

Abstract
Transgenic mice expressing the polyomavirus (PyV) middle T antigen (MT) develop multifocal mammary tumors which frequently metastasize to the lung. The potent transforming activity of PyV MT is correlated with its capacity to activate and associate with a number of signaling molecules, including the Src family tyrosine kinases, the 85-kDa Src homology 2 subunit of the phosphatidylinositol 3′ (PI-3′) kinase, and the Shc adapter protein. To uncover the role of these signaling proteins in MT-mediated mammary tumorigenesis, we have generated transgenic mice that express mutant PyV MT antigens decoupled from either the Shc or the PI-3′ kinase signaling pathway. In contrast to the rapid induction of metastatic mammary tumors observed in the strains expressing wild-type PyV MT, mammary epithelial cell-specific expression of either mutant PyV MT resulted in the induction of extensive mammary epithelial hyperplasias. The mammary epithelial hyperplasias expressing the mutant PyV MT defective in recruiting the PI-3′ kinase were highly apoptotic, suggesting that recruitment of PI-3′ kinase by MT affects cell survival. Whereas the initial phenotypes observed in both strains were global mammary epithelial hyperplasias, focal mammary tumors eventually arose in all female transgenic mice. Genetic and biochemical analyses of tumorigenesis in the transgenic strains expressing the PyV MT mutant lacking the Shc binding site revealed that a proportion of the metastatic tumors arising in these mice displayed evidence of reversion of the mutant Shc binding site. In contrast, no evidence of reversion of the PI-3′ kinase binding site was noted in tumors derived from the strains expressing the PI-3′ kinase binding site MT mutant. Tumor progression in both mutant strains was further correlated with upregulation of the epidermal growth factor receptor family members which are known to couple to the PI-3′ kinase and Shc signaling pathways. Taken together, these observations suggest that PyV MT-mediated tumorigenesis requires activation of both Shc and PI-3′ kinase, which appear to be required for stimulation of cell proliferation and survival signaling pathways, respectively.
Mammary epithelial cell-specific expression of the polyomavirus (PyV) middle T (MT) oncogene in transgenic mice results in the induction of multifocal metastatic mammary tumors involving 100% of the transgene carriers (19). The potent oncogenic properties of the PyV MT are due to its ability to associate with and activate a number of cellular signaling proteins. One class of cellular enzymes activated by PyV MT consists of members of the Src family tyrosine kinases (c-Src and c-Yes) (8, 12, 27, 30). Expression of PyV MT in mammary glands of Src-deficient mice rarely results in the induction of mammary tumors (20), suggesting that activation of Src by PyV MT is required for mammary tumorigenesis. Whereas activation of c-Src is required for the rapid induction of mammary tumors, this event is not sufficient, because expression of a constitutively active version of Src in the mammary glands of transgenic mice rarely leads to tumorigenesis. Instead, activated c-Src induces mammary epithelial hyperplasias that rarely progress to full malignancy (53).
One possible explanation for these observations is that in addition to activation of the Src tyrosine kinase, PyV MT must recruit additional cellular signaling pathways to effect malignant transformation of the mammary epithelial cell. Indeed, PyV MT is known to physically associate with and influence the activity of other cellular proteins known to be involved in proliferative signal transduction. In particular, PyV MT can associate with the 85-kDa regulatory subunit of the phosphaditylinositol 3′ (PI-3′) kinase, resulting in its enzymatic activation (11, 54). The association of PI-3′ kinase with PyV MT is thought to occur through the binding of p85 Src homology 2 domains with specific tyrosine phosphorylation sites (tyrosine residues 315 and 322) located in PyV MT (11, 54). More recently, specific complexes between the Shc adapter protein and PyV MT antigen have been reported (5, 14). These protein complexes occur through the binding of the Shc protein phosphotyrosine binding (PTB) domain to the tyrosine-phosphorylated NPTY motif located in PyV MT-coding sequences (MT residues 247 to 250). The importance of these PyV MT protein complexes in cellular transformation is supported by the observation that mutations that affect either tyrosine residue 250 or 315 and 322 in PyV MT-coding sequences interfere with binding of Shc or PI-3′ kinase, respectively, and result in a dramatic impairment of the transforming potential of the PyV MT oncogene in vitro (31). In addition to these PyV MT-associated proteins, stable complexes between protein phosphatase 2A (regulatory), protein phosphatase 2C (catalytic), phospholipase γ C (PLCγ), and 14-3-3 proteins have also been observed (36, 37, 47, 52). However, the significance of these associated proteins in PyV MT-mediated transformation is not known.
Previous studies with PyV MT mutants defective in their capacity to couple with either the Shc or PI-3′ kinase have indicated that recruitment of both of these signaling proteins is required for efficient transformation of established fibroblasts in vitro (31). Association of Shc with PyV MT results in tyrosine phosphorylation of Shc at tyrosine residues 239, 240, and 317, which in turn allows Shc to couple to a number of downstream signaling molecules (18, 50). In particular, tyrosine phosphorylation of Shc on tyrosine 317 leads to the recruitment of the Grb-2–SOS–Ras complex (42). Indeed, it has been demonstrated that activation of Ras is required for PyV MT-mediated transformation (22). Although tyrosine phosphorylation of Shc on tyrosines 239 and 240 also results in the recruitment of Grb-2, it has also been implicated in binding several other distinct tyrosine-phosphorylated proteins (50). In this regard, it has recently been reported that phosphorylation of Shc on tyrosines 239 and 240 may be involved in activating an antiapoptotic pathway acting independently of the Ras signaling pathway (18). However, the identities of these Shc-associated tyrosine-phosphorylated proteins are unclear (50).
Whereas binding of PyV MT to the Shc adapter protein leads to activation of the Ras signaling pathway, the interaction of PyV MT with the PI-3′ kinase through PyV MT tyrosine residues 315 and 322 results in stimulation of PI-3′ kinase activity, leading to the generation of 3′-phosphinositide lipid second messengers (11, 54). Moreover, activation of the PI-3′ kinase by PyV MT appears to be critical for tumorigenesis, since viral mutants lacking the MT PI-3′ kinase binding site are severely debilitated in their capacity to induce tumors in animals (16, 49). Recent studies have demonstrated that these phosphoinositide lipids lead to specific activation of Akt and S6 serine kinases, which may provide an important cell survival signal (13, 23, 26, 29). In addition, there is also evidence to suggest that activation of the PI-3′ kinase may lead to downstream activation of the Rac GTP binding protein (41). Therefore, activation of the PI-3′ kinase by PyV MT ultimately may influence cellular transformation by affecting both actin cystoskeleton and cell survival pathways (23, 24, 29, 41, 55).
Given that the role of certain signaling pathways, such as the c-Src pathway, is highly dependent on the tissue context (20, 25), the function of the PI-3′ kinase and Shc signaling molecules in PyV MT-mediated mammary tumorigenesis remains to be elucidated. To investigate the role of the PyV MT-coupled PI-3′ kinase or Shc signaling molecules in mammary tumorigenesis, transgenic mice carrying mutant PyV MT antigens decoupled from either Shc or PI-3′ kinase signaling molecules under the transcriptional control of the mouse mammary tumor virus (MMTV) long terminal repeat (LTR) were derived. Mammary gland-specific expression of either mutant PyV MT cDNA resulted in the induction of widespread mammary epithelial hyperplasias. Interestingly, the mammary epithelial hyperplasias derived from the mutant PyV MT defective in its ability to associate with the PI-3′ kinase are highly apoptotic. Although the initial phenotype exhibited by both classes of transgenic mice was global mammary epithelial hyperplasia, focal mammary tumors eventually arose in both PyV MT mutant strains with 100% penetrance. In a subset of mammary tumors arising in transgenic mice expressing the PyV MT Shc binding mutant, tumorigenesis involves reversion of the defective Shc binding site in the mutant PyV MT to its wild-type configuration. In contrast, tumorigenesis in transgenic mice expressing the PyV MT mutant decoupled from the PI-3′ kinase did not occur through reversion of the introduced mutations in the transgene. The progression of the mammary epithelial hyperplasias to tumors was also frequently associated with upregulation of both the ErbB-2 and ErbB-3 receptor tyrosine kinases, which also recruit the PI-3′ kinase and Shc signaling pathways. Taken together, these observations suggest that activation of Shc and PI-3′ kinase plays a critical role in mammary tumor progression by modulating both cell proliferation and apoptosis.
MATERIALS AND METHODS
DNA constructions.
To construct the MMTV/MT-Y315/22F mutant, PyV MT cDNA derived from plasmid pMMTV/MT was subcloned into the HindIII and EcoRI sites of Bluescript KS and subjected to standard M13 mutagenesis with oligonucleotides AB1712 (TTGGCATGAACTCCTCC) and AB1713 (TGTCCAAAAACAGATCC), resulting in conversion of wild-type MT tyrosine residues 315 and 322 to phenylalanine residues. The mutant sequences were confirmed by automated DNA sequence analyses. The mutant cDNA was subsequently cloned directionally into the HindIII-EcoRI sites of the MMTV expression vector (19).
To construct the pMMTV/MT-Y250F mutant, oligonucleotides AB3705 (CCAGCGGTTCTGCAGAATGCC), AB3706 (TCATAACAGAAAAGGTCGGG), AB3595 (GCCTAAGACTGCCGAGTCTTCTGAGCAACCCGACCTTTTCTGTTATG), and AB3596 (ATGAGCCCTCTGCAAATCCCGAAGAATCAGACCCTCCCATGG) were employed in a PCR-based strategy to generate a tyrosine-to-phenylalanine residue substitution at amino acid residue 250 of the PyV MT sequence. Briefly, matched sets of oligonucleotides harboring the necessary nucleotide changes were used to amplify sequences upstream (AB3705-AB3706) and downstream (AB3595-AB3596) of the target mutation site. These PCR products overlap and were subsequently PCR amplified with oligonucleotides AB3705 and AB3596 to generate a PCR product bearing the desired mutation. Automated DNA sequence analyses confirmed the presence of the nucleotide substitution allowing for the coding of a phenylalanine residue at MT site 250. Moloney murine leukemia virus (Mo-MuLV)-based MT and MT mutant expression cassettes were generated by subcloning the desired MT cDNA from the MMTV-derived plasmids via unique HindIII and EcoRI sites into the corresponding site of Mo-MuLV-based expression cassette pJ4-ω (a gift from B. Rowley).
The PyV MT antigen and PGK-1 ribonucleotide protection probe (riboprobe) pSP65mT (MTR) were generous gifts from M. Rudnicki and J. Hassell (19). The MTsn301 riboprobe used to distinguish between MT mutants MT-Y315/22F and MT-Y250F contains a 513-nucleotide fragment (bounded by nucleotides 727 to 1240) in the SphI and NcoI sites of plasmid vector pSL301 (Promega). The simian virus 40 (SV40) polyadenylation-specific riboprobe (SPA) contains the SV40 polyadenylation signals (SV40 nucleotides 2536 to 2773 and 4103 to 4713) in the BamHI and HindIII sites of Bluescript KS (Stratagene). After cleavage of MTR and SPA with HindIII, PGK-1 with EcoNI, and MTsn301 with XbaI, the antisense riboprobes was generated in vitro as described previously (34). All oligonucleotide syntheses and automated DNA sequencing were performed by Dinsdale Gooden and Brian Allore of the MOBIX Main Central Facility, McMaster University.
Generation and identification of transgenic mice.
DNA was prepared for microinjection by digestion with 4 U of SalI and SpeI per μg for 1.5 h. The DNA was electrophoresed through a 1% agarose gel and purified as described previously (43). Superovoluated FVB/N female mice (Taconic Farms, Germantown, Pa.) were mated with FVB/N males the night before injection. After isolation of the fertilized one-cell mouse embryos, the pronuclei of these zygotes were injected with 0.5 to 1 pl of DNA solution (5 μg/ml). Following microinjection, viable eggs were transferred to the oviducts of pseudopregnant Swiss-Webster mice (Taconic Farms).
To identify transgenic progeny, genomic DNA was extracted from 1.5-cm tail clippings as described by Muller et al. (34). The nucleic acid pellet was resuspended in 100 μl of distilled water at approximately 1 μg/ml, and 15 μl of the DNA solution was digested with 30 U of BamHI for 1.5 h. Following gel electrophoresis and Southern blot transfer (45) to GeneScreen filters (Dupont), the filters were hybridized with transgene radiolabeled with [α-32P]dCTP (Dupont) by random priming. Radiolabeled probes derived from the PyV MT cDNA were used to identify both MMTV/MT transgenic strains.
RNA analysis.
RNA was isolated from various tissues by using the guanidium isothiocyanate modification of CsCl gradient sedimentation described by Chirgwin et al. (10). Tissue was flash frozen in liquid nitrogen and stored at −80°C or immediately homogenized in 3 ml of guanidine isothiocyanate (GIT) (Bethesda Research Laboratories) solution (4 M GIT, 25 mM sodium citrate, and 0.1 M β-mercaptoethanol). The homogenate was layered onto 4 ml of 5.7 M CsCl containing 25 mM sodium acetate (pH 5.2), and RNA was pelleted by ultracentrifugation at 32,000 rpm and 20°C with an SW41Ti rotor (Beckman) for 24 h. GIT and CsCl layers were aspirated off, and the RNA pellet was resuspended in 500 μl of sterile water plus 300 mM sodium acetate (pH 5.2) and precipitated with 2 volumes of ice-cold ethanol. The RNA yield was determined by UV absorption at 260 nm (1 optical density unit = 40 mg of RNA per ml).
RNase protection assays were performed as described by Melton et al. (33), using 20 μg of total cellular RNA per tissue (except where noted) incubated with 1 ml of hybridization buffer [80% formamide, 40 mM piperazine-N,N′-bis(2-ethanesulfonic acid) (PIPES) (pH 6.4), 1 mM EDTA (pH 8.0), and 400 mM NaCl] at 85°C for 5 min. The hybridization reaction mixture was then allowed to anneal for at least 8 h at 50°C and subjected to RNase digestion as described previously (34). To detect pyrimidine mismatches or deletions, the hybridization reaction mixtures were digested with 120 mg of RNase A per ml in the absence of RNase T1 for 30 min at 37°C. Digests were terminated by addition of 20 to 30 μg of RNase-free tRNA and 500 μl of phenol-chloroform (1:1) followed by ethanol precipitation. RNA pellets were dried for 10 min under vacuum, resuspended in 10 μl of formamide loading buffer (80% formamide, 10 mM EDTA [pH 8.0], 1 mg of xylene cyanol FF per ml, 1 mg of bromophenol blue per ml) boiled for 7 min at 95°C, and resolved in a 6% urea–polyacrylamide gel (40% acrylamide–2% N,N′-methylene-bisacrylamide, 7 M urea, 0.001% ammonium persulfate, and 0.0005% N,N,N′,N′-tetramethylethylenediamine [TEMED]) electrophoresed at 60 to 80 A in 1× TBE running buffer (0.1 M Tris, 0.08 M boric acid, 0.002 M EDTA, pH 8.0). The gel was dried and exposed at −70°C to Kodak XAR-5 film in the presence of intensifying screens.
Antibodies.
Antibodies used include mouse monoclonal antibody pAb762 and rat monoclonal antibody pAb701 for PyV MT (a generous gift from S. Dilworth, ICRF London), rabbit polyclonal antibody N16 (Transduction Laboratories), and mouse monoclonal antibody 7D10 (Quality Biotech) for Src. Also used in these studies were antibodies specific for Shc (rabbit polyclonal and mouse monoclonal) (Transduction Laboratories), the p85 subunit of PI-3′ kinase (rabbit polyclonal) (Transduction Laboratories), and PLCγ1 (mouse monoclonal) (Upstate Biotechnology Inc.). Antibodies specific for ErbB-2 and ErbB-3 were obtained from Oncogene Sciences (AB-3) and Santa Cruz Biotechnology (C-17), respectively.
Protein extract preparation.
Tissue samples were flash frozen in liquid nitrogen and ground to a fine powder with a chilled mortar and pestle. Cells were lysed on ice for 30 min in TNE lysis buffer (20 mM Tris [pH 8.0], 150 mM NaCl, 1% Nonidet P-40, 2.5 mM EDTA, 1 mM sodium orthovanadate, 10 mM sodium fluoride, 10 μg of aprotinin per ml, and 10 μg leupeptin per ml) with constant agitation. Lysates were cleared by two consecutive centrifugations at 13,000 × g for 5 min each time. Supernatants were removed, and their protein concentrations were determined with the Bradford assay kit (Bio-Rad).
Immunoblot analyses.
Unless otherwise specified, a total of 100 μg of total protein lysate was used for each sample analyzed. An equal volume of 2× protein sample loading buffer (62.5 mM Tris [pH 6.8], 2% sodium dodecyl sulfate, 10% glycerol, 5% β-mercaptoethanol, 0.02% bromophenol blue) was added, and mixtures were boiled for 10 min at 95°C. Proteins were resolved in sodium dodecyl sulfate-polyacrylamide gels and transferred electrophoretically onto polyvinylidine difluoride membranes (Immobilon-P; Millipore). Membranes were incubated overnight in 3% powdered skim milk in TBS (20 mM Tris [pH 7.5], 150 mM NaCl, 5 mM KCl) or 3% bovine serum albumin (Sigma) in TBS for antiphosphotyrosine immunoblots. Membranes were subsequently incubated for 2 h at room temperature with antibodies (1:1,000 for all monoclonal antibody preparations and 1:250 for all polyclonal antibody preparations). After being washed four times (10 min each time) in TBS plus 0.01% Tween 20, membranes were incubated for 1 h at room temperature with the appropriate secondary antibody conjugated to horseradish peroxidase (Biocan Scientific) at 1:2,500. The membranes were washed four more times in TBS plus 0.01% Tween 20, and proteins were detected by using the enhanced chemiluminescence detection system (ECL; Amersham).
Immunoprecipitations.
Immunoprecipitations were performed by preincubating antigen-specific antibody (1 to 2 μg for monoclonal; 5 to 10 μg for polyclonal) with 30 to 40 μl of protein G-Sepharose Fast Flow (Pharmacia) in 800 μl of 1× phosphate-buffered saline (140 mM NaCl, 2.7 mM KCl, 4.3 mM Na2HPO4, 1.4 mM KH2PO4) for 2 to 12 h at 4°C on a rotating platform. Antibody-bound beads were washed once with 1 ml of phosphate-buffered saline and once with 1 ml of lysis buffer. Total protein lysate (500 μg to 1 mg) was added to a total volume of 700 μl and incubated with the prebound beads for 1 to 5 h at 4°C on a rotating platform. The beads were subsequently washed five times in lysis buffer, following which bound antigen could be analyzed.
Histological evaluation and in situ apoptosis assays.
Complete autopsies were performed, and both gross and microscopic examinations were done. Five mice from each time point (4, 8, 12, and 16 weeks) from transgenic strains MMTV/MT, MMTV/MT-Y250F, and MMTV/MT-Y315/22F and nontransgenic strain FVB/N were analyzed. Upper left mammary fat pad tissues were fixed in 4% paraformaldehyde, blocked in paraffin, sectioned at 5 μm, stained with hematoxylin and eosin, and examined. Whole-mount preparations were prepared with the upper right mammary fat pad as described by Vonderhaar and Greco (51). Briefly, resected tissue was spread out on glass slides and allowed to air dry overnight. After drying, the mammary glands were fixed by overnight incubation in acetone. The glands were then squeezed between glass slides and placed in fresh acetone the next morning. Harris modified hematoxylin was added for overnight staining of the glands. Destain solution (1% concentrated HCl in 75% ethanol) was added and discarded until the epithelial component of the mammary gland was seen in sharp contrast to the light background of the fat pad. The stain was fixed with a 30-s wash in 0.002% ammonium hydroxide. Slides were then transferred to 75% ethanol for 5 min and then to 100% ethanol for 3 h. Glands were cleared overnight in xylenes and mounted in Permount. In situ apoptosis assays were performed with the Apopttag In Situ Apoptosis Detection Kit (Oncor) as described previously (17). Analyses of apoptotic cell death in the 1A2 PyV MT-expressing mammary tumor cell line (1) was performed 72 h after exposure of the cells to either control adenoviral beta-galactosidase or Cre expression vectors (multiplicity of infection [MOI], 100).
RESULTS
Isolation and characterization of transgenic mice expressing PyV MT mutants defective in their ability to associate with either the PI-3′ kinase or Shc adapter molecule.
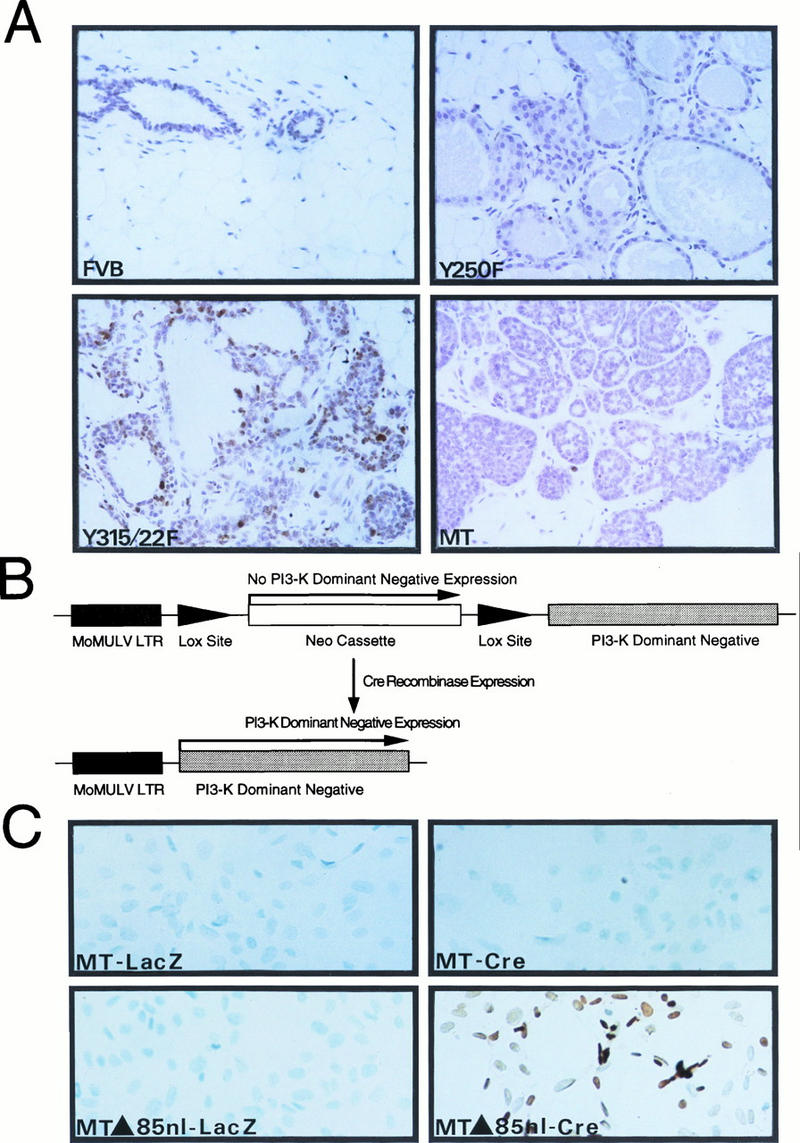
Although previous studies suggested that the association of either PI-3′ kinase or Shc proteins with PyV MT was crucial in inducing cellular transformation in established fibroblasts (31), the role of these signaling molecules in PyV MT-mediated mammary tumorigenesis is unclear. To assess the relative contributions of these signaling molecules to tumorigenesis effected by PyV MT, transgenic mice expressing a mutant lacking either the Shc binding site (MT-Y250F) or the PI-3′ kinase binding sites (MT-Y315/322F) in the mammary epithelium were generated. To accomplish this, the MT-Y315/322F and MT-Y250F mutant MT cDNAs were placed under the transcriptional control of the MMTV promoter-enhancer (Fig. 1A and C) and microinjected into one-cell mouse zygotes. This resulted in the generation of seven MT-Y315/322F transgenic and eight MT-Y250F transgenic founder animals.
FIG. 1.

Tissue specificity of transgene expression in MMTV/MT-Y250F transgenic mice. (A and C) Structures of the MMTV/MT-Y315/322F (A) and MMTV/MT-Y250F (C) transgenes. The Bluescript vector backbone is represented by a thin line on either side of the expression cassette, with the cross-hatched region corresponding to the MMTV LTR derived from plasmid pA9, the stippled portion corresponding to the MT-Y250F cDNA with phenylalanine substitutions at amino acids positions 315 and 322 or amino acid position 250, and the solid region corresponding to the transcriptional processing sequences derived from the SV40 early transcription unit. The transcription start site is indicated by the arrow. (B and D) RNA transcripts corresponding to the MMTV/MT-Y315/322F (B) and MMTV/MT-Y250F (D) transgenes in various organs of the MT-Y315/322F transgenic strain as assessed by RNase protection. Tissues were derived from a virgin tumor-bearing female, a virgin female, and a male. The antisense probe used in this RNase protection analysis (MTR) is complementary to a 203-nucleotide fragment corresponding to the amino terminus of PyV MT and is marked by MT-315/322F and an arrow. Also shown is an RNase protection analysis with identical RNA samples and an antisense probe directed against PGK, which protects a 124-nucleotide fragment indicated by PGK and an arrow. M.Gl., mammary gland.
To examine expression of the transgene in these strains, 20 μg of total cellular RNA was isolated from a variety of tissues and analyzed by RNase protection assays with an antisense riboprobe spanning the first 203 nucleotides of the PyV early region (19). Results from these analyses are summarized in Table 1 for MT-Y250F strains and in Table 2 for MT-Y315/322F strains. Results of representative RNase protection assays from the best-characterized lines from MT-Y315/322F (Db-5) and MT-Y250F (250-5) are shown in Fig. 1B and D, respectively. Consistent with previous studies with MMTV-driven transgenes (6), the highest levels of transgene transcript were detected in the mammary glands or mammary tumors (Fig. 1B, lanes 10 and 16, respectively, and Fig. 1D, lanes 1 and 15, respectively). However, lower levels of transgene expression could be detected in the salivary glands and male reproductive organs, particularly the epididymis and seminal vesicles (Fig. 1D, lanes 11, 12, and 14, for MT-Y250F strain 5a; Table 2 for MT-Y315/22 strain Db-5).
TABLE 1.
Transgene expression and onset of tumors in MMTV/MT-Y250F micea
| Line | Expression of transgeneb in:
|
Avg onset of mammary tumors (days)c | Tumor types and hyperplasias | ||||||
|---|---|---|---|---|---|---|---|---|---|
| M.Gl.T (female) | M.GL.N
|
Sal. | Epidid. | Sem. Ves. | Testes | ||||
| Female | Male | ||||||||
| 250-1 | NA | − | − | − | − | − | − | None | None |
| 250-2 | +++ | +++ | ND | +++ | ND | ND | ND | 136 (26) | M.Gl. adenocarcinomas, male reproductive abnormalities (vas deferens?), hypersensitivity to parasitic infections |
| 250-3 | − | − | − | − | − | − | − | None | None |
| 250-4 | − | − | − | − | − | − | − | None | None |
| 250-5(a) | +++ | +++ | − | ++ | + | ++ | − | 142 (74) | M.Gl. adenocarcinomas |
| 250-5(b) | +++ | ND | ND | ND | ND | ND | ND | 211 (23) | M.Gl. adenocarcinomas |
| 250-6 | NA | − | − | − | − | − | − | None | None |
| 250-7 | NA | − | − | − | − | − | − | None | None |
| 250-8 | NA | + | − | − | − | − | − | None | None |
RNase protection analysis was performed on 20 μg of total RNA isolated from a variety of organs in the MMTV/MT-Y250F strains as described in Materials and Methods.
Relative levels of transgene expression (as determined by quantitative phosphorimager analysis): −, not detected; +, low; ++, intermediate; +++, high. M.Gl.N, normal mammary gland; M.Gl.T, mammary gland tumor; Sal., salivary gland; Epidid., epididymus; Sem. Ves.; seminal vesicles; NA, not applicable; ND, not determined.
The number of animals analyzed is given in parentheses.
TABLE 2.
Transgene expression and onset of tumors in MMTV/MT-Y315/322F micea
| Line | Expression of transgeneb in:
|
Avg onset of mammary tumors (days)c | Tumor types and hyperplasias | ||||||
|---|---|---|---|---|---|---|---|---|---|
| M.Gl.T (female) | m.Gl.N
|
Sal. | Epidid. | Sem. Ves. | Testes | ||||
| Female | Male | ||||||||
| Db-1 | +++ | + | − | − | − | − | − | 255 (17) | M.Gl. adenocarcinomas |
| Db-2 (founder) | NA | ND | ND | ND | ND | ND | ND | None | None |
| Db-3 | +++ | +/− | − | ND | ND | ND | ND | 253 (15) | M.Gl. adenocarcinomas |
| Db-4 | +++ | +++ | − | ++ | − | − | − | 120 (70) | M.Gl. adenocarcinomas, salivary/parotid neoplasms?, early developmental failure to arborize fat pad |
| Db-5 | +++ | +++ | + | +++ | +++ | +++ | − | 107 (55) | M.Gl. adenocarcinomas, salivary/parotid neoplasms?, early developmental failure to arborize fat pad |
| Db-6 | +++ | + | − | − | − | − | − | ∼200 | M.Gl. adenocarcinomas |
| Db-7 (founder) | +++ | NA | NA | ND | NA | NA | NA | 65 (1) | M.Gl. adenocarcinomas |
RNase protection analysis was performed on 20 μg of total RNA isolated from a variety of organs in the MMTV/MT-Y315/322F strains as described in Materials and Methods.
Relative levels of transgene expression (as determined by quantitative phosphorimager analysis): −, not detected; +, low; ++, intermediate; +++, high. M.Gl.N, normal mammary gland; M.Gl.T, mammary gland tumor; Sal., salivary gland; Epidid., epididymus; Sem. Ves.; seminal vesicles; NA, not applicable; ND, not determined.
The number of animals analyzed is given in parentheses.
Female transgenic mice expressing elevated levels of either mutant PyV MT transgene (MT-Y250F or MT-Y315/322F) in the mammary epithelium were incapable of nursing their young. To examine whether this lactation defect was due to aberrant epithelial development, whole-mount analyses was conducted on 12-week-old virgin mammary fat pads derived from FVB/N, MMTV/MT-Y315/322F, and MMTV/MT-Y250F strains (Fig. 2). In contrast to the multifocal mammary tumors arising in the MMTV/wild-type MT strains (19), mammary gland expression of either the MT-Y315/322F or MT-Y250F transgene in 12-week-old virgin animals resulted in the global induction of mammary epithelial hyperplasias (Fig. 2). However, the mammary epithelial hyperplasias present in the MT-Y315/322F strains differed in several aspects from those exhibited by the MT-Y250F strains. For example, the MT-Y250F strains had well-defined alveolar hyperplasias, which resembled those exhibited by MMTV/transforming growth factor α strains (Fig. 2C and D) (32). In contrast, the mammary epithelial hyperplasias in the MT-Y315/322F strains were extremely cystic and dilated without defined alveolar structures (Fig. 2E and F).
FIG. 2.
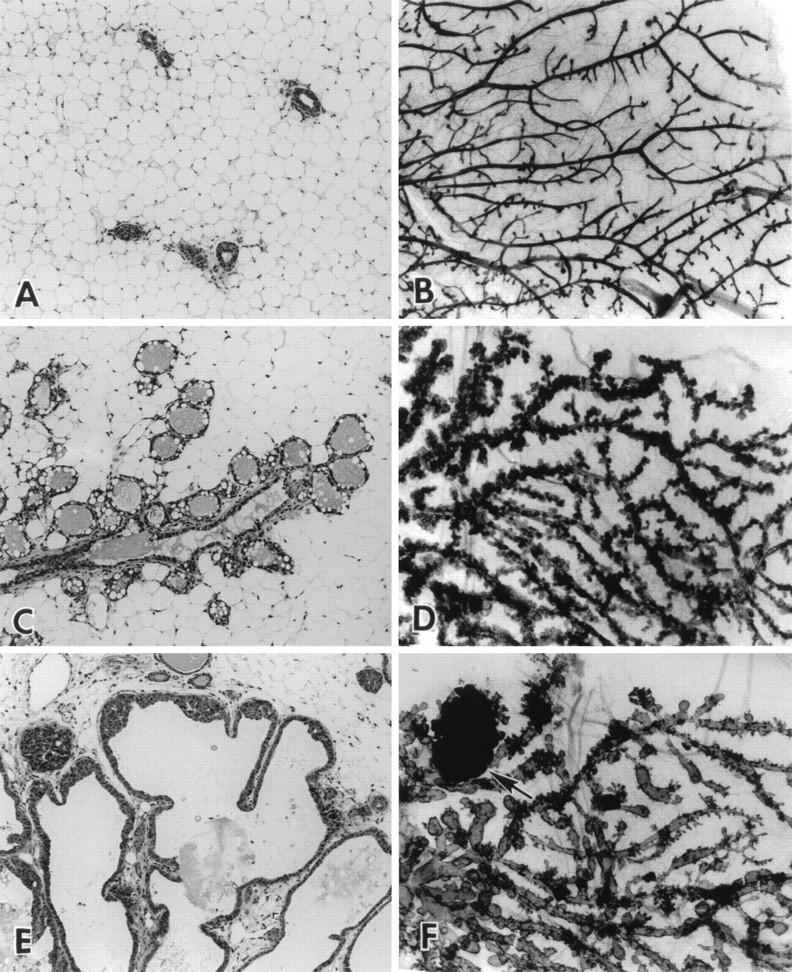
Histological analyses of mammary glands from FVB/N, MMTV/MT-Y250F, and MMTV/MT-Y315/22F transgenic animals. Photomicrographs comparing the histological (A, C, and E) (magnification, ×100) and whole-mount (B, D, and F) (magnification, ×10) appearances of virgin female FVB (A and B), MTY250F (C and D), and MT-Y315/322F (E and F) mice 12 weeks after birth are shown. Note that the mammary tree from the MT-Y250F mouse (C and D) has extensive formation of side buds along the major ducts. The MT-Y315/322F mammary tree has fewer side buds and a more dilated ductal system (E and F) with multilayered epithelium (hyperplasia) (E) and the formation of solid nests of dysplastic cells (F, arrow).
Mammary epithelial hyperplasias induced by the mutant PyV MT-Y315/322F transgene are highly apoptotic.
One unusual histological feature of the mammary epithelial hyperplasias derived from the MT-Y315/322F strains was the distended ductal development, which histologically exhibited features of apoptotic cell death. To test whether decoupling of the PI-3′ kinase from the mutant PyV MT resulted in increased rates of apoptosis, histological samples from age-matched 8-week-old transgenic animals were subjected to an in situ apoptosis assay, which scores for cells displaying extensive DNA fragmentation, a characteristic of cells undergoing apoptotic cell death (TUNEL [terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling] assay) (17, 48). The results of these analyses revealed that mammary epithelial hyperplasias expressing the mutant PyV MT-Y315/322F protein displayed extensive apoptotic cell death (Fig. 3A). In contrast, comparable age-matched mammary epithelial samples from either normal FVB/N, PyV MT-Y250F, or wild-type MT tissues failed to exhibit significant levels of apoptotic cell death (Fig. 3). Consistent with these analyses, similar elevated rates of apoptotic cell death were noted in mammary samples derived from another independently derived transgenic strain expressing the MT-Y315/322F mutant transgene (53a).
FIG. 3.
Activation of the PI-3′ kinase by PyV MT is involved in mammary tumor progression. (A) A panel of slide-mounted Mayer’s hematoxylin-stained mammary tissue sections from age-matched mice from nontransgenic FVB/N or transgenic MMTV-Y250F, MMTV/MT-Y315/322F, and wild-type MT strains. Cells were analyzed for apoptotic cell death as described previously (46). Digoxigenin-labeled DNA ends were detected with horseradish peroxidase-conjugated antidigoxigenin antibodies. Note the multiple apoptotic cells in the epithelial hyperplasias derived from the MT-Y315/322F strain and lack of comparable staining in mammary tissues from FVB/N, MT-Y250F, and wild-type MT mice. (B) Structure of Cre-inducible expression cassette carrying the dominant negative p85 inhibitor. The dark shaded box indicates the Mo-MuLV LTR; the arrows flanking the PGK-Neo cassette represent the LOX recombination sites. The unshaded box indicates the cDNA encoding the mutant p85 subunit of the PI-3′ kinase. (C) In situ apoptosis analyses (TUNEL) conducted with PyV MT mammary tumor cells infected with either a control adenovirus expressing a beta-galactosidase reporter (MT-LacZ) or an adenovirus vector expressing the Cre recombinase (MT-Cre). TUNEL analyses with PyV MT tumor cells possessing the inducible dominant negative p85 inhibitor infected with either the LacZ (MTΔ85nl-LacZ) or Cre adenovirus (MTΔ85nl-Cre) are also shown. Both sets of cells were infected at an MOI of 100. Note the presence of numerous cells undergoing apoptotic cell death in the Cre infection panel.
To further test the importance of the PI-3′ kinase signaling pathway in modulating apoptotic cell death, we derived several independent cell lines that inducibly express a dominant negative inhibitor of the PI-3′ kinase in an established mammary tumor cell line expressing the wild-type PyV MT antigen (1). The basis for the dominant negative action of the mutant PI-3′ kinase derives from a specific mutation in the Src homology 2-bearing p85 subunit, which prevents its association with the 110-kDa catalytic subunit. As a consequence of this mutation, the dominant negative inhibitor can occupy its binding site but is catalytically inert (28). Because stable expression of the p85 dominant negative mutant may not be compatible with cell viability, we isolated an inducible expression cassette in which the expression of the dominant negative inhibitor p85 can be activated by the Cre recombinase (Fig. 3B). The basis for the inducible nature of this mutant p85 derives from the presence of a phosphoglycerate kinase (PGK) promoter-neomycin expression transcription unit between the Mo-MuLV promoter and the p85-coding sequences. Because of the strong polyadenylation termination sequence in the PGK-Neo cassette upstream of the p85-coding sequence, transcription through p85-coding sequences will be substantially reduced. However, due to the presence of LOX recombination sites flanking the PGK-Neo cassette, this interfering sequence can be excised by transiently expressing Cre recombinase, leading to the expression of the dominant p85 inhibitor (Fig. 3B) (53a).
To test whether elevated expression of the p85 dominant negative inhibitor in these mammary tumor cells could induce apoptotic cell death, we derived several independent PyV MT mammary tumor cell lines that carried the inducible p85 dominant negative mutant. To induce expression of this dominant negative mutant, p85 dominant negative mutant-carrying cell lines were infected with either an adenovirus-Cre or control adenovirus–beta-galactosidase expression vector at an MOI of 100 (2) and subjected to TUNEL analyses. These experiments revealed that transient expression of Cre in the p85-carrying cells resulted in the extensive induction of apoptotic cell death (Fig. 3C, MT-Δ85nl-Cre). In contrast, transfection of the control adenovirus-LacZ expression vector failed to induce significant apoptosis in these cell lines (Fig. 3C, MT-Δ85nl-LacZ). In addition, adenoviral expression of either Cre recombinase or LacZ in the parental MT cells failed to induce a comparable apoptotic response (Fig. 3C, MT-LacZ and MT-Cre). Transient transfection of the Cre expression plasmid resulted in similar induction of apoptotic cell death, albeit at lower levels, which directly correlated with lower transfection efficiencies (53a). Thus, abrogation of the PI-3′ kinase signaling pathway in PyV MT-transformed mammary tumor cells results in the induction of apoptotic cell death. Taken together with the TUNEL analyses with the MT-Y315/322F transgenic strains, these data suggest that activation of the PI-3′ kinase by PyV MT may be required to prevent apoptotic cell death during mammary tumor progression.
Biochemical characterization of mammary tumors expressing the mutant PyV MT antigens.
Transgenic mice expressing the wild-type PyV MT rapidly develop multifocal mammary tumors without evidence of a hyperplastic precursor lesion (median age at which tumors were palpable [T50] = 53 days [Fig. 4]) (19). In contrast, transgenic mice expressing either of the mutant PyV MTs developed extensive mammary epithelial hyperplasias. However, both of these strains eventually developed mammary tumors with 100% penetrance (Fig. 4). Indeed, whole-mount analyses of the mammary glands derived from 12-week virgin animals revealed the presence of focal dysplastic lesions in the MT-Y315/322F strains arising next to the mammary epithelial tissues (Fig. 2F). Comparison of the onset of palpable tumors for wild-type MT relative to those for the mutants (Fig. 4) revealed that there was a significant delay in the ability of both MT mutants to induce tumors in vivo. The MT-Y250F-bearing mice demonstrated the longest delay in tumor formation, with a T50 of 145 days, whereas the MT-Y315/22F strain developed mammary tumors at a slightly earlier age (T50 = 123 days). In addition, the tumors that arose in these strains were focal in origin, in contrast to the global multifocal phenotype displayed by the strains expressing wild-type PyV MT (19). Histological examination of these tumors revealed gross differences in both the cellular architecture and differentiation status of the tumors (Fig. 4). Tumors derived from the MMTV/MT-Y315/322F strains appeared to be less differentiated than tumors expressing either wild-type PyV MT or MT-Y250F oncogenes (Fig. 4). The tumors derived from the MT-Y250F strains possessed a large stromal component which resembled that of the primary tumors expressing an activated c-src gene (Fig. 4B) (53), suggesting the possibility that these tumor cells may be acting in a paracrine manner to stimulate stromal cell proliferation.
FIG. 4.

Kinetics of mammary tumor occurrence and histopathology of mammary tumors derived from MMTV/wild-type MT, MMTV/MT-Y250F, and MMTV/MT-Y315/22F transgenic animals. (A to D) Photomicrographs comparing the histology of invasive malignancies from a wild-type PyV MT (A), an MT-Y250F tumor (B), and an MT-Y315/322F tumor (D) (magnification, ×250) with that of a noninvasive dysplasia (C) (magnification, ×100). The preinvasive MT-Y315/322F dysplastic nodule (C) is found at the end of dialated ducts but is a more extreme form of the hyperplasia observed in Fig. 2E and represents the solid nest in Fig. 2F. (E) Age at which a mammary tumor is first palpable in each transgenic strain. Also shown are the number of animals analyzed for each strain (n) and the median age at which tumors are palpable (T50).
Because association of PyV MT with Src family members is critical in effecting transformation (20), it was important to assess whether either PyV MT mutant was impaired in its associated kinase activity. To this end, tumor extracts from both wild-type MT and MT mutants were immunoprecipitated with PyV MT-specific antisera, and in vitro kinase activity was assessed by utilizing acid-denatured enolase as an exogenous substrate (Fig. 5A). Although the levels of PyV MT protein differed between tumors (as measured by 125I immunoblot analysis), careful quantitation of the data indicated that associated kinase activities of the mutant PyV MT proteins were comparable to that of wild-type MT (Fig. 5B). Taken together, these observations strongly argue that the delayed tumorigenesis observed in both mutant PyV MT transgenic strains was not a consequence of the inability to complex and functionally activate c-Src tyrosine kinase family members.
FIG. 5.

PyV MT-associated in vitro kinase activities in mammary tumors of the various PyV MT mutants. (A) PyV MT-associated tyrosine kinase activity in tumors induced in the MT-Y315/322F, MT-Y250F, and wild-type MT strains. Mammary tumor lysates derived from MT-Y315/322F, MT-Y250F, wild-type MT, and control MMTV/Neu (N202) (20) strains were subjected to immunoprecipitation with PyV MT-specific antisera and incubated with γ-32P-labeled ATP in the presence of exogenous enolase substrate. (B) Phosphorimager quantitation of enolase phosphorylation, is normalized to the levels of PyV MT protein. (C) PyV MT-associated PI-3′ kinase activity in tumors induced in the MT-Y315/322F, MT-Y250F, and wild-type MT strains. Mammary tumor lysates derived from MT-Y315/322F, MT-Y250F, wild-type MT, and control MMTV/Neu (N202) strains (21) were subjected immunoprecipitation with PyV MT-specific antisera and incubated with γ-32P-labeled ATP in the presence of exogenous PI lipid. (D) Phosphorimager quantitation of the phosphorylated PI-3′ lipid, normalized to the levels of PyV MT protein.
In addition to activation of the Src family tyrosine kinases by PyV MT, the activity of PI-3′ kinase is dramatically elevated following formation of specific complexes of the PI-3′ kinase with PyV MT (11, 54). To ascertain whether the various mutant MT antigens expressed in the mammary tumors were still capable of activating the PI-3′ kinase, tumor extracts were subjected to immunoprecipitation analyses with PyV MT-specific antisera. The immunoprecipitates were then incubated in vitro with PI lipid and [γ-32P]ATP, and the phosphorylated lipid products were subjected to thin-layer chromatography (Fig. 5C). After normalization for the levels of PyV MT protein in the tumor lysates, quantitative analyses revealed that the levels of PyV MT-associated PI-3′ kinase activity were severely impaired in tumor lysates derived from the MT-Y315/322F strains compared to tumor lysates derived from the wild-type MT strains (Fig. 5D). Interestingly, the level of PI-3′ MT-associated kinase activity observed in the MT-Y250F-derived tumors was also reduced, albeit only twofold.
To ascertain whether the levels of PyV MT PI-3′ kinase-associated activities correlated with the capacity of PyV MT to associate with the 85-kDa subunit of the PI-3′ kinase, tumor extracts were subjected to reciprocal immunoprecipitation-immunoblot analyses with PyV MT-specific antisera and antibodies specific to the PI-3′ kinase p85 regulatory subunit (Fig. 6B). The results of these analyses revealed that the 85-kDa PI-3′ kinase subunit could be detected in PyV MT immunoprecipitates of extracts of the MT-Y250F- and wild-type MT-derived tumor tissues (Fig. 6B, lanes 2 to 6, 9, and 10). In contrast, the mutant PyV MT derived from the MT-Y315/322F tumor lysates bound the mutant PyV MT antigen poorly (Fig. 6B, lanes 7 and 8). The observed differences in p85 binding between the various mutant PyV MTs were not due to differences in the levels of PyV MT, because immunoblot analyses revealed comparable levels of PyV MT protein in the samples (Fig. 6C). Interestingly, these analyses revealed that two of the MT-Y250F tumor samples were expressing a PyV MT protein that migrated with faster mobility than wild-type MT protein (Fig. 6C, lanes 5 and 6). Taken together, these observations suggest that the defect in MT-associated PI-3′ kinase activity observed in the MT-Y315/322F tumors is due to the inability of this mutant to bind the 85-kDa subunit of the PI-3′ kinase.
FIG. 6.

Binding properties of Shc and the p85 subunit of the PI-3′ kinase with the various mutant PyV MTs expressed in mammary tumors. (A) Shc immunoblot analysis of MT-specific immunoprecipitates (IP) isolated from MT-Y250F (lanes 2 to 4), reverted MT-Y250F (dl MT) (lanes 5 and 6), MT-Y315/22F (lanes 7 and 8), and wild-type MT (lanes 9 and 10) mammary tumors. As a nonspecific control, a protein lysate derived from an MMTV/Neu mammary tumor (lane 1) was also included. The MT-associated 52-kDa Shc protein is indicated by the arrow. (B) The same PyV MT immunoprecipitates were subjected to p85 immunoblot analyses. As a nonspecific control, a protein lysate from an MMTV/Neu tumor (lane 1) was also included (18). The p85 kDa subunit of the PI-3′ kinase is indicated by the arrow. (C) PyV MT immunoblot analysis of MT-specific immunoprecipitates isolated from the same sets of mammary tumor samples. As a nonspecific control, mammary tumor protein lysate derived from an MMTV/Neu tumor was included. Indicated by the arrows are the wild-type 56-kDa PyV MT and deleted MT (dlMT) mutant proteins.
Tyrosine phosphorylation of MT residue 250 creates a high-affinity binding site for the PTB domain of Shc (38). To determine whether Shc binding was affected in the tumors induced by the MT-Y250F transgene, immunoprecipitation-immunoblot analyses with either PyV MT or Shc-specific antisera were conducted on tumor lysates derived from both mutant and wild-type MT strains. As expected, complexes of Shc and PyV MT were detected in tumor lysates derived from either the mutant MT-Y315/322F or wild-type MT lysates (Fig. 6A, lanes 7 to 10). However, no detectable complexes between PyV MT and Shc were observed in the tumor lysates from three MT-Y250F lysates (Fig. 6A, lanes 2 to 4). However, coimmunoprecipitation analyses of two other MT-Y250F tumor samples that expressed the altered PyV MT (Fig. 6C, lanes 5 and 6) revealed PyV MT-associated Shc (Fig. 6A, lanes 5 and 6). Reprobing of the same blot with MT-specific antibody pAB701 revealed that the differences in Shc binding between these samples could not be due to differences in the levels of PyV MT (Fig. 6C). These observations argue that in certain cases the mutant PyV MT-Y250F protein has reacquired the capacity to associate with Shc, perhaps through the occurrence of somatic mutations in PyV MT-coding sequences.
Tumor progression in the MT-Y250F strains involves the reacquisition of a functional Shc binding site.
Our biochemical and genetic analyses of tumors derived from the MT-Y250F strains revealed that the PyV MTs of certain tumors were capable of binding to Shc (Fig. 6A, lanes 5 and 6), seemingly as a result of a deletion occurring in PyV MT-coding sequences (Fig. 6C, lanes 5 and 6). Because somatic mutation and consequent reversion of the transgene could potentially account for the ability of PyV MT to bind to Shc, we designed a method with which to screen large numbers of mammary tumors and derived lung metastases for potential reversion of the mutant PyV MT cDNAs. To accomplish this, an RNase protection probe corresponding to this region of wild-type PyV MT-coding sequences was created, and RNase protection conditions were modified so as to allow the detection of single-base-pair mismatches or deletions (see Materials and Methods). Due to the stringent RNase digestion conditions used in these assays, multiple protected fragments (Fig. 7A, MT-Y250F) were observed, rather than just the two expected protected fragments corresponding to the cleavage at the single-nucleotide mismatch at the phenylalanine substitution at tyrosine residue 250. As shown in Fig. 7A, these RNase protection analyses revealed that two of the seven primary MT-Y250F mammary tumors exhibited a pattern of protected fragments that differed from the other MT-Y250F tumor samples (lanes 5 and 7). One set of protected fragments migrated at a position expected for wild-type MT (Fig. 7A, lane 7) whereas the other sets of protected fragments were consistent with the occurrence of a deletion in MT-Y250F-coding sequences (Fig. 7A, lane 5). Of the 57 primary tumor RNA samples analyzed by this approach, a total of four samples exhibited RNase protection patterns corresponding to either wild-type or deleted forms of PyV MT (Fig. 7B). Interestingly, animals displaying these altered transcripts developed extensive lung metastases which also gave protected species characteristic of the primary tumors (Fig. 7A, lanes 8 and 11). Analyses of further sets of metastatic mammary tumors from different MT-Y250F animals demonstrated that an additional lung metastasis also displayed an RNase-protected fragment characteristic of the wild-type PyV MT transgene (Fig. 7A, lane 10). Using a similar RNase protection approach, we also tested whether a comparable number of MT-Y315/322F tumors displayed any evidence for reversion. In contrast to the case for the MT-Y250F samples, RNase protection analyses of the MT-Y315/322F tumor RNA samples failed to exhibit evidence for reversion of the mutant PI-3′ kinase binding site (53a).
FIG. 7.
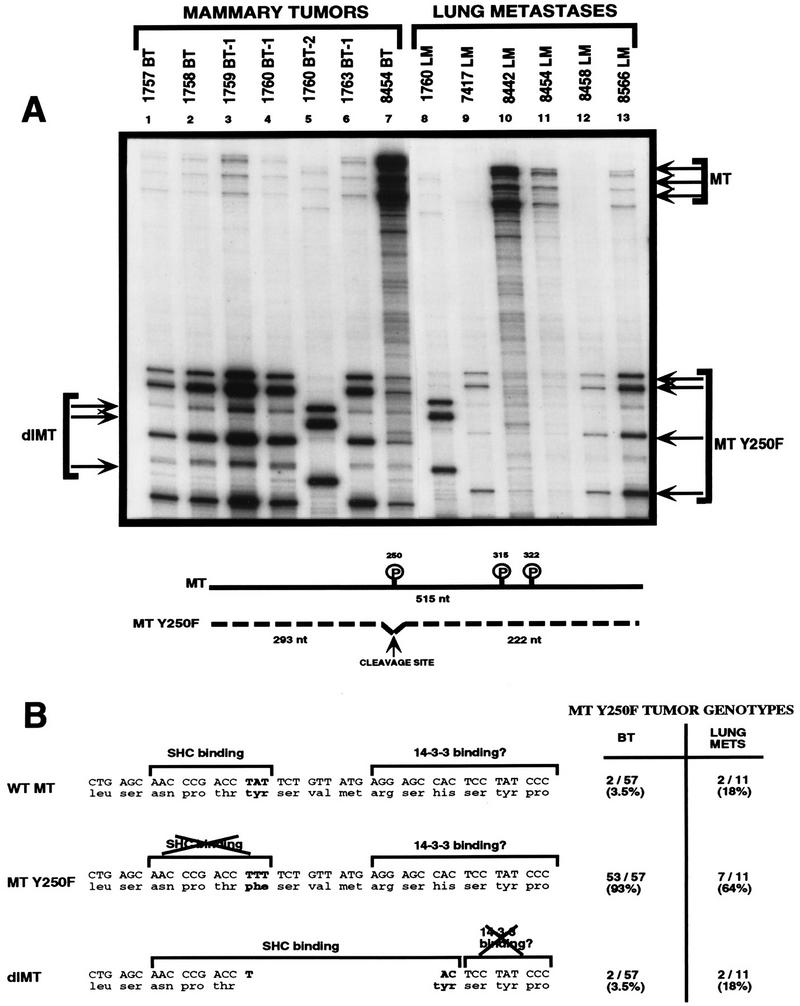
Restoration of Shc binding in the MT-Y250F mutant can occur through somatic mutations in the transgene during tumor progression. (A) Stringent RNase protection analyses on RNA samples derived from either primary mammary tumors (lanes 1 to 7) or lung metastases (lanes 8 to 13) arising in the MT-Y250F strains. Thirty micrograms of total RNA was hybridized with an antisense riboprobe (MTsn301) spanning the wild-type MT Shc binding site. The arrows indicate the expected protected bands for a wild-type MT (MT) or MT-Y250F transcript. Also shown is expected cleavage occurring at tyrosine residue 250. Note that the 1760 RNA sample (lane 5) displays an RNase protection profile that is deleted relative to the expected MT-Y250F pattern. (B) DNA sequence analyses of the transcripts detected in the MT-Y250F samples. Sequence analysis of cloned RT-PCR products from these tumors confirmed the presence of a phenylalanine residue substitution at site 250 for RNA tumor samples exhibiting the expected MT-Y250F RNase protection profile (lanes 1 to 4, 6, 9, 12, and 13 in panel A). DNA sequence analyses of the tumor samples derived from tumor samples displaying wild-type MT RNase protection profile (lanes 7, 10, and 11 in panel A) revealed the presence of a T-to-A mutation leading to conversion of the phenylalanine to tyrosine. Sequence analyses of the RT-PCR product derived from deleted transcript revealed the presence of an 18-nucleotide in-frame deletion spanning the binding site tyrosine residue (nucleotides 730 to 783). Also indicated are critical amino acid residues implicated in Shc and 14-3-3 binding. Note that the 18-nucleotide deletion restores the Shc binding consensus NPXY. The relative incidence of either the point mutation (WT MT) or the deletion (dlMT) is indicated in both primary tumors (BT) and lung metstases (LUNG METS).
To investigate the precise structures of these altered transcripts, RNA samples derived from these tissues were subjected to reverse transcription-PCR (RT-PCR) with oligonucleotides flanking this region of the PyV MT cDNA followed by direct DNA sequence analyses. The results of these experiments revealed that the altered transcripts encoded either a wild-type MT-coding sequence as a result of a single nucleotide substitution or a deletion that removed the phenylalanine residue at position 250 and five adjacent amino acids (Fig. 7B). The union of DNA following the deletion creates a tyrosine residue regenerating the Shc binding core sequence NPTY (Fig. 7B). A similar analysis of metastatic tumors in arising in these strains revealed that 36% (n = 11) of these had acquired either one of these reversions (Fig. 7B).
To further test the possibility that the MT deletion was responsible for the induction of the mammary tumor metastases, the corresponding deletion (dlMT) was engineered into a wild-type MT cDNA in mammalian expression cassette J4-ω, and its transforming activity was assessed in Rat-1 fibroblasts. The results revealed that the dlMT mutant displayed a transforming activity comparable to that of wild-type PyV MT (93.4% ± 11.6% of that of wild-type MT). In contrast, neither of the parental PyV MT mutants was able to efficiently transform Rat-1 cells (53a). These observations suggest that tumorigenesis in the MT-Y250F strain in certain tumors involves the occurrence of somatic mutations in the transgene that restore Shc binding.
Tumor progression in the mutant PyV MT strains involves the coordinate upregulation of ErbB-2 and ErbB-3 growth factor receptor tyrosine kinases.
Whereas reversion of the Shc binding site can account for tumorigenesis in 7% of the primary tumors in the MT-Y250F strains, the majority of the tumors that arise in either mutant PyV MT strain appear to retain the mutant transgene configuration. One possible explanation for these observations is that tumor progression in these strains involves the indirect recruitment of the PI-3′ kinase and Shc signaling pathways through activation of specific growth factor receptors. For example, for both murine and human mammary tumors there is compelling evidence to suggest that elevated expression of the ErbB-2 and ErbB-3 members of the epidermal growth factor receptor (EGFR) family are functionally involved in tumor induction (6). Significantly, this heterodimer combination of EGFR family members has been demonstrated to result in the recruitment and activation of both the PI-3′ kinase and Shc signaling molecules (39, 44). To explore this possibility, we examined the levels of ErbB-2 and ErbB-3 receptor tyrosine kinases in both hyperplasias and tumors from either the MT-Y315/322F or MT-Y250F strains with antibodies specific to either ErbB-2 or ErbB-3. As shown in Fig. 8, comparison of the levels of ErbB-2 and ErbB-3 in tumors derived from the MT-Y315/322F strain revealed a dramatic upregulation in the levels of both ErbB-2 and ErbB-3 proteins in three of the four tumors compared to those in mammary epithelial hyperplasias (Fig. 8A, compare lanes 6 to 8 with lanes 1 to 4). Analyses of a larger sample of tumors derived from these strains (n = 10) revealed that 80% of the tumors expressed elevated levels of both of these EGFR family members. The differences in levels of expression of ErbB-2 and ErbB-3 between the hyperplasias and the tumors could not be accounted for by differences in transgene expression, since both hyperplasias and tumors expressed equivalent amounts of PyV MT transcript (Fig. 1B, compare lanes 10 and 16; Fig. 1D, compare lanes 1 and 15). These observations suggest that the progression of the mammary epithelial hyperplasias to tumors in the MT-Y315/322F strains is correlated with the upregulation of ErbB-2 and ErbB-3 expression.
FIG. 8.
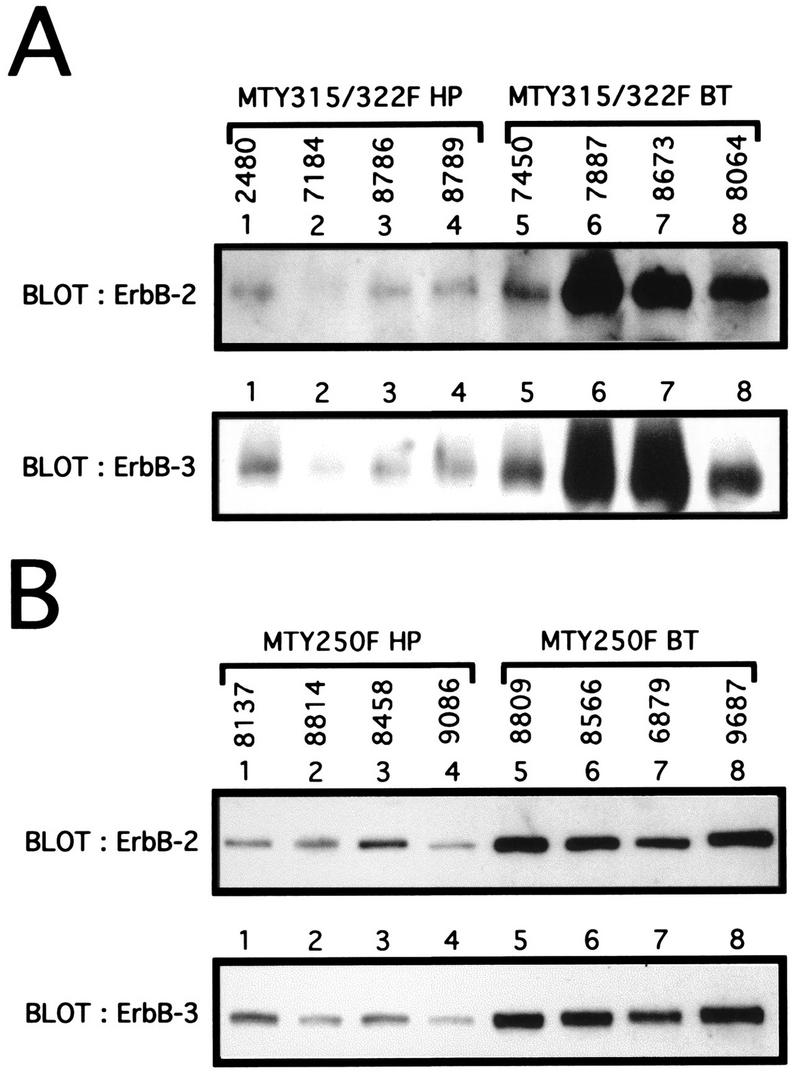
Elevation of ErbB-2 and ErbB-3 expression during tumor progression in the mutant PyV MT strains. (A) Immunoblot analyses of mammary epithelial hyperplasias (HP) or breast tumors (BT) from the MT-Y315/322F strain. One hundred micrograms of total protein lysate for four different hyperplasias (2480, 7184, 8786, and 8789) and tumors (7450, 7887, 8673, and 8064) was subjected to immunoblot analyses with either ErbB-2- or ErbB-3-specific antibodies. (B) Immunoblot analyses of mammary epithelial hyperplasias or breast tumors from the MT-Y250F strain. Sixty micrograms of total protein lysate for four different hyperplasias (8137, 8814, 8458, and 9086) or breast tumors (8809, 8566, 6879, and 9687) was subjected to immunoblot analyses with either ErbB-2 or ErbB-3.
To determine whether elevated expression of ErbB-2 and ErbB-3 could also be detected in the tumors derived from the MT-Y250F strains, protein extracts derived from either mammary epithelial hyperplasias or mammary tumors were subjected to immunoblot analyses with either ErbB-2- or ErbB-3-specific antisera (Fig. 8B). The tumors derived from these sets of samples failed to display any evidence of reversion of the mutant Shc binding site. The results of these analyses revealed that like the MT-Y315/322F-derived tumors, the MT-Y250F-derived tumors expressed elevated levels of both the ErbB-2 and ErbB-3 growth factor receptors compared to those in hyperplastic mammary epithelium. Taken together, these observations argue that upregulation of these growth factor receptor signaling pathways plays an important role in tumor progression in both of these mutant PyV MT transgenic strains.
DISCUSSION
The PyV MT oncogene provides an excellent model to allow identification of the important signaling pathways involved in mammary tumorigenesis. Given the potent transforming activity of the PyV MT oncogene in the mammary epithelia of transgenic mice (19, 20), we sought to exploit this system to elucidate the relative contributions of the PyV MT coupled signaling pathways in PyV MT-mediated mammary tumorigenesis. To elucidate the roles of Shc and PI-3′ kinase in PyV MT-mediated tumorigenesis, we have derived transgenic mice that express mutant PyV MT oncogenes defective in their capacity to bind to these signaling proteins. Consistent with previous studies of MMTV-driven transgenes, analyses of the tissue-specific patterns of expression of both PyV mutant MT cDNAs revealed that the primary site of expression was the mammary gland, with secondary sites noted in the male reproductive tissues and salivary glands (Fig. 1 and Tables 1 and 2). In both sets of transgenic strains, the initial phenotype exhibited by female mice was the inability to lactate. Whole-mount analyses of the mammary epithelia of virgin females from either the MT-Y250F or MT-Y315/322F strains showed that they displayed extensive epithelial hyperplasias that were histologically distinct (Fig. 2).
The observation that the mammary epithelial hyperplasias derived from the MT-Y315/322F strains exhibit elevated rates of apoptosis (Fig. 3A) suggests that activation of PI-3′ kinase plays a critical role in promoting cell survival. Moreover, expression of a dominant negative inhibitor of PI-3′ kinase in mammary tumor cells expressing wild-type PyV MT resulted in the rapid induction of programmed cell death (Fig. 3C). These observations are consistent with the emerging concept that activation of the PI-3′ kinase signaling pathway is involved in the regulation of apoptosis in a number of different cell types. For example, abrogation of growth factor-mediated activation of the PI-3′ kinase signaling pathway through administration of either specific PI-3′ kinase inhibitors or expression of mutant growth factor receptors decoupled from the PI-3′ kinase in a number cell types results in the induction of apoptotic cell death (23, 55). More recently, it has been demonstrated that activation of the PI-3′ kinase blocks c-Myc- or UVB-induced apoptosis in fibroblasts (23, 29). In addition, recent studies demonstrate that activation of the Akt kinase, which is immediately downstream of the PI-3′ kinase (15), can also prevent c-Myc-induced apoptosis (23, 29). Taken together with our observations, these data suggest that activation of the PI-3′ kinase signaling pathway may be required to promote cell survival.
Although the initial phenotypes observed with the PyV MT mutant strains were mammary epithelial hyperplasias, females derived from several independent strains developed mammary tumors with 100% penetrance. However, in contrast to parental wild-type PyV MT strains, which developed multifocal mammary tumors, the tumors in the mutant PyV MT mutant strains were focal in origin, arising adjacent to hyperplastic mammary tissue (Fig. 2 and 4). Moreover, the tumors arose with delayed onset in comparison to the case for wild-type strains (Fig. 4). Measurement of associated tyrosine kinase activity of mutant PyV MT species isolated from mammary hyperplasias or tumors revealed that association with and consequent activation of c-Src was unaffected compared to that for wild-type PyV MT (Fig. 5). Thus, the increased latency with which mammary tumors arise in both PyV MT mutant transgenic strains is not due to an inability to complex with and activate the Src family of tyrosine kinases. Although recent studies have suggested that tyrosine 322 in PyV MT is also capable of binding PLCγ in certain cell types (47), we and others have failed to detect evidence of comparable complexes of murine PyV MT and PLCγ in tumors induced by the PyV MT oncogene (3, 53a). Although we cannot formally preclude the potential involvement of other PTB domain- or SH2-containing signaling molecules in these mutant PyV phenotypes, the failure of PyV MT to bind the corresponding associated cellular protein (i.e., Shc in MT-Y250F mice and PI-3′ kinase in MT-Y315/322F transgenic mice) is likely responsible for the delayed onset of mammary tumors observed in these mutant PyV MT strains.
The data generated suggest that expression of the PyV MT mutants is not sufficient for mammary tumorigenesis and requires additional genetic events. In contrast to these observations, it has recently been reported that inactivation of the Shc binding site does not interfere with the ability of PyV to induce a variety of tumors in animals (4). More recently, another independent group reported that an Shc binding site PyV mutant displayed an altered tumor spectrum (56). However, in both these reports the incidence of mammary tumors was unaffected by the introduction of the Shc binding site mutant. The difference between these observations and ours may be due to the fact that in the other studies a functional PyV large T antigen is also expressed (4, 56) and may compensate for inability of PyV MT to associate with and activate Shc. Alternatively, unlike that of other tissues, transformation of the mammary epithelial cell may require activation of Shc. Indeed, it has been demonstrated that the requirement for Src in PyV MT-mediated tumorigenesis is highly dependent on the tissue context (20, 25). Whatever the explanation, our observations strongly suggest that activation of Shc is required for PyV MT-mediated mammary tumorigenesis.
One important clue to the nature of these additional events derives from observations that in 7% of the tumors arising in the MT-Y250F strains, the mutant PyV MT had reacquired the capacity to bind Shc through somatic mutations occurring in the transgene. DNA sequence analyses of these alterations revealed that restoration of Shc binding can occur either through simple point mutation at the substituted phenylalanine codon or through an in-frame deletion of 18 bp occurring immediately downstream of the phenylalanine codon (Fig. 7B). Another interesting aspect of these reversions is that they occurred at a much higher frequency in lung metastases (Fig. 7B). Indeed, of the 11 metastatic lesions examined, 36% possessed either reversion event, suggesting that there is a selective pressure for restoration of Shc binding during PyV MT-mediated metastatic progression. Because Shc can recruit the Grb-2–Sos–Ras complex to PyV MT (14, 42, 50), the selection for Shc binding during metastasis reflects an essential role for activation of the Ras pathway during metastatic progression. In this regard, we have recently demonstrated that ectopic expression of Grb-2 in the mammary epithelium of the MT-Y250F strains can result in dramatic acceleration of growth of mammary tumors (40). Alternatively, the requirement for Shc binding may reflect its ability to recruit other signaling pathways. Indeed, it has recently been demonstrated that tyrosine phosphorylation of tyrosine residues 239 and 240 in Shc is involved in the generation of an antiapoptotic signal involving activation of the Myc transcription factor (17). Determination of the prevalent signaling pathways involved in this phenotype will provide important insight into the molecular basis of metastatic progression.
In contrast to those from MT-Y250F transgenic animals, tumors derived from the MT-Y315/322F strains failed to demonstrate reversion at either tyrosine phosphorylation site in the transgene responsible for binding the p85 subunit of the PI-3′ kinase. It is conceivable that tumorigenesis in these mutant strains requires recruitment of PI-3′ kinase but that this occurs in an indirect manner. Activation of growth factor receptors in an autocrine or paracrine manner during tumor progression in these strains could lead to the indirect recruitment of the PI-3′ kinase signaling pathway. In this regard, we have demonstrated that the ErbB-3 and ErbB-2 EGFR family member which specifically couples to the PI-3′ kinase and Shc signaling pathways (39, 44) is upregulated during the transition of mammary epithelial hyperplasias to the tumor phenotype in both these mutant strains (Fig. 8). Consistent with these observations, other studies have demonstrated that recruitment of PI-3′ kinase by activated growth factors such as platelet-derived growth factor and insulin growth factor-2 is required to provide a survival signal to prevent cells from undergoing apoptosis (35, 55). However, the identification of potential signaling pathways that may be involved in tumor progression in these mutant MT strains awaits further investigation.
Activation of PI-3′ kinase-coupled growth factor receptors during tumor progression in these various transgenic strains may reflect the requirement for the generation of an antiapoptotic signal (Fig. 3). Indeed, in the insulin promoter-SV40 large T antigen transgenic mice there is suppression of apoptotic cell death during tumor progression (35). In addition to suppressing apoptotic cell death, activation of the PI-3′ kinase by PyV MT may influence other important steps involved in tumor progression. In this regard, we and our collaborators have recently demonstrated that mammary tumor cells expressing the MT-Y315/322F mutant showed a marked decrease in the induction of angiogenic blood supply that was further correlated with a 10-fold decrease in metastatic potential compared to that for a mammary tumor cell line expressing wild-type PyV MT (9). Consistent with these observations, we have found that only 36% of tumor-bearing female animals carrying the mutant MT-Y315/322F develop metastatic lesions (53a), whereas 100% of those carrying the parental wild-type MT strains develop lung mestastases (19). More recently, a transforming homolog of the PI-3′ kinase has been described, suggesting that activation of the PI-3′ kinase can directly result in the induction of tumors (7). Taken together, these observations suggest that recruitment of the PI-3′ kinase by PyV MT may play multiple roles in tumor progression.
Given that activation of Ras appears to be a common component of receptor tyrosine kinase signaling cascades, the observation that the PyV MT mutant defective in its capacity to associate with Shc–Grb-2 is also impaired in its ability to induce tumors suggests that direct recruitment of the Ras pathway is critical for tumor progression. Indeed, it has been demonstrated that PyV MT requires Ras function to transform fibroblasts in vitro (22). However, despite the defect in tumor induction, expression of the MT-Y250F mutant is still capable of inducing extensive epithelial hyperplasias. It is conceivable that the mitogenic response of mammary epithelial cells to the PyV MT-Y250F mutant reflects its capacity to signal in a Ras-independent fashion. Indeed, the Raf serine kinase can be activated by c-Src through a Ras-independent mechanism (46). Alternatively, the MT-Y250F mutant may stimulate cell proliferation by indirectly activating the Ras signaling pathway.
The studies described above have important implications in elucidating the molecular basis for the oncogene-mediated induction of metastatic mammary tumors. Our studies suggest that recruitment of both Shc and PI-3′ kinase signaling molecules to PyV MT plays a critical role in the transition from mammary hyperplasias to metastatic mammary tumors. These observations further argue that mammary tumorigenesis in these transgenic mouse models is dependent on the concerted activation of both cell-proliferative and survival-coupled pathways. The results of this study also have general implications with respect to the roles that growth factor receptor-coupled signal transduction pathways play in tumor progression. Because tumor progression in these mutant PyV MT strains is dependent on the genetic events that complement the defects in these signaling pathways, these transgenic tumor models can serve as a powerful genetic system to dissect the importance of various signaling molecules in mammary tumor progression. Further studies with these mutant PyV MT transgenic mice will provide important insight into the nature of events involved in the progression of mammary epithelial hyperplasias to metastatic mammary tumors.
ACKNOWLEDGMENTS
This work was supported by a Canadian Breast Cancer Initiative grant awarded to W.J.M. This work was also partially supported by grants from the Medical Research Council of Canada, by National Cancer Institute of Canada (NCIC) awards to F.L.G. and J.A.H., and by National Cancer Institute grant RO1-CA S4285 awarded to R.D.C. M.A.W. and C.G.T. were supported by studentships from the Cancer Research Society, and J.N.H. and M.J.R. were supported by studentships from the National Science and Engineering Research Council (NSERC). W.J.M. is a Medical Research Council of Canada Scientist. F.L.G. is a Terry Fox Research Scientist of the NCIC.
REFERENCES
- 1.Addison C, Braciak T, Ralston R, Muller W, Gauldie J, Graham F L. Intratumoral injection of an adenovirus expressing interleukin-2 induces regression and immunity in a murine breast cancer model. Proc Natl Acad Sci USA. 1995;92:8522–8526. doi: 10.1073/pnas.92.18.8522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anton M, Graham F. Site-specific recombination mediated by an adenovirus vector expressing the Cre recombinase protein: a molecular switch for control of gene expression. J Virol. 1995;69:4600–4606. doi: 10.1128/jvi.69.8.4600-4606.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brizuela L, Ulug E T, Jones M A, Courtneidge S A. Induction of interleukin-2 transcription by the hamster polyoma virus middle T antigen: a role for Fyn in T cell signal transduction. Eur J Immunol. 1995;25:385–393. doi: 10.1002/eji.1830250212. [DOI] [PubMed] [Google Scholar]
- 4.Bronson R, Dawe C, Carrol J, Benjamin T. Tumor induction by a transformation-defective polyoma virus mutant blocked in signaling through Shc. Proc Natl Acad Sci USA. 1997;94:7954–7958. doi: 10.1073/pnas.94.15.7954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Campbell K S, Ogris E, Burke B, Su W, Auger K R, Druker B J, Schaffhausen B S, Roberts T M, Pallas D C. Polyoma middle tumor antigen interacts with SHC protein via the NPTY (Asn-Pro-Thr-Tyr) motif in middle tumor antigen. Proc Natl Acad Sci USA. 1994;91:6344–6348. doi: 10.1073/pnas.91.14.6344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cardiff R D, Muller W J. Transgenic mouse models of mammary tumorigenesis. Cancer Surv. 1993;16:97–113. [PubMed] [Google Scholar]
- 7.Chang H W, Akoi M, Fruman D, Auger K R, Bellacosa A, Tsi P N, Cantley L W, Roberts T M, Vogt P K. Transformation of chicken cells by the gene encoding the catalytic subunit of PI-3-kinase. Science. 1997;276:1848–1850. doi: 10.1126/science.276.5320.1848. [DOI] [PubMed] [Google Scholar]
- 8.Cheng S H, Harvey R, Espino P C, Semba K, Yamamota T, Toyoshima K, Smith A E. Peptide antibodies to the human c-fyn gene product demonstrate pp59 c-fyn is capable of complex formation with the middle-T antigen of polyomavirus. EMBO J. 1988;7:3845–3855. doi: 10.1002/j.1460-2075.1988.tb03270.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cheung A T W, Young L J T, Chao C Y, Chien P C Y, Ndoye A, Barry P A, Muller W J, Cardiff R D. Microcirculation and metstasis in a new mouse mammary tumor model system. Int J Oncol. 1997;11:69–77. [PubMed] [Google Scholar]
- 10.Chirgwin J M, Przybyla A E, MacDonald R J, Rutter W J. Isolation of biologically active ribonucleic acid from sources rich in ribonuclease. Biochemistry. 1979;18:5294–5299. doi: 10.1021/bi00591a005. [DOI] [PubMed] [Google Scholar]
- 11.Courtneidge S A, Hebner A. An 81 kDa protein complexed with middle T antigen and pp60c-src: a possible phosphatidylinositol kinase. Cell. 1987;50:1031–1037. doi: 10.1016/0092-8674(87)90169-3. [DOI] [PubMed] [Google Scholar]
- 12.Courtneidge S A, Smith A E. Polyomavirus transforming protein associates with the product of the c-src cellular gene. Nature. 1983;303:435–439. doi: 10.1038/303435a0. [DOI] [PubMed] [Google Scholar]
- 13.Dahl J, Freund R, Blenis J, Benjamin T J. Studies of partially transforming polyomavirus mutants establish a role for the phosphaditylinositol-3′-kinase in activation of the pp70 S6 kinase. Mol Cell Biol. 1996;16:2728–2735. doi: 10.1128/mcb.16.6.2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dilworth S M, Brewster C E P, Jones M D, Lanfrancone L, Pelicci G, Pelicci P G. Transformation by polyomavirus middle T-antigen involves the binding and tyrosine phosphorylation of Shc. Nature. 1994;367:87–90. doi: 10.1038/367087a0. [DOI] [PubMed] [Google Scholar]
- 15.Franke T F, Yang S-I, Chan T O, Datta K, Kazlauskas A, Morrison D K, Kaplan D R, Tsichlis P N. The protein encoded by the Akt proto-oncogene is a target of the PDGF-activated phosphatidylinositol 3-kinase. Cell. 1995;81:727–736. doi: 10.1016/0092-8674(95)90534-0. [DOI] [PubMed] [Google Scholar]
- 16.Freund R, Dawe C J, Carrol J P, Benjamin T L. Changes in frequency, morphology and behaviour of tumors induced in mice by a Polyomavirus mutant with a specifically altered oncogene. Am J Pathol. 1992;141:1409–1425. [PMC free article] [PubMed] [Google Scholar]
- 17.Gaviereli Y S, Ben-Sasson S A. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol. 1992;119:493–501. doi: 10.1083/jcb.119.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gotoh N, Tojo A, Shibuya M. A novel pathway from phosphorylation of tyrosine residues 239/240 of Shc, contributing to suppress apoptosis by IL-3. EMBO J. 1996;15:6197–6204. [PMC free article] [PubMed] [Google Scholar]
- 19.Guy C T, Cardiff R D, Muller W J. Induction of mammary tumors by expression of polyomavirus middle T oncogene: a transgenic mouse model for metastatic disease. Mol Cell Biol. 1992;12:954–961. doi: 10.1128/mcb.12.3.954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guy C T, Muthuswamy S K, Cardiff R A, Soriano P, Muller W J. Activation of the c-Src tyrosine kinase is required for the induction of mammary tumors in transgenic mice. Genes Dev. 1994;8:23–32. doi: 10.1101/gad.8.1.23. [DOI] [PubMed] [Google Scholar]
- 21.Guy C T, Webster M A, Schaller M, Parsons T J, Cardiff R D, Muller W J. Expression of the neu protooncogene in the mammary epithelium of transgenic mice induces metastatic disease. Proc Natl Acad Sci USA. 1992;89:10578–10582. doi: 10.1073/pnas.89.22.10578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jelinek M A, Hassell J A. Reversion of middle T antigen-transformed Rat-2 cells by Krev-1: implications for the role of p21 c-ras in polyomavirus-mediated transformation. Oncogene. 1992;7:1687–1698. [PubMed] [Google Scholar]
- 23.Kennedy S G, Wagner A J, Conzen S D, Jordan J, Bellacosa A, Tsichilis P N, Hay N. The PI 3′ kinase/Akt signaling pathway delivers an anti-apoptotic signal. Genes Dev. 1997;11:701. doi: 10.1101/gad.11.6.701. [DOI] [PubMed] [Google Scholar]
- 24.Khwaja A, Rodriguez-Viciana P, Wennstrom S, Warne P H, Downward J. Matrix adhesion and Ras transformation both activate a phosphoinositide 3-OH kinase and protein kinase B/Akt cellular survival pathway. EMBO J. 1997;16:2783–2793. doi: 10.1093/emboj/16.10.2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kiefer F, Anhauser I, Soriano P, Aguzzi A, Courtneidge S A, Wagner E F. Endothelial cell transformation by polyomavirus middle T antigen in mice lacking Src-related kinases. Curr Biol. 1994;4:100–109. doi: 10.1016/s0960-9822(94)00025-4. [DOI] [PubMed] [Google Scholar]
- 26.Klippel A, Kavanaugh M, Pot D, Williams L T. A specific product of phosphahadityl 3′-kinase directly activates the protein kinase Akt through its pleckstrin homology domain. Mol Cell Biol. 1997;17:338–344. doi: 10.1128/mcb.17.1.338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kornbluth S, Sudol M, Hanafusa H. Association of the polyomavirus middle T antigen with the c-yes protein. Nature. 1987;325:171–173. doi: 10.1038/325171a0. [DOI] [PubMed] [Google Scholar]
- 28.Kotani K, Yonezawa K, Hara K, Ueda H, Kitmura Y, Sakaue H, Calas B, Grigorescu F, Nishiyama M, Waterfield M D, Kasuga M. Involvement of phosphoinositide 3-kinase in insulin- or IGF-1-induced membrane ruffling. EMBO J. 1994;13:2213–2221. doi: 10.1002/j.1460-2075.1994.tb06515.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kulik G, Klippel A, Weber M J. Antiapoptotic signalling by the insulin-like growth factor I receptor, phosphadityl 3-kinase, and Akt. Mol Cell Biol. 1997;17:1595–1606. doi: 10.1128/mcb.17.3.1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kypta R M, Hemming A, Courtneidge S A. Identification and characterization of p59 fyn (a Src like protein kinase) in normal and polyomavirus transformed cells. EMBO J. 1988;7:3837–3844. doi: 10.1002/j.1460-2075.1988.tb03269.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Markland W, Smith A E. Mutants of polyomavirus middle-T antigen. Biochim Biophys Acta. 1987;907:299–321. doi: 10.1016/0304-419x(87)90011-4. [DOI] [PubMed] [Google Scholar]
- 32.Matsui Y, Halter S, Holt J T, Hogan B L, Coffey R J. Development of mammary hyperplasia and neoplasia in MMTV-TGFα transgenic mice. Cell. 1990;61:1147–1155. doi: 10.1016/0092-8674(90)90077-r. [DOI] [PubMed] [Google Scholar]
- 33.Melton D A, Krieg P A, Rebagliati M R, Maniatis T, Zinn K, Green M R. Efficient in vitro synthesis of biologically active RNA and RNA hybridization probes from plasmids containing a bacteriophage SP6 promoter. Nucleic Acids Res. 1984;12:7035–7056. doi: 10.1093/nar/12.18.7035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Muller W J, Sinn E, Pattengale P K, Wallace R, Leder P. Single-step induction of mammary adenocarcinoma in transgenic mice bearing the activated c-neu oncogene. Cell. 1988;54:105–115. doi: 10.1016/0092-8674(88)90184-5. [DOI] [PubMed] [Google Scholar]
- 35.Naik P, Karrin J, Hanahan D. The rise and fall of apoptosis during multistage tumorigenesis: down-modulation contributes to tumor progression from angiogenic progenitors. Genes Dev. 1996;10:2105–2116. doi: 10.1101/gad.10.17.2105. [DOI] [PubMed] [Google Scholar]
- 36.Pallas D C, Fu H, Haehnel L C, Weller W, Collier R J, Roberts T M. Association of the polyomavirus middle tumor antigen with 14-3-3 proteins. Science. 1994;265:535–537. doi: 10.1126/science.8036498. [DOI] [PubMed] [Google Scholar]
- 37.Pallas D C, Shahrik L K, Martin B L, Jasper S, Miller T B, Brautigan D L, Roberts T M. Polyoma small and middle T antigens and SV40 small t antigen form stable complexes with protein phosphatase 2A. Cell. 1990;60:167–176. doi: 10.1016/0092-8674(90)90726-u. [DOI] [PubMed] [Google Scholar]
- 38.Pawson T. Protein modules and signalling networks. Nature. 1995;373:573–580. doi: 10.1038/373573a0. [DOI] [PubMed] [Google Scholar]
- 39.Pringent S A, Gullick W J. Identification of the c-erbB-3 binding sites for phosphatidylinositol 3′-kinase and SHC using an EGF receptor/c-erbB-3 chimera. EMBO J. 1994;13:2831–2841. doi: 10.1002/j.1460-2075.1994.tb06577.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rauh, M. J., C. Tortorice, R. Daly, and W. J. Muller. 1997. Unpublished observations.
- 41.Rodriguez-Viciana P, Warne P H, Khaja A, Marte B M, Pappin D J, Das P, Waterfield M, Ridley A, Downward J. Role of the phosphoinositide 3-OH kinase in cell transformation and control of the actin cytoskelton by Ras. Cell. 1997;89:457–468. doi: 10.1016/s0092-8674(00)80226-3. [DOI] [PubMed] [Google Scholar]
- 42.Rozakis-Adcock M, McGlade J, Mbamalu G, Pelicci G, Daly R, Li W, Batzer A, Thomas S, Brugge J, Pelicci P G, Schlessinger J, Pawson T. Association of the Shc and Grb2/sem5 SH2-containing proteins is implicated in activation of the Ras pathway by tyrosine kinases. Nature. 1992;360:689–692. doi: 10.1038/360689a0. [DOI] [PubMed] [Google Scholar]
- 43.Sinn E, Muller W, Pattengale P, Tepler I, Wallace R, Leder P. Coexpression of MMTV/v-Ha-ras and MMTV-c-myc genes in transgenic mice: synergistic action of oncogenes in vivo. Cell. 1987;49:465–475. doi: 10.1016/0092-8674(87)90449-1. [DOI] [PubMed] [Google Scholar]
- 44.Soltoff S, Carraway III P K, Pringent S A, Gullick W, Cantley L. ErbB3 is involved in activation of phosphaditylinositol 3-kinase by epidermal growth factor. Mol Cell Biol. 1994;14:3550–3558. doi: 10.1128/mcb.14.6.3550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Southern E M. Detection of specific sequences among different DNA fragments separated by gel electrophoresis. J Mol Biol. 1975;98:503–517. doi: 10.1016/s0022-2836(75)80083-0. [DOI] [PubMed] [Google Scholar]
- 46.Stokoe D, McCormick F. Activation of c-Raf-1 by Ras and Src through different mechanisms: activation in vivo and in vitro. EMBO J. 1997;16:2384–2396. doi: 10.1093/emboj/16.9.2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Su W, Liu W, Schaffhausen B S, Roberts T M. Association of polyomavirus middle tumor antigen with phospholipase Cγ1. J Biol Chem. 1995;270:12331–12334. doi: 10.1074/jbc.270.21.12331. [DOI] [PubMed] [Google Scholar]
- 48.Surh C D, Sprent J. T-cell apoptosis detected in situ during positive and negative selection in the thymus. Nature. 1994;372:73–77. doi: 10.1038/372100a0. [DOI] [PubMed] [Google Scholar]
- 49.Talmage D A, Freund R, Young D T, Dahl J, Benjamin T. Phosphorylation of middle T by pp60c-src: a switch for binding of the phosphadityl inositol 3-kinase and optimal tumorigenesis. Cell. 1989;59:55–66. doi: 10.1016/0092-8674(89)90869-6. [DOI] [PubMed] [Google Scholar]
- 50.van der Geer P, Wiley S, Gish G D, Pawson T. The Shc adaptor protein is highly phosphorylated at conserved twin tyrosine residues (Y239/240) that mediate protein-protein interactions. Curr Biol. 1996;6:1435–1444. doi: 10.1016/s0960-9822(96)00748-8. [DOI] [PubMed] [Google Scholar]
- 51.Vonderhaar B, Greco A E. Lobulo-alveolar development of mouse mammary glands is regulated by thyroid hormones. Endocrinology. 1979;2:409–418. doi: 10.1210/endo-104-2-409. [DOI] [PubMed] [Google Scholar]
- 52.Walter G, Ruediger R, Slaughter C, Mumby M. Association of protein phosphatase 2A with polyoma virus medium tumor antigen. Proc Natl Acad Sci USA. 1990;87:2521–2525. doi: 10.1073/pnas.87.7.2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Webster M A, Cardiff R D, Muller W J. Induction of mammary epithelial hyperplasias and mammary tumors in transgenic mice expressing a mammary murine tumor virus/activated c-src fusion gene. Proc Natl Acad Sci USA. 1995;92:7849–7853. doi: 10.1073/pnas.92.17.7849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53a.Webster, M. A., J. N. Hutchinson, and W. J. Muller. 1997. Unpublished observations.
- 54.Whitman M, Kaplan D R, Schaffhausen B, Cantley L, Roberts T M. Association of phosphatidylinositol kinase activity with polyoma middle-T competent for transformation. Nature. 1985;315:239–242. doi: 10.1038/315239a0. [DOI] [PubMed] [Google Scholar]
- 55.Yao R, Cooper G M. Requirement for phosphaditylinositol-3 kinase in prevention of apoptosis by nerve growth factor. Science. 1995;267:2003–2006. doi: 10.1126/science.7701324. [DOI] [PubMed] [Google Scholar]
- 56.Yi X, Peterson J, Freund R. Transformation and tumorigenic properties of a mutant polyomavirus containing a middle T antigen defective in Shc binding. J Virol. 1997;71:6279–6286. doi: 10.1128/jvi.71.9.6279-6286.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]