1.0, and GCN2 was immunoprecipitated from whole-cell extracts containing 150 μg of total protein with polyclonal antibodies against GCN2. Immune complexes were incubated in kinase reaction buffer in the presence of [γ-32P]ATP for 20 min at 30°C. Radiolabeled proteins were separated by SDS-PAGE with 4 to 16% gradient gels, transferred to PVDF membranes, and visualized by autoradiography (top panels). Immunodetection of GCN2 was performed on the same membranes with anti-GCN2 polyclonal antibodies and ECL to visualize the immune complexes (bottom panels). GCN2∼P, radiolabeled autophosphorylated GCN2.
Phosphoamino acid analysis of wild-type GCN2 following autophosphorylation in vitro in the presence of [32P]ATP revealed the presence of only phosphothreonine (Fig. 11A). In contrast, autophosphorylation of the Ser-882,Ser-887 double-mutant protein produced only phosphoserine (Fig. 11B). Autophosphorylation of the Thr-882,Ser-887 and Ser-882,Thr-887 singly substituted mutant proteins gave rise to both phosphothreonine and phosphoserine (Fig. 11C and D). In both cases, phosphoserine was less abundant than phosphothreonine, but this difference was especially pronounced for the Thr-882,Ser-887 mutant protein (Fig. 11D). These results indicate that Thr-882 and Thr-887 are both used as autophosphorylation sites and that these are the only sites of autophosphorylation detectable under the conditions of our in vitro kinase assays.
FIG. 11.

Analysis of phosphoamino acids produced by autophosphorylation of GCN2 in vitro. Transformants of strain H1894 bearing the indicated GCN2 alleles on high-copy-number plasmids were grown in liquid SD medium, and GCN2 was immunoprecipitated from whole-cell extracts in 5 or 10 reactions, each containing 200 to 400 μg of total yeast protein, with polyclonal antibodies against GCN2. Immune complexes were incubated in kinase reaction buffer in the presence of [γ-32P]ATP for 20 min at 30°C. Radiolabeled GCN2 proteins in the samples were separated by SDS-PAGE with 6% gels, transferred to PVDF membranes, and visualized by autoradiography. Strips of membranes containing GCN2 were treated with HCl to hydrolyze the protein, and soluble amino acids were resolved by thin-layer chromatography as described in Materials and Methods. Labeled phosphoamino acids were detected by autoradiography. The positions of unlabeled phosphoamino acid standards resolved in parallel and visualized by ninhydrin staining are indicated on the left as S (phosphoserine), T (phosphothreonine), and Y (phosphotyrosine).
DISCUSSION
Importance of autophosphorylation in the activation loop of PKR for full eIF2α kinase activity.
PKR exists in a latent form in mammalian cells and becomes activated during virus infection, presumably by dsRNA produced in the course of viral replication. There is evidence that dsRNA binding increases the affinity of the kinase domain for ATP (3, 16) and stimulates PKR autokinase activity. The level of PKR autophosphorylation has been correlated with its ability to recognize exogenous substrates (16), and autophosphorylation seems to lock the enzyme into an active conformation that is maintained in the absence of dsRNA (26). There is also evidence that dimerization is required for the activation of PKR (26, 28, 38) and that the dsRBMs mediate dimerization in addition to binding the dsRNA activators (11, 33, 34, 47). These findings and the fact that PKR can autophosphorylate in trans (44) lead to the notion that dimerization is required to promote trans-autophosphorylation events crucial for activating the eIF2α kinase function of PKR (42).
The N-terminal regulatory region seems to inhibit the function of the kinase domain in a manner that is overcome by dsRNA binding (47, 51). Accordingly, the binding of dsRNA may release the N-terminal domain from an inhibitory interaction with the kinase domain and also may promote dimerization and trans-autophosphorylation (48). We showed previously that PKR autophosphorylates in vitro at Ser-242, Thr-255, and Thr-258, located between the dsRBMs and the kinase domain, and that Thr-258 is required to achieve wild-type PKR activity in vitro and in vivo (42). We suggested that residues 242 to 258 reside in a hinge region between the dsRNA-binding and kinase domains and that the phosphorylation of Thr-258 alters the interaction between these two domains in a way that promotes the active conformation of the enzyme (42). This idea helps explain why the autophosphorylated form of PKR remains active in the absence of dsRNA.
Several protein kinases are activated by phosphorylation of one or more residues in the activation loop between kinase subdomains VII and VIII (19). It is believed that the phosphorylation of these residues alters the conformation of the activation loop in a way that stimulates one or more steps in the phosphorylation of peptide substrates, including the binding of ATP (1), the binding of peptide substrates (12, 50), or the phosphoryl transfer reaction itself (1). We found that Ala substitutions of Thr-446 and Thr-451 in the activation loop of PKR led to a substantial reduction (position 446) or complete inactivation (position 451) of the autokinase and eIF2α kinase activities of PKR both in vivo and in vitro. Both mutations led to more severe reductions in kinase activity than were observed for Ala substitutions at the previously identified phosphorylation sites at Ser-242, Thr-255, and Thr-258. The fact that Ser and Asp substitutions at these positions in PKR led to substantially (Ser) or slightly (Asp) higher PKR activities than were seen for the corresponding Ala substitutions is consistent with the idea that autophosphorylation at both sites is important for PKR activation. We obtained biochemical evidence that both Thr-446 and Thr-451 were phosphorylated by PKR in synthetic peptides corresponding to the PKR activation loop. Little or no phosphorylation occurred at Ser-448 and Ser-456 in the synthetic peptides, suggesting that Thr-446 and Thr-451 were preferentially phosphorylated. MS analysis of peptides derived from PKR purified from yeast confirmed that Thr-446 is an autophosphorylation site in vivo. By analogy with other protein kinases that are activated by phosphorylation between subdomains VII and VIII, we propose that the autophosphorylation of Thr-446 in PKR alters the conformation of the activation loop in a way that stimulates phosphoryl transfer or the binding of ATP or peptide substrates, including eIF2α or segments of PKR bearing other autophosphorylation sites.
Phosphorylation of Thr-451 was not detected by our MS analysis. The failure to detect a phosphopeptide by this technique could be explained in several ways. One possibility, of course, is that Thr-451 is not an autophosphorylation site. In this event, mutations of this residue would interfere with a critical aspect of catalysis, since the Ala-451 mutant is completely defective for both autophosphorylation and substrate phosphorylation. In accordance with this interpretation, the position corresponding to Thr-451 in PKR is occupied by Thr or Ser in almost every known serine/threonine protein kinase, with the exception of Wee1 and SplA (19). In addition, crystallographic analysis showed that the residue corresponding to Thr-451 in PKA (Thr-201) is involved in anchoring the peptide substrate to the large lobe of the catalytic domain. The side-chain —OH group of Thr-201 hydrogen bonds to the —NH3 group of Lys-168, which in turn hydrogen bonds to the serine —OH group in the peptide substrate (29). An alternative explanation is that the autophosphorylation of Thr-451 is the final event required to achieve full kinase activity and that only a fraction of PKR in yeast cells exists in this fully activated state. Even if much of the PKR were phosphorylated at Thr-451 in vivo, it is possible that dephosphorylation of this residue occurred during isolation or that peptides which contained phosphothreonine at residue 451 were unstable or insoluble. There is precedence for the idea that different phosphorylated residues in a protein can exhibit widely different sensitivities to phosphatases, as recombinant PKA autophosphorylates at four different residues but only Thr-197 (in the activation loop) and Ser-338 are stable against phosphatase treatment (8, 41). In fact, we did not detect the phosphorylation of Ser-242, Thr-255, and Thr-258 in the PKR preparation used to obtain the data shown in Table 2, despite the fact that these sites were autophosphorylated in vitro in PKR purified from mammalian cells (42). In accordance with the latter study, however, our MS analysis did reveal the phosphorylation of multiple Thr and Ser residues at positions 88 to 97, located between the dsRBMs (data not shown). It appears, therefore, that the phosphorylation of residues 446 and 88 to 97 is either more extensive in vivo or less labile in vitro than is the phosphorylation of residues 242, 255, and 258.
Substitution of Asp-227 with Asn in the yeast MAP kinase kinase FUS3 partially suppressed the low-level activation of FUS3 resulting from inadequate phosphorylation of its critical activation loop residues Thr-180 and Tyr-182 by the MAP kinase STE7 (5). Because Asp-227 is predicted to be in the G helix and located in the vicinity of the Tyr residue in the activation loop of FUS3 that is phosphorylated by STE7 (10), the Asn-227 substitution could alter interactions between the G helix and the activation loop to increase kinase activity in the hypophosphorylated form of FUS3. We found that the loss of kinase activity in the PKR Ala-451 mutant was partially suppressed by the replacement of Glu-490 with Gln. Considering that Glu-490 in PKR corresponds closely in location to Asp-227 in FUS3, this finding suggests that interactions between the activation loop and a negatively charged residue in the vicinity of the G helix in subdomain X are conserved features of kinases that require phosphorylation of the activation loop.
The fact that the Ala-446 mutant retained both autophosphorylation and eIF2α kinase activities at reduced levels shows that partial activation of PKR can be achieved without autophosphorylation at position 446. The same conclusion was reached previously for Thr-258 (42), although autophosphorylation at this site appears to be less important than that at position 446. Combining Ala substitutions at both position 258 and position 446 led to a drastic reduction in kinase activity relative to that seen for the Ala-446 single mutant (Fig. 2 and 3), suggesting that autophosphorylation at one or the other of these two residues is critically required for high-level kinase function. To account for the pronounced additive effect of Ala substitutions at positions 258 and 446, we propose that an inhibitory conformation of the activation loop is stabilized by interactions with the N-terminal regulatory region of PKR. In this event, achieving the altered conformation required for eIF2α phosphorylation might require dissociation of the N-terminal segment from the activation loop. Such dissociation could be promoted by autophosphorylation of Thr-258 in the N-terminal domain in addition to autophosphorylation of Thr-446 in the activation loop. Once Thr-446 is phosphorylated, Thr-258 could be dephosphorylated without reversing kinase activation.
The T258A,T446A double-mutant and the S242A,T255A,T258A,T446A quadruple-mutant proteins retained very low levels of eIF2α kinase activity in vivo (Fig. 2), raising the possibility that partial activation of PKR can be achieved through autophosphorylation of residues in addition to those present at positions 242, 255, 258, and 446. As mentioned above, there was previous evidence for PKR autophosphorylation in vitro at residues 88 to 97 (42) that we confirmed by MS analysis of PKR expressed in yeast. Thus far, Ala substitutions at several of these sites have had little effect on PKR function (42a), even when combined with Ala substitutions at positions 242, 255, 258 and 446 (data not shown). Therefore, additional autophosphorylation sites that make important contributions to the activation of PKR by dsRNA may yet be identified.
Autophosphorylation of conserved Thr residues in the activation loop of GCN2 is required for full kinase function.
GCN2 contains two Thr residues at exactly the same positions relative to subdomain VIII as those occupied by Thr-446 and Thr-451 in PKR. It was striking that Ala substitutions at Thr-882 and Thr-887 in GCN2 had relative effects on kinase function similar to those of the Ala-446 and Ala-451 substitutions in PKR. Because Thr-882 and Thr-887 appear to be the only sites phosphorylated by GCN2 in vitro, it was possible to provide strong biochemical evidence through phosphoamino acid analysis that both residues are autophosphorylated. Recent findings from MS analysis of purified GCN2 indicate that the tryptic peptide containing Thr-882 and Thr-887 is phosphorylated in vivo (15a). Considering that GCN2 and PKR belong to the same small cluster of closely related kinases, our findings on the autophosphorylation of GCN2 provide another reason for suspecting that PKR may autophosphorylate at Thr-451 in addition to Thr-446. Additional experimentation will be required to resolve this question. Like PKR in mammalian cells, GCN2 is present in yeast cells in a latent form and becomes activated by uncharged tRNA that accumulates in amino acid-starved cells and binds to a regulatory domain in GCN2 related to histidyl-tRNA synthetases (45, 46). There is some evidence suggesting that GCN2 oligomerizes in vivo (15), so it is conceivable that the two protomers in a GCN2 dimer undergo trans-autophosphorylation in response to tRNA binding as a means of activating the enzyme in amino acid-starved cells. Not all protein kinases require phosphorylation of the activation loop for full kinase activity (21). The fact that GCN2 and PKR require autophosphorylation at one or both Thr residues at the same locations relative to subdomain VIII suggests that there may be additional similarities in the molecular mechanisms involved in converting them from latent enzymes to fully activated eIF2α kinases.
ACKNOWLEDGMENTS
We thank Makiko Kobayashi and Tom Dever for their generous gift of a His-tagged PKR construct, Graham Pavitt for strain GP3299, Lisa Bianco for skilled assistance with the animal studies, Ron Wek for the bacterial strain expressing eIF2α, and Jinsheng Dong for the gift of the purified eIF2α protein. We thank Tom Dever for helpful comments on the manuscript and Bobbie Felix for help with manuscript preparation.
This study was supported in part by grant AI 34552 from the National Institute of Allergy and Infectious Diseases to M.B.M.
REFERENCES
- 1.Adams J A, McGlone M L, Gibson R, Taylor S S. Phosphorylation modulates catalytic function and regulation in the cAMP-dependent protein kinase. Biochemistry. 1995;34:2447–2454. doi: 10.1021/bi00008a007. [DOI] [PubMed] [Google Scholar]
- 2.Barber G N, Wambach M, Wong M L, Dever T E, Hinnebusch A G, Katze M G. Translational regulation by the interferon-induced double-stranded RNA-activated 68-kDa protein kinase. Proc Natl Acad Sci USA. 1993;90:4621–4625. doi: 10.1073/pnas.90.10.4621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bischoff J R, Samuel C E. Mechanism of interferon action: the interferon-induced phosphoprotein P1 possesses a double-stranded RNA-dependent ATP-binding site. J Biol Chem. 1985;260:8237–8239. [PubMed] [Google Scholar]
- 4.Boyle W J, van der Geer P, Hunter T. Phosphopeptide mapping and phosphoamino acid analysis by two-dimensional separation on thin-layer cellulose plates. Methods Enzymol. 1991;201:110–149. doi: 10.1016/0076-6879(91)01013-r. [DOI] [PubMed] [Google Scholar]
- 5.Brill J A, Elion E A, Fink G R. A role for autophosphorylation revealed by activated alleles of FUS3, the yeast MAP kinase homolog. Mol Biol Cell. 1994;5:297–312. doi: 10.1091/mbc.5.3.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bushman J L, Foiani M, Cigan A M, Paddon C J, Hinnebusch A G. Guanine nucleotide exchange factor for eukaryotic translation initiation factor 2 in Saccharomyces cerevisiae: interactions between the essential subunits GCD2, GCD6, and GCD7 and the regulatory subunit GCN3. Mol Cell Biol. 1993;13:4618–4631. doi: 10.1128/mcb.13.8.4618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Canagarajah B J, Khokhlatchev A, Cobb M H, Goldsmith E J. Activation mechanism of the MAP kinase ERK2 by dual phosphorylation. Cell. 1997;90:859–869. doi: 10.1016/s0092-8674(00)80351-7. [DOI] [PubMed] [Google Scholar]
- 8.Chiu Y S, Tao M. Autophosphorylation of rabbit skeletal muscle cyclic AMP-dependent protein kinase I catalytic subunit. J Biol Chem. 1978;253:7145–7148. [PubMed] [Google Scholar]
- 9.Chong K L, Feng L, Schappert K, Meurs E, Donahue T F, Friesen J D, Hovanessian A G, Williams B R G. Human p68 kinase exhibits growth suppression in yeast and homology to the translational regulator GCN2. EMBO J. 1992;11:1553–1562. doi: 10.1002/j.1460-2075.1992.tb05200.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cobb M H, Goldsmith E J. How MAP kinases are regulated. J Biol Chem. 1995;270:1483–1486. doi: 10.1074/jbc.270.25.14843. [DOI] [PubMed] [Google Scholar]
- 11.Cosentino G P, Venkatesan S, Serluca F C, Green S R, Mathews M B, Sonenberg N. Double-stranded-RNA-dependent protein kinase and TAR RNA-binding protein form homo- and heterodimers in vivo. Proc Natl Acad Sci USA. 1995;92:9445–9449. doi: 10.1073/pnas.92.21.9445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.De Bondt H L, Rosenblatt J, Jancarik J, Jones H D, Morgan D O, Kim S H. Crystal structure of cyclin-dependent kinase 2. Nature. 1993;363:595–602. doi: 10.1038/363595a0. [DOI] [PubMed] [Google Scholar]
- 13.Dever T E, Chen J J, Barber G N, Cigan A M, Feng L, Donahue T F, London I M, Katze M G, Hinnebusch A G. Mammalian eukaryotic initiation factor 2α kinases functionally substitute for GCN2 in the GCN4 translational control mechanism of yeast. Proc Natl Acad Sci USA. 1993;90:4616–4620. doi: 10.1073/pnas.90.10.4616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dever T E, Feng L, Wek R C, Cigan A M, Donahue T D, Hinnebusch A G. Phosphorylation of initiation factor 2α by protein kinase GCN2 mediates gene-specific translational control of GCN4 in yeast. Cell. 1992;68:585–596. doi: 10.1016/0092-8674(92)90193-g. [DOI] [PubMed] [Google Scholar]
- 15.Diallinas G, Thireos G. Genetic and biochemical evidence for yeast GCN2 protein kinase polymerization. Gene. 1994;143:21–27. doi: 10.1016/0378-1119(94)90599-1. [DOI] [PubMed] [Google Scholar]
- 15a.Dong, J., X. Zhang, A. G. Hinnebusch, and J. Qin. Unpublished observations.
- 16.Galabru J, Hovanessian A. Autophosphorylation of the protein kinase dependent on double-stranded RNA. J Biol Chem. 1987;262:15538–15544. [PubMed] [Google Scholar]
- 17.Galabru J, Katze M G, Robert N, Hovanessian A G. The binding of double-stranded RNA and adenovirus VAI RNA to the interferon-induced protein kinase. Eur J Biochem. 1989;178:581–589. doi: 10.1111/j.1432-1033.1989.tb14485.x. [DOI] [PubMed] [Google Scholar]
- 18.Green S R, Mathews M B. Two RNA-binding motifs in double-stranded RNA-activated protein kinase DAI. Genes Dev. 1992;6:2478–2490. doi: 10.1101/gad.6.12b.2478. [DOI] [PubMed] [Google Scholar]
- 19.Hanks S K, Hunter T. The eukaryotic protein kinase superfamily. In: Hardie G, Hanks S, editors. The protein kinase facts book. San Diego, Calif: Academic Press, Inc.; 1995. pp. 7–47. [Google Scholar]
- 20.Hinnebusch A G. Translational control of GCN4: gene-specific regulation by phosphorylation of eIF2. In: Hershey J W B, Mathews M B, Sonenberg N, editors. Translational control. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1996. pp. 199–244. [Google Scholar]
- 21.Johnson L N, Noble M E M, Owen D J. Active and inactive protein kinases: structural basis for regulation. Cell. 1996;85:149–158. doi: 10.1016/s0092-8674(00)81092-2. [DOI] [PubMed] [Google Scholar]
- 22.Kamps M P. Determination of phosphoamino acid composition by acid hydrolysis of protein blotted to immobilon. Methods Enzymol. 1991;201:21–27. doi: 10.1016/0076-6879(91)01005-m. [DOI] [PubMed] [Google Scholar]
- 23.Knighton D R, Zheng J, Ten Eyck L F, Xuong N H, Taylor S S, Sowadski J M. Crystal structure of the catalytic subunit of cyclic adenosine monophosphate-dependent protein kinase. Science. 1991;253:407–414. doi: 10.1126/science.1862342. [DOI] [PubMed] [Google Scholar]
- 24.Koerner T J, Hill J E, Myers A M, Tzagoloff A. High-expression vectors with multiple cloning sites for construction of trpE fusion genes: pATH vectors. Methods Enzymol. 1991;194:477–490. doi: 10.1016/0076-6879(91)94036-c. [DOI] [PubMed] [Google Scholar]
- 25.Koromilas A E, Roy S, Barber G N, Katze M G, Sonenberg N. Malignant transformation by a mutant of the IFN-inducible dsRNA-dependent protein kinase. Science. 1992;257:1685–1689. doi: 10.1126/science.1382315. [DOI] [PubMed] [Google Scholar]
- 26.Kostura M, Mathews M B. Purification and activation of the double-stranded RNA-dependent eIF-2 kinase DAI. Mol Cell Biol. 1989;9:1576–1586. doi: 10.1128/mcb.9.4.1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Laemmli U. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 28.Langland J O, Jacobs B L. Cytosolic double-stranded RNA-dependent protein kinase is likely a dimer of partially phosphorylated Mr=66,000 subunits. J Biol Chem. 1992;267:10729–10736. [PubMed] [Google Scholar]
- 29.Madhusudan E, A T, Xuong N-H, Adams J A, ten Eyck L F, Taylor S S, Sowadski J M. cAMP-dependent protein kinase: crystallographic insights into substrate recognition and phosphotransfer. Protein Sci. 1994;3:176–187. doi: 10.1002/pro.5560030203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mathews M B. Viral evasion of cellular defense mechanisms: regulation of the protein kinase DAI by RNA effectors. Semin Virol. 1993;4:247–257. [Google Scholar]
- 31.Meurs E F, Galabru J, Barber G N, Katze M G, Hovanessian A G. Tumor suppressor function of the interferon-induced double-stranded RNA-activated protein kinase. Proc Natl Acad Sci USA. 1993;90:232–236. doi: 10.1073/pnas.90.1.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Parent S A, Fenimore C M, Bostian K A. Vector systems for the expression, analysis and cloning of DNA sequences in S. cerevisiae. Yeast. 1985;1:83–138. doi: 10.1002/yea.320010202. [DOI] [PubMed] [Google Scholar]
- 33.Patel R C, Stanton P, McMillan N M J, Williams B R G, Sen G C. The interferon-inducible double-stranded RNA-activated protein kinase self-associates in vitro and in vivo. Proc Natl Acad Sci USA. 1995;92:8283–8287. doi: 10.1073/pnas.92.18.8283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Patel R C, Stanton P, Sen G C. Specific mutations near the amino terminus of double-stranded RNA-dependent protein kinase (PKR) differentially affect its double-stranded RNA binding and dimerization properties. J Biol Chem. 1996;271:25657–25663. doi: 10.1074/jbc.271.41.25657. [DOI] [PubMed] [Google Scholar]
- 35.Pavitt G D, Yang W, Hinnebusch A G. Identification of amino acids in homologous segments of 3 subunits of the guanine nucleotide exchange factor eIF2B required for translational regulation by phosphorylation of eukaryotic translation initiation factor 2. Mol Cell Biol. 1996;17:1298–1313. doi: 10.1128/mcb.17.3.1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Qin J, Fenyo D, Zhao Y, Hall W W, Chao D M, Wilson C J, Young R A, Chait B T. A strategy for rapid, high-confidence protein identification. Anal Chem. 1997;69:3995–4001. doi: 10.1021/ac970488v. [DOI] [PubMed] [Google Scholar]
- 37.Ramirez M, Wek R C, Vazquez de Aldana C R, Jackson B M, Freeman B, Hinnebusch A G. Mutations activating the yeast eIF-2α kinase GCN2: isolation of alleles altering the domain related to histidyl-tRNA synthetases. Mol Cell Biol. 1992;12:5801–5815. doi: 10.1128/mcb.12.12.5801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Romano P R, Green S R, Barber G N, Mathews M B, Hinnebusch A G. Structural requirements for double-stranded RNA binding, dimerization, and activation of the human eIF-2α kinase DAI in Saccharomyces cerevisiae. Mol Cell Biol. 1995;15:365–378. doi: 10.1128/mcb.15.1.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sambrook J, Fritsch E F, Maniatis T. Molecular cloning: a laboratory manual. 2nd ed. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory; 1989. [Google Scholar]
- 40.Santoyo J, Alcalde J, Mendez R, Pulido D, de Haro C. Cloning and characterization of a cDNA encoding a protein synthesis initiation factor-2α (eIF-2α) kinase from Drosophila melanogaster. J Biol Chem. 1997;272:12544–12550. doi: 10.1074/jbc.272.19.12544. [DOI] [PubMed] [Google Scholar]
- 41.Shoji S, Titani K, Demaille J, Fischer E H. Sequences of two phosphorylated sites in the catalytic subunit of bovine cardiac muscle adenosine 3′:5′-monophosphate-dependent protein kinase. J Biol Chem. 1979;254:6211–6214. [PubMed] [Google Scholar]
- 42.Taylor D R, Lee S B, Romano P R, Marshak D R, Hinnebusch A G, Esteban M, Mathews M B. Autophosphorylation sites participate in the activation of the double-stranded RNA-activated protein kinase PKR. Mol Cell Biol. 1996;16:6295–6302. doi: 10.1128/mcb.16.11.6295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42a.Taylor, D. R., and M. B. Mathews. Unpublished observations.
- 43.Thomis D C, Samuel C E. Mechanism of interferon action: autoregulation of RNA-dependent P1/eIF-2α protein kinase (PKR) expression in transfected mammalian cells. Proc Natl Acad Sci USA. 1992;89:10837–10841. doi: 10.1073/pnas.89.22.10837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Thomis D C, Samuel C E. Mechanism of interferon action: characterization of the intermolecular autophosphorylation of PKR, the interferon-inducible, RNA-dependent protein kinase. J Virol. 1995;69:5195–5198. doi: 10.1128/jvi.69.8.5195-5198.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wek R C, Ramirez M, Jackson B M, Hinnebusch A G. Identification of positive-acting domains in GCN2 protein kinase required for translational activation of GCN4 expression. Mol Cell Biol. 1990;10:2820–2831. doi: 10.1128/mcb.10.6.2820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wek S A, Zhu S, Wek R C. The histidyl-tRNA synthetase-related sequence in the eIF-2α protein kinase GCN2 interacts with tRNA and is required for activation in response to starvation for different amino acids. Mol Cell Biol. 1995;15:4497–4506. doi: 10.1128/mcb.15.8.4497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu S, Kaufman R J. Double-stranded (ds) RNA binding and not dimerization correlates with the activation of the dsRNA-dependent protein kinase (PKR) J Biol Chem. 1996;271:1756–1763. doi: 10.1074/jbc.271.3.1756. [DOI] [PubMed] [Google Scholar]
- 48.Wu S, Kaufman R J. A model for the double-stranded RNA (dsRNA)-dependent dimerization and activation of the dsRNA-activated protein kinase PKR. J Biol Chem. 1997;272:1291–1296. doi: 10.1074/jbc.272.2.1291. [DOI] [PubMed] [Google Scholar]
- 49.Yon J, Fried M. Precise gene fusion by PCR. Nucleic Acids Res. 1988;17:4895. doi: 10.1093/nar/17.12.4895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang F, Strand A, Robbins D, Cobb M H, Goldsmith E J. Atomic structure of the MAP kinase ERK2 at 2.3 A resolution. Nature. 1994;367:704–710. doi: 10.1038/367704a0. [DOI] [PubMed] [Google Scholar]
- 50a.Zhang, X., C. Herring, P. R. Romano, J. Szczepanowska, H. Brzeska, A. G. Hinnebusch, and J. Qin. Submitted for publication.
- 51.Zhu S, Romano P R, Wek R C. Ribosome targeting of PKR is mediated by two double-stranded RNA-binding domains and facilitates in vivo phosphorylation of eukaryotic initiation factor-2. J Biol Chem. 1997;272:14434–14441. doi: 10.1074/jbc.272.22.14434. [DOI] [PubMed] [Google Scholar]
- 52.Zhu S, Sobolev A Y, Wek R C. Histidyl-tRNA synthetase-related sequences in GCN2 protein kinase regulate in vitro phosphorylation of eIF-2. J Biol Chem. 1996;271:24989–24994. doi: 10.1074/jbc.271.40.24989. [DOI] [PubMed] [Google Scholar]