

Abstract
MEK kinase 1 (MEKK1) is a 196-kDa protein that, in response to genotoxic agents, was found to undergo phosphorylation-dependent activation. The expression of kinase-inactive MEKK1 inhibited genotoxin-induced apoptosis. Following activation by genotoxins, MEKK1 was cleaved in a caspase-dependent manner into an active 91-kDa kinase fragment. Expression of MEKK1 stimulated DEVD-directed caspase activity and induced apoptosis. MEKK1 is itself a substrate for CPP32 (caspase-3). A mutant MEKK1 that is resistant to caspase cleavage was impaired in its ability to induce apoptosis. These findings demonstrate that MEKK1 contributes to the apoptotic response to genotoxins. The regulation of MEKK1 by genotoxins involves its activation, which may be part of survival pathways, followed by its cleavage, which generates a proapoptotic kinase fragment able to activate caspases. MEKK1 and caspases are predicted to be part of an amplification loop to increase caspase activity during apoptosis.
It has become evident in recent years that chemotherapeutic drugs that induce DNA damage or inhibit essential biosynthetic pathways induce apoptosis of cancer cells (21, 26). Animal studies and experiments with numerous cultured cancer cell lines have demonstrated that apoptosis is the primary death response to the major classes of drugs used to clinically treat human cancer (17, 42, 43). It is believed that these drugs induce damage to the cell but also activate signal transduction pathways that commit the cell to cellular suicide. Obviously, an understanding of the pathways committing a cancer cell to apoptosis is extremely important for advances in chemotherapy.
Proteases of the ICE/Ced-3 family (caspases) (1) are activated during the apoptotic response, including that activated by chemotherapeutic drugs, and cleave specific protein substrates. It is believed that activation of the caspases is a final commitment step for apoptosis. Several caspase substrates have been identified; these include poly(ADP-ribose) polymerase (35), U1 small nuclear ribonucleoprotein (10), lamin (36), D4-GDI (44), fodrin (13), protein kinase Cδ (18), p21-activated kinase 2 (51), sterol regulatory element binding protein (58), retinoblastoma protein (2), DNA-dependent protein kinase (9), MDM2 (a negative regulator of p53) (19), the Alzheimer-associated presenilins PS1 and PS2 (31), and the proteases themselves (48).
At least two caspase activities appear to be necessary for the apoptotic response; each has a specific substrate selectivity. ICE (caspase-1)-like proteases have a specificity for proteins encoding the four-amino-acid sequence YVAD (28), while CPP32 (caspase-3)-like proteases have a preference for the sequence DEVD (45). Both groups of proteases cleave at the carboxy-terminal aspartic acid residue of the recognition sequence. Several viruses encode proteins that are specific inhibitors of the caspases. Most notably, CrmA is a poxvirus protein that inhibits ICE and FLICE (caspase-8) (66), and p35 is a baculovirus protein that has broad inhibitory activity toward caspases (12, 22). The expression of CrmA and p35 inhibits the apoptotic response to many different stimuli, demonstrating the requirement for caspases during programmed cell death (5, 40).
In addition to caspases, it is becoming increasingly clear that signal transduction pathways involving specific protein kinases are involved in mediating apoptosis. Specifically, the c-Jun kinases (JNKs) and p38 kinases have been proposed to mediate apoptosis (57, 62, 64). However, a number of reports have challenged the notion that the activation of JNKs and/or p38 kinases is sufficient to induce apoptosis (29, 30, 34, 38, 39, 49, 53, 56). Thus, it appears that other signal pathways are required for apoptosis. However, the integration and balance of the JNK and p38 pathways probably do contribute to commitment to apoptosis (23, 62).
Members of our laboratory have cloned several protein serine-threonine kinases, referred to as MEK kinases (MEKKs), that are members of sequential protein kinase pathways regulating MAP kinases, including JNKs and ERKs (6, 24, 32, 33, 63). In our experiments, MEKKs have not significantly activated p38 kinases (6, 24). Of the four MEKK members that we have characterized, MEKK1 has been found to have the unique property of being a strong stimulator of apoptosis (34, 62). The kinase domain of MEKK1 is only 50% conserved relative to the kinase domains of MEKK2, MEKK3, and MEKK4, consistent with MEKK1 having unique substrate recognition properties and catalytic activity involved in mediating the apoptotic response. MEKK1 is a 196-kDa protein that encodes a protease cleavage sequence for caspase-3-like proteases. We demonstrate in this report that UV irradiation and DNA-damaging chemicals activate MEKK1 kinase function and induce the proteolytic cleavage of MEKK1. An inhibitory mutant of MEKK1 blocks apoptosis in response to these agents. These findings demonstrate that MEKK1 is an integral component of the apoptotic response to genotoxins.
MATERIALS AND METHODS
Cells.
Human embryonic kidney 293 cells (HEK293) stably expressing the EBNA-1 protein from Epstein-Barr virus (Invitrogen) were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 100 U each of penicillin and streptomycin per ml and containing 10% bovine calf serum. The cells were transfected with Lipofectamine (Gibco).
Plasmids.
The full-length cDNA-encoded mouse MEKK1 was modified by addition of the hemagglutinin (HA) tag sequence (MGYPYDVDYAS) at its NH2 terminus and inserted into the expression plasmids pCEP4 and pcDNA3 (Invitrogen), resulting in plasmids MEKK1.cp4 and MEKK1.dn3, respectively. MEKK1k.cp4 has been described elsewhere (61). The MEKK1 sequences DTVD (amino acids 871 to 874) and DEVE (amino acids 857 to 860) in MEKK1.cp4 were substituted with alanines by a PCR strategy. The resulting plasmids were named DTVD_A.cp4 and DEVE_A.cp4, respectively. The DTVD→A MEKK1 mutant was also subcloned in pcDNA3 (plasmid DTVD_A.dn3). The cDNAs for CrmA (50) and p35 (8) were subcloned in pCEP_ (a pCEP4-derived vector from which the hygromycin resistance gene was removed), resulting in plasmids CrmA.cp_ and p35.cp_, respectively. Plasmid pcDNA_3.cp4 is the result of the ligation of pCEP4 and pcDNA3. MEKK1k.MT4 has been described elsewhere (60). The kinase-inactive form of HA-tagged MEKK1 [MEKK1 K(1253)→M] was generated by PCR and subcloned in pcDNA3. The resulting plasmid was named MEKK1(−).dn3. The HA-tagged 91-kDa C-terminal portion of MEKK1 (amino acids 875 to 1493 [fragment C]) was generated by PCR and subcloned in pcDNA3, resulting in plasmid G875.dn3.
In vitro kinase assays.
Lysis buffer (70 mM β-glycero-phosphate, 1 mM EGTA, 100 μM Na3VO4, 1 mM dithiothreitol, 2 mM MgCl2, 0.5% Triton X-100, 20 μg of aprotinin per ml) was added to cells 15 to 24 h after transfection. Cellular debris was removed by centrifugation at 8,000 × g for 5 min. Protein concentration was normalized by the Bradford assay with bovine serum albumin as a standard.
JNK.
JNK activity was measured by a solid-phase kinase assay in which glutathione S-transferase (GST)–c-Jun1–79 (GST-Jun) bound to glutathione–Sepharose 4B beads was used to affinity purify JNK from cell lysates as described previously (23, 25). Alternatively, JNK1 or JNK2 was immunoprecipitated with isoform-specific antibodies (Santa Cruz Biotechnology) and GST-Jun was used as a substrate in an in vitro kinase assay (25). Quantitation of the phosphorylation of GST-Jun was performed with a PhosphorImager.
ERK.
ERK2 was immunoprecipitated as described above for the JNK isoforms with ERK2 (C-14) antibody (Santa Cruz Biotechnology). The beads were washed twice with 1 ml of lysis buffer and twice with 1 ml of lysis buffer without Triton X-100. Thirty-five microliters of the last wash was left in the tube, mixed with 20 μl of kinase 2× mix (50 mM β-glycero-phosphate, 100 μM Na3VO4, 20 mM MgCl2, 200 μM ATP, 1 μCi of [γ-32P]ATP per μl, 400 μM epidermal growth factor receptor peptide [residues 662 to 681], 50 μg of inhibitory peptide [IP]-20 per μl, 1 mM EGTA), incubated for 20 min at 20°C, and spotted on Whatman P81 paper. The samples were washed three times for 5 min each time in 75 mM phosphoric acid and once for 2 min in acetone and air dried, and their radioactivity was determined with a β counter.
SEK1 K→M phosphorylation. (i) Method 1.
MEKK1 was immunoprecipitated from cell lysates (200 to 500 μg) with antibodies raised against specific sequences of MEKK1 or antibody 12CA5, which recognizes the HA tag sequence. The immunoprecipitates were used in an in vitro kinase assay with recombinant kinase-inactive SEK1 (SEK1 K→M) as previously described (6). (ii) Method 2. For the experiment shown in Fig. 1C, the cells were lysed in 20 mM Tris (pH 7.5)–1% Triton X-100–0.5% Nonidet P-40–150 mM NaCl–20 mM NaF–200 μM Na3VO4–1 mM EDTA–1 mM EGTA–1 mM phenylmethylsulfonyl fluoride (lysis buffer II). After clearing at 8,000 × g, 1 mg of the lysate was rotated at 4°C with antiserum 12851 (1/100 dilution) in a final volume of 500 μl. Fifteen microliters of a 1:1 slurry of protein A-Sepharose was added, and the mixture was rotated for 1 h at 4°C. The beads were washed three times with 1 ml of lysis buffer II and twice with 1 ml of PAN [10 mM piperazine-N,N′-bis(2-ethanesulfonic acid) (PIPES), 100 mM NaCl, 20 μg of aprotinin per ml]. Thirty-five microliters was left in the tubes after the last wash. The beads were incubated with 4 μl of 10× universal kinase buffer (200 mM PIPES, 100 mM MnCl2, 20 μg of aprotinin per ml)–2 μl of [γ-32P]ATP (1 μCi/μl)–1 μl of SEK1 K→M (∼1 μg/μl) for 20 min at 30°C. The reaction was run on an 8 to 10% polyacrylamide gel. After drying, the radioactivity of the bands corresponding to the SEK1 K→M protein was quantitated with a PhosphorImager.
FIG. 1.
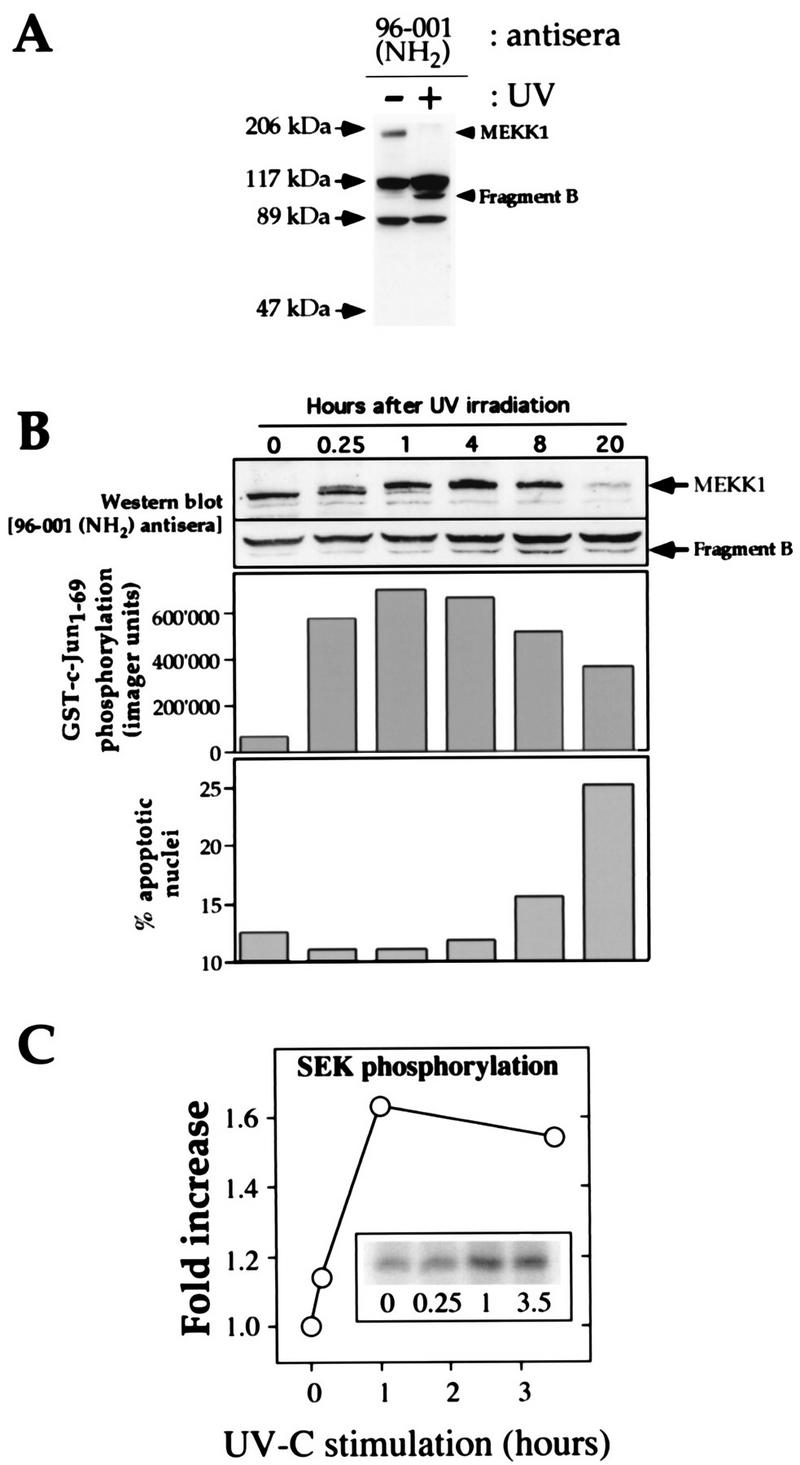
UV induces rapid phosphorylation of endogenous MEKK1 in HEK293 cells, followed by the cleavage of MEKK1 into smaller fragments. (A) HEK293 cells were irradiated or were not irradiated with UV (100 J/m2) and incubated for 16 h in DMEM containing 0.1% serum. The cells were then lysed and subjected to Western blot analysis with the MEKK1 NH2-terminus-specific antibody 96-001. The positions of full-length MEKK1 and the UV-generated NH2-terminal fragment (fragment B) are indicated. (B) The cells were treated with UV (100 J/m2) and incubated for the indicated times in DMEM containing 0.1% serum. (Top panel) Cell lysates were analyzed as described in the legend to Fig. 1A. For clarity, only the portions of the gel containing the 196-kDa MEKK1 protein and the immunoreactive MEKK1-derived fragment are shown. (Middle panel) Activation of the JNK pathway was measured with Sepharose-bound GST-Jun as a substrate. (Bottom panel) Propidium iodide-stained nuclei were analyzed with a fluorescence-activated cell sorter (46). The percentage of nuclei with an altered shape (apoptotic nuclei) was plotted as a function of time. (C) The cells were treated as in panel B. The kinase activity of the endogenous MEKK1 protein in response to UV-C irradiation was measured as described in Materials and Methods. The activation of MEKK1 in response to UV-C irradiation temporally correlated with its gel shift and with the activation of the JNK pathway.
MEKK1 staining and TdT-mediated incorporation of fluorescent dUTP.
Cells were grown on glass coverslips and transfected with Lipofectamine. Two days after transfection, the medium was removed and the cells were fixed in 2% paraformaldehyde–3% sucrose in phosphate-buffered saline (PBS) for 10 min at room temperature. Following three washes with PBS, the cells were permeabilized for 10 min with 0.2% Triton X-100 in PBS. After three PBS washes, the cells were blocked with filtered cultured medium for 15 min. The coverslips were incubated for 1 h in terminal deoxytransferase (TdT) reaction mix (200 mM potassium cacodylate, 25 mM Tris-HCl [pH 6.6], 250 μg of bovine serum albumin per ml, 5 mM CoCl2, 0.25 U of TdT [Boehringer] per μl, 10 μM biotin-dUTP [Boehringer]) at 37°C in a humidified atmosphere. After three PBS washes, the coverslips were incubated for 1 h at room temperature with a 1/500 dilution in filtered culture medium of an affinity-purified rabbit antiserum directed at peptide DRPPSRELLKHPVFR of mouse MEKK1 (amino acids 1476 to 1490) (33). The coverslips were washed six times over a 30-min period with PBS and incubated for 1 h at room temperature with a 1/1,000 dilution in filtered culture medium of a donkey anti-rabbit indocarbocyanine (Cy3)-conjugated antibody (Jackson Immunological) mixed with 5 μg of fluorescein isothiocyanate (Jackson Immunological)-conjugated streptavidin per ml. The coverslips were washed six times with PBS and incubated overnight in PBS before being mounted in 20 mg of o-phenyldiamine dihydrochloride (Sigma) per ml in 0.1 M Tris (pH 8.5)–90% glycerol. Images were taken with a Leica DMRXA microscope and analyzed with SlideBook version 2.0 software (Intelligent Imaging Innovations, Denver, Colo.). The subcellular localization of endogenous MEKK1 observed with the anti-COOH-terminal MEKK1 antibody was identical to that observed with a second antibody recognizing the NH2-terminal portion of the MEKK1 protein (data not shown).
Immunoblots.
Cell lysate protein (200 to 400 μg) was subjected to sodium dodecyl sulfate (SDS)–9% polyacrylamide gel electrophoresis (PAGE) and transferred to nitrocellulose membranes. Blotting was performed exactly as described previously (59). To detect HA-tagged proteins, mouse monoclonal antibody 12CA5 (Babco) was used as the primary antibody, followed by a rabbit anti-mouse antibody (Cappel). Horseradish peroxidase-conjugated protein A at a 1/5,000 dilution (Zymed) and 125I-protein A at a 1/500 dilution (Dupont NEN) were used for enhanced chemiluminescence (ECL) detection and for quantitation with a PhosphorImager, respectively. To detect MEKK1, three different polyclonal antisera were used as primary antibodies, followed by ECL detection with horseradish peroxidase-conjugated protein A (see above). These sera were generated by injecting rabbits with GST proteins fused with different portions of the MEKK1 protein (see Fig. 7A).
FIG. 7.

Mutation of the mouse MEKK1 sequence 871DTVD874 blocks p35-inhibited MEKK1 cleavage. (A) Schematic representation of the HA-tagged mouse MEKK1 protein showing the regions (the numbers correspond to the positions of the amino acids) used to generate the indicated antibodies. Also shown is the sequence (one-letter code) between amino acids 853 and 888 where the tetrapeptides DEVE and DTVD (in bold type) are replaced with alanine residues in mutants DEVE→A and DTVD→A, respectively. (B) Western blot analysis with the antibodies shown in panel A of lysates derived from HEK293 cells transfected with 0.5 μg of pcDNA_3.cp4, MEKK1.cp4, DEVE_A.cp4, or DTVD_A.cp4. Letters A to D indicate the same cleavage products as those shown in Fig. 6A. (C) Full-length activated MEKK1 and fragment C have similar SEK1 K→M phosphorylation activities. HEK293 cells were transfected with 4 μg of MEKK1 in pcDNA3 (MEKK1k.dn3 plasmid) or fragment C in pcDNA3 (G875.dn3 plasmid). At 18 h after transfection, 5 mg of cell lysate proteins was immunoprecipitated with antibody 12CA5 (recognizing the NH2-terminal HA tag). Serial dilutions of the immunoprecipitates (1:1, 1:2, 1:4, and 1:8) were then analyzed by Western blotting with antibody 95-012 (recognizing the COOH terminus of MEKK1) or analyzed for their ability to phosphorylate the SEK1 K→M substrate. (D) Schematic representation of p35-inhibited and p35-insensitive cleavage in the mouse MEKK1 protein. Letters A to D indicate the names of the cleavage products. The molecular masses were calculated from the migration of the markers in at least two different experiments, such as the one presented in panel B.
Preparation of lysates from apoptotic Jurkat cells.
Jurkat cells were incubated with 1 μg of anti-Fas immunoglobulin M antibodies (Upstate Biotechnology no. 05-201) per ml in PBS for 20 to 30 min on ice. The cells were washed twice with PBS, resuspended in RPMI 1640 (catalog no. 31800-022; Life Technologies, Inc.) supplemented with 100 U each of penicillin and streptomycin per ml and containing 10% fetal bovine serum, and incubated for 1 h at 37°C in 5% CO2. When caspase inhibitors were used, they were incubated with Jurkat cells both during the incubation with anti-Fas antibodies and during the incubation at 37°C. The cells were lysed in the lysis buffer used to measure caspase activities (see below).
Measurements of caspase activities.
Transfected cells were lysed in 50 mM Tris (pH 7.4)–1 mM EDTA–10 mM EGTA–10 μM digitonin for 10 min at 37°C. Lysate proteins (60 μg) were incubated with 5 μM DEVE-AMC (Bachem) in 1 ml of 50 mM Tris (pH 7.4)–1 mM EDTA–10 mM EGTA for 20 min at 37°C. Fluorescence was monitored with an excitation wavelength of 380 nm and an emission wavelength of 460 nm. Fluorescence of the substrate alone was subtracted in each case.
In vitro translation.
Proteins were translated in vitro with the TNT T7-coupled reticulocyte lysate system (Promega) in accordance with the manufacturer’s instructions. The plasmids used were MEKK1.dn3 and DTVD_A.dn3. The cleavage assay for the in vitro-translated proteins was performed with the buffer used to measure DEVD-directed caspase activity (see above).
PP-2A treatment.
MEKK1 was immunoprecipitated from cell lysates (200 to 500 μg) with the 96-001 (NH2) antisera and washed twice with 1 ml of extraction buffer (1% Triton X-100, 10 mM Tris [pH 7.4], 50 mM NaCl, 50 mM NaF, 5 mM EDTA), twice with 1 ml of TC (50 mM Tris [pH 7.0], 0.1 mM CaCl2), and once with 1 ml of TC containing 60 mM β-mercaptoethanol–1 mM MgCl2. Thirty-five microliters of the last wash was left in the tube, and 0.5 U of protein phosphatase 2A (PP-2A) (Upstate Biotechnology) was added for 30 to 45 min. The phosphatase reaction was terminated by the addition of 1 μl of 200 mM Na3VO4. For in vitro kinase assays, the immunoprecipitates were washed three more times with 1 ml of PAN before being mixed with the SEK1 K→M substrate and [γ-32P]ATP.
RESULTS
UV irradiation of HEK293 cells induces rapid phosphorylation and subsequent cleavage of the endogenous MEKK1 protein.
The ability of UV irradiation to induce apoptosis is well defined (11, 61). We examined the regulation of endogenous MEKK1 in response to UV irradiation. HEK293 cells were either left untreated or irradiated with UV (100 J/m2) and incubated for 16 h in low-serum media. The presence of MEKK1 was then determined with a MEKK1 NH2-terminus-specific antibody (antibody 96-001). Figure 1A shows that upon UV irradiation, full-length MEKK1 disappeared and was replaced by a new immunoreactive fragment recognized by the amino-terminus-specific antibody. This fragment of about 113 kDa was named fragment B (see below).
A time course experiment was performed to determine the effects of UV-C irradiation on the endogenous MEKK1 protein, activation of the JNK pathway, and the extent of apoptosis. Figure 1B (top panel) shows that at 15 min after UV irradiation, a MEKK1 species that was shifted upward in the gel compared to the MEKK1 species detected before exposure to UV irradiation was generated. At 1 h after irradiation, most of the full-length MEKK1 protein was shifted upward in the gel. The gel shift of MEKK1 was temporally associated with an increase in its kinase activity, as assessed by the ability of immunoprecipitated MEKK1 from UV-C-irradiated cells to phosphorylate the kinase-inactive SEK1 K→M substrate (Fig. 1C). At 8 h after irradiation, the amount of the gel-shifted MEKK1 started to decrease, and at 20 h after UV treatment, only a trace amount of full-length MEKK1 was observed.
As shown in Fig. 1A, the 113-kDa MEKK1-derived fragment B was generated as a result of UV irradiation. This fragment was first detected at 1 h after UV irradiation, but its maximal production occurred after 8 h of UV irradiation (Fig. 1B, top panel). JNK activation after UV irradiation paralleled the extent of the MEKK1 gel shift (Fig. 1B, middle panel). Apoptosis, as assessed by morphological changes in propidium iodide-stained nuclei, was first detectable at 8 h after UV irradiation and was most apparent after 20 h (Fig. 1B, lower panel).
To determine whether the upward gel shift of MEKK1 was due to phosphorylation, lysates of epitope-tagged MEKK1-transfected cells were immunoprecipitated with antibody 12CA5 and incubated with or without PP-2A. In cells overexpressing MEKK1, two or three MEKK1-immunoreactive bands are usually detected in the 200-kDa region of polyacrylamide gels (for examples, see Fig. 2A and 7B). Figure 2A shows that phosphatase treatment converted the upper, gel-shifted band to the lower band, demonstrating that the gel shift was a phosphorylation-dependent event. To determine whether phosphorylation of MEKK1 was required for its activity, the ability of immunoprecipitated MEKK1 to phosphorylate one of its substrates, SEK1 K→M, was assessed after pretreatment with PP-2A. Figure 2B shows that immunoprecipitated MEKK1 phosphorylated SEK1 K→M but, when treated with phosphatase, lost autophosphorylation and the phosphorylation activity toward SEK1 K→M. These results, together with those of other studies (15, 52), indicate that phosphorylation of MEKK1 is required for its activation and that, in transfected cells, a fraction of MEKK1 is constitutively phosphorylated and activated. The fact that UV irradiation induces a gel shift and the activation of endogenous MEKK1 before the appearance of a MEKK1 fragment (Fig. 1B and C) indicates that the activation of MEKK1 precedes its cleavage in response to UV irradiation. This activation of MEKK1 parallels the extent of JNK activation.
FIG. 2.
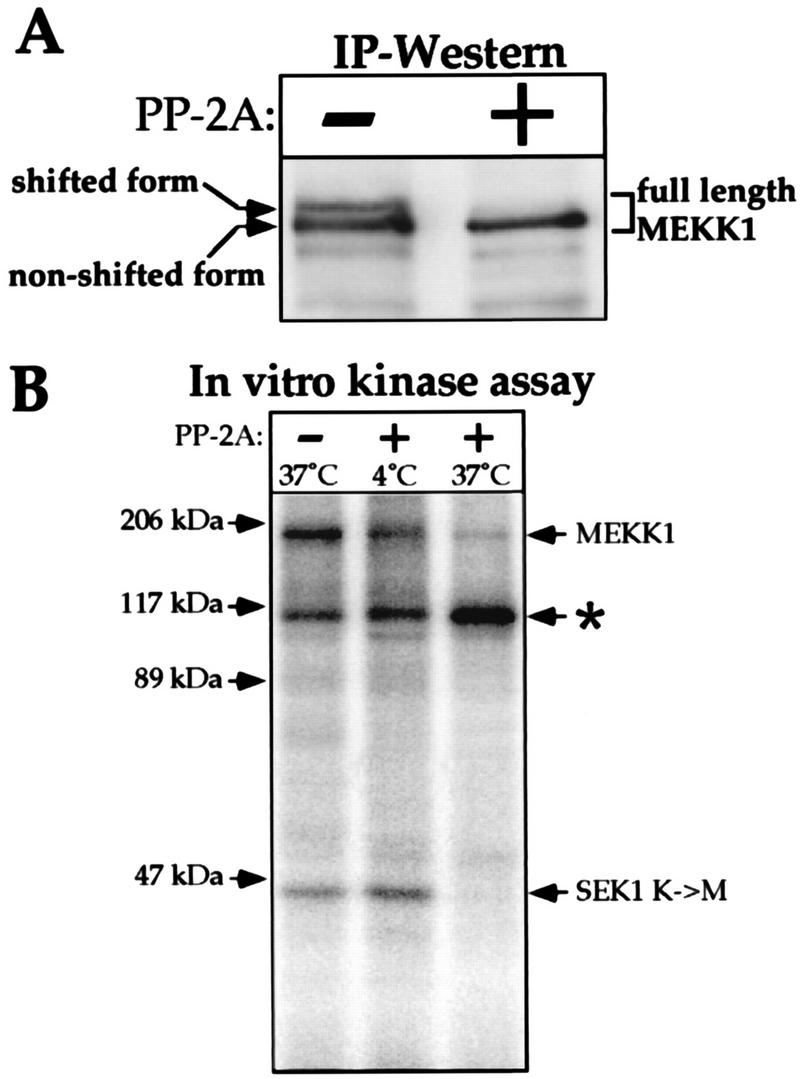
The gel-shifted form of MEKK1 corresponds to an active phosphorylated kinase. (A) Cell lysates from HEK293 cells were transfected with 1 μg of MEKK1.cp4, carrying the HA-tagged full-length MEKK1, immunoprecipitated with antibody 12CA5, and incubated for 45 min with or without PP-2A at 37°C as described in Materials and Methods. The immunoprecipitates (IP) were then subjected to Western blot analysis with antibody 96-001. Arrows indicate the positions of the nonshifted and shifted forms of MEKK1. (B) Cell lysates from MEKK1-transfected cells were immunoprecipitated and incubated with PP-2A at 4 or 37°C or left untreated as described in panel A. After several washes, the immunoprecipitates were incubated with recombinant SEK1 K→M substrate and [γ-32P]ATP. Phosphorylated proteins were resolved by SDS-PAGE. The identity of the protein indicated by an asterisk is unknown. Note that when the immunoprecipitates were incubated with PP-2A at 4°C and washed, SEK1 K→M phosphorylation still occurred, showing that the absence of SEK1 K→M phosphorylation by immunoprecipitates treated with PP-2A at 37°C was not due to residual PP-2A activity that would not have been eliminated during the washing steps.
Cleavage of MEKK1 is induced by genotoxins.
Several additional genotoxic stress stimuli were applied to HEK293 cells, and their effect on the MEKK1 protein was assessed. Figure 3 shows that UV irradiation, cisplatin, etoposide, and mitomycin induced the loss of full-length MEKK1 and the appearance of a lower-molecular-weight fragment (fragment B). While no full-length MEKK1 protein remained after UV and cisplatin treatments, a small amount of full-length MEKK1 shifted upward in the gel was detected in etoposide- and mitomycin-treated cells. Thus, chemicals capable of forming DNA adducts induce the phosphorylation of MEKK1 before its cleavage.
FIG. 3.

Genotoxins induce the cleavage of MEKK1. HEK293 cells were either left untreated, stimulated with UV at 100 J/m2, or incubated with 50 μM cisplatin, 100 μM etoposide, or 30 μM mitomycin in DMEM containing 0.1% fetal bovine serum. After 18 h, the cells were lysed and subjected to Western blot analysis with antibody 96-001. For clarity, only the unphosphorylated and gel-shifted full-length MEKK1 proteins and the NH2-terminal MEKK1-derived fragment (fragment B) are shown.
To determine if MEKK1 was necessary for the apoptotic response to genotoxic stress stimuli, HEK293 cells that stably expressed a kinase-inactive mutant of MEKK1 were isolated. In this mutant, the active lysine residue of the ATP binding pocket is substituted with a methionine residue. Figure 4A shows that, in contrast to the wild-type MEKK1 protein, the mutant did not activate the JNK pathway when overexpressed in cells, demonstrating that it is truly kinase inactive. In stable clones 7 and 11, kinase-inactive MEKK1 was present as the 196-kDa full-length protein and as a fragment of approximately 134 kDa (named fragment A; see below), which was larger than fragment B generated in genotoxin-treated cells (Fig. 1A). The JNK activity induced by transfection of a plasmid expressing wild-type MEKK1 was reduced by ∼60% in clones stably expressing the kinase-inactive MEKK1 protein compared to control clones (data not shown), consistent with the known ability of the kinase-inactive MEKK1 mutant to block JNK activity in several cell types (7, 62). The JNK activity measured 1 h after UV irradiation (100 J/m2) or following a 4-h stimulation with 100 μM etoposide was only partially inhibited (<30%) in HEK293 clones expressing the kinase-inactive MEKK1 protein, indicating that JNK activation in response to these stimuli involves other kinases in addition to MEKK1. Nonetheless, Fig. 4C shows that the two HEK293 clones expressing kinase-inactive MEKK1 had a markedly diminished apoptotic response to each of the genotoxic agents. Thus, a competitive inhibitory kinase-inactive MEKK1 strongly suppresses apoptosis in response to genotoxins. If the regulation of MEKK1 were secondary and a consequence of, rather than a necessity for, the induction of apoptosis, the kinase-inactive mutant would not have suppressed the cell death response to genotoxic agents.
FIG. 4.

MEKK1 is involved in genotoxin-induced apoptosis. (A) HEK293 cells were transfected with 4 μg of empty pcDNA3 vector or pcDNA3 containing wild-type MEKK1 (plasmid MEKK1.dn3) or the MEKK1 K(1253)→M mutant [plasmid MEKK1(−).dn3]. After 18 h, the cells were lysed and the MEKK1 proteins were immunoprecipitated with antibody 12CA5, recognizing the NH2-terminal HA tag. The immunoprecipitates were then analyzed by Western blotting (WB) with antibody 12CA5. Alternatively, the JNK activity of the cell lysates was determined as described in Materials and Methods. Despite similar levels of expression, only the wild-type MEKK1 protein was able to induce the activity of the JNK pathway. (B) Clones of HEK293 cells stably transfected with pcDNA3 (clones V1 and V4) or pcDNA3 expressing the kinase-inactive MEKK1 K(1253)→M protein [plasmid MEKK1(−).dn3] (clones 7 and 11) were lysed, and the expression of MEKK1 was determined by Western blot analysis with antibody 12CA5. The positions of the full-length kinase-inactive MEKK1 and fragment A are indicated (see the text for details about the generation of fragments A and B). (C) The clones shown in panel B were stimulated as described in the legend to Fig. 3. Apoptotic cells were scored after acridine orange staining (41). Data are the mean ± standard error of the mean for duplicate determinations. (D) Clone 11 (described in panel B) was incubated with etoposide or irradiated with UV as described in Fig. 3. The cells were lysed and then analyzed by Western blotting with anti-HA antibody 12CA5. The positions of the full-length kinase-inactive MEKK1 and fragment A are indicated.
We then assessed whether the catalytic activity of overexpressed MEKK1 was required for phosphorylation and the cleavage events induced by apoptotic stimuli. A stable clone expressing the kinase-inactive mutant form of MEKK1 (clone 11) was subjected to etoposide treatment or UV irradiation, and the effects on the mutant MEKK1 protein were determined by Western blot analysis. Figure 4D shows that the catalytically inactive mutant MEKK1 protein was not cleaved into fragment B in response to etoposide treatment or UV irradiation, in contrast to the endogenous MEKK1 protein in untransfected cells (Fig. 1B). This result indicates that the kinase activity of MEKK1 is required for its cleavage in response to genotoxins and further suggests an important role of MEKK1 in genotoxin-induced apoptosis.
Expression of the 196-kDa MEKK1 protein by gene transfection induces apoptosis.
Expression of just the kinase domain of MEKK1 (ΔMEKK1) induces cell death by apoptosis (7, 34, 61, 62). To assess whether the full-length protein had the same effect, HEK293 cells were transfected with a plasmid encoding the mouse MEKK1 and stained 2 days later for MEKK1 expression with an antibody directed to the COOH terminus of the protein. Cell death was measured by monitoring DNA fragmentation as TdT-mediated incorporation of fluorescent dUTP. The apoptotic response quantitated by the TdT assay was verified by morphological changes, including cytoplasmic shrinkage and nuclear condensation. Figure 5A (left panel) shows that a large proportion of HEK293 cells expressing MEKK1 (red staining) had fragmented DNA (green staining). The MEKK1-expressing cells characteristically became round and began to lift off the coverslips (Fig. 5B, upper left panel; note that the untransfected cells and MEKK1-expressing cells did not lie on the same focal plane due to the morphological changes associated with apoptosis). MEKK1 also induced chromatin condensation, and the nuclei in these cells often dissociated from the surrounding cytoplasm (Fig. 5B, upper panels). Quantitation of green (DNA fragmentation) and red (MEKK1 expression) cells revealed that about 30% of MEKK1-expressing cells were apoptotic after 48 h (Fig. 5C). This amount is an underestimate because the apoptotic cells eventually detached from the coverslips and often lost their nuclei. Thus, expression of the 196-kDa MEKK1 protein by gene transfection induced cell death characteristic of apoptosis and similar to that observed for genotoxins. Longer exposure to MEKK1 induced the death of >95% of the cells, as assessed by a colony-formation assay (60).
FIG. 5.
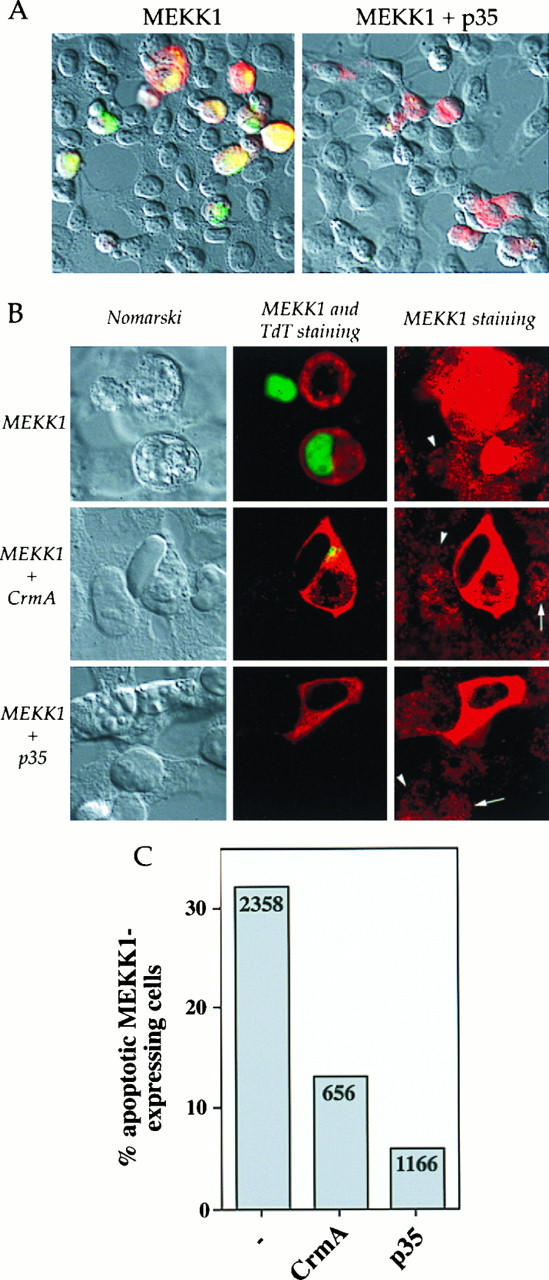
p35 and CrmA inhibit MEKK1-induced DNA fragmentation in HEK293 cells. Cells were transfected with 0.5 μg of MEKK1.cp4 alone or in combination with 2 μg of either p35.cp_ or CrmA.cp_. Two days later, the cells were stained for MEKK1 expression and for DNA fragmentation. (A) Nomarski views (magnification, ×40) of HEK293 cells transfected with MEKK1 or with MEKK1 and p35 and overlaid with fluorescent staining for MEKK1 expression (red staining) and for DNA fragmentation (green staining). (B) Views (magnification, ×160) of HEK293 cells transfected with MEKK1 alone or in combination with either CrmA or p35. (Left panels) Nomarski views. (Middle panels) Fluorescent staining for MEKK1 expression (red staining) and DNA fragmentation (green staining). (Right panels) Fluorescent staining for MEKK1 expression. In these last views, an exposure longer than that in the middle panels was used to visualize the localization of endogenous human MEKK1. The arrows indicate granular cytoplasmic localization, while the arrowheads indicate nuclear localization. (C) Quantitation of the percentage of MEKK1-transfected cells, in the presence or in the absence of the indicated proteins, that showed DNA fragmentation. The numbers in the columns indicate the number of cells transfected with MEKK1 and counted on at least four coverslips from at least two different experiments.
MEKK1-induced DNA fragmentation is inhibited by p35 and CrmA.
Inhibition of caspases by the baculovirus p35 protein or by the poxvirus CrmA protein has been shown to protect cells from apoptosis in response to diverse stimuli (5, 12). Cotransfection of HEK293 cells with MEKK1 and p35 inhibited the DNA fragmentation seen with the expression of MEKK1 alone (Fig. 5A, right panel). Cotransfection of MEKK1 with CrmA also inhibited DNA fragmentation, but to a lesser extent. While only about 5% of the cells cotransfected with MEKK1 and p35 showed some DNA fragmentation, this proportion was about 15% in MEKK1- and CrmA-cotransfected cells (Fig. 5C). A small area of fragmented DNA was typically seen in the nuclei of these cells (Fig. 5B, middle panels). These findings demonstrate that viral caspase inhibitors prevent MEKK1-induced apoptosis.
CrmA and p35 inhibit cleavage of the 196-kDa MEKK1 protein and the generation of an active kinase fragment.
When MEKK1 was expressed by transfection of HEK293 cells, two additional immunoreactive polypeptides besides the full-length protein (fragments A and B; Fig. 6A, left panel) were detected by Western blotting with an antibody directed to the HA tag of MEKK1 (antibody 12CA5). Antibody 12CA5 recognizes the first 11 amino acids at the NH2 terminus of the tagged MEKK1 protein, indicating that fragments A and B must be the result of proteolysis of the full-length MEKK1 protein and cannot have arisen from other potential translation sites. When an antibody directed to the COOH terminus of MEKK1 was used (antibody 95-012), additional immunoreactive fragments were also detected (Fig. 6A, right panel). Based on their apparent molecular masses and on their patterns of recognition by different MEKK1-specific antibodies (see Fig. 7A), two of these fragments, named C and D, should be the corresponding moieties of cleavage products B and A, respectively (for a discussion, see below). It is also important to note that further proteolytic processing of fragment C might generate fragment D. Based on its behavior in the SDS gel, the band marked with an asterisk in Fig. 6A might be a dimer of D or a modified form of C.
FIG. 6.

CrmA and p35 inhibit the generation of a MEKK1-derived kinase-active cleavage product. Cells were transfected as described in the legend to Fig. 5. (A) Western blot analysis of lysates with antibodies 12CA5 and 95-012. The immunoreactive proteins were detected by ECL. Fragments A, B, C, and D correspond to MEKK1 cleavage products, and the band marked with an asterisk may correspond to a dimer of fragment D (see the text). (B) A kinase-inactive MEKK1 mutant (MEKK1 K→M) is not cleaved into fragments B and C. Cells were transfected with 1 μg of vector alone (pcDNA3), HA-tagged MEKK1 in pcDNA3 (plasmid MEKK1.dn3), or HA-tagged MEKK1 K(1253)→M in pcDNA3 [plasmid MEKK1(−).dn3]. At 18 h after transfection, the cells were lysed and the presence of MEKK1 species was detected by Western blot analysis with antibody 12CA5. The positions of fragments A and B and full-length MEKK1 are indicated. (C) In vitro SEK1 K→M phosphorylation assay performed on cell lysates immunoprecipitated with the indicated antibodies. The positions of MEKK1, fragments C and D, and SEK1 K→M are indicated.
The observation that MEKK1 can be proteolyzed to very specific fragments prompted us to determine whether p35 or CrmA could inhibit the generation of fragments A, B, C, and D. Figure 6A shows that p35 almost totally and CrmA partially inhibited the appearance of fragments B and C. Quantitation of the fragments in six independent experiments revealed that CrmA and p35, while leaving the relative proportion of fragment A unchanged, diminished the relative proportion of fragment B by 50 and 90%, respectively. This result indicates that these protease inhibitors prevented the formation of fragments B and C but had no effect on the proteolytic activity that cleaves MEKK1 into fragment A. Since the cleavage of MEKK1 into fragment A was unaffected by CrmA and p35, it was surprising to find that the amount of fragment D, the corresponding moiety of fragment A, was reduced in the presence of the inhibitors (Fig. 6A, right panel). However, since fragment D could be derived from fragment C, blocking the generation of fragment C would result in less fragment D. Moreover, because the amounts of fragments A and B formed in MEKK1-transfected cells were not significantly different from one another, the observation that there was far less fragment D than fragment C (Fig. 6A, right panel, lane MEKK1) suggested that fragment D might be unstable and rapidly degraded. Since it has not been formally proven that fragments A and D are the products of a single cleavage event, an alternative explanation may be that fragment D is generated independently from fragment A, but in a less efficient way.
The immunoblots also showed that there were smaller amounts of total MEKK1-immunoreactive species (fragments and full-length MEKK1) when the cleavage generating fragments B and C was blocked. This result might be explained by our observation that expression of the kinase domain of MEKK1 stimulates the activity of the cytomegalovirus promoter (unpublished observation). Figure 6B shows that the kinase activity of MEKK1 was required for stimulation of its cleavage to generate fragment B. Kinase-inactive MEKK1 was neither cleaved to fragment B nor expressed to the same level as wild-type MEKK1. Wild-type MEKK1 displayed a slightly reduced mobility compared to kinase-inactive MEKK1. This result was due to phosphorylation of the kinase, leading to its activation (see above).
To assess whether MEKK1 fragments had kinase activity, cells were transfected with HA-tagged MEKK1 alone or in combination with CrmA or p35. Immunoprecipitation with antibody 12CA5 recovered the full-length protein and N-terminal fragments A and B. Immunoprecipitation of the lysates with an antibody directed to the COOH-terminal moiety of MEKK1 (antibody 95-012) recovered the full-length protein as well as C-terminal fragments C and D. The immunoprecipitates were then incubated with a MEKK1 substrate (SEK1 K→M) and [γ-32P]ATP. When the MEKK1 protein was immunoprecipitated with antibody 12CA5, it showed measurable autophosphorylation and activity toward SEK1 K→M (Fig. 6C, left panel). No phosphorylation of fragments A and B was observed. When MEKK1 was immunoprecipitated with antibody 95-012, a stronger SEK1 K→M phosphorylation signal was detected (Fig. 6C, right panel, lane MEKK1). The full-length MEKK1 protein and fragments C and D were immunoprecipitated with similar efficiencies (data not shown). The increased phosphorylation of SEK1 K→M was due to the presence of fragments C and D in the immunoprecipitates. This phosphorylation was reduced in the presence of CrmA. In the presence of p35, antibody 12CA5 immunoprecipitated the same amount of kinase activity as in the absence of p35, indicating that the kinase activity of the full-length protein was unaffected by coexpression of this protein. Phosphorylation of fragments C and D was also detected in immunoprecipitates obtained with antibody 95-012 (Fig. 6C, right panel). This phosphorylation was reduced by CrmA and almost completely abolished by p35, as expected from the effects of these inhibitors on the expression of MEKK1 and the generation of fragments C and D (Fig. 6A).
p35-inhibited cleavage occurs at position 871DTVD874 in the mouse MEKK1 protein.
The p35-inhibited cleavage of MEKK1 generated a COOH-terminal fragment of about 90 kDa and an NH2-terminal fragment of about 110 kDa (Fig. 6A; see also Fig. 7D), indicating that the cleavage occurred between residues 820 and 900. Two tetrapeptide sequences that are found in this region of MEKK1 closely resemble the caspase-3 cleavage site, DEVD (45). These sequences are 857DEVE860 and 871DTVD874 (see Fig. 7A). The proteases inhibited by p35 have been shown to be cysteine proteases cleaving after the aspartic acid residue in the fourth position of the consensus cleavage sequence (28, 45); therefore, only the DTVD sequence should be a cleavage site for the caspase-3-like protease. Two mutants that have either the DEVE or the DTVD sequence replaced with alanine residues were generated (see Fig. 7A). These mutants were transfected into HEK293 cells, and the presence of MEKK1 and MEKK1-derived fragments was detected by immunoblot analysis with antibody 12CA5 and the three MEKK1-specific antibodies shown in Fig. 7A. Figure 7B shows that when transfected into HEK293 cells, the DEVE→A mutant, like the wild-type protein, was cleaved into fragments A, B, C, and D. In contrast, the DTVD→A mutant was cleaved only into fragments A and D. Thus, fragments B and C are not generated in cells expressing the DTVD→A mutant or in cells expressing MEKK1 and p35. The results indicate that p35-inhibited cleavage occurs at position 871DTVD874 in the mouse MEKK1 sequence.
To compare the kinase activities of the 196-kDa full-length MEKK1 protein and the 91-kDa kinase fragment (fragment C), lysates from cells transfected with the HA-tagged version of each protein were immunoprecipitated with anti-HA antibodies. Serial dilutions of the immunoprecipitates were then analyzed by Western blotting with an antibody specific for the COOH terminus of MEKK1 or analyzed for their ability to phosphorylate a MEKK1 substrate (SEK1 K→M). Comparison of the Western blot analysis and the in vitro kinase assay revealed that both proteins had similar kinase specific activities toward SEK1 (Fig. 7C). Also, both proteins had autophosphorylation activities. Thus, when these proteins are overexpressed in cells, they become phosphorylated and activated; the cleavage of activated MEKK1 therefore does not detectably increase its kinase activity because the full-length protein is already activated when overexpressed in cells.
Based on the results presented above, Fig. 7D shows a model of the MEKK1 cleavage events occurring in transfected cells. In this model, the overexpression of MEKK1 induces deregulated cleavage events generating two sets of fragments (A and D; B and C). The kinase activity of MEKK1 is required for the generation of fragments that are no longer associated with the NH2-terminal regulatory domain (Fig. 4B and D and 6B). Fragment C encodes the catalytic domain of MEKK1. Caspases are responsible for the cleavage of MEKK1 into fragments B and C because this cleavage can be inhibited by p35 and CrmA. Mutagenesis revealed that the cleavage site generating fragments B and C is 871DTVD874. Fragment C can probably be further processed into a smaller polypeptide (fragment D). It is possible that the proteolytic activity which generates fragment D is part of a regulatory mechanism involved in the termination of the response induced by the cleavage of MEKK1 into active fragment C.
The finding that the kinase activity of MEKK1 was required for its proteolysis suggested that MEKK1 stimulated protease activity. Cells were transfected with pCEP4, MEKK1k.cp4, or MEKK1.cp4 or left untreated. At 24 h after transfection, caspase activities were measured as described in Materials and Methods. Caspase activities, expressed as the fold increase compared to that in untreated cells (set at 1.0) (mean ± standard deviation for three independent experiments), for pCEP4, ΔMEKK1, and MEKK1 were 1.1 ± 0.1, 6.0 ± 1.3, and 2.5 ± 0.3, respectively, indicating that expression of ΔMEKK1 and full-length MEKK1 stimulated caspase-3-like activity. The truncated form of MEKK1 stimulates this activity more than did the full-length protein, but this result likely was due to the higher level of expression of the former. The substrate for MEKK1 that results in caspase activation is presently unknown.
The DTVD sequence in MEKK1 is a caspase-3-like cleavage site.
Lysates from Fas-activated Jurkat cells have proven to be a good system for the assay of DEVD-directed caspases (55). To determine whether caspases activated in apoptotic cells could cleave MEKK1, in vitro-translated [35S]methionine-labeled MEKK1 was incubated with lysates from control Jurkat cells or with lysates from Jurkat cells stimulated with Fas in the presence or absence of caspase inhibitors. Figure 8A shows that buffer alone (control) or lysates from untreated Jurkat cells did not induce the cleavage of MEKK1. In contrast, lysates from apoptotic Jurkat cells cleaved MEKK1 into two fragments that correspond to fragments B and C, according to their apparent molecular masses. The cleavage of MEKK1 was blocked when lysates from caspase inhibitor-treated Jurkat cells were used, indicating again that the cleavage of MEKK1 is a caspase-dependent event. In contrast to the wild-type protein, the DTVD→A mutant was not cleaved by lysates prepared from apoptotic cells (Fig. 8B) or when incubated with purified CPP32 enzyme (Fig. 8C). This result demonstrates that MEKK1 is a DEVD-directed caspase substrate.
FIG. 8.

The DTVD sequence in MEKK1 is a caspase-3-like cleavage site. Wild-type MEKK1 and the DTVD→A mutant were translated in vitro as described in Materials and Methods. (A) In vitro-translated wild-type MEKK1 was incubated for 2 h with 6 μg of lysates from Jurkat cells stimulated with 1 μg of anti-Fas immunoglobulin M antibodies per ml for 1 h in the presence or absence of a 20 μM concentration of the caspase inhibitor Ac-YVAD-CMK (Bachem) or Z-DEVD-FMK (Enzyme Systems Products). The control lane indicates untreated in vitro-translated MEKK1. (B) In vitro-translated wild-type (wt) MEKK1 and DTVD→A mutant were incubated for 2 h with 6 μg of lysates from control Jurkat cells (−) or Jurkat cells stimulated with anti-Fas antibodies (+) as described in panel A. (C) In vitro-translated wild-type (wt) MEKK1 and DTVD→A mutant were left untreated (−) or incubated with 200 ng of purified caspase-3 (CPP32) (Pharmingen) for 1 h at 37°C (+). In each panel, the positions of full-length MEKK1 and fragments B and C are indicated.
The DTVD→A mutant has a reduced ability to promote DNA fragmentation in HEK293 cells.
We next determined whether the DTVD→A mutant induces DNA fragmentation when expressed in HEK293 cells. Figure 9A shows that expression of the DEVE→A mutant or the wild-type MEKK1 protein induced DNA fragmentation. In contrast, cells expressing the DTVD→A mutant MEKK1 protein showed little DNA fragmentation (Fig. 9A). Quantitation of the response revealed that the number of DTVD→A-expressing cells that showed DNA fragmentation was reduced by 65% compared to the number of cells transfected with the wild-type MEKK1 protein or the DEVE→A mutant MEKK1 protein (Fig. 5C and 9B). The observation that the caspase-resistant MEKK1 mutant had a diminished ability to induce apoptosis indicates that the cleavage of MEKK1 into fragments B and C enhances the cell death resulting from MEKK1 activation.
FIG. 9.
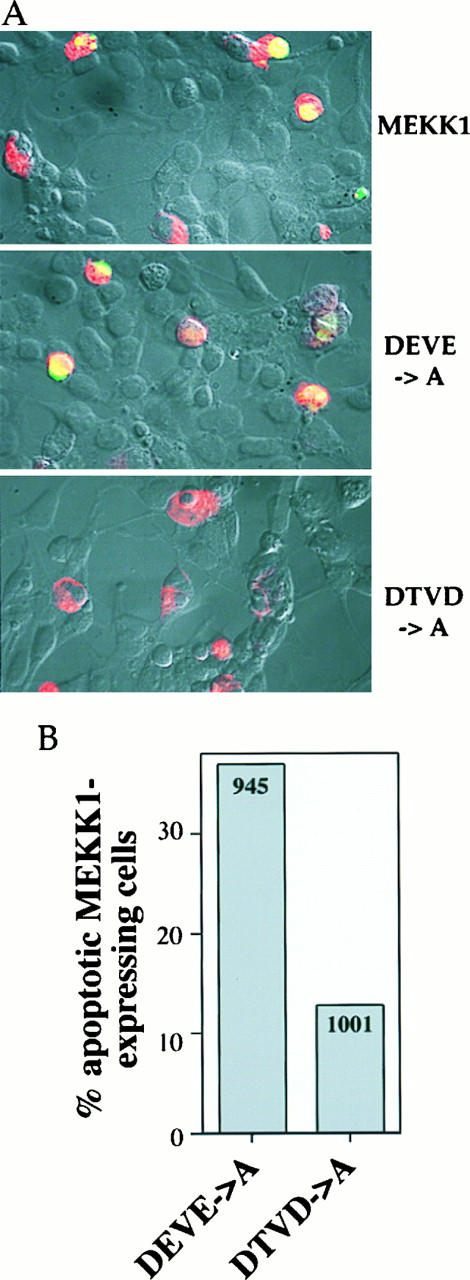
The DTVD→A mutant has a reduced ability to promote DNA fragmentation in HEK293 cells. HEK293 cells were transfected with 1 μg of MEKK1.cp4, DEVE_A.cp4, or DTVD_A.cp4 and processed as described in the legend to Fig. 5. (A) Nomarski views (magnification, ×40) of HEK293 cells transfected with wild-type MEKK1 or the indicated mutants and overlaid with fluorescent staining for MEKK1 expression (red staining) and for DNA fragmentation (green staining). (B) Quantitation of the percentage of MEKK1 mutant-transfected cells that showed DNA fragmentation. The numbers in the columns indicate the number of cells transfected with the MEKK1 mutants and counted on at least four coverslips from at least two different experiments.
p35 inhibits ΔMEKK1-induced apoptosis.
The kinase domain of MEKK1 (ΔMEKK1) is a strong inducer of apoptosis (34, 62). ΔMEKK1 and the 91-kDa carboxy-terminal MEKK1 fragment C have the same ability to induce cell death when overexpressed in cells (data not shown) (61). Since p35 inhibits programmed cell death induced by most, if not all, apoptotic stimuli (12), we wished to determine whether this inhibitor could also block ΔMEKK1-induced apoptosis. Figure 10A (upper panel) shows that ΔMEKK1 induced DNA fragmentation when expressed in HEK293 cells. This effect was inhibited by the coexpression of p35 (Fig. 10A, lower panel). Quantitation showed that 40% of cells expressing ΔMEKK1 showed DNA breaks; coexpression of p35 and ΔMEKK1 reduced this number to 10% (Fig. 10B). The number of ΔMEKK1-expressing cells appeared to be increased when p35 was present (compare upper and lower panels of Fig. 10A), suggesting that less cell death occurred when ΔMEKK1 and p35 were coexpressed. Even if the cotransfected cells showed less DNA fragmentation than the cells transfected with ΔMEKK1 alone, they were clearly affected by the expression of ΔMEKK1 and were rounded, and most showed some membrane blebbing (Fig. 10A, lower panel). This effect differed from the effect of p35 on full-length MEKK1-transfected cells, where the inhibitor appeared to better protect the cells from DNA fragmentation and obvious morphological changes (compare Fig. 5A and the lower panels of Fig. 5B with the lower panel of Fig. 10A), the predicted result if a cleavage product of MEKK1 were proapoptotic. These results indicate that p35 inhibits the cleavage of MEKK1 into a proapoptotic kinase fragment and events downstream of the MEKK1 cleavage. Thus, MEKK1 induces caspase-3-like activity and is a substrate for caspases.
FIG. 10.
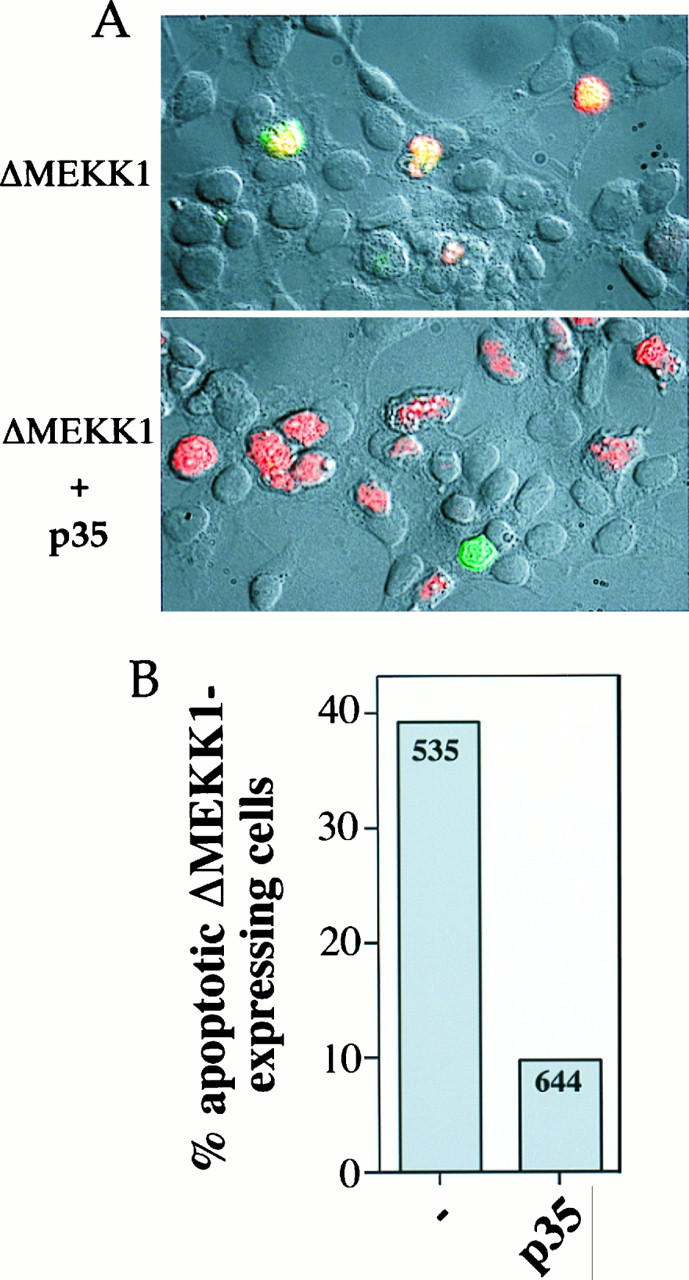
p35 inhibits ΔMEKK1-induced DNA fragmentation. HEK293 cells were transfected with 0.1 μg of MEKK1k.cp4 alone or in combination with 2 μg of p35.cp_ and stained 2 days later for MEKK1 expression and DNA fragmentation. (A) Nomarski views (magnification, ×40) of cells overlaid with fluorescent staining for MEKK1 expression (red staining) and for DNA fragmentation (green staining). (B) Quantitation of the percentage of ΔMEKK1-transfected cells that showed DNA fragmentation. The numbers in the columns indicate the number of cells expressing ΔMEKK1 and counted on four coverslips from two different experiments.
Activation of the ERK and JNK pathways is not correlated with MEKK1-induced DNA fragmentation.
Since activation of the JNK pathway has been proposed to induce apoptosis (57), we wished to determine whether p35 or CrmA inhibition of MEKK1-induced apoptosis was correlated with an effect on JNK or ERK activation. Figure 11A shows that p35 or CrmA had little, if any, effect on the activation of ERK2 or JNK by MEKK1. The DEVE→A and DTVD→A mutants activated JNK to the same level as wild-type MEKK1 (Fig. 11A, lower panel). Transfection of MEKK1 in HEK293 cells did not activate the p38 kinase (60). The in vitro SEK1 K→M phosphorylation ability of transfected MEKK1 was reduced when cleavage at position 874 was blocked (see above). Under the same conditions, no reduction of JNK activation was observed (Fig. 11A). This finding is explained in part by the fact that the assay measuring JNK activity is more sensitive than the assay assessing SEK1 K→M phosphorylation activity (Fig. 11B). Cumulatively, these results show that under conditions where MEKK1-induced DNA fragmentation is inhibited (i.e., when p35 is cotransfected with MEKK1 or when the DTVD→A mutant is expressed), the ERK and JNK pathways are still activated to an extent similar to that found in MEKK1-transfected cells. Thus, neither the ERK nor the JNK pathway is sufficient to promote or inhibit the cell death pathway induced by MEKK1.
FIG. 11.
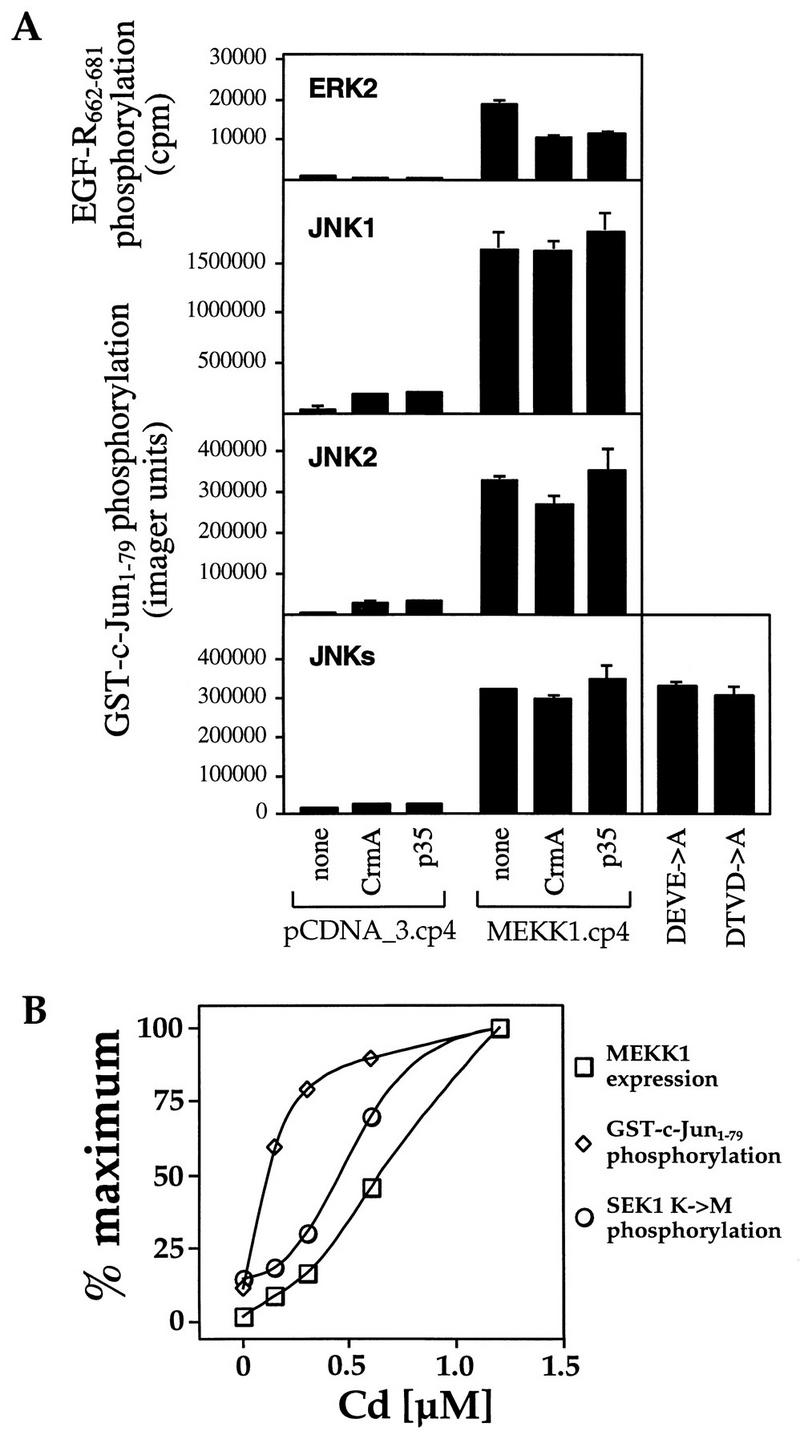
Lack of correlation between ERK or JNK pathway activation and MEKK1-induced DNA fragmentation. (A) HEK293 cells were transfected with 0.5 μg of the vector pcDNA_3.cp4 or with MEKK1.cp4 alone or in combination with 2 μg of CrmA.cp_ or p35.cp_. Alternatively, the cells were transfected with 2 μg of DEVE_A.cp4 or DTVD_A.cp4. The activation of ERK2, JNK1, JNK2, or JNK isoforms (JNKs) was then measured as described in Materials and Methods. EGF-R662–681, epidermal growth factor receptor peptide (residues 662 to 681). (B) Cells were transfected with 1 μg of a pCEP4-derived plasmid in which HA-tagged ΔMEKK1 was placed under the control of the metallothionein promoter (MEKK1k.MT4). Cadmium was added at the indicated concentrations at the time of serum addition in the transfection protocol. After 18 h, ΔMEKK1 expression (□), activation of JNK (GST–c-Jun1–79 phosphorylation) (◊), and the ability of ΔMEKK1 to phosphorylate SEK1 K→M (○) were measured. Data were normalized to the maximal responses.
DISCUSSION
Genotoxic stress, a balance between rescue and suicide with MEKK1 as a switch.
Our results show that DNA-damaging agents induce rapid phosphorylation and activation of MEKK1. The rapid JNK response observed with genotoxins could actually contribute to a protective response against cell death. The JNK pathway has been proposed to mediate the antiapoptotic effect of CD40 in B cells (54, 56) and to inhibit methyl methanesulfonate-induced 3T3 cell apoptosis (38). The activation of NF-κB in response to stresses, including UV irradiation and genotoxic chemicals, would also be a protective response (4); MEKK1 has been shown to be involved in the activation of NF-κB (4, 27, 37). We suggest that MEKK1 could function as a switch point, regulated by a proteolytic event controlled by caspases, that contributes to cell fate determination in response to a stress stimulus. MEKK1 cleavage appears to be involved in the apoptotic response elicited by diverse stimuli that activate caspases.
Figure 12 shows a model defining the involvement of MEKK1 in apoptosis. The 196-kDa MEKK1 protein can be activated by many extracellular stimuli, including those mediated by tyrosine kinase-encoded growth factor receptors, G protein-coupled receptors (3), and genotoxins. The activation of MEKK1 correlates with its phosphorylation. It is unclear at present if MEKK1 phosphorylation involves autophosphorylation or additional kinases. MEKK1 activated independently of its proteolysis is capable of regulating the JNK pathway and the ERK pathway. Both of these pathways can stimulate antiapoptotic responses (23, 38, 47, 62). Moreover, MEKK1 can lead to NF-κB activation, which can be a strong inhibitor of apoptosis (4, 27, 37).
FIG. 12.

Mechanistic model of MEKK1 regulation of apoptosis. See the text for details.
With appropriate protease activation, MEKK1 is cleaved to generate a 91-kDa kinase domain that is a strong inducer of apoptosis. The substrates for the 91-kDa kinase may include caspases, proteins that will induce such proteases, or even proteins such as Bcl-2 family members. The truncated form of MEKK1 can induce caspase activity, which in turn stimulates more MEKK1 cleavage. MEKK1 and caspases are thus predicted to be part of an amplification loop for increasing caspase activity during apoptosis.
Genotoxins versus anoikis and the regulation of MEKK1.
MEKK1 is involved in the apoptotic response to survival factor withdrawal (62), loss of adherence (anoikis) (7), and genotoxins (this study). Several critical differences in the regulation of apoptosis induced by genotoxins and loss of adherence are, however, evident from our studies. Cardone et al. (7) showed that the loss of adherence induced the cleavage of MEKK1 and the subsequent activation of the carboxy-terminal cleavage fragment. The reason why full-length MEKK1 is not activated by anoikis could be to avoid the activation of protective pathways (Fig. 12) that would hamper efficient death of detached endothelial cells. The genotoxic response is quite different and has significant implications for cell fate. Unlike the loss of adherence, genotoxins activate MEKK1 by a mechanism involving its phosphorylation, and this response occurs before MEKK1 cleavage. Thus, MEKK1 signals as a full-length protein early after exposure to genotoxins and, according to our model, could stimulate survival pathways. However, exposure of cells to high doses of genotoxins eventually activates caspases, and MEKK1 becomes cleaved into a potent proapoptotic carboxy-terminal product that participate in the cell death process.
MEKK1-mediated apoptosis requires both kinase activity and proteolytic cleavage.
We showed previously that the kinase activity of MEKK1 is required for its apoptotic activity, because kinase-inactive ΔMEKK1 is unable to promote apoptosis (34). Here we show that there is an integration of kinase and protease activities in the MEKK1-induced apoptotic response. Proteases are required for MEKK1-induced apoptosis at least at two levels in the transduction pathway. The first level corresponds to the cleavage of MEKK1 at position 871 or 874 in the mouse MEKK1 sequence (7), and the second level corresponds to downstream cleavage events mediating the structural alteration leading to cell death. When cleavage is prevented in the DTVD→A MEKK1 mutant, apoptosis is impaired. Caspases are required for this cleavage to occur, since the viral inhibitors CrmA and p35 inhibit the cleavage. Our data demonstrate that caspase-3 or a caspase-3-like enzyme directly cleaves MEKK1, at least at position 874, because purified caspase-3 cleaved MEKK1 in vitro (Fig. 8). The recognition site for the protease at position 874 in mouse MEKK1 is DTVD, a sequence that closely resembles the DEVD recognition site of the caspase-3 substrate poly(ADP-ribose) polymerase (45). The human MEKK1 sequence at this site is the same as the mouse MEKK1 sequence (unpublished observation), while the corresponding rat sequence is DTLD (63). These data indicate that the cleavage site is conserved among mouse, rat, and human MEKK1 proteins and further support the importance of caspase cleavage in MEKK1 function.
Differential activation of the JNK and caspase pathways by full-length MEKK1 and its cleaved 91-kDa carboxy-terminal fragment.
Both full-length MEKK1 and the carboxy-terminal caspase cleavage product of MEKK1 (fragment C) efficiently activate the JNK MAP kinase pathway. However, the cleavage of MEKK1 is not required for the activation of JNK, since (i) kinetic experiments showed that maximal JNK activation correlates with activation of the endogenous full-length MEKK1 but does not correlate with the appearance of cleavage products (Fig. 1), (ii) a cleavage-resistant MEKK1 mutant activates the JNK pathway as efficiently as its wild-type counterpart (Fig. 11), and (iii) blockage of MEKK1 cleavage does not impair its ability to activate the JNK pathway (Fig. 11). In contrast, activation of the proapoptotic pathway requires the cleavage of MEKK1 into the 91-kDa carboxy-terminal fragment (Fig. 9) (7).
Cellular location could be the basis for the differential effects of full-length MEKK1 and the 91-kDa fragment of MEKK1 on the JNK and apoptotic pathways. We have observed, using cellular fractionation, that the endogenous MEKK1 protein is exclusively associated with membranes and is not detected in the cytosolic fraction (28a), consistent with the granular staining observed for HEK293 cells with MEKK1-specific antibodies (see arrows in Fig. 5B). When MEKK1 is overexpressed in HEK293 cells, full-length MEKK1 is present in both the membrane and the cytosolic fractions (at about a 1:1 ratio), indicating that overexpression may saturate the MEKK1 docking sites on membranes. Remarkably, the 91-kDa fragment generated in cells overexpressing MEKK1 is exclusively found in the cytosolic fraction (51a). It is thus possible that, at physiological levels of expression, full-length MEKK1 is exclusively membrane associated and that, upon caspase activation, a 91-kDa active carboxy-terminal MEKK1 fragment is generated and translocates to the cytosol, where it could activate a new set of substrates involved in apoptosis.
We recently showed that 14-3-3 proteins can bind to the full-length MEKK1 protein but not to the 91-kDa carboxy-terminal fragment (20). 14-3-3 proteins could thus be responsible for the differential locations of full-length MEKK1 and its fragments. 14-3-3 proteins have indeed been shown to be able to sequester proapoptotic proteins away from their intracellular sites of action. For example, when Bad, a proapoptotic Bcl2-related protein, is phosphorylated by enzymes involved in cell survival, such as Akt (14, 16), it is bound by 14-3-3 proteins and removed from mitochondrial membranes (65), where Bad exerts its proapoptotic action. It is thus possible that 14-3-3 proteins play a similar role in the regulation of the function of MEKK1 in apoptosis. The observation that about 50% of overexpressed MEKK1 is found in the cytosol, in contrast to the endogenous protein, could be the basis of the deregulation of MEKK1 function in transfected cells, leading to uncontrolled activation and cleavage events and resulting in the activation of a cell death program.
ACKNOWLEDGMENTS
We thank Thomas Schlesinger for performing the experiment shown in Fig. 4A.
C.W. is the recipient of grant 823A-042980 from the Swiss National Science Foundation. P.G. is supported by the Fullbright Commission; the Wennergren Foundation; the Karolinska Institute; and the Swedish Medical Research Council, Cancer Foundation, Society of Medicine, and Institute. This work was supported by NIH grants DK37871, DK48845, CA58157, and GM30324.
REFERENCES
- 1.Alnemri E S, Livingston D J, Nicholson D W, Salvesen G S, Thornberry N A, Wong W W, Yuan J. Human ICE/CED-3 protease nomenclature. Cell. 1996;87:171. doi: 10.1016/s0092-8674(00)81334-3. [DOI] [PubMed] [Google Scholar]
- 2.An B, Dou Q P. Cleavage of retinoblastoma protein during apoptosis: an interleukin 1β-converting enzyme-like protease as candidate. Cancer Res. 1996;56:438–442. [PubMed] [Google Scholar]
- 3.Avdi N J, Winston B W, Russel M, Young S K, Johnson G L, Worthen G S. Activation of MEKK by formyl-methionyl-leucyl-phenylalanine in human neutrophils. Mapping pathways for mitogen-activated protein kinase activation. J Biol Chem. 1996;271:33598–33606. doi: 10.1074/jbc.271.52.33598. [DOI] [PubMed] [Google Scholar]
- 4.Baeuerle P A, Baltimore D. NF-(kappa)B: ten years after. Cell. 1996;87:13–20. doi: 10.1016/s0092-8674(00)81318-5. [DOI] [PubMed] [Google Scholar]
- 5.Beidler D R, Tewari M, Friesen P D, Poirier G, Dixit V M. The baculovirus p35 protein inhibits Fas- and tumor necrosis factor-induced apoptosis. J Biol Chem. 1996;270:16526–16528. doi: 10.1074/jbc.270.28.16526. [DOI] [PubMed] [Google Scholar]
- 6.Blank J L, Gerwins P, Elliott E M, Sather S, Johnson G L. Molecular cloning of mitogen-activated protein/ERK kinase kinases (MEKK) 2 and 3. Regulation of sequential phosphorylation pathways involving mitogen-activated protein kinase and c-Jun kinase. J Biol Chem. 1996;271:5361–5368. doi: 10.1074/jbc.271.10.5361. [DOI] [PubMed] [Google Scholar]
- 7.Cardone M, Salvesen G S, Widmann C, Johnson G L, Frisch S M. The regulation of anoikis: MEKK-1 activation requires cleavage by caspases. Cell. 1997;90:315–323. doi: 10.1016/s0092-8674(00)80339-6. [DOI] [PubMed] [Google Scholar]
- 8.Cartier J L, Hershberger P A, Friesen P D. Suppression of apoptosis in insect cells stably transfected with baculovirus p35: dominant interference by N-terminal sequences p351–76. J Virol. 1994;68:7728–7737. doi: 10.1128/jvi.68.12.7728-7737.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Casciola-Rosen L A, Anhalt G J, Rosen A. DNA-dependent protein kinase is one of a subset of antoantigens specifically cleaved early during apoptosis. J Exp Med. 1995;182:1625–1634. doi: 10.1084/jem.182.6.1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Casciola-Rosen L A, Miller D K, Anhalt G J, Rosen A. Specific cleavage of the 70-kDa protein component of the U1 small nuclear ribonucleoprotein is a characteristic biochemical feature of apoptotic cell death. J Biol Chem. 1994;269:30757–30760. [PubMed] [Google Scholar]
- 11.Chen Y-R, Wang X, Templeton D, Davis R J, Tan T-H. The role of c-Jun N-terminal kinase (JNK) in apoptosis induced by ultraviolet C and gamma irradiation. Duration of JNK activation may determine cell death and proliferation. J Biol Chem. 1996;271:31929–31936. doi: 10.1074/jbc.271.50.31929. [DOI] [PubMed] [Google Scholar]
- 12.Clem R J, Hardwick J M, Miller L K. Anti-apoptotic genes of baculoviruses. Death Differ. 1996;3:9–16. [PubMed] [Google Scholar]
- 13.Cryns V L, Bergeron L, Zhu H, Li H, Yuan J. Specific cleavage of α-fodrin during Fas- and tumor necrosis factor-induced apoptosis is mediated by an interleukin-1β-converting enzyme/Ced-3 protease distinct from the poly(ADP-ribose) polymerase protease. J Biol Chem. 1996;271:31277–31282. doi: 10.1074/jbc.271.49.31277. [DOI] [PubMed] [Google Scholar]
- 14.Datta S R, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg M E. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- 15.Deak J C, Templeton D J. Regulation of the activity of MEK kinase 1 (MEKK1) by autophosphorylation within the kinase activation domain. Biochem J. 1997;322:185–192. doi: 10.1042/bj3220185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.del Peso L, Gonzalez-Garcia M, Page C, Herrera R, Nunez G. Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt. Science. 1997;278:687–689. doi: 10.1126/science.278.5338.687. [DOI] [PubMed] [Google Scholar]
- 17.Dive C, Hickman J A. Drug-target interactions: only the first step in the commitment to a programmed cell death? Br J Cancer. 1991;64:192–196. doi: 10.1038/bjc.1991.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Emoto Y, Manome Y, Meinhardt G, Kisaki H, Kharbanda S, Robertson M, Ghayur T, Wong W W, Kamen R, Weichselbaum R, Kufe D. Proteolytic activation of protein kinase C δ by an ICE-like protease in apoptotic cells. EMBO J. 1995;14:6148–6156. doi: 10.1002/j.1460-2075.1995.tb00305.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Erhardt P, Tomaselli K J, Cooper G M. Identification of the MDM2 oncoprotein as a substrate for CPP32-like apoptotic proteases. J Biol Chem. 1997;272:15049–15052. doi: 10.1074/jbc.272.24.15049. [DOI] [PubMed] [Google Scholar]
- 20.Fanger G R, Widmann C, Porter A C, Sather S, Johnson G L, Vaillancourt R R. 14-3-3 proteins interact with specific MEK kinases. J Biol Chem. 1998;273:3476–3483. doi: 10.1074/jbc.273.6.3476. [DOI] [PubMed] [Google Scholar]
- 21.Fisher D E. Apoptosis in cancer therapy: crossing the threshold. Cell. 1994;78:539–542. doi: 10.1016/0092-8674(94)90518-5. [DOI] [PubMed] [Google Scholar]
- 22.Fraser A, Evan G. A license to kill. Cell. 1996;85:781–784. doi: 10.1016/s0092-8674(00)81005-3. [DOI] [PubMed] [Google Scholar]
- 23.Gardner A M, Johnson G L. Fibroblast growth factor-2 suppression of tumor necrosis factor α-mediated apoptosis requires Ras and the activation of mitogen-activated protein kinase. J Biol Chem. 1996;271:14560–14566. doi: 10.1074/jbc.271.24.14560. [DOI] [PubMed] [Google Scholar]
- 24.Gerwins P, Blank J L, Johnson G L. Cloning of a novel mitogen-activated protein kinase kinase kinase, MEKK4, that selectively regulates the c-Jun amino terminal kinase pathway. J Biol Chem. 1997;272:8288–8295. doi: 10.1074/jbc.272.13.8288. [DOI] [PubMed] [Google Scholar]
- 25.Hibi M, Lin A, Smeal T, Minden A, Karin M. Identification of an oncoprotein- and UV-responsive protein kinase that binds and potentiates the c-Jun activation domain. Genes Dev. 1993;7:2135–2148. doi: 10.1101/gad.7.11.2135. [DOI] [PubMed] [Google Scholar]
- 26.Hickman J A, Potten C S, Merritt A J, Fisher T C. Apoptosis and cancer chemotherapy. Philos Trans R Soc London Ser B. 1994;345:319–325. doi: 10.1098/rstb.1994.0112. [DOI] [PubMed] [Google Scholar]
- 27.Hirano M, Osada S-I, Aoki T, Hirai S-I, Hosaka M, Inoue J-I, Ohno S. MEK kinase is involved in tumor necrosis factor α-induced NF-kB activation and degradation of IkB-α. J Biol Chem. 1996;271:13234–13238. doi: 10.1074/jbc.271.22.13234. [DOI] [PubMed] [Google Scholar]
- 28.Howard A D, Kostura M J, Thornberry N, Ding G J-F, Limjuco G, Weidner J, Salley J P, Hogquist K A, Chaplin D D, Mumford R A, Schmidt J A, Tocci M J. IL-1-converting enzyme requires aspartic acid residues for processing of the IL-1β precursor at two distinct sites and does not cleave 31-kDa IL-1α. J Immunol. 1991;147:2964–2969. [PubMed] [Google Scholar]
- 28a.Jarpe, M. Unpublished observations.
- 29.Juo P, Kuo C J, Reynolds S E, Konz R F, Raingeaud J, Davis R J, Biemann H-P, Blenis J. Fas activation of the p38 mitogen-activated protein kinase signalling pathway requires ICE/CED-3 family proteases. Mol Cell Biol. 1997;17:24–35. doi: 10.1128/mcb.17.1.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Khwaja A, Downward J. Lack of correlation between activation of Jun-NH2-terminal kinase and induction of apoptosis after detachment of epithelial cells. J Cell Biol. 1997;139:1017–1023. doi: 10.1083/jcb.139.4.1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim T-W, Pettingell W H, Jung Y-K, Kovacs D M, Tanzi R E. Alternative cleavage of Alzheimer-associated presenilins during apoptosis by a caspase-3 family protease. Science. 1997;277:373–376. doi: 10.1126/science.277.5324.373. [DOI] [PubMed] [Google Scholar]
- 32.Lange-Carter C A, Johnson G L. Ras-dependent growth factor regulation of MEK kinase in PC12 cells. Science. 1994;265:1458–1461. doi: 10.1126/science.8073291. [DOI] [PubMed] [Google Scholar]
- 33.Lange-Carter C A, Pleiman C M, Gardner A M, Blumer K J, Johnson G L. A divergence in the MAP kinase regulatory network defined by MEK kinase and Raf. Science. 1993;260:315–319. doi: 10.1126/science.8385802. [DOI] [PubMed] [Google Scholar]
- 34.Lassignal Johnson N, Gardner A M, Diener K M, Lange-Carter C A, Gleavy J, Jarpe M B, Minden A, Karin M, Zon L I, Johnson G L. Signal transduction pathways regulated by mitogen-activated/extracellular response kinase kinase kinase induce cell death. J Biol Chem. 1996;271:3229–3237. doi: 10.1074/jbc.271.6.3229. [DOI] [PubMed] [Google Scholar]
- 35.Lazebnik Y A, Kaufmann S H, Desnoyers S, Poirier G G, Earnshaw W C. Cleavage of poly(ADP-ribose) polymerase by a proteinase with properties like ICE. Nature. 1994;371:346–347. doi: 10.1038/371346a0. [DOI] [PubMed] [Google Scholar]
- 36.Lazebnik Y A, Takahashi A, Moir R D, Goldman R D, Poirier G G, Kaufmann S H, Earnshaw W C. Studies of the lamin proteinase reveal multiple parallel biochemical pathways during apoptotic execution. Proc Natl Acad Sci USA. 1995;92:9042–9046. doi: 10.1073/pnas.92.20.9042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee F S, Hagler J, Chen Z J, Maniatis T. Activation of the I-kappa-Bα kinase complex by MEKK1, a kinase of the JNK pathway. Cell. 1997;88:213–222. doi: 10.1016/s0092-8674(00)81842-5. [DOI] [PubMed] [Google Scholar]
- 38.Liu Z-G, Baskaran R, Lea-Chou E T, Wood L D, Chen Y, Karin M, Wang J Y J. Three distinct signalling responses by murine fibroblasts to genotoxic stress. Nature. 1996;384:273–276. doi: 10.1038/384273a0. [DOI] [PubMed] [Google Scholar]
- 39.Liu Z-G, Hsu H, Goeddel D V, Karin M. Dissection of TNF receptor 1 effector functions: JNK activation is not linked to apoptosis while NF-kB activation prevents cell death. Cell. 1996;87:565–576. doi: 10.1016/s0092-8674(00)81375-6. [DOI] [PubMed] [Google Scholar]
- 40.Los M, Van de Craen M, Penning L C, Schenk H, Westendorp M, Baeuerle P A, Dröge W, Krammer P H, Fiers W, Schulze-Osthoff K. Requirement of an ICE/CED-3 protease for Fas/APO-1-mediated apoptosis. Nature. 1995;375:81–83. doi: 10.1038/375081a0. [DOI] [PubMed] [Google Scholar]
- 41.Mercille S, Massie B. Induction of apoptosis in nutrient-deprived cultures of hybridoma and myeloma cells. Biotechnol Bioeng. 1994;44:1140–1154. doi: 10.1002/bit.260440916. [DOI] [PubMed] [Google Scholar]
- 42.Meyn R E, Stephens L C, Hunter N R, Milas L. Apoptosis in murine tumors treated with chemotherapy agents. Anticancer Drugs. 1995;6:443–450. doi: 10.1097/00001813-199506000-00013. [DOI] [PubMed] [Google Scholar]
- 43.Moreira L F, Naomoto Y, Hamada M, Kamikawa Y, Orita K. Assessment of apoptosis in oesophageal carcinoma preoperatively treated by chemotherapy and radiotherapy. Anticancer Res. 1995;15:639–644. [PubMed] [Google Scholar]
- 44.Na S, Chuang T-H, Cunningham A, Turi T G, Hanke J H, Bokoch G M, Danley D E. D4-GDI, a substrate of CPP32, is proteolyzed during Fas-induced apoptosis. J Biol Chem. 1996;271:11209–11213. doi: 10.1074/jbc.271.19.11209. [DOI] [PubMed] [Google Scholar]
- 45.Nicholson D W, Ali A, Thornberry N A, Vaillancourt J P, Ding C K, Gallan M, Gareau Y, Griffin P R, Labelle M, Lazebnik Y A, Munday N A, Raju S M, Smulson M E, Yamin T-T, Yu V L, Miller D K. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature. 1995;376:37–43. doi: 10.1038/376037a0. [DOI] [PubMed] [Google Scholar]
- 46.Nicoletti I, Miglioratti G, Pagliacci M C, Grignani F, Riccardi C. A rapid and simple method for measuring thymocyte apoptosis by propidium iodide staining and flow cytometry. J Immunol Methods. 1991;139:271–279. doi: 10.1016/0022-1759(91)90198-o. [DOI] [PubMed] [Google Scholar]
- 47.Nishina H, Fischer K D, Radvanyi L, Shahinian A, Hakem R, Rubie E A, Bernstein A, Mak T W, Woodgett J R, Penninger J M. Stress-signalling kinase Sek1 protects thymocytes from apoptosis mediated by CD95 and CD3. Nature. 1997;385:350–353. doi: 10.1038/385350a0. [DOI] [PubMed] [Google Scholar]
- 48.Orth K, O’Rourke K, Salvesen G S, Dixit V M. Molecular ordering of apoptotic mammalian CED-3/ICE-like proteases. J Biol Chem. 1996;271:20977–20980. doi: 10.1074/jbc.271.35.20977. [DOI] [PubMed] [Google Scholar]
- 49.Park D S, Stefanis L, Yan C Y I, Farinelli S E, Greene L A. Ordering the cell death pathway. Differential effects of Bcl2, an interleukin-1β-converting enzyme family protease inhibitor, and other survival agents of JNK activation in serum/nerve growth factor-deprived PC12 cells. J Biol Chem. 1996;271:21898–21905. doi: 10.1074/jbc.271.36.21898. [DOI] [PubMed] [Google Scholar]
- 50.Pickup D J, Ink B S, Hu W, Ray C A, Joklik W K. Hemorrhage in lesions caused by cowpox virus is induced by a viral protein that is related to plasma protein inhibitors of serine proteases. Proc Natl Acad Sci USA. 1986;83:7698–7702. doi: 10.1073/pnas.83.20.7698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rudel T, Bokoch G M. Membrane and morphological changes in apoptotic cells regulated by caspase-mediated activation of PAK2. Science. 1997;276:1571–1574. doi: 10.1126/science.276.5318.1571. [DOI] [PubMed] [Google Scholar]
- 51a.Schlesinger, T. Unpublished observations.
- 52.Siow Y L, Kalmar G B, Sanghera J S, Tai T, Oh S S, Pelech S L. Identification of two essential phosphorylated threonine residues in the catalytic domain of Mekk1. Indirect activation by Pak3 and protein kinase C. J Biol Chem. 1997;272:7586–7594. doi: 10.1074/jbc.272.12.7586. [DOI] [PubMed] [Google Scholar]
- 53.Smith A, Ramos-Morales F, Ashworth A, Collins M. A role for JNK/SAPK in proliferation, but not apoptosis, of IL-3-dependent cells. Curr Biol. 1997;7:893–896. doi: 10.1016/s0960-9822(06)00380-0. [DOI] [PubMed] [Google Scholar]
- 54.Sumimoto S-I, Heike T, Kanazashi S-I, Shintaku N, Jung E-Y, Hata D, Katamura K, Mayumi M. Involvement of LFA-1/intracellular adhesion molecule-1-dependent cell adhesion in CD40-mediated inhibition of human B lymphoma cell death induced by surface IgM crosslinking. J Immunol. 1994;153:2488–2496. [PubMed] [Google Scholar]
- 55.Toyoshima F, Moriguchi T, Nishida E. Fas induces cytoplasmic apoptotic responses and activation of the MKK7-JNK/SAPK and MKK6-p38 pathways independent of CPP32-like proteases. J Cell Biol. 1997;139:1005–1015. doi: 10.1083/jcb.139.4.1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tsubata T, Wu J, Honjo T. B-cell apoptosis induced by antigen receptor crosslinking is blocked by a T-cell signal through CD40. Nature. 1993;364:645–648. doi: 10.1038/364645a0. [DOI] [PubMed] [Google Scholar]
- 57.Verheij M, Bose R, Lin X H, Yao B, Jarvis W D, Grant S, Birrer M J, Szabo E, Zon L I, Kyriakis J M, Haimovitz-Friedman A, Fuks Z, Kolesnick R N. Requirement for ceramide-initiated SAPK/JNK signalling in stress-induced apoptosis. Nature. 1996;380:75–79. doi: 10.1038/380075a0. [DOI] [PubMed] [Google Scholar]
- 58.Wang X, Zelenski N G, Yang J, Sakai J, Brown M S, Goldstein J L. Cleavage of sterol regulatory element binding proteins (SREBPs) by CPP32 during apoptosis. EMBO J. 1996;15:1012–1020. [PMC free article] [PubMed] [Google Scholar]
- 59.Widmann C, Dolci W, Thorens B. Agonist-induced internalization and recycling of the glucagon-like peptide-1 receptor in transfected fibroblasts and in insulinomas. Biochem J. 1995;310:203–214. doi: 10.1042/bj3100203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Widmann, C., P. Gerwins, N. Lassignal Johnson, and G. L. Johnson. MEKK1 and MEKK4 activate CPP32-like caspases, but differ in their ability to induce apoptosis. Submitted for publication.
- 61.Widmann C, Lassignal Johnson N, Gardner A M, Smith R J, Johnson G L. Potentiation of apoptosis by low dose stress stimuli in cells expressing activated MEK kinase 1. Oncogene. 1997;15:2439–2447. doi: 10.1038/sj.onc.1201421. [DOI] [PubMed] [Google Scholar]
- 62.Xia Z, Dickens M, Raingeaud J, Davis R J, Greenberg M E. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- 63.Xu S, Robbins D J, Christerson L B, English J M, Vanderbilt C A, Cobb M H. Cloning of rat MEK kinase 1 cDNA reveals an endogenous membrane-associated 195-kDa protein with a large regulatory domain. Proc Natl Acad Sci USA. 1996;93:5291–5295. doi: 10.1073/pnas.93.11.5291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yang X, Khosravi-Far R, Chang H Y, Baltimore D. Daax, a novel Fas-binding protein that activates JNK and apoptosis. Cell. 1997;89:1067–1076. doi: 10.1016/s0092-8674(00)80294-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zha J, Harada H, Yang E, Jockel J, Korsmeyer S J. Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14-3-3 not Bcl-XL. Cell. 1996;87:619–628. doi: 10.1016/s0092-8674(00)81382-3. [DOI] [PubMed] [Google Scholar]
- 66.Zhou Q, Snipas S, Orth K, Muzio M, Dixit V M, Salvesen G S. Target protease specificity of the viral serpin CrmA. Analysis of five caspases. J Biol Chem. 1997;272:7797–7800. doi: 10.1074/jbc.272.12.7797. [DOI] [PubMed] [Google Scholar]