

Abstract
Purine-rich enhancers are exon sequences that promote inclusion of alternative exons, usually via activation of weak upstream 3′ splice sites. A recently described purine-rich enhancer from the caldesmon gene has an additional activity by which it directs selection of competing 5′ splice sites within an alternative exon. In this study, we have compared the caldesmon enhancer with another purine-rich enhancer from the chicken cardiac troponin T (cTNT) gene for the ability to regulate flanking splice sites. Although similar in sequence and length, the two enhancers demonstrated strikingly different specificities towards 5′ splice site choice when placed between competing 5′ splice sites in an internal exon. The 32-nucleotide caldesmon enhancer caused effective usage of the exon-internal 5′ splice site, whereas the 30-nucleotide cTNT enhancer caused effective usage of the exon-terminal 5′ splice site. Both enhancer-mediated splicing pathways represented modulation of the default pathway in which both 5′ splice sites were utilized. Each enhancer is multipartite, consisting of two purine-rich sequences of a simple (GAR)n repeat interdigitated with two enhancer-specific sequences. The entire enhancer was necessary for maximal splice site selectivity; however, a 5- to 7-nucleotide region from the 3′ end of each enhancer dictated splice site selectivity. Mutations that interchanged this short region of the two enhancers switched specificity. The portion of the cTNT enhancer determinative for 5′ splice site selectivity was different than that shown to be maximally important for activation of a 3′ splice site, suggesting that enhancer environment can have a major impact on activity. These results are the first indication that individual purine-rich enhancers can differentiate between flanking splice sites. Furthermore, localization of the specificity of splice site choice to a short region within both enhancers indicates that subtle differences in enhancer sequence can have profound effects on the splicing pathway.
Sequences within exons in addition to splice sites have emerged in the last several years as powerful determinants of splicing efficiency (2, 13, 24). Of these sequences, the purine-rich exon enhancers containing the generic core sequence (GAR)n (R = G or A) (42) have received the most attention (4, 6, 9, 17, 20, 23, 25, 32, 41–43). Even short purine-rich enhancers can have major effects on the efficiency of exon inclusion. Most characterized purine-rich enhancers reside in alternative exons and have been shown to be essential sequence elements for exon inclusion, usually via the activation of weak 3′ splice sites (2, 14, 26). Exon enhancers are often interchangeable in their ability to activate weak 3′ splice sites, not only between genes (8, 17, 20, 36, 41–43), but also between species (13), suggesting that either most enhancers bind the same factors or the bound factors have interchangeable activities.
Purine-rich enhancers bind to members of the arginine-serine-rich class of splicing factors (29, 44), the S/R proteins (reviewed in references 10 and 24). Different enhancers demonstrate binding preferences for individual members of this family of proteins (20, 23, 25, 31–34, 41). These preferences correlate with the ability of different S/R proteins to affect in vitro splicing of enhancer-containing exons. Raising the in vivo level of S/R proteins via expression of cDNAs coding for individual members of the family has been shown to increase inclusion of an exon containing a purine-rich enhancer (3, 39). Furthermore, disruption of the gene coding for one S/R protein, ASF/SF2, has been demonstrated to be lethal in cultured cells (40). Cumulatively, the available data suggests that individual purine-rich enhancers bind a preferred subset of S/R proteins during exon recognition.
Although purine-rich enhancers are frequently associated with cassette exons, they have not been routinely associated with splicing choices involving alternative recognition of competing splice sites within a single exon. The only two reported examples of enhancers regulating this latter type of alternative splicing occur in the caldesmon gene (12, 16, 17) and the Drosophila fruitless gene (28). Exon 5 of the caldesmon gene is a large internal exon containing two competing 5′ splice sites. An extensive region of purine repeats, consisting of five copies of a 32-nucleotide purine-rich repeat, resides between the two splice sites. In vivo, the purine elements are necessary for maximal exon inclusion and proper regulation of splice site choice (15). A monomer repeat unit is sufficient to mediate both exon inclusion and 5′ splice site regulation. The caldesmon regulatory sequence has properties of both an enhancer and a silencer in that it stimulates inclusion of an exon without itself being included in the spliced product RNA. In a heterologous exon without competing splice sites, the caldesmon enhancer behaves as a simple splicing enhancer (15), suggesting that it is best considered a complicated member of the purine-rich exon enhancer family despite its activation of an upstream 5′ splice site.
Here, we compare the caldesmon enhancer to a more standard purine-rich enhancer from the chicken cardiac troponin T (cTNT) gene and report the surprising result that the two enhancers direct different splicing events both in vivo and in vitro when positioned between two competing 5′ splice sites in an internal exon. This difference is observed despite considerable sequence similarity in the two enhancers. The caldesmon 32-nucleotide enhancer stimulated usage of the upstream, exon-internal 5′ splice site, whereas the 30-nucleotide cTNT enhancer stimulated usage of the downstream, exon-terminal site. The two enhancers directed opposite choices both in vivo in a modified internal exon from the caldesmon gene and in vitro in an artificial exon containing strong constitutive splice sites derived from adenovirus. In the absence of any enhancer or in the presence of nonspecific exon sequences, both 5′ splice sites in the tested internal exons were utilized, indicating that both enhancers modulated default splice site utilization, albeit in opposite directions. The sequences responsible for the differences in 5′ splice site specificity were localized to a short region near the 3′ end of the enhancers in which the sequence of both enhancers diverged from a simple GAR repeat. Although this short region determined splice site selectivity, it was not sufficient; other sequences within both enhancers were also necessary for enhancer function. These results emphasize the multipartite nature of exon enhancers and suggest considerable complexity in the nature of recognition of exon enhancers by splicing factors. Perhaps most importantly, our results show that regions of an enhancer important for 5′ splice site selectivity may not be the same sequence required for 3′ splice site activation, indicating that the activity of an enhancer can be environmentally determined.
MATERIALS AND METHODS
In vivo and in vitro constructs.
The in vivo constructs used were based on a mini-gene containing the caldesmon alternative exon. In this mini-gene, the natural human caldesmon exon 5 and its adjoining intron sequences replaced exon 2 of the mouse metallothionein II gene driven by the Rous sarcoma virus (RSV) promoter. This mini-gene directs exon 5 inclusion via the exon-internal 5′ splice site in most cell lines tested (15). The basic mini-gene contained the natural caldesmon exon 5, in which the region between the two 5′ splice sites (687 nucleotides) within the exon contained the natural purine-rich enhancer consisting of five copies of the 32-nucleotide repeat (see Fig. 1A). Derivatives in which 357 nucleotides containing all copies of the purine-rich repeat between the two 5′ splice sites in exon 5 were replaced with 48 to 52 nucleotides of heterologous sequence were constructed. These sequences consist of one copy of the caldesmon 32-nucleotide repeat, one copy of the natural exon sequences (30 nucleotides [see sequence in Fig. 1B]) from exon 5 of the chicken cTNT gene, one copy of an “up-mutant” (25) of the cTNT exon 5 enhancer (sequence AAGAGGAAGAAGAAGAAGAGGAAGAC-GACG), and a neutral cDNA sequence (GTTATGCTCGTTATGCGCGTTATGCTCGTTATGGTCG).
FIG. 1.

Caldesmon and cTNT enhancers regulate in vivo 5′ splice site selection and stimulate exon inclusion. (A) Diagram of the alternative exons (black boxes) of the human caldesmon and chicken cTNT genes containing the purine-rich enhancers studied (circle and triangle, respectively). The two purine-regions in each enhancer (gray boxes) and the enhancer-specific sequences that separate the two purine-rich regions (italic) are indicated. An up-mutant of cTNT that promotes exon inclusion of cTNT exon 5 more efficiently than the wild-type element (25) (triangle with plus sign). Five copies of the 32-nucleotide caldesmon enhancer from the natural exon are shown (five circles). (B) RT-PCR analysis of spliced RNA produced upon transfection of CHO cells with mini-genes containing the indicated enhancer sequences placed between the competing 5′ splice sites of the natural caldesmon exon 5, diagrammed below the gel. The 5′ splice site upstream of the enhancer is termed the exon-internal site (IS), and the 5′ splice site downstream of the enhancer is termed the exon-terminal site (TS). For the constructs used in lanes 2 to 6, 357 nucleotides of exon including the natural enhancer were replaced with 48 to 52 nucleotides of heterologous sequence. RNA phenotypes were assessed by low-cycle quantitative RT-PCR (15) using PCR primers located in the exons flanking the alternative exon. The internal exon utilized in lane 1 is the natural caldesmon exon and is not drawn to scale. Product RNA resulting from use of the exon-terminal site in this construct is not observed (15) and would be much larger than the spliced RNAs produced in the other constructs. The constructs in lanes 2 to 6 had internal exons that did not significantly differ in length; therefore, the PCR products produced from each of these constructs resulting from usage of the exon-terminal 5′ splice site were indistinguishable in length in the gel system used. These constructs have significantly shorter second exons than the natural caldesmon gene. Shortening reduces the dependence of the exon on the enhancer for inclusion and permits examination of effects on splice site specificity only. The construct (lane 2) in which the natural enhancer was deleted (no element) produced PCR products of 973, 643, and 240 nucleotides resulting from exon inclusion via the exon-terminal splice site, exon inclusion via the exon-internal splice site, and exon skipping, respectively. The constructs containing a single copy of the caldesmon enhancer (lane 3), nonspecific sequences (lane 4), cTNT enhancer (lane 5), or improved cTNT enhancer containing a purine spacer (lane 6) produced PCR products of 1,111 to 1,115, 643, and 240 nucleotides resulting from exon inclusion via the exon-terminal splice site, exon inclusion via the exon-internal splice site, and exon skipping, respectively. The identities of individual RNA species within each band were confirmed by sequencing of PCR products.
The in vitro constructs used to prepare precursor RNA for in vitro splicing were derived from adenovirus (27, 35). All splice sites in the constructs (see Fig. 2) are from the second exon of adenovirus. Sequences were placed in between the 5′ splice sites of the middle exon by using small sequence cassettes (30 to 37 nucleotides) of information, including the caldesmon enhancer monomer, the wild-type cTNT exon 5 enhancer, the cTNT up-mutant enhancer, a nonspecific sequence, and the first half of the caldesmon unit enhancer (the left 16 nucleotides of the enhancer shown in Fig. 1B) or the second half of the caldesmon unit enhancer (the right 16 nucleotides of the enhancer shown in Fig. 1B). Insertions were added at an XbaI site introduced 139 nucleotides upstream of the exon-terminal 5′ splice site. Chimeric enhancers were created synthetically as described below and were inserted at the above-mentioned XbaI site. The identities of all constructs were verified by sequencing.
FIG. 2.

Caldesmon and cTNT enhancers promote differential utilization of identical flanking 5′ splice sites in an in vitro precursor RNA. (A) Pathway of splicing of two intron precursor RNAs with a second exon containing two 5′ splice sites flanking either the caldesmon or the cTNT enhancer. The precursor contains duplicated splice sites derived from adenovirus exon 2. The diagram indicates the preferred path of splicing exhibited by all in vitro precursors used in this study in which intron 1 is removed prior to intron 2. nt, nucleotides; IS and TS, exon-internal and exon-terminal sites, respectively. (B) Denaturing 8 and 5% acrylamide gels of 45-min reaction products enhance visualization of lariat species. Note that if a pathway in which intron 2 was removed first via the exon-internal 5′ splice site had been used, a diagnostic product band of 363 nt would have been produced. A similar pathway of initial intron 2 removal using the exon-terminal 5′ splice site would produce a diagnostic released exon 1 band of 485 nt. Neither of these bands was observed, indicating little processing by a pathway removing intron 2 before intron 1. Therefore, all splicing via the alternative 5′ splice sites in exon 2 can be visualized in the product bands TS and IS. Lanes M, HpaII-digested pBR322, which is the marker for all subsequent figures. Band sizes are indicated in nucleotides. (C) The caldesmon enhancer (circle), cTNT enhancer (triangle), improved cTNT enhancer (triangle with plus sign), or nonspecific sequence (striped box) was placed in the in vitro precursor RNA diagrammed below the gel. Splicing reactions were performed for 0, 25, or 45 min under standard conditions. Products resulting from intron 1 removal, double splicing using the exon-terminal 5′ splice site (TS), or double splicing using the exon-internal 5′ splice site (IS) are indicated.
In vivo RNA determination.
RNA splicing phenotypes were derived for whole-cell RNA by the use of a low-cycle reverse transcription-PCR (RT-PCR) described and quantified previously (15). PCR primers were from exon 3 of the metallothionein mini-gene backbone and from the RSV promoter region of exon 1. This primer pair could not amplify endogenous metallothionein mRNA. The identities of spliced products were verified by direct sequencing of RT-PCR products as described previously (15).
In vitro splicing.
In vitro splicing assays (25 μl) using HeLa nuclear extract were performed as described previously (25, 27). Maximal observation of differential phenotypes of the utilized enhancers took place with final concentrations of 2.0 mM MgCl2 and 1% polyethylene glycol. Splicing reaction products were quantified with a Molecular Dynamics PhosphorImager. The exon-internal splice site efficiencies were calculated as percent internal splice site usage [IS/(IS + TS), where IS is the exon-internal splice site and TS is the exon-terminal splice site]. Each reported efficiency represents the mean of at least four independent experiments with calculated standard deviations (as shown in Tables 1 and 2).
TABLE 1.
Enhancer domains and chimeras
 |
Caldesmon sequences are shown in uppercase.
cTNT sequence are shown in lowercase. L and R, 6-nucleotide half-domains (domain 2). Symbols are as for the figures.
TABLE 2.
Enhancer-specific sequences within domain 3B
 |
Caldesmon sequences are shown in uppercase.
cTNT sequences are shown in lowercase. XX and UU, point mutations made by deleting the two C’s in domain 3B or changing them to U’s, respectively. Symbols are as for the figures.
RESULTS
The caldesmon and cTNT enhancers regulate in vivo 5′ splice site selection and stimulate exon inclusion.
It has been shown previously that the purine-rich sequences residing between the two competing 5′ splice sites within alternative exon 5 of the caldesmon gene (Fig. 1A) are necessary for both exon inclusion and modulation of 5′ splice site utilization (15). In their presence, the exon-internal 5′ splice site (site upstream of the enhancer) is dominantly used; in their absence or when they are replaced by nonspecific cDNA sequences, exon inclusion levels fall and RNA is produced by using both 5′ splice sites. Therefore, the exon 5 enhancer both stimulates exon inclusion via a positive effect and regulates inclusion via recognition of a 5′ splice site positioned upstream of the enhancer. This form of splice site activation effectively positions the enhancer outside the exon whose inclusion is being stimulated. The enhancer must be within an exon to be effective in modulating 5′ splice site choice (15), indicating that it can be considered an exon element even though it stimulates usage of an upstream 5′ splice site.
Most importantly, previous results indicated that usage of the exon-internal splice site is an active rather than a passive choice because mutation of the exon-internal 5′ splice site does not activate usage of the exon-terminal site. Instead, RNA is produced via activation of a normally silent cryptic 5′ splice site upstream of the enhancer in an element-dependent manner (15). Thus, the caldesmon enhancer has both positive and negative characteristics in that it stimulates the inclusion of upstream sequences without stimulating inclusion of the region of the pre-mRNA containing the enhancer. This property of the enhancer distinguishes it from other purine-rich exon enhancers.
A single 32-nucleotide repeat of the caldesmon enhancer is sufficient for both exon inclusion and regulation of splice site choice (15). The caldesmon enhancer sequence is shown in Fig. 1A (the caldesmon minimal 32-nucleotide enhancer monomer is indicated in Fig. 1 to 3). Figure 1A also shows the purine-rich enhancer from alternative exon 5 of the cTNT gene (the cTNT 30-nucleotide enhancer is indicated in Fig. 1 to 3). Both enhancers are multipartite, consisting of two purine-rich sequences with the consensus (GAR)n (R = G or A) interdigitated with enhancer-specific sequences. Both enhancers activate inclusion of a heterologous gene dependent upon purine-rich enhancers for maximal splicing (15). To compare the abilities of the two enhancers to direct exon inclusion and modulate 5′ splice site choice in the caldesmon gene, mini-genes for in vivo transfection studies containing the natural caldesmon alternative exon 5 were constructed. The large natural caldesmon enhancer residing between the competing 5′ splice sites was replaced with minimal enhancers or nonspecific sequences. This replacement significantly shortened the exon and reduced the need for an enhancer for exon inclusion. (In the natural gene, replacement of the enhancer with nonspecific sequences raises exon skipping to 65 to 99%, whereas in constructs with shorter exons, replacement of the enhancer causes only 13% skipping [15].) Thus, the utilized constructs assay the ability of the enhancer sequence to regulate 5′ splice site utilization with a minimal effect on exon inclusion.
FIG. 3.

Specificity of the caldesmon enhancer requires the entire enhancer monomer. Precursor RNAs similar to those diagrammed in Fig. 2 containing the entire caldesmon enhancer, the 5′ half of the enhancer (nucleotides 1 to 16 of the sequence shown in Fig. 1A), or the 3′ half of the enhancer (nucleotides 17 to 32 of the sequence shown in Fig. 1A) were prepared. Reaction products resulting from splicing using the exon-internal (IS) or exon-terminal (TS) 5′ splice site are indicated. The gel used for this experiment has a different cross-linking ratio than that in Fig. 2, causing lariat species to migrate just above the TS band representing double splicing using the terminal 5′ splice site.
Figure 1B shows the RNAs produced from transfection with these mini-genes using a quantitative RT-PCR assay described previously (15). Inclusion of the natural caldesmon exon containing five enhancer monomers was efficient, and all of the RNA that included exon 5 was produced via utilization of the exon-internal 5′ splice site (Fig. 1B, lane 1). When there was no purine-rich sequence between the two competing 5′ splice sites or when the natural purine-rich sequence was replaced with a short nonspecific cDNA sequence, both 5′ splice sites were used to direct mRNA synthesis (lanes 2 and 4). These results indicated that a purine-rich sequence was necessary for majority utilization of the exon-internal 5′ splice site.
When a single 32-nucleotide copy of the caldesmon enhancer was present, exon inclusion levels were high and RNA was produced predominantly via usage of the exon-internal 5′ splice site (Fig. 1B, lane 3). In contrast, when the cTNT enhancer was present, spliced RNA was produced predominantly via usage of the exon-terminal 5′ splice site (Fig. 1B, lane 5). Therefore, despite the similarity in enhancer length and sequence, the caldesmon and cTNT enhancers directed opposite 5′ splice site selection when placed in the same environment. A mutant cTNT enhancer in which the central enhancer domain was replaced with a GAR repeat afforded a slight increase in utilization of the exon-terminal 5′ splice site (Fig. 1B, lane 6). Thus, a mutation in the cTNT element that improves exon inclusion in the cTNT environment (25) also improves 5′ splice site selectivity when positioned in the caldesmon exon. These results show that the caldesmon and cTNT enhancers effectively stimulate usage of splice sites lying upstream or downstream of the enhancer sequence, respectively. Such differential selectivity is unusual among purine-rich enhancers and presents an optimal test system with which to investigate enhancer function.
The caldesmon and cTNT enhancers differentially regulate in vitro utilization of competing identical strong 5′ splice sites.
The alternative exon tested in Fig. 1 is derived from the natural caldesmon exon from which the caldesmon enhancer was isolated. This exon is heterologous to the cTNT enhancer. To test both enhancers in a heterologous environment and to determine whether the two enhancers regulate 5′ splice site choice in vitro, we utilized a three-exon in vitro precursor RNA containing a middle exon with two 5′ splice sites. The precursor, diagrammed in Fig. 2, was constructed from adenovirus sequences (27, 35) and contains identical 5′ splice sites (i.e., the two 5′ splice sites within exon 2 are identical and identical to the 5′ splice site in exon 1). Similarly, the 3′ splice sites in exons 2 and 3 are identical. Therefore, any observed preferential splice site utilization cannot be due to inherent differences between the competing splice sites. The design of the test constructs was chosen to place the enhancers within an internal exon between competing 5′ splice sites. Use of an internal exon insulates the enhancers from exon definition effects caused by cap-binding proteins (11) and places the tested enhancers in an exon environment structured similarly to the alternative caldesmon exon. Use of a strong adenovirus precursor RNA provided enhancer-independent removal of intron 1 to permit assessment of the ability of the sequences to regulate 5′ splice site recognition without an accompanying requirement for activation of an upstream 3′ splice site.
Short cassettes (30 to 37 nucleotides) of nonspecific sequence, the caldesmon enhancer, or the cTNT enhancer (wild type and improved) were inserted between the competing 5′ splice sites within exon 2 of these constructs. All tested precursor RNAs were spliced efficiently, producing products in which both introns 1 and 2 were removed (Fig. 2A and B). No evidence for exon skipping was observed, reflecting the strength of the splice sites flanking exon 2. Intron 1 was removed at equal frequencies in all tested constructs. In addition, intron 1 was removed before noticeable removal of intron 2 via usage of either 5′ splice site flanking the tested enhancer (Fig. 2A). Thus, all usage of the alternative 5′ splice sites within exon 2 could be monitored by observation of the levels of doubly spliced product RNAs (IS and TS species in all figures). Constructs differed as to the preference of utilization of alternative 5′ splice sites within exon 2. Therefore, these substrates provided a measure of the ability of each enhancer to affect the specificity of utilization of flanking 5′ splice sites without interference from effects on overall exon 2 inclusion.
When a precursor RNA containing nonspecific sequences between the competing 5′ splice sites was spliced in vitro, RNA was produced via relatively equivalent utilization of the two 5′ splice sites (Fig. 2C, lanes 10 to 12). A similar result was observed when no additional sequence was placed between the two competing 5′ splice sites (data not shown). When the 32-nucleotide caldesmon enhancer was placed between the two 5′ splice sites, splicing proceeded almost exclusively from the exon-internal 5′ splice site (Fig. 2B and C, lanes 1 to 3). This preference was observed over a long period with various batches of HeLa nuclear extract. An average of 14 experiments indicated that the internal site was used to generate (79.4 ± 8.1)% of the doubly spliced RNA (quantification in Table 1). Thus, when the caldesmon monomer enhancer was present, splicing occurred preferentially at the 5′ splice site located upstream of the enhancer sequence even when the splice sites were strongly constitutive. In contrast, the 30-nucleotide cTNT enhancer switched the 5′ splice site preference to the exon-terminal 5′ splice site (Fig. 2B and C, lanes 4 to 6) so that only (19.1 ± 7.9)% of the doubly spliced product RNA resulted from usage of the internal 5′ splice site. The improved cTNT enhancer was even more effective in directing splice site utilization to the exon-terminal 5′ splice site, so that essentially all of the product RNA resulted from usage of the exon-terminal 5′ splice site (Fig. 2C, lanes 7 to 9). Thus, the cTNT enhancer directed splicing using a 5′ splice site located downstream of the enhancer sequence both in vivo and in vitro.
These results strongly indicate that the two enhancers, despite their sequence similarity, cause utilization of 5′ splice sites lying on opposite sides of the enhancer sequence in a fashion independent of other sequences within the regulated exon. To our knowledge, this is the first example of two purine-rich enhancers with relatively similar sequences demonstrating opposite splice site specificities.
Specificity of the caldesmon enhancer requires the entire enhancer monomer.
Both the caldesmon and the cTNT enhancers have multipartite structures in which two segments of purine-rich sequence are separated by a spacer sequence. Previous work indicated that the cTNT enhancer functions to stimulate exon inclusion in its natural environment primarily through the upstream purine-rich region (25). To determine whether the multipartite nature of the caldesmon enhancer was required for its ability to regulate 5′ splice site choice, versions of the in vitro precursor RNA used in the experiment whose results are shown in Fig. 2 in which the monomer 32-nucleotide caldesmon enhancer was replaced by either the right or the left half of the enhancer (16 nucleotides each) were prepared (Fig. 3). This splitting of the enhancer created a 5′-half enhancer containing only purines and a 3′-half enhancer containing both purines and pyrimidines.
When assayed in vitro, the precursor RNA containing either half of the caldesmon enhancer spliced efficiently. Specificity of 5′ splice site utilization, however, was lost, so that considerable product was generated when the exon-terminal 5′ splice site with precursor RNAs containing either half of the caldesmon enhancer was used (Fig. 3, lanes 4 to 9), with 56 to 59% of the product RNA resulting from usage of the exon-internal site (Table 1). These values are different than the 47% average we observed for nonspecific sequences (Table 1), but it should be noted that there was a considerable standard deviation associated with 5′ splice site usage patterns when neutral or inactivated enhancer sequences were analyzed. We have therefore chosen to set a threshold of 60% 5′ splice site preference for minimum specificity. Enhancer sequences failing to meet this criteria are considered neutral sequences. By these criteria, the full enhancer containing two purine-rich regions was required to direct 5′ splice site usage to the exon-internal site (Fig. 3, lanes 1 to 3).
Splice site specificity of the caldesmon and cTNT enhancers requires all regions of each enhancer and is strongly influenced by sequences in the 3′-terminal segment.
To determine which sequences within each enhancer are required for 5′ splice site selectivity, each enhancer was arbitrarily divided into three domains. As shown in Fig. 4A, domain 1 is a 5′ purine-rich region with a simple (GAR)3 sequence, domain 2 is a region that is variable in sequence and length in the two enhancers, and domain 3 begins with (GAR)2 and terminates with 5 to 7 nucleotides of enhancer-specific sequence. Chimeric enhancers were created by interchanging each of the three domains between the two enhancers. Swapping any of the three regions between the enhancers produced an alteration in 5′ splice site utilization. The magnitude of the effect was different for individual regions. Table 1 lists the chimeras created for this study and indicates the percentage of splicing via utilization of the exon-internal 5′ splice site of exon 2 for each construct.
FIG. 4.

Sequences within domain 3 of the enhancers provide 5′ splice site specificity. (A) The caldesmon and cTNT sequences were divided into three domains as diagrammed for the purpose of domain interchange experiments. Purine repeats of the sequence GAR are indicated (shaded). Each domain is represented by a black (caldesmon) or white (cTNT) box (heterologous sequence is represented by a striped box in panel C). (B to D) In vitro splicing of precursor RNAs containing wild-type or chimeric enhancers. Reactions were performed for 45 min. Reaction products are identified as for Fig. 2. Sizes are indicated in nucleotides. In panel C, several different chimeras containing alternate domain 2 were made to control for differences in length of domain 2 between the two enhancers. When caldesmon domain 2 sequences were inserted into the cTNT enhancer, either the entire domain (12 nucleotides) (lane 5) or 5′ or 3′ half-domains (6 nucleotides each [boxes L and R]) (lanes 7 and 8) were used. When cTNT domain 2 sequences were inserted into the caldesmon enhancer, either one copy (6 nucleotides, represented by one white box) or two copies (two white boxes) were used.
Most of the ability to affect 5′ splice site choice resided within sequences in the right third of the enhancers (domain 3 in Table 1 and Fig. 4B). Replacement of domain 3 of the caldesmon enhancer with the corresponding region from the cTNT enhancer (13 nucleotides replaced 11 nucleotides) reduced usage of the exon-internal 5′ splice site from 79.4 to 37.2%, effectively converting the caldesmon enhancer splice site selection pattern to 70% of that of the cTNT enhancer (Fig. 4B, lane 3). Conversely, replacement of domain 3 of the cTNT enhancer with that from the caldesmon enhancer effectively converted the cTNT enhancer splice site selection to 79% of that of the caldesmon enhancer, and usage of the exon-internal site increased from 19.1 to 66.7% (Fig. 4B, lane 4).
Although interchanging domain 3 of the two enhancers had a pronounced effect on 5′ splice site choice, the resulting chimeric enhancers had only 70 to 80% of the full specificity of the parent enhancers, suggesting that domains 1 and 2 of each enhancer also played a role in splice site selection. Furthermore, as shown in Fig. 3, the right half of the caldesmon enhancer was not sufficient to direct splice site selectivity when present alone, underscoring the need for other regions of the enhancer for specificity.
To analyze the importance of the domain 2 regions of the two enhancers, which are very different from each other in sequence, the spacers were swapped between the two enhancers (Fig. 4C). Interchange of the domain 2 regions of the enhancers had a reproducible but minimal effect on splice site choice that accounted for 15 to 30% of the selectivity (Fig. 4C, lanes 4 and 5, and Table 1). Domain 2 of the caldesmon enhancer is twice the length of domain 2 from the cTNT enhancer, raising the possibility that spacing between the two purine-rich regions could be important. To address this possibility, domain 2 from the caldesmon enhancer (12 nucleotides) was replaced by two copies of domain 2 from the cTNT enhancer (total of 12 nucleotides) (Fig. 4C, lane 6), and domain 2 from the cTNT enhancer was replaced by either half (AAAAGG or GCAGCA) of domain 2 from the caldesmon enhancer (lanes 7 and 8, respectively). These alterations had similar and relatively minimal impacts on splice site choice, suggesting that both the sequence and the length of domain 2 play a role in splice site choice, but are not determinative.
Previous studies of the cTNT enhancer suggested that the GAR repeat sequences in the 5′ portion of the enhancer were important for enhancer function and S/R protein binding (25). To test the importance of these sequences for enhancer specificity, chimeric enhancers interchanging domain 1 were analyzed (Fig. 4D, lanes 3 and 4). Interchange of these sequences had minimal impact on splice site selectivity. It should be noted, however, that the two sequences are relatively similar. Coupled with the inability of the 3′ half of the enhancers to direct splice site selectivity, the data suggests that domain 1 may be essential for enhancer function but nondeterminative for specificity. Thus, when both enhancers were dissected for functional elements that were required for alternative recognition of flanking splice sites, each enhancer was revealed to be a complex multipartite element.
Short enhancer-specific sequences at the 3′ end of the cTNT and caldesmon enhancers provide splice site specificity.
To identify the nucleotides within domain 3 that are important for enhancer specificity, domain 3 was further subdivided into two domains (3A and 3B) (Fig. 5 and Table 2). Domain 3A consists of two GAR repeats, and domain 3B contains 5 to 7 nucleotides of enhancer-specific sequence. It seemed unlikely, given the sequence similarity in domain 3A between the two enhancers, that specificity determinants were present in this domain. Therefore, analysis concentrated on domain 3B. Domain 3B is quite short, consisting of 5 nucleotides (AGGCA) in the caldesmon enhancer and 7 nucleotides (GACGACG) in the cTNT enhancer. Mutations were made within domain 3B, and the mutants were analyzed for specificity and overall splicing efficiency (Fig. 5).
FIG. 5.
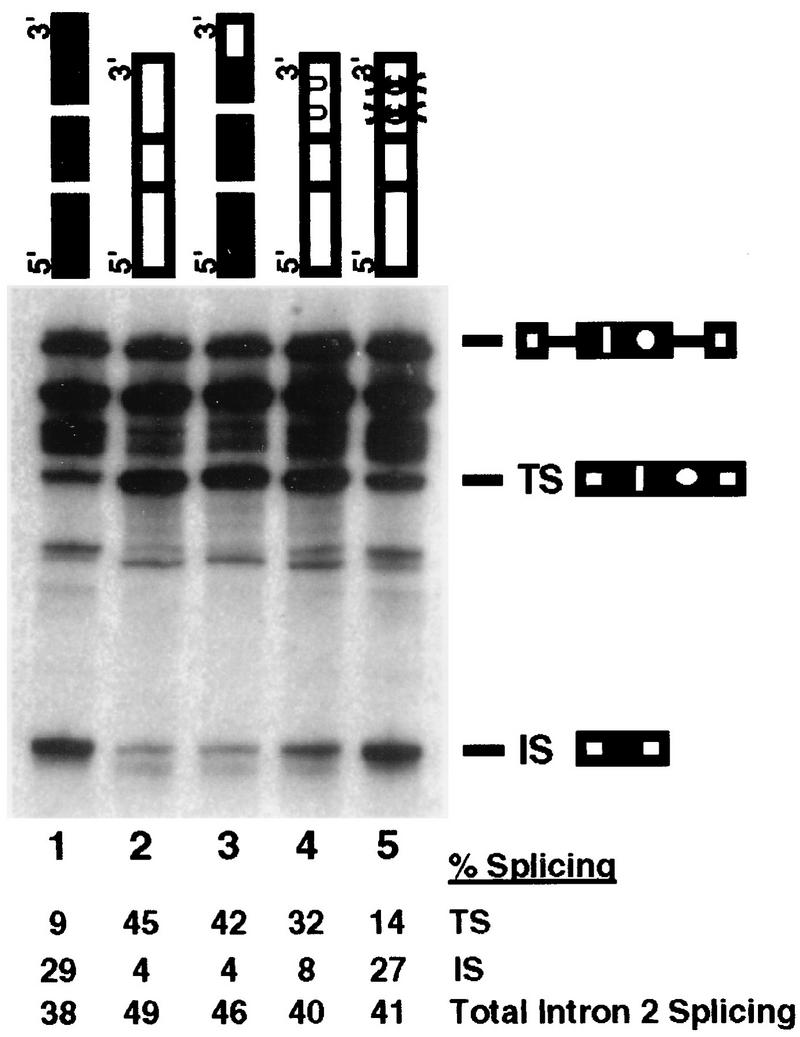
Sequences within domain 3B dictate 5′ splice site specificity. For this experiment, the two enhancers were considered to contain four domains. Compared to the domains identified in Fig. 4, the extra domain arises by subdivision of domain 3 into domain 3A, which contains the first 6 nucleotides of domain 3, with the sequence (GAR)2 and domain 38, which contains the terminal enhancer-specific. Caldesmon (black boxes) and cTNT (white boxes) domains are indicated. One mutant enhancer was prepared by replacing domain 3B of the caldesmon enhancer with domain 3B from the cTNT enhancer (lane 3). Two point mutations were made by altering the two C’s in the cTNT domain 3B to U’s (UU) (lane 4) or deleting both C’s (XX) (lane 5). Splicing reactions were performed for 45 min. Product RNAs are identified as for Fig. 2. Quantification of the indicated product RNAs is shown beneath the gel. The amounts of products are shown in arbitrary PhosphorImager units as a percentage of total RNA precursor and products, normalized for uridine content.
In the first mutant, the heptanucleotide GACGACG from the cTNT enhancer domain 3B replaced domain 3B of the caldesmon enhancer. This replacement strongly affected splice site choice, activating usage of the exon-terminal splice site in vitro, so that 82% of the product RNA was now spliced via the exon-terminal 5′ splice site versus 21% for the recipient caldesmon enhancer (Fig. 5, lane 3, and Table 2). Strong activation of usage of the exon-terminal splice site with this small sequence suggests that domain 3B is determinative for splice site selectivity.
Two additional mutants with alterations in the specific sequence within domain 3B of the cTNT enhancer were created. The first mutation altered the two C nucleotides of the GAC repeat to GAU. This alteration had minimal impact on splice site selectivity (Fig. 5, lane 4, and Table 2). A second mutant in which the two Cs in each GAC repeat were simultaneously deleted was created (Fig. 5, lane 5, and Table 2). This deletion effectively converts domain 3 of the cTNT enhancer to a sequence almost identical to the equivalent region of the caldesmon enhancer (GAGGAAGACGACG converted to GAGGAAGAGAG, compared to the caldesmon domain 3 sequence of GAGGAGAGGCA). The deletion caused activation of splicing via the exon-internal site. As shown in Table 2, 70% of the product RNA was spliced via usage of the exon-internal site versus 19% via usage of the parental cTNT enhancer, i.e., the cTNT enhancer was strongly converted to the caldesmon enhancer with respect to splice site selectivity. Thus, small alterations in domain 3B located at the very 3′ terminus of both enhancers strongly activated opposite splice site selectivity. The sequences implicated for exon-terminal or exon-internal 5′ splice site activation by these experiments are GACGACG and AGGCA, respectively.
DISCUSSION
Exon splicing enhancers have been shown to be important elements in the efficiency of exon recognition (2, 14, 26). One major class of exon enhancers are the purine-rich enhancers, exemplified by the caldesmon and cTNT enhancers compared in this study (4, 5, 8, 9, 15, 20, 22, 25, 31–34, 36, 38, 41–43). The caldesmon and cTNT enhancers are similar in both sequence and length, consisting of two blocks of purine repeats interdigitated with enhancer-specific sequences. Both enhancers stimulate splicing of a single-intron heterologous pre-mRNA (15, 42). However, when placed between two competing 5′ splice sites in an internal exon bearing two sites, the enhancers demonstrated opposite phenotypes. Both increased exon inclusion levels in vivo compared to exons lacking an enhancer; however, the two enhancers directed inclusion by different 5′ splice sites within the alternative exon. Both shifted the 5′ splice site usage pattern away from the roughly equal usage of the two 5′ splice sites observed in the absence of an enhancer between the sites. The caldesmon enhancer caused usage of the exon-internal 5′ splice site, the site upstream of the enhancer, both in the natural caldesmon exon and in a heterologous exon containing strong adenovirus-derived constitutive 5′ splice sites. In contrast, the cTNT enhancer caused usage of the exon-terminal 5′ splice site, the site downstream of the enhancer, in both the caldesmon exon and the adenovirus exon. Thus, despite their sequence similarity, the presence of the two enhancers resulted in opposite splice site choices. These results suggest that subtle differences in exon sequence can strongly affect splicing choices.
Multipartite enhancer structure.
Analysis of the domains within each enhancer that are important for function indicated that each enhancer could be considered as a tetrapartite sequence made up of a 5′ domain consisting of a simple purine repeat, (GAR)3; a second domain of enhancer-specific sequence and length; a third domain consisting of (GAR)2; and a fourth differentiating short domain with the sequence AGGCA (caldesmon) or GACGACG (cTNT). Full-length enhancers were required for specificity. However, interchange of regions 1 and 2 had only 10 to 30% effect on splice site specificity, indicating that although these regions of the enhancer are necessary, the sequence differences between domains 1 and 2 played a minor role in specificity. It should be noted, however, that the two enhancers are very similar in domain 1.
In contrast, interchange of the last 5 to 7 nucleotides of the enhancers strongly altered enhancer specificity, almost converting the specificity of each enhancer to that of the other. The sequence of this region of each enhancer is unique, suggesting that the binding of enhancer-specific factors to this region of the enhancer regulates specificity. Thus, although the multipartite nature of each enhancer was required for splice site specificity, a short stretch of nucleotides near the 3′ end of the enhancers was found to be determinative for splice site choice when present within the whole enhancer.
Positive versus negative regulation.
The caldesmon exon enhancer is a very unusual splicing regulatory sequence in that it causes recognition of a 5′ splice site lying upstream of the enhancer within the exon containing the enhancer. Thus, the enhancer does not become incorporated into product RNA resulting from usage of the upstream splice site. This raises the question of whether the enhancer functions to cause alternative splice site recognition via activation of the upstream splice site or repression of the downstream splice site. Although some aspects of repression are certainly possible, we have no evidence suggesting that the enhancer is inhibitory to 5′ splice site recognition. Placement of the enhancer in a weak exon activates splicing when the exon is 3′ terminal (15) or internal (unpublished data). In these contexts, however, the caldesmon enhancer is never as powerful as the cTNT enhancer.
In its natural gene, the enhancer is absolutely required for significant inclusion of exon 5 via recognition of the exon-internal 5′ splice site. Usage of the internal 5′ splice site effectively places the enhancer outside the enhanced exon, suggesting that the enhancer has the ability to activate flanking sequences (15). Replacement of the enhancer with nonspecific sequences causes exon skipping, not inclusion via the exon-terminal splice site. A model in which the enhancer repressed usage of the external 5′ splice site would have predicted inclusion via the terminal splice site in the absence of a functional enhancer.
Perhaps more revealing, however, is the phenotype observed when the upstream splice site is mutated in the natural caldesmon gene. If the enhancer caused silencing of the downstream 5′ splice site, it might be predicted that mutation of the upstream 5′ splice site would induce exon skipping. Instead, exon inclusion levels remain high, and normally silent cryptic splice sites are activated (15). Therefore, we prefer an interpretation in which the caldesmon enhancer is viewed as a complicated enhancer-silencer activating neighboring sequences for inclusion by causing an overall preference for binding of splicing factors to an upstream splice site versus a downstream site. Such activation at a distance with concomitant internal silencing for splicing is reminiscent of the mechanism of activation of inclusion of the alternative 3′-terminal exon in the calcitonin/calcitonin gene-related peptide gene by an intron-located enhancer containing wild-type splice sites and binding splicing factors but not itself used for splicing (21).
Regulation of 5′ splice sites by enhancers.
Exon enhancers have usually been shown to affect exon inclusion by activating weak 3′ splice sites, via the interaction of enhancer-bound S/R proteins and the 35-kDa subunit of U2AF. In the constructs examined in this study, the enhancers regulate 5′ splice site utilization, presumably through an effect on the binding of U1 snRNPs. Indeed, exon enhancers can replace a 5′ splice site during early exon recognition and have been shown to activate U1 snRNP binding in reconstituted in vitro reactions (19, 31). Furthermore, S/R proteins have recently been shown to provide 5′ splice site recognition in the absence of U1 snRNPs (7, 37).
What, if any, differences exist between the mechanisms utilized by S/R proteins to activate 3′ splice sites and 5′ splice sites? Our study suggests that there may be at least some differences, because regions of the cTNT enhancer characterized as important for 5′ splice site selectivity were different than those shown to be maximally important for activation of a 3′ splice site. The cTNT enhancer used in this study has been subjected to exhaustive mutagenesis with respect to its ability to support exon inclusion in its natural 30-nucleotide internal exon from the cTNT gene (5, 6, 42). This analysis revealed the importance of domains 1 and 2 of the enhancer for both default and regulated exon inclusion. Mutations within domain 1 reduced exon inclusion in nonmuscle and muscle cells from 26 and 73% to 2 and 4%, respectively. Mutation of domain 3B, containing the sequences important for 5′ splice site selectivity, in this study reduced inclusion to only 6 and 30% in the same cell lines. Thus, domain 3B was less important than domain 1 for maximal exon inclusion. This difference in sequence requirements in the two test situations suggests that environment can influence enhancer activity and the binding of specific proteins. This result may be similar to the observation that an exon purine enhancer becomes inhibitory if positioned close to the binding site of U2AF (22).
Enhancer-binding proteins.
The sequences present in the specificity domains of each enhancer suggest little about the identity of the proteins binding to these sequences and affecting 5′ splice site specificity. Neither represents a sequence selected by iterative selection as a preferred binding site for a known S/R protein. The cTNT enhancer had been shown to bind SRp75, SRp55, SRp40, and ASF/SF2 in vitro, but not SC35 (25). Binding of these proteins, however, was strongly affected by sequences within domains 1 and 2 of the cTNT enhancer. Preliminary in vitro experiments with the caldesmon enhancer indicate that it binds SC35 better than SRp40. In addition, both enhancers can be UV cross-linked to unique proteins with molecular weights not characteristic of known S/R proteins.
Addition of SRp40, but not SC35, to an S100 in vitro splicing extract activates splicing of the natural cTNT exon in an enhancer-dependent fashion (25). Addition of individual S/R proteins (SRp55, SRp40, or SC35) did not cause enhancer-mediated alterations in 5′ splice site utilization in the constructs used in this study (data not shown). In fact, addition of any S/R protein or a magnesium pellet enriched for a mixture of S/R proteins caused dominant usage of the exon-terminal 5′ splice site in any substrate with two 5′ splice sites in exon 2 regardless of the presence or absence of an enhancer between the splice sites (data not shown). Furthermore, replacement of the enhancer with a dimer with a sequence selected by SRp40 by iterative selection did not result in 5′ splice site selectivity (data not shown). Thus, at the moment, we do not know the identity of the trans-acting factors responsible for enhancer specificity. Given the complexity of the enhancer sequences revealed in this study and the inability of individual S/R proteins to affect 5′ splice site utilization in an enhancer-responsive fashion, regulation may require the binding of multiple proteins to multiple domains within the enhancer.
Our results suggest that exon enhancers, even relatively short ones like the 30-nucleotide cTNT and caldesmon enhancers, may have complicated multipartite structures. In this regard, the cTNT and caldesmon enhancers resemble the complicated exon regulatory sequences within certain human immunodeficiency virus exons in which short exon silencers are closely juxtaposed to short exon enhancers (1, 30). Full regulatory potential in both situations requires the entire multipartite structure, but short regions can have a determinative effect. Given the high degree of similarity of subdomains within the caldesmon and cTNT enhancers, it seems likely that the two enhancers bind some common factors, implying that an individual factor can be involved in both positive and negative splicing decisions, depending upon the identity of the factors also bound to the enhancers.
ACKNOWLEDGMENTS
We thank R. Sierra for technical assistance.
This research was supported by the American Cancer Society (T.A.C.) and by PHS grant GM38526 and the Robert A. Welch Foundation (S.M.B.). T.A.C. is an Established Investigator of the American Heart Association.
REFERENCES
- 1.Amendt B A, Si Z H, Stoltzfus C M. Presence of exon splicing silencers within human immunodeficiency virus type 1 tat exon 2 and tat-rev exon 3: evidence for inhibition mediated by cellular factors. Mol Cell Biol. 1995;15:4606–4615. doi: 10.1128/mcb.15.8.4606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Black D L. Finding splice sites within a wilderness of RNA. RNA. 1995;1:763–771. [PMC free article] [PubMed] [Google Scholar]
- 3.Caceres J F, Stamm S, Helfman D M, Krainer A R. Regulation of alternative splicing in vivo by overexpression of antagonistic splicing factors. Science. 1994;265:1706–1709. doi: 10.1126/science.8085156. [DOI] [PubMed] [Google Scholar]
- 4.Caputi M, Casari G, Guenz S, Tagliabue R, Sidoli A, Melo C A, Baralle F E. A novel bipartite splicing enhancer modulates the differential processing of the human fibronectin EDA exon. Nucleic Acids Res. 1994;22:1018–1022. doi: 10.1093/nar/22.6.1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cooper T A. In vitro splicing of cardiac troponin T precursors—exon mutations disrupt splicing of the upstream intron. J Biol Chem. 1992;267:5330–5338. [PubMed] [Google Scholar]
- 6.Cooper T A, Ordahl C P. Nucleotide substitutions within the cardiac troponin T alternative exon disrupt pre-mRNA alternative splicing. Nucleic Acids Res. 1989;17:7905–7921. doi: 10.1093/nar/17.19.7905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Crispino J D, Blencowe B J, Sharp P A. Complementation by SR proteins of pre-mRNA splicing reactions depleted of U1 snRNP. Science. 1994;265:1866–1869. doi: 10.1126/science.8091213. [DOI] [PubMed] [Google Scholar]
- 8.Dirksen W P, Hampson R K, Qiang S, Rottman F M. A purine-rich exon sequence enhances alternative splicing of bovine growth hormone pre-mRNA. J Biol Chem. 1994;269:6431–6436. [PubMed] [Google Scholar]
- 9.Dominski Z, Kole R. Identification of exon sequences involved in splice site selection. J Biol Chem. 1994;269:23590–23596. [PubMed] [Google Scholar]
- 10.Fu X-D. The superfamily of arginine/serine-rich splicing factors. RNA. 1993;1:663–680. [PMC free article] [PubMed] [Google Scholar]
- 11.Gamberi C, Izurralde E, Beisel C, Mattaj I W. Interaction between the human nuclear cap-binding protein complex and hnRNP F. Mol Cell Biol. 1997;17:2575–2597. doi: 10.1128/mcb.17.5.2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hayashi K, Yano H, Hashida T, Takeuchi R, Takeda O, Asada K, Takahashi E, Kato I, Sobue K. Genomic structure of the human caldesmon gene. Proc Natl Acad Sci USA. 1992;89:12122–12126. doi: 10.1073/pnas.89.24.12122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hedley M L, Maniatis T. Sex-specific splicing and polyadenylation of dsx pre-mRNA requires a sequence that binds specifically to tra-2 protein in vitro. Cell. 1991;65:579–586. doi: 10.1016/0092-8674(91)90090-l. [DOI] [PubMed] [Google Scholar]
- 14.Hodges D, Bernstein S I. Genetic and biochemical analysis of alternative splicing. Adv Genet. 1994;31:207–281. doi: 10.1016/s0065-2660(08)60399-5. [DOI] [PubMed] [Google Scholar]
- 15.Humphrey M B, Bryan J, Cooper T A, Berget S M. A 32-nucleotide exon-splicing enhancer regulates usage of competing 5′ splice sites in a differential internal exon. Mol Cell Biol. 1995;15:3979–3988. doi: 10.1128/mcb.15.8.3979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Humphrey M B, Bryan J. Abstracts of the American Society of Cell Biology Thirty-Second Annual Meeting. Bethesda, Md: American Society of Cell Biology; 1992. Human caldesmon isoforms are generated by an unusual alternative splicing mechanism, abstr. 234. [Google Scholar]
- 17.Humphrey M B, Herrera-Sosa H, Gonzalez G, Lee R, Bryan J. Cloning of cDNAs encoding human caldesmons. Gene. 1992;112:197–204. doi: 10.1016/0378-1119(92)90376-z. [DOI] [PubMed] [Google Scholar]
- 18.Kanopka A, Muhlemann O, Akusjarvi G. Inhibition by SR proteins of splicing of a regulated adenovirus pre-mRNA. Nature. 1996;381:535–538. doi: 10.1038/381535a0. [DOI] [PubMed] [Google Scholar]
- 19.Kohtz J D, Jamison S F, Will C L, Zuo P, Luhrmann R, Garcia-Blanco M A, Manley J L. Protein-protein interactions and 5′ splice site recognition in mammalian mRNA precursors. Nature. 1994;368:119–124. doi: 10.1038/368119a0. [DOI] [PubMed] [Google Scholar]
- 20.Lavigueur A, La Branche H, Kornblihtt A R, Chabot B. A splicing enhancer in the human fibronectin alternate ED1 exon interacts with S/R proteins and stimulates U2 snRNP binding. Genes Dev. 1993;7:2405–2417. doi: 10.1101/gad.7.12a.2405. [DOI] [PubMed] [Google Scholar]
- 21.Lou H, Gagel R F, Berget S M. An intron enhancer recognized by splicing factors activates polyadenylation. Genes Dev. 1996;10:208–219. doi: 10.1101/gad.10.2.208. [DOI] [PubMed] [Google Scholar]
- 22.Lynch K W, Maniatis T. Synergistic interactions between two distinct elements of a regulated splicing enhancer. Genes Dev. 1995;9:284–293. doi: 10.1101/gad.9.3.284. [DOI] [PubMed] [Google Scholar]
- 23.Lynch K W, Maniatis T. Synergistic interactions between two distinct elements of a regulated splicing enhancer. Genes Dev. 1995;9:284–293. doi: 10.1101/gad.9.3.284. [DOI] [PubMed] [Google Scholar]
- 24.Manley J L, Tacke R. SR proteins and splicing control. Genes Dev. 1996;10:1569–1579. doi: 10.1101/gad.10.13.1569. [DOI] [PubMed] [Google Scholar]
- 25.Ramchatesingh J, Zahler A M, Neugebauer K M, Roth M B, Cooper T. A subset of S/R proteins activates splicing of the cardiac troponin T alternative exon by direct interactions with an exonic enhancer. Mol Cell Biol. 1995;15:4898–4907. doi: 10.1128/mcb.15.9.4898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reed R. Initial splice-site recognition and pairing during pre-mRNA splicing. Curr Opin Genet Dev. 1996;6:215–220. doi: 10.1016/s0959-437x(96)80053-0. [DOI] [PubMed] [Google Scholar]
- 27.Robberson B L, Cote G J, Berget S M. Exon definition may facilitate splice site selection in RNAs with multiple exons. Mol Cell Biol. 1990;10:84–94. doi: 10.1128/mcb.10.1.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ryner L C, Goodwin S F, Castrillon D H, Anand A, Villella A, Baker B S, Hall J C, Taylor B J, Wasserman S A. Control of male sexual orientation in Drosophila by the fruitless gene. Cell. 1990;87:1079–1090. doi: 10.1016/s0092-8674(00)81802-4. [DOI] [PubMed] [Google Scholar]
- 29.Screaton G R, Caceres J F, Mayeda A, Bell M V, Plebanski M, Jackson D G, Bell J I, Krainer A R. Identification and characterization of three members of the human SR family of pre-mRNA splicing factors. EMBO J. 1995;14:4336–4349. doi: 10.1002/j.1460-2075.1995.tb00108.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Staffa A, Cochrane A. Identification of positive and negative splicing regulatory elements within the terminal tat-rev exon of human immunodeficiency virus type 1. Mol Cell Biol. 1995;15:4597–4605. doi: 10.1128/mcb.15.8.4597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Staknis D, Reed R. S/R proteins promote the first specific recognition of pre-mRNA and are present together with the U1 small nuclear ribonucleoprotein particle in a general splicing enhancer complex. Mol Cell Biol. 1994;14:7670–7682. doi: 10.1128/mcb.14.11.7670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sun Q, Mayeda A, Hampson R K, Krainer A R, Rottman F M. General splicing factor SF2/ASF promotes alternative splicing by binding to an exonic splicing enhancer. Genes Dev. 1993;7:2598–2608. doi: 10.1101/gad.7.12b.2598. [DOI] [PubMed] [Google Scholar]
- 33.Tacke R, Manley J L. The human splicing factors ASF/SF2 and SC35 possess distinct, functionally significant RNA binding specificities. EMBO J. 1995;14:3540–3551. doi: 10.1002/j.1460-2075.1995.tb07360.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tacke R, Manley J L. Sequence-specific binding by an SR protein requires RS domain phosphorylation: creation of an SRp40-specific splicing enhancer. Proc Natl Acad Sci USA. 1997;94:1148–1153. doi: 10.1073/pnas.94.4.1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Talerico M, Berget S M. Effect of 5′ splice site mutations on splicing of the preceding intron. Mol Cell Biol. 1990;10:6299–6305. doi: 10.1128/mcb.10.12.6299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tanaka K, Watakabe A, Shimura Y. Polypurine sequences within a downstream exon function as a splicing enhancer. Mol Cell Biol. 1994;4:1347–1354. doi: 10.1128/mcb.14.2.1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tarn W Y, Steitz J A. SR protein can compensate for the loss of U1 snRNP function in vitro. Genes Dev. 1994;8:2704–2717. doi: 10.1101/gad.8.22.2704. [DOI] [PubMed] [Google Scholar]
- 38.van Oers C C M, Adema G J, Zandberg H, Moen T C, Baas P D. Two different sequence elements within exon 4 are necessary for calcitonin-specific splicing of the human calcitonin/calcitonin gene-related peptide I pre-mRNA. Mol Cell Biol. 1994;14:951–960. doi: 10.1128/mcb.14.2.951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang J, Manley J L. Overexpression of the S/R proteins ASF/SF2 and SC35 influences alternative splicing in vivo in diverse ways. RNA. 1995;1:335–346. [PMC free article] [PubMed] [Google Scholar]
- 40.Wang J, Takagaki Y, Manley J L. Targeted disruption of an essential vertebrate gene: ASF/SF2 is required for cell viability. Genes Dev. 1996;10:2588–2599. doi: 10.1101/gad.10.20.2588. [DOI] [PubMed] [Google Scholar]
- 41.Watakabe A, Tanaka K, Shimura Y. The role of exon sequences in splice site selection. Genes Dev. 1993;7:407–418. doi: 10.1101/gad.7.3.407. [DOI] [PubMed] [Google Scholar]
- 42.Xu R, Teng J, Cooper T A. The cardiac troponin T alternative exon contains a novel purine-rich positive splicing element. Mol Cell Biol. 1993;13:3660–3674. doi: 10.1128/mcb.13.6.3660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yeakley J M, Hedjran F, Morfin J-P, Merillat N, Rosenfeld M G, Emerson R B. Control of calcitonin/calcitonin gene-related peptide pre-mRNA processing by constitutive exon and intron elements. Mol Cell Biol. 1993;13:5999–6011. doi: 10.1128/mcb.13.10.5999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zahler A M, Lane W S, Stolk J A, Roth M B. S/R proteins—a conserved family of pre-messenger-RNA splicing factors. Genes Dev. 1992;6:837–847. doi: 10.1101/gad.6.5.837. [DOI] [PubMed] [Google Scholar]