

Abstract
Using complementation tests and nucleotide sequencing, we showed that the rad58-4 mutation was an allele of the MRE11 gene and have renamed the mutation mre11-58. Two amino acid changes from the wild-type sequence were identified; one is located at a conserved site of a phosphodiesterase motif, and the other is a homologous amino acid change at a nonconserved site. Unlike mre11 null mutations, the mre11-58 mutation allowed meiosis-specific double-strand DNA breaks (DSBs) to form at recombination hot spots but failed to process those breaks. DSB ends of this mutant were resistant to lambda exonuclease treatment. These phenotypes are similar to those of rad50S mutants. In contrast to rad50S, however, mre11-58 was highly sensitive to methyl methanesulfonate treatment. DSB end processing induced by HO endonuclease was suppressed in both mre11-58 and the mre11 disruption mutant. We constructed a new mre11 mutant that contains only the phosphodiesterase motif mutation of the Mre11-58 protein and named it mre11-58S. This mutant showed the same phenotypes observed in mre11-58, suggesting that the phosphodiesterase consensus sequence is important for nucleolytic processing of DSB ends during both mitosis and meiosis.
The genes of the RAD52 epistasis group in Saccharomyces cerevisiae are necessary for repair of double-strand DNA breaks (DSBs) during mitosis and meiosis (35). Mutants resulting from mutations of these genes are classified into two subgroups according to their recombination abilities and meiotic DSB formation properties. One subgroup comprises rad51, -52, -54, -55, and -57 mutants. These are defective in mating-type switching and both mitotic and meiotic recombination (34, 35, 43). In these mutants, meiosis-specific DSBs form at recombination hot spots but are left unrepaired with extensive processing (34, 42, 47). Mutants resulting from mutations in these genes are also defective in viable spore formation, and spore inviability is not alleviated by introducing an additional spo13 mutation, which eliminates meiotic reductional division (24). The other subgroup consists of mre11, xrs2, and rad50 null mutants, which are proficient in mating-type switching and show spontaneous recombination at a high frequency during mitosis (1, 14, 18, 30). In rad50 and xrs2 null mutants, processing of DSB ends is reduced and formation of recombinant is delayed (18, 20, 45). During meiosis, however, these three mutants are deficient in formation of meiosis-specific DSBs, induction of meiotic recombination, and viable spore formation (1, 5, 18, 21), and their viable spore formation deficiency is alleviated by the introduction of a spo13 mutation (35). A mutant resulting from a non-null mutation of RAD50, called rad50S, accumulates unprocessed DSBs, and its spore inviability is not rescued by introducing a spo13 mutation (3). Therefore, MRE11, XRS2, and RAD50 appear to be involved in two distinct processes: (i) DSB repair during mitosis and (ii) DSB formation and processing from DSB ends during meiosis (5, 18, 21).
Recently, a new mutation with phenotypes similar to those of the mre11, rad50, or xrs2 null mutant in mitosis was reported (6). This mutation, called rad58-4, caused high gamma ray sensitivity, allowed proficient mating-type switching, and caused high-frequency spontaneous recombination. In meiosis, however, this mutant was found to be deficient in meiotic recombination and viable spore formation. The spore inviability of this mutant was not alleviated by a spo13 mutation, a phenotype shared with the rad50S mutant. The rad58-4 locus was mapped on the right arm of chromosome XIII between CEN13 and ADE4, 48 cM from ADE4 (25), very close to the MRE11 gene locus, which was also mapped to a similar location (21). This prompted us to investigate whether rad58-4 is allelic to the MRE11 gene.
In this study, we showed that rad58-4 is a novel mutant allele of the MRE11 gene, and we propose that this allele should be renamed mre11-58. The mre11-58 mutant was proficient in formation of DSBs in meiosis but defective in DSB end processing, as observed for rad50S mutants, and end processing of DSBs induced by the HO endonuclease during mitosis was reduced in this mutant. In view of these characteristics of the mre11-58 mutant, we propose that the Mre11 protein is involved in exonucleolytic processing from the ends of DSBs, which are produced at both HO cutting sites and meiotic recombination hot spots.
MATERIALS AND METHODS
Plasmids.
Plasmids were constructed by standard procedures (39). pKJ1112-S (21) and pΔRAD51 (42) were used for MRE11 and RAD51 gene disruption, respectively. The genes of both these plasmids were disrupted by inserting the hisG-URA3-hisG fragment from pNKY51 (2). The pHT62 plasmid was constructed by cloning a 4.3-kb BamHI fragment from pKJ1101 (21) at the BamHI site of YCp50 (37). The same BamHI fragment was cloned at the BamHI site of pRS316 (44), and then the unique NruI site was filled with the Klenow fragment, where the XhoI linker was ligated to make pHO5. pHT139 was an mre11 mutant version of pHO5 whose MRE11 gene was replaced with the mre11-58S mutant gene (see site-directed mutagenesis below for details). The pNKY291 (5) plasmids were used as the sources of probes for detecting DSBs for the HIS4LEU2 locus (5). This plasmid contained a 1.5-kb PstI-EcoRI fragment downstream of the HIS4 gene and could be liberated by PstI-BglII digestion. The pHT46 plasmid carried an EcoRI-HindIII fragment containing MATa on YCplac22 (15), and pHT51 was based on pJH283 (a gift from J. E. Haber). It contained the HO gene, which was under the control of a galactose promoter, and was constructed by cloning a HindIII-ClaI fragment containing the THR4 gene with its PvuII site in the open reading frame (ORF) inserted with a 35-mer HO recognition sequence, the sequence of which is gtcgactttagtttcagctttccgcaacagtataa (SalI site is embedded on the left), into the EcoRI site of pJH283.
Yeast strains.
The yeast strains used in this study are listed in Table 1. The 20B-D3142 and p192 strains were gifts from V. G. Korolev. The HTY231 to HTY703 strains (except HTY553, -666, and -693), OSY052, OSY053, HTY1114, and HTY1115 were derivatives of the SK1 strain, which enters into meiosis in a highly synchronous manner (11). HTY666 was produced by mating two rad58-4 haploids derived from HTY553 by crossing three times with an SK1 strain, HTY703 was derived from HTY553 after crossing four times with an SK1 strain, and HTY665 was produced by mating NKY276 with NKY278, both of which were gifts from N. Kleckner. Gene disruption was carried out by the one-step gene disruption method (38). The MRE11 and RAD51 disruption strains were constructed by displacing chromosomal genes with BamHI fragments prepared from pKJ1112-S (21) and pΔRAD51 (42), respectively. The RAD52 disruption strain was produced by transformation with pANHUH after SalI and EcoRI digestion, and rad50S-KI81::URA3 (a gift from N. Kleckner) was introduced with pNKY349 after EcoRI and BamHI digestion. The mre11-58S mutant strain was constructed by transforming OSY053 with BamHI-digested pHT139 and selecting Ura− colonies on synthetic complete medium containing 5-FOA (5-fluoroorotic acid; U.S. Biological, Swampscott, Mass.).
TABLE 1.
Strains used
| Strain | Genotype |
|---|---|
| CG379 | MATα ura3-52 leu2-3,112 trp1-289 ade5-1 his7-2 |
| 20B-D3142 | MATa ade2-192 leu2-3,112 ura3-160,188 trp1 rad58-4 (mre11-58) |
| p192 | MATa ade2-192 (a parental strain of 20B-D3142) |
| NKY1003 | MATa ho::LYS2 lys2 ura3 rad50S-KI81::URA3 |
| HTY231 | CG379 but mre11::hisGURA3hisG |
| HTY464 | MATa ho::LYS2 lys2 ura3 leu2::hisG his4B::LEU2 arg4-bgl |
| HTY477 | HTY464 but mre11::hisGURA3hisG |
| HTY525 | MATa ho::LYS2 lys2 ura3 his4B::LEU2 arg4-bgl |
| MATα ho:LYS2 lys2 ura3 his4X::LEU2 arg4-nsp | |
| HTY533 | HTY525 but homozygous for rad51::hisGURA3hisG |
| HTY553 | MATα rad58-4 (mre11-58) trp1 leu2-3,112 ura3 |
| HTY603 | HTY525 but homozygous for rad50S-KI81::URA3 |
| HTY665 | MATa ho::LYS2 lys2 ura3 |
| MATαho::LYS2 lys2 ura3 | |
| HTY666 | HTY665 but homozygous for mre11-58 |
| HTY693 | HTY464 but mre11-58 |
| HTY703 | HTY525 but homozygous for mre11-58 |
| HTY925 | CG379 with pHT51 |
| HTY927 | HTY925 but mre11::hisGURA3hisG |
| HTY929 | HTY925 but rad52::hisGURA3hisG |
| HTY931 | HTY553 with pHT51 |
| HTY933 | HTY925 but rad50S-KI81::URA3 |
| OSY052 | MATα ho::LYS2 lys2 ura3 his4X::LEU2 arg4-nsp trp1 mre11::URA3 |
| OSY053 | MATa ho::LYS2 lys2 ura3 his4B::LEU2 arg4-bgl trp1 mre11::URA3 |
| HTY1114 | MATa ho::LYS2 lys2 ura3 his4B::LEU2 arg4-bgl trp1 mre11-58S |
| HTY1115 | OSY052 × HTY1114 |
Site-directed mutagenesis.
About 1 ng of pHO5 was amplified with a set of oligonucleotides listed below as primers with 2.5 U of LA Taq (TAKARA). PCR conditions were 25 cycles of 98°C for 20 s and 65°C for 7 min, followed by 10 min at 72°C. The sample was then treated with 20 U of DpnI at 37°C to digest the template DNA. The amplified plasmid was used for transforming DH5α. MRE11 genes on amplified plasmids were sequenced to confirm that the mutation had been introduced at the right place and that no additional mutation occurred during the PCR process. One of the clones was named pHT139. The sequences of oligonucleotides used are aatttaatgtgcgtctatcaaaatcatacagg and tgtatgattttgatagacgcacattaaacc.
Media.
All of the media used in this study are described elsewhere (21). YPLac contained 1% yeast extract, 2% Bacto Peptone, and 2% sodium lactate (pH 5.5), and YPGal was identical to YPLac, except that it contained 2% galactose instead of lactate.
Determination of mutation sites of mre11-58.
Genomic DNAs of 20B-D3141 and p192 were used for PCR amplification with the following pairs of oligonucleotide primers: F1 and R1, F1 and R2, F3 and R3, and F4 and R4. The nucleotide sequences of these primers are as follows: F1, cgcggatccgtaaggaagacaatgtgg; F3, cgcggatcctgttccccatttgaggcc; F4, cgcggatccgaacagatgatgcagagg; R1, cgcggatcccgaacagcggctaatccg; R2, cgcggatcctcctcattagcgtcgcgg; R3, cgcggatccctttagggggagtcggcc; and R4, cgcggatccgttcgcgaaggcaagccc. The amplified fragments were digested with BamHI, cloned at the BamHI site of pUC118, and sequenced from both ends. More than two independent clones were sequenced per set, and the altered sequences common to independent clones were considered real changes, not the changes produced by the PCR process.
Return-to-growth experiment and DSB detection.
Synchronous entry of cells into meiosis and return-to-growth experiments were as described previously (5, 21), and meiosis-specific DSBs were detected as described elsewhere (21). Genomic DNAs were extracted from cells harvested at the required times during meiosis, cut with an appropriate restriction enzyme, fractionated with a 0.7% agarose gel, and subjected to Southern blotting. To examine DSBs at the HIS4LEU2 locus (5), genomic DNA was cut with PstI and a 1.5-kb EcoRI-BglII fragment from pNKY291 was used as a probe (see Fig. 3).
FIG. 3.

DSB formation at the HIS4LEU2 locus in mre11-58. The physical map of the HIS4LEU2 locus is shown in the upper panel. Horizontal arrows indicate the major two sites of DSBs, called sites I and II. Genomic DNAs were prepared from cells collected at various times after entering meiosis and cut with PstI, and the fragments corresponding to the parent (12.6 kb), site I (3.7 kb), and site II (6.0 kb) were detected by Southern blotting with the 1.5-kb PstI-EcoRI fragment of pNKY291, which contains the PstI-BglII fragment shown in the upper panel, as a probe. The lower panel represents the images of DSBs at the HIS4LEU2 locus, and the numbers above the images indicate the times (in hours) after entry into meiosis. Strains: HTY525 (wild type), HTY603 (rad50S), HTY533 (rad51Δ), and HTY703 (mre11-58).
Lambda exonuclease treatment of genomic DNA in meiosis.
Genomic DNAs from 3 × 108 cells (mre11-58 cells at 8 h and wild-type cells at 3 h after entering into meiosis), added with or without the addition of 10 pg of the PstI-EcoRI fragments from pNKY291, were treated with 8 U of lambda exonuclease (GIBCO BRL) for 2 h at 37°C in a mixture containing 1× NEB3 buffer (New England Biolabs, Inc. Beverly, Mass.). Exonuclease was inactivated at 65°C for 15 min. Samples were then digested with PstI for 3 h at 37°C. Samples were separated on a 0.7% agarose gel and Southern blotted with a PstI-EcoRI fragment from pNKY291 as a probe.
Induction and detection of HO breaks.
Strains transformed with pHT51 were suspended in 2 ml of synthetic complete medium lacking tryptophan (SD-TRP) medium, and the suspension was incubated for 24 h at 30°C. The culture was then centrifuged and washed with distilled water, and the cells were resuspended in 200 ml of SD-TRP medium. After incubation at 30°C for a further 24 h, the cells were harvested and resuspended in 200 ml of YPLac, and the suspensions were incubated at 30°C for 12 h, after which an aliquot of cells was removed as an initial sample (time zero). The rest of the culture was harvested, resuspended in 200 ml of YPGal and incubated for 1 h and another aliquot of cells was harvested. The rest of the culture was harvested and resuspended in 200 ml of YPD, and aliquots of cells were taken after the required incubation times up to a total culture period of 6 h. To detect DSBs on pHT51, genomic DNAs were cut with HindIII, and a 2.4-kb PvuII-PvuII fragment of pJH283 was used as a probe. Mating-type switching was studied by cutting genomic DNAs with StyI and using a 1.0-kb NdeI-HindIII fragment of pHT46 as a probe.
RESULTS
Complementation analysis of the rad58-4 and mre11 disruption mutations.
The locus of the new gene RAD58 was mapped very close to the MRE11 gene; the mitotic properties of rad58-4 were found to be very similar to those of the mre11 null mutant. Both mutants showed high sensitivity to methyl methanesulfonate (MMS) and an elevated frequency of spontaneous recombination in comparison with the parental wild-type strain (6, 21, 25). These similarities prompted us to examine whether these two loci are allelic. First, we performed a complementation test. A haploid rad58-4 strain (20B-D3142, obtained from V. G. Korolev) was mated with the mre11 disruption mutant (HTY231) and a wild-type strain (CG379), and the sensitivities to MMS of the diploids obtained were tested. The survival fraction of the diploid containing the rad58-4 and mre11 alleles was 0.02% in the presence of 0.02% MMS, whereas no survival reduction was observed with the diploid containing heteroalleles rad58-4 and RAD58 under these conditions (Fig. 1A). This suggests that rad58-4 and mre11 are allelic. We noticed, however, that the diploid containing rad58-4 and mre11 alleles showed a biphasic survival curve as the MMS concentration increased; in the presence of MMS at concentrations of less than 0.01%, the majority of the diploid cells showed high sensitivity to MMS, but at concentrations of more than 0.02%, about 0.02% of the cells showed almost the same sensitivity as the wild-type strain (Fig. 1A). Among the colonies of the cells that survived in the presence of 0.02% MMS, 14 colonies were subcultured, and their MMS sensitivities were tested. They were all as resistant, as were the wild-type cells. Since the mre11 mutant used in this experiment is an insertion disruptant, the resistance was probably due to formation of the wild-type MRE11 gene by recombination between the heteroalleles.
FIG. 1.

(A) Lack of complementation for MMS sensitivity between the rad58-4 mutant and the mre11 disruptant. Diploids obtained by mating rad58-4 (20B-D3142) with an mre11 (HTY231) (solid circles) strain and with a wild-type strain (CG379) (open circles) were grown in yeast-peptone-dextrose (YPD) liquid medium, diluted appropriately, and spread on YPD plates containing various concentrations of MMS, and the plates were incubated at 30°C for 3 days. Colonies growing on each plate were counted. (B) Complementation of the repair defect of the rad58-4 mutant with the cloned MRE11 gene. The rad58-4 mutant (HTY553) carrying the MRE11 gene on a single-copy vector, pHT62 (open circles), or the vector alone, YCp50 (solid circles), was tested for MMS sensitivity. Each transformant was grown in SD-URA, diluted appropriately, and spread on SD-URA plates containing various concentrations of MMS.
The original rad58-4 haploid strain (20B-D3142) exhibited extraordinarily slow growth. To obtain a derivative of 20B-D3142 which grew at a reasonable rate, we mated this strain with a wild-type haploid strain (CG379) and chose one haploid (HTY553) segregant which retained MMS sensitivity. The sensitivity of this new strain to MMS was almost the same as those of the original rad58-4 strain and the mre11 disruptant (data not shown). When the complementation test was repeated with this new strain, a biphasic survival curve similar to that described above was observed (data not shown). When this strain was mated to a wild-type haploid strain and sporulated, MMS-sensitive and MMS-resistant spores always segregated at 2:2, and the MMS-sensitive phenotype was linked to the slow-growth phenotype. Therefore, HTY553 was thought to carry the original rad58-4 mutation and not to carry an extragenic suppressor. The HTY553 strain was transformed with a plasmid containing the MRE11 gene (pHT62) or vector alone (YCp50). HTY553 containing pHT62 was resistant to MMS, whereas HTY553 containing YCp50 was sensitive to MMS (Fig. 1B). We concluded that the rad58-4 mutation is located in the MRE11 gene and renamed it mre11-58.
Mutation sites of the mre11-58 allele.
The mre11-58 mutant and its wild-type genes were cloned from the original strains, 20B-D3142 and p192, respectively (obtained from V. G. Korolev). We amplified both genes as four overlapping fragments, respectively, by PCR with four pairs of oligonucleotide primers (Materials and Methods). The amplified fragments were subjected to sequencing after being cloned on plasmids. The MRE11 wild-type gene of p192 showed six nucleotide differences from the nucleotide sequence of the standard strain, S288C (D11463). The changes were as follows: C1138T, C1137T, C1681T, A1716T, A1891G, and C1975T (numbers indicate the distances from the 5′ end of the ORF). These changes resulted in four amino acid substitutions: Pro380Ser, Pro561Ser, Asn631Asp, and Pro659Ser, respectively. Since p192 is resistant to DNA damage and produces viable spores, these amino acid changes must not affect the functions of the Mre11 protein. We regard these changes as reflecting polymorphism between the p192 and S288C strains. On the other hand, the mre11-58 mutant had three additional mutations. These were C636T, C637T, and T673A. The three nucleotide changes resulted in two amino acid substitutions: His213Tyr and Leu225Ile (Fig. 2A). His213 is one of the well-conserved amino acids among five known Mre11 protein homologs (Fig. 2A) and is located in the fourth of five phosphoesterase consensus motifs proposed by B. Baum (3a) (Fig. 2B). Therefore, the, His213-to-Tyr change was expected to affect Mre11 function. The other change, Leu225 to Ile, is a homologous amino acid change, and Ile is a normal constituent at the corresponding site of the human and mouse Mre11 proteins (Fig. 2A). To confirm this suggestion, we constructed a mutant strain carrying a single mutation, His213 to Tyr. The properties of the new mutant, named mre11-58S (separated), were indistinguishable from those of the parental mre11-58 mutant as described below.
FIG. 2.

(A) Comparison of amino acid sequences of Mre11 homologs around the mutant sites of mre11-58; (B) comparison of amino acid sequences of various phosphoesterases. The motif sequence is shown at the top (from reference 3a). Shading, identical amino acids; boxes, similar amino acids. Sc, S. cerevisiae; Sp, Schizosaccharomyces pombe; Ec, E. coli; Mm, Mus musculus; Hs, Homo sapiens; Dro, Drosophila; Ce, Caenorhabditis elegans. Sc Mre11, 1513065; Ec sbcD, 1586770; Ec apaH, 1003022; Ec cpdB, 67263; Ec ushA, 137173; Sc Dbr1, 171382; Sc Pph1, 319859; Sc Pph21, 4203; Dro rdcC, 158238; Hs ASM, 179095; and Hs TR-AP, 130722. Numbers indicate NCBI sequence identity.
A spo13 mutation fails to alleviate the spore lethality of mre11-58.
The spore lethality of some S. cerevisiae mutants that are defective in the early stage of meiotic recombination can be overcome by introducing a spo13 mutation (35). Null mutants of mre11, rad50, and xrs2 belong to this class (1, 18, 27, 29). However, rad58-4 spo13 double mutants were reported to form inviable dyads (6). To verify this result in the SK1 background, the spo13 mre11-58 double mutant was produced. The spore viability of this mre11-58 spo13 homozygous diploid (HTY699) was less than 1.7%, and those of the control strains HTY703 (mre11-58), HTY722 (spo13), HTY724 (mre11Δ spo13), and the wild type (HTY525) were less than 0.83, 65, 78, and 98%, respectively (Table 2). Therefore, the spo13 mutation did not alleviate the spore lethality of the mre11-58 mutant.
TABLE 2.
Spore viability of mre11-58
| Strain | Genotype | No. of asci dissected | No. of viable spores
|
Spore viability (%) | ||||
|---|---|---|---|---|---|---|---|---|
| 4+ | 3+ | 2+ | 1+ | 0+ | ||||
| HTY525 | RAD+ | 30 | 28 | 2 | 0 | 0 | 0 | 98 |
| HTY703 | mre11-58 | 30 | 0 | 0 | 0 | 0 | 30 | <0.83 |
| HTY722 | RAD+ spo13 | 30 | 14 | 11 | 5 | 65 | ||
| HTY699 | mre11-58 spo13 | 30 | 0 | 0 | 30 | <1.7 | ||
| HTY724 | mre11Δspo13 | 30 | 20 | 7 | 3 | 78 | ||
Accumulation of meiosis-specific DSBs in the mre11-58 mutant.
As shown above, the spore lethality of the mre11-58 mutant was not overcome by a spo13 mutation. This phenomenon was interpreted to be due to a defect occurring after the initiation step of meiotic recombination (27, 28). Therefore, mre11-58 was expected to show impaired DSB repair. To test this, DSB formation was examined at an artificial recombination hot spot, HIS4LEU2 (5). In the wild-type strain, DSBs reached maximum levels by 4 h and disappeared by 8 h. In the mre11-58 mutant, DSBs accumulated and did not disappear, persisting through 12 h (Fig. 3). To provide a comparison, the rad51 deletion (rad51Δ) mutant, in which DSB ends undergo extensive processing without being repaired, and rad50S, which forms discrete DSB ends that are not processed, were examined simultaneously (Fig. 3). In the mre11-58 mutant, the patterns of the accumulated DNA fragments produced by DSBs at sites I and II were similar to those produced by the rad50S mutant, but not to those of the rad51Δ and wild-type strains (Fig. 3). This shows that the fragments produced by the mre11-58 mutant were not processed from their ends. We observed the same type of DSB accumulation in mre11-58 at a native recombination hot spot, YCR47c/48w (16) (data not shown).
Resistance of meiotic DSB ends in mre11-58 to lambda exonuclease.
In the rad50S mutant, DSBs formed during meiosis contained tightly bound protein at their 5′ ends (9, 23, 26) and were resistant to lambda exonuclease treatment (26). The DSB-associated protein was identified as Spo11, which was suggested as the catalytic subunit of the meiotic DNA cleavage activity (26). To see if these DSB ends of meiotic DNA from mre11-58 are also protected as are those from rad50S, genomic DNA was prepared from mre11-58 cells 8 h after entry into meiosis, when meiotic DSBs were fully formed. As an internal control, 1.5-kb DNA fragments with PstI and EcoRI-digested ends were added. The DSBs formed in both rad50S (HTY603) and mre11-58 (HTY703) were resistant to digestion by lambda exonuclease, while the DNA fragment with the ends created by restriction enzymes was completely degraded (Fig. 4). These results suggest that the DSB ends of mre11-58 are protected as well as those of rad50S, possibly by the Spo11 protein.
FIG. 4.

Lambda exonuclease digestion of mre11-58 meiotic DNA. Genomic DNAs from meiotic cells were prepared when DSBs were fully formed and were treated with lambda exonuclease. After inactivating exonuclease, genomic DNAs were cut with PstI and subjected to Southern hybridization after agarose gel electrophoresis as shown in Fig. 3. A DNA fragment cut produced by PstI EcoRI double digestion was added before exonuclease treatment as an internal control. Lanes 1 to 4, rad50S DNA; lanes 5 to 8, mre11-58 DNA.
Lack of meiotic recombination in the mre11-58 mutant.
In the mre11-58 mutant, DSBs occurred and remained unrepaired during meiosis. To ascertain whether the accumulated DSB ends produced by meiosis-specific DSBs in mre11-58 were recombinogenic in mitosis, the formation of recombinants at two heteroalleles, his4X/his4B and arg4-bgl/arg4-nsp, was monitored in a return-to-growth experiment (10, 41). No increase in recombinants at either of these loci was observed in the mre11-58 mutant (HTY703), not even after incubation in a sporulation medium for 24 h (Fig. 5A), whereas the rad50S mutant (HTY603) showed an approximately 10-fold increase above the frequency of spontaneous mitotic recombination (Fig. 5B). At 0 h, the frequencies of recombination were 2.4 and 5.8 times higher than those at the HIS4 and ARG4 loci, respectively, of the wild-type strain (average values of two independent experiments). The survival fractions of both mutant cells decreased gradually as meiosis proceeded, and approximately 10% of the input cells survived the 24-h incubation period (Fig. 5A and B). These results show that despite being returned to mitosis, the DSBs that accumulated during meiosis in mre11-58 cells did not promote recombination.
FIG. 5.

Meiotic recombination deficiency of mre11-58. Diploid strains were introduced synchronously into meiosis, as described in Materials and Methods. Surviving fractions were obtained by dividing the numbers of CFU on complete medium (MYPD) after each incubation time by those at 0 h. Recombinant fractions at two sets of heteroalleles, his4X/his4B and arg4-nsp/arg4-bgl, were obtained. Each value plotted is the ratio of the number of HIS+ or ARG+ CFU to the total number of CFU at each time point. (A) Open circles, mre11-58 (HTY703) surviving fractions; open triangles, ARG+ fractions; open squares, HIS+ fractions; (B) Open symbols, wild type (HTY525); solid symbols, rad50S (HTY603); circles, surviving fractions; squares, HIS+ fractions; triangles, ARG+ fractions. SPM, sporulation medium.
Retardation of mitotic DSB repair in mre11-58.
Mitotic repair of DSB formed by HO endonuclease is retarded in null mutants of xrs2 and rad50 (19, 20, 45). To establish whether the mre11-58 and the mre11 disruption mutants show similar phenotypes, the efficiencies of DSB repair at two locations, on the chromosome and on a plasmid, were monitored (Fig. 6A [i and ii]).
FIG. 6.

Kinetics of repair of HO endonuclease-induced DSBs. Haploid strains were transformed with pHT51 carrying the HO gene under the control of the galactose promoter. HO endonuclease expression was induced by suspending the cells in a galactose-containing medium for 1 h and was then repressed by suspending the cells in a glucose-containing medium, and the DSB repair kinetics were observed as described in Materials and Methods. (A) Physical maps used for DSB repair detection. (i) The MATα locus. The indicated probe (a 1.0-kb NdeI-HindIII fragment) was used to detect the 2.2-kb MAT distal, 1.8-kb MATα, and 0.7-kb HO-cut fragments after StyI digestion. HO endonuclease produces a 0.7-kb HO-cut fragment from the 1.8-kb MATα fragment, and this 0.7-kb fragment is replaced by a 0.9-kb MATa fragment when the mating type switches from MATα to MATa. (ii) pHT51. The indicated probe (a 2.4-kb PvuII-PvuII fragment obtained from pJH283) was used to detect 5.7-kb parental and 3.3-kb HO-cut fragments after HindIII digestion. Stippled and solid rectangles, the homologous regions of pHT51 and the genome and the 35-mer HO recognition sequence, respectively. (B) Kinetics of DSB repair at the MATα locus. (C) Kinetics of DSB repair on pHT51. Strains: HTY925 (rad+), HTY927 (mre11Δ), HTY929 (rad52Δ), HTY931 (mre11-58), and HTY933 (rad50S).
HO endonuclease expression was controlled by a GAL1 promoter. HO endonuclease induction for 1 h resulted in DSB formation at the Yα/Z junction of the MATα locus and generated a 0.7-kb fragment containing Z1 and Z2 (Fig. 6A [i] and B). Upon further incubation of a wild-type strain under conditions of glucose repression, the DSB band disappeared rapidly within 3 h. A new 0.9-kb MATa-specific band indicative of the mating-type conversion from MATα to MATa by recombination appeared within an hour of repression (8, 20, 46). A 0.7-kb fragment was produced by mre11-58 in a manner similar to that of the wild-type strain, but this fragment remained unrepaired during further incubation without HO gene expression, and a 0.9-kb band began to appear only after 3 h. Thus, mating-type conversion was delayed by about 1.5 h compared to that for the wild-type strain. A similar delay of repair during mating-type switching was observed with the mre11 disruptant. The kinetics of the disappearance of the HO-cut fragment band and the appearance of the MATa band in the rad50S mutant were identical to those of the wild-type strain, whereas in the rad52 mutant, the 0.7-kb band from MATα appeared and disappeared in a manner similar to that observed with the wild-type strain. The 0.9-kb band, however, did not appear at all in the rad52 mutant, indicating that mating-type switching was abolished in this mutant, as shown previously (46). The DSB ends in rad52 appeared to be undergoing exonucleolytic processing without repair during gene conversion.
A similar experiment was performed in which a DSB was produced on a plasmid, pHT51, in which an HO cutting site was inserted in the THR4 gene (Fig. 6C). A DSB induced on this plasmid can be repaired with the THR4 gene on the genome by homologous recombination. HO-induced DSBs on pHT51 generated 3.3-kb fragments from the 5.7-kb parental fragments after HindIII digestion. Fragments generated in the wild-type, rad50S, and rad52Δ strains disappeared completely within 2 h, but those generated in mre11-58 and mre11 disruption mutants remained for more than 4 h after glucose repression.
In both cases, the disappearance of the fragments generated by HO endonuclease would have been brought about by two processes: repair of DSBs and degradation from DSB ends. In the wild-type strain and the rad50S and rad52 mutants, the amounts of the fragments produced by HO endonuclease decreased and showed the same kinetics, regardless of the appearance of the recombinants, indicating that the time the cut fragments persists during incubation depends on the efficiency of exonucleolytic processing (19, 20, 45, 46). The cut fragments generated in the mre11-58 and mre11 disruption mutants persisted for longer than those generated in the wild-type strain, indicating that exonucleolytic processing of the DSB ends was impaired in these two mutants.
mre11-58 and mre11-58S have the same phenotype.
The mre11-58 allele had two mutations, His213 to Tyr and Leu225 to Ile. Although Leu225 to Ile was a normal constituent at the corresponding site of the human and mouse Mre11 protein, His213 was located in the phosphoesterase consensus sequence and expected to be responsible for mre11-58 phenotypes. To confirm this expectation, we compared MMS sensitivities and DSB formation between mre11-58 and mre11-58S and tested the protection of DSB ends in mre11-58S.
The MMS survival curves of both mutant strains are shown in Fig. 7. mre11-58 (HTY693) and mre11-58S (HTY1114) strains showed the same sensitivities against MMS and were indistinguishable from each other.
FIG. 7.

The mre11-58S mutant is as sensitive to MMS as mre11-58. Haploid strains HTY1114 (mre11-58S), HTY693 (mre11-58), HTY1075 (mre11 null), and NKY1003 (rad50S), and a wild-type strain (HTY464) were grown in YPD liquid medium, diluted appropriately, spread on MYPD plates containing various concentrations of MMS, the plates were incubated at 30°C for 5 days, and the numbers of colonies growing on each plate were counted.
DSB formation in mre11-58 (HTY703) and mre11-58S (HTY1115) strains was examined. Meiotic DSBs were formed and accumulated similarly in both strains (data not shown). To compare the frequencies of DSBs in both mutant strains, meiotic DNA was prepared at 8 h after entering meiosis, when the amount of DSBs reached a maximum level, and DSBs occurring at the his4LEU2 recombination hot spot were measured by densitometry (Fig. 8). Totals of 4 and 9% of total DNA in mre11-58 and 3 and 9% of total DNA in mre11-58S were accumulated at site I and site II, respectively. Therefore, both mutants were almost identical in DSB formation.
FIG. 8.
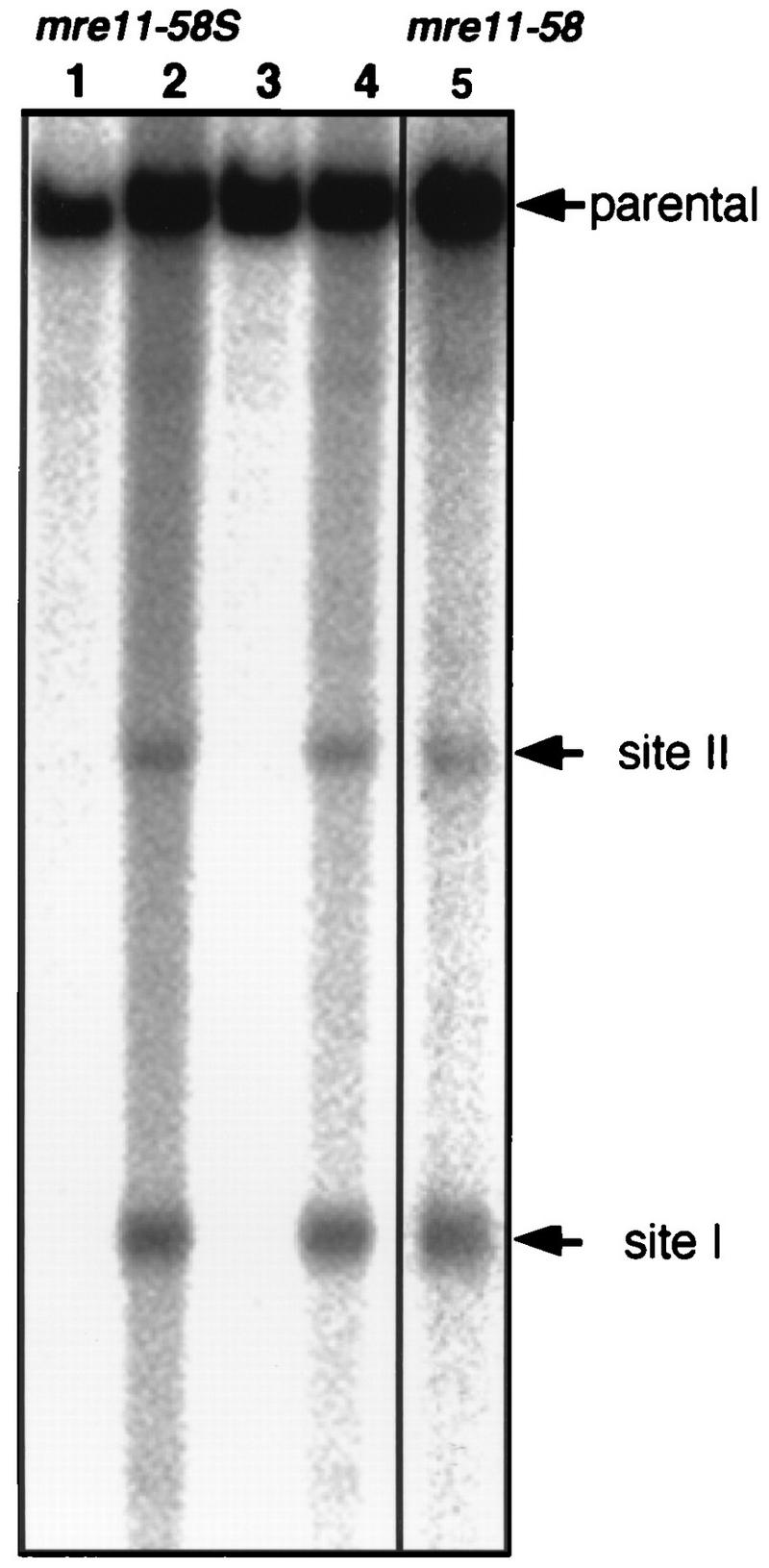
DSB formation at the HIS4LEU2 locus in mre11-58S. Horizontal arrows, major two sites of DSBs, called sites I and II. Genomic DNAs from mre11-58S (HTY1115) and mre11-58 (HTY703) were prepared from cells collected at 0 and 8 h (only 8 h for mre11-58) after entering meiosis, and Southern blotting was performed to detect fragments corresponding to the parent (12.6 kb), site I (3.7 kb), and site II (6.0 kb) as described in the legend to Fig. 3. Lanes 1 and 3, 0-h sample; lanes 2 and 4, 8-h sample. Lanes 1 and 2 and 3 and 4 are the results of independent clones of HTY1115. Lane 5, mre11-58 (HTY703).
To test whether the meiotic DSB ends of DNA from mre11-58S are protected, the genomic DNA of mre11-58S used for Fig. 8 was subjected to lambda exonuclease. As an internal control, 1.5-kb DNA fragments with PstI- and EcoRI-digested ends were added. The DSBs formed in mre11-58S were resistant to the processing of lambda exonuclease when the DNA fragments with the ends created by restriction enzymes were completely degraded (Fig. 9, lanes 9 to 12). In sharp contrast, meiotic DSBs formed in wild-type cells suffered degradation by lambda exonuclease like that of the restriction enzyme-cut fragment (Fig. 9, lanes 5 to 8). No DSBs were observed in genomic DNAs from mitotic cells (Fig. 9, lanes 1 to 4). These results showed that the DSB ends of mre11-58S are protected to the same extent as those formed in mre11-58.
FIG. 9.

Lambda exonuclease digestion of mre11-58S meiotic DNA. Genomic DNA of the mre11-58S strain was isolated from meiotic cells at 8 h after entering meiosis when DSBs were fully formed, and that of the wild type was isolated from mitotic cells and from cells at 3 h after entering meiosis. The genomic DNAs were treated with lambda exonuclease. After inactivation of exonuclease, genomic DNAs were digested with PstI and subjected to Southern hybridization after agarose gel electrophoresis as shown in Fig. 3. DNA fragments cut with PstI and EcoRI double digestion were added before exonuclease treatment as an internal control. Lanes 1 to 4, wild-type DNA (0 h); lanes 5 to 8, wild-type DNA (3 h); lanes 9 to 12, mre11-58S DNA (8 h). Strains used were HTY525 (wild type), and HTY1115 (mre11-58S).
DISCUSSION
During mitosis, the mre11-58 mutant showed an elevated frequency of spontaneous recombination and high sensitivity to MMS in comparison with those of the wild-type strain. DSB repair during mitosis was retarded; fragments produced by HO endonuclease persisted longer in the mre11-58 strain and the mre11 disruptant than in the rad52 strain, indicating that the efficiency of DSB end processing was reduced in the mre11-58 and mre11 disruptants. All mitotic phenotypes of the mre11-58 mutant were almost identical to those of the mre11 disruptant. During meiosis, the mre11-58 mutant showed defective induction of recombination and accumulated DSBs at recombination hot spots, and these meiotic properties are similar to those of rad50S.
The Mre11 complex in meiosis.
The 5′ ends of DSBs in rad50S are protected by a certain protein through a covalent linkage (9, 23, 26). Recently, this protein was shown to be Spo11, a member of a novel type II topoisomerase family, implicated to be a catalytic subunit of DSB endonuclease (4, 22). When either the Spo11 or the Mre11 protein is absent, no DSBs occur, suggesting that both of these proteins are required for DSB formation, presumably as components of a complex. Cytological observations revealed that during meiosis, Mre11, Xrs2, and Rad50 proteins colocalized specifically at the same foci in the rad50S mutant and that the number of foci increased with the incubation time as the amount of DSBs increased (45a), supporting the complex formation of Mre11 (Xrs2 and Rad50) and Spo11.
A mutation in a phosphoesterase consensus sequence affects DSB processing activity.
The mre11-58 mutant showed a defect in processing from the ends of the HO endonuclease-induced breaks and meiotic DSBs. This finding strongly suggests that the Mre11 complex is involved in the exonucleolytic process during both meiosis and mitosis. Recently, the Mre11 and Rad50 proteins were found to have homology with SbcD and SbcC proteins in Escherichia coli (40), respectively, lending support to this idea. Mre11 and SbcD proteins both have a phosphoesterase consensus sequence (3a, 40), and SbcD and SbcC form a tight complex, which retains single-strand endonuclease and ATP-dependent double-strand exonuclease activities (7). We identified a mutation site of the mre11-58 allele at the well-conserved amino acid of this consensus sequence (3a). Our observations and the site of the mutation suggest that the processing reaction after DSB formation during both mitosis and meiosis is dependent on the putative phosphodiesterase activity of the Mre11 protein. On the other hand, another non-null mutant strain of MRE11, called the mre11S strain, also accumulates unprocessed DSBs during meiosis but shows resistance to MMS. This phenotype is very similar to that of rad50S, in which we showed that unprocessed DSBs accumulated during meiosis but that no such defect was observed in mitotic processing. The amino acid changes of mre11S, Pro84 to Ser and Thr188 to Ile, were not located in the phosphodiesterase consensus region (33), suggesting that the Mre11S protein retains phosphodiesterase activity. The repair proficiency of this mutant also supports this possibility. The Mre11S and Rad50S proteins may have defects in, for example, interaction with other proteins that assist meiosis-specific DSB processing.
The process of removing protein from the 5′ ends of meiotic DSBs.
In the mre11-58 mutant, appearance of recombinants after DSB formation was impaired specifically during meiosis, not mitosis. The Spo11 protein is suggested as presenting a barrier to meiotic DSBs becoming committed to homologous recombination (9, 23, 26). If this is so, the meiotic recombination defect of mre11-58 could be due to failure to remove Spo11 from DSB ends. The observation that the DSB ends formed in this mutant are protected from degradation by lambda exonuclease strongly supports this idea. Removal of the Spo11 protein covalently bound to the 5′ end of the meiotic DSBs may be catalyzed by the Mre11 protein complex through its putative phosphodiesterase activity. This process possibly requires an additional protein(s), such as Sae2/Com1, the null mutant of which also accumulated unprocessed DSBs during meiosis (31, 36).
Meiotic recombination was induced to a certain extent in rad50S and mre11S, but not in mre11-58 (Fig. 5) (36). Kinetic experiments in mitosis showed that the rad50S mutation does not affect repair of HO endonuclease-induced DSBs (Fig. 6B and C). The defect of rad50S seems to be specific to processing of meiotic DSBs with protected ends. The Mre11 complex consisting of Rad50S or Mre11S is presumed to retain phosphoesterase activity because the phosphoesterase consensus is not altered in these mutants. The meiosis-specific defect of these strains can be explained by assuming a lack of interaction between the Mre11 complex and Sae2/Com1. Even without Sae2/Com1 proteins, the Mre11 complex could slowly process Spo11-bound DSB ends, or the Mre11 complex containing Rad50S or Mre11S could interact weakly with Sae2/Com1. By contrast, since mre11-58S mutation is in the phosphodiestrase consensus sequence, which is essential for exonuclease activity, it is natural that the Mre11 complex lose this activity almost completely.
Mitotic recombination in mre11-58 mutants.
The mre11-58 mutant is proficient in mitotic recombination but not in meiotic recombination. Spontaneous recombination occurs at a frequency higher than that in a wild-type strain (Fig. 5), and mating-type switching also occurs at the same level as that for a wild-type strain, although mating-type conversion was delayed by about approximately 1.5 h in comparison with that for the wild-type strain (Fig. 6B and C). This characteristic suggests the presence of another exonuclease which can process DSB ends in mitosis. The activity of the exonuclease to lead recombinant formation should be significantly lower in rate than that in the Mre11 complex but should be enough to attain a wild-type level at least. One of the candidate genes of the exonuclease may be the EXO1 gene (12, 17), which is a multicopy suppressor of the MMS sensitivities of mre11 and rad50 deletions (45a). Why does another exonuclease not substitute for the meiotic recombination defect of the mre11-58 mutation? It may be because protection of the 5′ ends of DSBs blocks other exonucleases in the mre11-58 as well as rad50S mutants. Therefore, the removal of covalently attached Spo11 at the 5′ ends of the DSBs should be a critical step in the process of meiotic recombination.
The defect in repair of MMS damage in the mre11-58 mutant.
The mre11-58 mutant showed high MMS sensitivity, although it was proficient in mitotic recombination. This is also true of mre11, rad50, and xrs2 null mutants. Assuming that homologous recombination is not sufficient for repairing MMS-damaged DNA, how is the Mre11 complex involved in the repair process? One possibility is that certain DSBs can be repaired only with the aid of the exonuclease activity of the Mre11 complex. Unlike HO endonuclease, MMS may cause a wide variety of DNA lesions, some with chemical adducts at the ends of strand breaks (13). Such adducts may block entry of other types of exonuclease, such as ExoI, and would be removed only by the activity of Mre11. Alternatively, the Mre11 complex may have another activity in DSB repair, i.e., DSB end-to-end joining. Moore and Haber showed that DSBs in S. cerevisiae are repaired by nonhomologous end joining as well as by homologous recombination (32).
In conclusion, the meiotic process in which Mre11 is involved can be separated into three steps. First is DSB formation, in which the Mre11 complex, including Spo11 and probably other meiotic proteins, induces DSB formation at recombination hot spots. This reaction is catalyzed by Spo11 (22). The second step is a meiosis-specific process necessary for removing Spo11 bound to the 5′ ends of DSBs. The third step is nucleolytic processing of DSB ends, which also occurs during mitosis. During this process, the free 5′ ends of the DSBs are resected, producing a long stretch of 3′-ended, single-stranded DNA.
ACKNOWLEDGMENTS
We thank J. Tomizawa, T. Ogawa, A. Shinohara, H. Masukata, and T. Nakagawa for helpful discussion and critical reading of the manuscript; D. K. Bishop and M. Lichten for critical reading of the manuscript; H. Oshiumi for providing strains and plasmids; M. Sekiguchi for technical assistance; and members of the Ogawa laboratory for helpful discussion. We also thank N. Kleckner and J. E. Haber for providing yeast strains and plasmids.
This work was supported by a grant-in aid for Specially Promoted Research (06101003) from the Ministry of Education, Science, Sports and Culture of Japan, by the Howard Hughes Medical Institute and by the Human Frontier Science Program.
REFERENCES
- 1.Ajimura M, Leem S-H, Ogawa H. Identification of new genes required for meiotic recombination in Saccharomyces cerevisiae. Genetics. 1993;133:51–66. doi: 10.1093/genetics/133.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alani E, Cao L, Kleckner N. A method for gene disruption that allows repeated use of URA3 selection in the construction of multiply disrupted yeast strains. Genetics. 1987;116:541–545. doi: 10.1534/genetics.112.541.test. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alani E, Padmore R, Kleckner N. Analysis of wild-type and rad50 mutants of yeast suggests an intimate relationship between meiotic chromosome synapsis and recombination. Cell. 1990;61:419–436. doi: 10.1016/0092-8674(90)90524-i. [DOI] [PubMed] [Google Scholar]
- 3a.Baum B. Mre11 and S. pombe Rad32 are phosphoesterases. Yeast Update Newsl. 1995;3.7:1. [Google Scholar]
- 4.Bergerat A, de Massy B, Gadelle D, Varoutas P-C, Nicolas A, Forterre P. An atypical topoisomerase II from archaea with implications for meioric recombination. Nature. 1997;386:414–417. doi: 10.1038/386414a0. [DOI] [PubMed] [Google Scholar]
- 5.Cao L, Alani E, Kleckner N. A pathway for generation and processing of double-strand breaks during meiotic recombination in S. cerevisiae. Cell. 1990;61:1089–1101. doi: 10.1016/0092-8674(90)90072-m. [DOI] [PubMed] [Google Scholar]
- 6.Chepurnaya O V, Kozhin S A, Peshekhonov V T, Korolev V G. RAD58 (XRS4)—a new gene in the RAD52 epistasis group. Curr Genet. 1995;28:274–279. doi: 10.1007/BF00309787. [DOI] [PubMed] [Google Scholar]
- 7.Connelly J C, Leach D R F. The sbcC and sbcD genes of Escherichia coli encode a nuclease involved in palindrome inviability and genetic recombination. Genes Cells. 1996;1:285–291. doi: 10.1046/j.1365-2443.1996.23024.x. [DOI] [PubMed] [Google Scholar]
- 8.Connolly B, White C I, Haber J E. Physical monitoring of mating type switching in Saccharomyces cerevisiae. Mol Cell Biol. 1988;8:2342–2349. doi: 10.1128/mcb.8.6.2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de Massy B, Rocco V, Nicolas A. The nucleotide mapping of DNA double-strand breaks at the CYS3 initiation site of meiotic recombination in Saccharomyces cerevisiae. EMBO J. 1995;14:4589–4598. doi: 10.1002/j.1460-2075.1995.tb00138.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Esposito R E, Esposito M S. Genetic recombination and commitment to meiosis in Saccharomyces. Proc Natl Acad Sci USA. 1974;71:3172–3176. doi: 10.1073/pnas.71.8.3172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fast D. Sporulation synchrony of Saccharomyces cerevisiae grown in various carbon sources. J Bacteriol. 1973;116:925–930. doi: 10.1128/jb.116.2.925-930.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fiorentini P, Huang K N, Tishkoff D X, Kolodner R D, Symington L S. Exonuclease I of Saccharomyces cerevisiae functions in mitotic recombination in vivo and in vitro. Mol Cell Biol. 1997;17:2764–2773. doi: 10.1128/mcb.17.5.2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Friedberg E C, Walker G C, Siede W. DNA repair and mutagenesis. Washington, D.C: ASM Press; 1995. [Google Scholar]
- 14.Game J C, Mortimer R K. A genetic study of X-ray sensitive mutants in yeast. Mutat Res. 1974;24:281–292. doi: 10.1016/0027-5107(74)90176-6. [DOI] [PubMed] [Google Scholar]
- 15.Gietz R D, Sugino A. New yeast-Escherichia coli shuttle vectors constructed with in vitro mutagenized yeast genes lacking six-base pair restriction sites. Gene. 1988;74:527–534. doi: 10.1016/0378-1119(88)90185-0. [DOI] [PubMed] [Google Scholar]
- 16.Goldway M, Sherman A, Zenvirth D, Arbel T, Simchen G. A short chromosomal region with major roles in yeast chromosome III meiotic disjunction, recombination and double strand breaks. Genetics. 1993;133:159–169. doi: 10.1093/genetics/133.2.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang K N, Symington L S. A 5′-3′ exonuclease from Saccharomyces cerevisiae is required for in vitro recombination between linear DNA molecules with overlapping homology. Mol Cell Biol. 1993;13:3125–3134. doi: 10.1128/mcb.13.6.3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ivanov E L, Korolev V G, Fabre F. XRS2, a DNA repair gene of Saccharomyces cerevisiae, is needed for meiotic recombination. Genetics. 1992;132:651–664. doi: 10.1093/genetics/132.3.651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ivanov E L, Sugawara N, Fishman-Lobell J, Haber J E. Genetic requirements for the single-strand annealing pathway of double-strand break repair in Saccharomyces cerevisiae. Genetics. 1995;142:693–704. doi: 10.1093/genetics/142.3.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ivanov E L, Sugawara N, White C I, Fabre F, Haber J E. Mutations in XRS2 and RAD50 delay but do not prevent mating-type switching in Saccharomyces cerevisiae. Mol Cell Biol. 1994;14:3414–3425. doi: 10.1128/mcb.14.5.3414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johzuka K, Ogawa H. Interaction of Mre11 and Rad50: two proteins required for DNA repair and meiosis-specific double-strand break formation in Saccharomyces cerevisiae. Genetics. 1995;139:1521–1532. doi: 10.1093/genetics/139.4.1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Keeney S, Giroux C N, Kleckner N. Meiosis-specific DNA double-strand breaks are catalyzed by Spo11, a member of a widely conserved protein family. Cell. 1997;88:375–384. doi: 10.1016/s0092-8674(00)81876-0. [DOI] [PubMed] [Google Scholar]
- 23.Keeney S, Kleckner N. Covalent protein-DNA complexes at the 5′ strand termini of meiosis-specific double-strand breaks in yeast. Proc Natl Acad Sci USA. 1995;92:11274–11278. doi: 10.1073/pnas.92.24.11274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Klapholz S, Esposito R E. Isolation of spo12-1 and spo13-1 from a natural variant of yeast that undergoes a single meiotic division. Genetics. 1980;96:567–588. doi: 10.1093/genetics/96.3.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kozhin S A, Chepurnaya O V, Korolev V G. The RAD58 (XRS4) gene: map position on the right arm of chromosome XIII. Yeast. 1995;11:1211–1213. doi: 10.1002/yea.320111211. [DOI] [PubMed] [Google Scholar]
- 26.Liu J, Wu T C, Lichten M. The location and structure of double-strand DNA breaks induced during yeast meiosis: evidence for a covalently linked DNA-protein intermediate. EMBO J. 1995;14:4599–4608. doi: 10.1002/j.1460-2075.1995.tb00139.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Malone R E. Multiple mutant analysis of recombination in yeast. Mol Gen Genet. 1983;189:405–412. doi: 10.1007/BF00325902. [DOI] [PubMed] [Google Scholar]
- 28.Malone R E, Bullard S, Hermiston M, Rieger R, Cool M, Galbraith A. Isolation of mutants defective in early steps of meiotic recombination in the yeast Saccharomyces cerevisiae. Genetics. 1991;128:79–88. doi: 10.1093/genetics/128.1.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Malone R E, Esposito R E. Recombinationless meiosis in Saccharomyces cerevisiae. Mol Cell Biol. 1981;1:891–901. doi: 10.1128/mcb.1.10.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Malone R E, Ward T, Lin S, Waring J. The RAD50 gene, a member of the double strand break repair epistasis group, is not required for spontaneous mitotic recombination in yeast. Curr Genet. 1990;18:111–116. doi: 10.1007/BF00312598. [DOI] [PubMed] [Google Scholar]
- 31.McKee A H, Kleckner N. A general method for identifying recessive diploid-specific mutations in Saccharomyces cerevisiae, its application to the isolation of mutants blocked at intermediate stages of meiotic prophase and characterization of a new gene SAE2. Genetics. 1997;146:797–816. doi: 10.1093/genetics/146.3.797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moore J K, Haber J E. Cell cycle and genetic requirements of two pathways of nonhomologous end-joining repair of double-strand breaks in Saccharomyces cerevisiae. Mol Cell Biol. 1996;16:2164–2173. doi: 10.1128/mcb.16.5.2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nairz K, Klein F. mre11S—a yeast mutation that blocks double-strand-break processing and permits nonhomologous synapsis in meiosis. Genes Dev. 1997;11:2272–2290. doi: 10.1101/gad.11.17.2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ogawa T, Shinohara A, Nabetani A, Ikeya T, Yu X, Egelman E H, Ogawa H. RecA-like recombination proteins in eukaryotes: functions and structures of RAD51 genes. Cold Spring Harbor Symp Quant Biol. 1993;58:567–576. doi: 10.1101/sqb.1993.058.01.063. [DOI] [PubMed] [Google Scholar]
- 35.Petes T D, Malone R E, Symington L S. Recombination in yeast. In: Broach J, Pringle J, Jones E, editors. The molecular and cellular biology of the yeast Saccharomyces cerevisiae: genome dynamics, protein synthesis and energetics. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory; 1991. pp. 407–521. [Google Scholar]
- 36.Prinz S, Amon A, Klein F. Isolation of COM1, a new gene required to complete meiotic double-strand break-induced recombination in Saccharomyces cerevisiae. Genetics. 1997;146:781–795. doi: 10.1093/genetics/146.3.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rose M D, Novick P, Thomas J H, Botstein D, Fink G R. A Saccharomyces cerevisiae genomic plasmid bank based on a centromere-containing shuttle vector. Gene. 1987;60:237–243. doi: 10.1016/0378-1119(87)90232-0. [DOI] [PubMed] [Google Scholar]
- 38.Rothstein R J. One-step gene disruption in yeast. Methods Enzymol. 1983;101:202–211. doi: 10.1016/0076-6879(83)01015-0. [DOI] [PubMed] [Google Scholar]
- 39.Sambrook J, Fritsch E F, Maniatis T. Molecular cloning: a laboratory manual. 2nd ed. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory; 1989. [Google Scholar]
- 40.Sharples G J, Leach D R. Structural and functional similarities between the SbcCD proteins of Escherichia coli and the RAD50 and MRE11 (RAD32) recombination and repair proteins of yeast. Mol Microbiol. 1995;17:1215–1217. doi: 10.1111/j.1365-2958.1995.mmi_17061215_1.x. [DOI] [PubMed] [Google Scholar]
- 41.Sherman F, Roman H. Evidence for two types of allelic recombination in yeast. Genetics. 1963;48:255–261. doi: 10.1093/genetics/48.2.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shinohara A, Ogawa H, Ogawa T. Rad51 protein involved in repair and recombination in S. cerevisiae is a RecA-like protein. Cell. 1992;69:457–470. doi: 10.1016/0092-8674(92)90447-k. [DOI] [PubMed] [Google Scholar]
- 43.Shinohara A, Ogawa T. Homologous recombination and the roles of double-strand breaks. Trends Biochem Sci. 1995;20:387–391. doi: 10.1016/s0968-0004(00)89085-4. [DOI] [PubMed] [Google Scholar]
- 44.Sikorski R S, Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sugawara N, Haber J E. Characterization of double-strand break-induced recombination: homology requirements and single-stranded DNA formation. Mol Cell Biol. 1992;12:563–575. doi: 10.1128/mcb.12.2.563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45a.Tsubouchi, H., and H. Ogawa. Unpublished data.
- 45b.Usui, T., and H. Ogawa. Unpublished observations.
- 46.White C I, Haber J E. Intermediates of recombination during mating type switching in Saccharomyces cerevisiae. EMBO J. 1990;9:663–673. doi: 10.1002/j.1460-2075.1990.tb08158.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xu L, Weiner B M, Kleckner N. Meiotic cells monitor the status of the interhomolog recombination complex. Genes Dev. 1997;11:106–118. doi: 10.1101/gad.11.1.106. [DOI] [PubMed] [Google Scholar]