

Abstract
Members of the INK4 protein family specifically inhibit cyclin-dependent kinase 4 (cdk4) and cdk6-mediated phosphorylation of the retinoblastoma susceptibility gene product (Rb). p16INK4A, a prototypic INK4 protein, has been identified as a tumor suppressor in many human cancers. Inactivation of p16INK4A in tumors expressing wild-type Rb is thought to be required in order for many malignant cell types to enter S phase efficiently or to escape senescence. Here, we demonstrate another mechanism of tumor suppression by implicating p16INK4A in a G1 arrest checkpoint in response to DNA damage. Calu-1 non-small cell lung cancer cells, which retain Rb and lack p53, do not arrest in G1 following DNA damage. However, engineered expression of p16INK4A at levels compatible with cell proliferation restores a G1 arrest checkpoint in response to treatment with γ-irradiation, topoisomerase I and II inhibitors, and cisplatin. A similar checkpoint can be demonstrated in p53−/− fibroblasts that express p16INK4A. DNA damage-induced G1 arrest, which requires the expression of pocket proteins such as Rb, can be abrogated by overexpression of cdk4, kinase-inactive cdk4 variants capable of sequestering p16INK4A, or a cdk4 variant incapable of binding p16INK4A. After exposure to DNA-damaging agents, there was no change either in overall levels of p16INK4A or in amounts of p16INK4A found in complex with cdks 4 and 6. Nonetheless, p16INK4A expression is required for the reduction in cdk4- and cdk6-mediated Rb kinase activity observed in response to DNA damage. During tumor progression, loss of p16INK4A expression may be necessary for cells with wild-type Rb to bypass this G1 arrest checkpoint and attain a fully transformed phenotype.
p16INK4A is a specific inhibitor of cyclin dependent kinase 4 (cdk4) and cdk6, which participate in the cyclin D-dependent phosphorylation of the retinoblastoma susceptibility gene product, Rb (31). Hyperphosphorylation of Rb inactivates its growth-suppressive properties, allowing cells to enter S phase. Several lines of evidence indicate that p16INK4A is a tumor suppressor. First, its gene maps to 9p21, a chromosomal locus deranged in many human cancers (15). Second, p16INK4A is commonly deleted, mutated, or hypermethylated and transcriptionally silenced in tumors that retain wild-type Rb, and ectopic expression of p16INK4A in these cells at high levels results in G1 arrest (17, 19, 22, 30, 33, 35). Furthermore, p16INK4A-deficient mice are susceptible to several types of malignancies (32), and germ line mutations of p16INK4A in humans are associated with familial syndromes involving malignant melanoma and pancreatic cancer (8, 14, 16, 40).
The precise mechanism by which p16INK4A exerts its tumor-suppressive effects is less clear. One straightforward suggestion is that inactivation of p16INK4A is required for malignant cells to enter S phase efficiently. However, many normal cells express p16INK4A throughout G1 and are able to proliferate, suggesting that other mechanisms of tumor suppression must be operating. An alternative mechanism involves the recently identified link between p16INK4A expression and cellular senescence (1, 10, 28, 32). As fibroblasts or epithelial cells age, p16INK4A levels increase dramatically, and it has been proposed that loss of p16INK4A expression is required for cells to escape senescence during their progression to malignancy.
Another possibility is that p16INK4A plays a role in the maintenance of genome integrity (34). Frequently, following DNA damage normal cells arrest their proliferation at cell cycle checkpoints, the most prominent of which occur at the G1-S and G2-M boundaries. Arrest allows time for repair prior to continued cell cycle progression. One G1 arrest checkpoint is controlled by p53 (5, 18). In response to DNA damage, p53 levels increase by a posttranscriptional mechanism, resulting in the transcriptional activation of p21WAF1, a universal inhibitor of cyclin-dependent kinases, which can mediate G1 arrest (6, 11, 42). Inactivation of p53 is the most common genetic event in human cancer, suggesting that loss of a DNA damage-induced G1 checkpoint is an essential step in tumor progression. This allows damaged DNA to be replicated, which leads to the accumulation of additional mutations and the eventual emergence of a malignant clone.
DNA damage also induces alterations in cyclin D1-cdk4 activity. For example, UV irradiation can lead to decreases in cyclin D1 levels and to inhibitory phosphorylation of cdk4 (24, 27, 37). Such perturbations may contribute to G1 arrest following DNA damage. In the present study, we have investigated whether p16INK4A may also be involved in the response to DNA damage. We have used non-small cell lung cancer (NSCLC) cells, which lack p53 and do not arrest in G1 following DNA damage. When these cells are engineered to express p16INK4A at levels compatible with proliferation, the ability to arrest in G1 in response to DNA damage is restored. Furthermore, we demonstrate that p53−/− fibroblasts maintain a similar G1 arrest checkpoint in response to DNA damage, which correlates with the level of p16INK4A they express. Although neither overall p16INK4A levels nor the amount complexed to cdk4 and cdk6 changes following DNA damage, the presence of p16INK4A causes a decrease in cdk4- and cdk6-mediated Rb kinase activity and results in G1 arrest, even in the absence of p53.
MATERIALS AND METHODS
Cell lines.
Calcium phosphate precipitation (4) was used to transfect Bing packaging cells (provided by Warren Pear, Massachusetts Institute of Technology, Cambridge, Mass.) with pBPSTR1 (25) or pBPSTR1 into which a cDNA encoding full-length p16INK4A had been cloned (31). Viral supernatants were used to infect Calu-1 cells, which were then selected in puromycin (0.5 μg/ml). A mass population of cells infected with the pBPSTR1 virus was isolated, as were several individual clones arising from pBPSTR1-p16INK4A infection. Cells were maintained in tetracycline HCl (2 μg/ml) and deprived of tetracycline for 24 h to induce higher levels of p16INK4A expression. These lines were subsequently transfected with expression vectors encoding cdk4, cdk4 variants, and human papillomavirus (HPV) E7, and mass populations were selected for resistance to G418. Calu-1 cells were obtained from the American Type Culture Collection (Rockville, Md.) and primary embryo fibroblasts from p53−/− mice were obtained from Tyler Jacks (Massachusetts Institute of Technology). Early (passage 5)- and late-passage samples of these cells were provided by David Fisher (Dana-Farber Cancer Institute). Normal human bronchial epithelial cells were purchased from Clonetics Corp. (San Diego, Calif.) and maintained in the growth factor-supplemented medium recommended by the supplier.
Construction of cdk4R24C-HA.
A pBlueScript plasmid containing a cDNA encoding cdk4R24C (41) was obtained from David Beach (Cold Spring Harbor Laboratories, Cold Spring Harbor, N.Y.). An NdeI-BamHI fragment from the 3′ region of a hemagglutinin (HA)-tagged wild-type cdk4 (in pCMV-neo) (7) was used to replace the analogous region of pBlueScript-cdk4R24C, and the resulting HA-tagged cdk4R24C cDNA was cloned into pcDNA3.
Immune precipitations.
Cells were metabolically radiolabeled with [35S]cysteine and [35S]methionine, and lysed in Nonidet P-40 (NP-40)-containing lysis buffer (50 mM Tris HCl [pH 8], 150 mM NaCl, 1.0% NP-40, and 1 mM phenylmethylsulfonyl fluoride). Lysate from a 10-cm-diameter plate was subjected to immune precipitation using an anti-p16INK4A monoclonal antibody (ZJ11) raised against a glutathione S-transferase (GST)–p16INK4A fusion protein (a gift from James DeCaprio, Dana-Farber Cancer Institute) or with rabbit antisera raised against peptides derived from the C-terminal domains of cdk4 (Clontech, Palo Alto, Calif.) or cdk6 (Santa Cruz Biotechnology, Santa Cruz, Calif.). Double immune precipitations for p16INK4A, cdk4, and cdk6 were performed as described previously (33, 38). For immune depletion experiments, lysates were subjected to five rounds of immune precipitation using ZJ11 prior to analysis.
Immune blotting.
Cells were lysed in cold NP-40-containing lysis buffer, and 100 to 150 μg of cellular protein was fractionated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Protein was electrophoretically transferred to Immobilon-P membranes (Millipore, Danvers, Mass.) in 10 mM 3-(cyclohexylamino)-1-propane sulfonic acid (pH 11) and 15% methanol. Nonspecific binding sites were blocked by incubating the membrane in Tris-buffered saline (TBS)–10% nonfat dried milk. Primary antibody incubations were carried out in TBS–1% milk with the following: anti-human p16INK4A, anti-murine p16INK4A, and anti-cdk4 (Santa Cruz Biotechnology), anti-Rb (clone G3-245; PharMingen, San Diego, Calif.), anti-p21WAF1 (Oncogene Research Products, Cambridge, Mass.), anti-HA (BAbCO, Richmond, Calif.), anti-E7 (a gift from James DeCaprio, Dana-Farber Cancer Institute), and anti-cyclin D1 (Neomarkers, Fremont, Calif., or Upstate Biotechnology, Inc., Lake Placid, N.Y.). After being washed membranes were incubated in horseradish peroxidase-conjugated secondary antibodies and developed with enhanced chemiluminescence substrate (Amersham, Arlington Heights, Ill.).
DNA damage treatments.
Prior to treatment, Calu-1 cells were plated at 105 cells/ml and fibroblasts from p53−/− mice were plated at 5 × 104 cells/ml. In both cases, this provided cultures that were 50 to 70% confluent at the time of treatment. Twenty-four hours after plating, Calu-1 cells were treated with the following concentrations of chemotherapeutic agents for 48 h: 0.035-μg/ml adriamycin (ADR), 2.5 μM etoposide, 5 μM camptothecin, and 2.6 μg/ml cisplatin. In some experiments, cells were cultured for an additional 36 h in the same concentration of drug along with nocodazole (0.4 μg/ml). For γ-irradiation of Calu-1 cells, 10 Gy of irradiation was delivered by a 137Cs source at 118 cGy/min. This dose was adequate to induce a G2 arrest in parental Calu-1 cells. Twenty-four hours after plating, fibroblasts from p53−/− mice were treated for 24 h with ADR (0.03 μg/ml), etoposide (2 μM), and cisplatin (1 μg/ml). In some experiments, cells were cultured in the same concentration of drug along with nocodazole (0.4 μg/ml) for an additional 16 h. For γ-irradiation, cells were treated with 20 Gy of irradiation. This dose was required in order to induce a G2 arrest in late-passage p53−/− embryo fibroblasts.
Fluorescence-activated cell sorting (FACS) analysis.
Cells were collected by trypsinization, washed, and resuspended in 1 ml phosphate-buffered saline. An additional 1 ml of 80% ethanol was added, and cells were fixed overnight at 4°C. Fixed cells were centrifuged and resuspended in 0.5 ml of 500-μg/ml RNase A and incubated for 45 min at 37°C. Cells were centrifuged and resuspended in 0.5 ml of 69 μM propidium iodide in 38 mM sodium citrate and incubated at room temperature for a minimum of 30 min. Cells were then analyzed for DNA content by flow cytometry (Becton Dickinson, Hialeah, Fla.).
Rb kinase assays.
Cells were lysed for 1 h at 4°C in 50 mM HEPES (pH 7.2)–150 mM NaCl–1 mM EDTA–2.5 mM EGTA–1 mM dithiothreitol–0.1% Tween 20 supplemented with 10% glycerol–1 mM NaF–0.5 mM sodium orthovanadate–aprotinin (1 μg/ml)–leupeptin (1 μg/ml)–10 mM β-glycerophosphate–phenylmethylsulfonyl fluoride (100 μg/ml). Lysates were clarified by centrifugation at 10,000 × g for 10 min, and the supernatants were precleared with rabbit immunoglobulin G prior to immune precipitation using 300 ng each of rabbit antisera against cdk4 and cdk6 (described above). Immune precipitates were collected by using protein A-Sepharose beads equilibrated with lysis buffer containing 4% bovine serum albumin. Beads were washed four times with lysis buffer and then twice with kinase buffer containing 50 mM HEPES (pH 7.2), 10 mM MgCl2, 5 mM MnCl2, and 1 mM dithiothreitol. After the final wash, 25 μl of kinase reaction mix was added, consisting of kinase buffer with 20 μM ATP, 10 μCi of [γ-32P]ATP, and 1 μg of GST-Rb (amino acids 792 to 928). Samples were incubated at 37°C for 30 min with occasional mixing, boiled in SDS-PAGE sample buffer, and fractionated by electrophoresis through 10% polyacrylamide gels. Proteins were electrophoretically transferred to nitrocellulose, and phosphorylated GST-Rb was visualized by autoradiography. The nitrocellulose filter was then subjected to Western blotting using cdk4- and cdk6-specific antibodies, to ensure that equivalent amounts had been immune precipitated for each kinase assay.
To prepare the GST-Rb substrate, a culture of Escherichia coli transformed with pGEX-Rb (792-928) was induced with IPTG (isopropyl-β-d-thiogalactopyranoside) and lysed as previously described (21). Fusion protein was captured on glutathione-Sepharose 4B and released by incubation with reduced glutathione. The concentration of the soluble fusion protein was estimated by Coomassie blue staining of electrophoresed protein in comparison to protein standards of known concentration.
RESULTS
Engineered expression of p16INK4A in Calu-1 cells.
Calu-1 cells are NSCLC cells which have wild-type Rb, deleted p53, and transcriptionally silent and hypermethylated p16INK4A loci (3, 33). Ectopic expression of p16INK4A at high levels in these and other cell types that retain wild-type Rb produces G1 arrest (17, 19, 22, 30, 33, 35). In the present experiments, we generated Calu-1 cells expressing levels of p16INK4A compatible with proliferation by infection with a tetracycline-suppressible retroviral vector, pBPSTR1 (25), encoding human p16INK4A (31). Figure 1A shows that several independently isolated clones expressed p16INK4A in the presence of tetracycline and that expression could be enhanced three- to fourfold by tetracycline withdrawal. Levels of p16INK4A expression in the absence of tetracycline were comparable to those seen in WI38 diploid human fibroblasts and somewhat higher than those seen in normal human bronchial epithelial (NHBE) cells (Fig. 1A). Levels of p16INK4A expressed by engineered Calu-1 cells in the presence of tetracycline were similar to those in NHBE cells. Although ectopically expressed p16INK4A bound to cdk4 and cdk6 (Fig. 1B), the levels of p16INK4A expressed in these cells did not affect growth rates (Fig. 2). Immune depletion of p16INK4A from extracts of these cells revealed populations of free cdk4 and cdk6 (see Fig. 10), and depletion of these cdks removed all detectable p16INK4A (data not shown). Thus, cdk4 and cdk6 are present in excess over p16INK4A in these cells, resulting in sufficient Rb phosphorylation to allow S-phase entry.
FIG. 1.

Characterization of Calu-1 cells engineered to express p16INK4A. Calu-1 cells were infected with the retrovirus pBPSTR1 or the same retrovirus into which a cDNA encoding human p16INK4A had been cloned. (A) To the left is shown an immunoblot of p16INK4A and Rb in a mass population of Calu-1 cells infected with pBPSTR1 and in three pBPSTR1-p16INK4A clones (clones 6, 14, and 18) in the presence (+) and absence (−) of tetracycline. To the right is shown an immunoblot analysis of p16INK4A expression in three pBPSTR1-p16INK4A clones in the absence of tetracycline compared to extracts from WI38 diploid human fibroblasts and NHBE cells. Blots were stripped and reprobed for proliferating-cell nuclear antigen as a loading control. (B) p16INK4A-expressing clones were radiolabeled with [35S]methionine, and extracts were subjected to immune precipitation with anti-p16INK4A. Immune precipitates were dissociated (33, 38), divided into three aliquots, and precipitated separately with anti-p16INK4A, anti-cdk4, and anti-cdk6. SDS-PAGE and autoradiographic analysis of these second precipitates demonstrate that cdk4 and cdk6 were present in the initial p16INK4A immune precipitates.
FIG. 2.

Growth curves. Calu-1 cells were plated at ∼106 cells/10-cm-diameter dish and counted daily. Solid symbols, presence of tetracycline; open symbols, absence of tetracycline; ▪ and □, mass culture of Calu-1 cells infected with pBPSTR1; • and ○, clone 6; ⧫ and ◊, clone 14; ▴ and ▵, clone 18.
FIG. 10.

Effect of DNA-damaging agents on expression of p16INK4A, p21WAF1, and cdk4 and distribution of p16INK4A. (A) Calu-1 cells infected with pBPSTR1 or clone 6 cells expressing p16INK4A were grown in the presence or absence of tetracycline (+tet or −tet, respectively) and were left untreated (NT) or were treated with ADR (A) or etoposide (E) for 48 h as described in the legend to Fig. 3. Cell lysates were analyzed by immunoblotting for p16INK4A, p21WAF1, and cdk4. (For p16INK4A and cdk4, 100 μg of total cell lysate protein was analyzed, but because of the low abundance of p21WAF1 in these cells, 150 μg of protein was analyzed and exposure times to X-ray film were prolonged.) (B) Late- and early-passage primary fibroblasts from p53−/− mice were untreated or were treated with γ-irradiation (γ), ADR, or etoposide as described in the legend to Fig. 6. Cell lysates were analyzed by immunoblotting for p16INK4A. (C) Calu-1 cells expressing p16INK4A (clone 14) were left untreated or were treated with ADR, and 44 to 48 h later cells were radiolabeled with [35S]methionine. Some cell lysates were depleted of p16INK4A by several rounds of immune precipitation. Control lysates and those depleted of p16INK4A were then analyzed by immune precipitation for p16INK4A, cdk4, and cdk6. The persistence of cdk4 and cdk6 in extracts depleted of p16INK4A indicates that the cdks were present in stoichiometric excess over p16INK4A. Furthermore, the amounts of free cdk4 and cdk6 after ADR treatment were approximately the same as those before treatment (as determined by phosphorescence image analysis), indicating that ADR treatment did not result in an increased proportion of p16INK4A bound to cdk4 and cdk6.
Response of p16INK4A-expressing Calu-1 cells to DNA-damaging agents.
When control Calu-1 cells that do not express p16INK4A were treated with ADR or etoposide for 48 h, nearly all cells arrested in G2 (Fig. 3A). On average, 3% of these cells had a G1 DNA content after ADR treatment, and 10% had a G1 DNA content after etoposide treatment (Fig. 3B). In contrast, cells expressing p16INK4A responded to ADR or etoposide treatment by growth arrest with a significantly larger proportion of cells in G1 (Fig. 3A). After ADR treatment, on average, 12 and 27% of cells had a G1 DNA content in the presence and absence of tetracycline, respectively (Fig. 3B). After etoposide treatment, the G1 DNA proportions were 21 and 32% in the presence and absence of tetracycline, respectively. Other chemotherapeutic agents, such as camptothecin and cisplatin (Fig. 4), as well as γ-irradiation (Fig. 4 and Table 1) also induced G1 arrest in Calu-1 cells only when they expressed p16INK4A.
FIG. 3.

Response of Calu-1 cells to DNA damage induced by ADR or etoposide. (A) DNA content histograms for Calu-1 cells infected with pBPSTR1 and one clone (clone 6) of Calu-1 cells expressing p16INK4A. Cells were grown in the presence or absence of tetracycline (+Tet or −Tet, respectively) and treated with ADR or etoposide for 48 h. Cells were then stained with propidium iodide and analyzed by FACS. (pBPSTR1 cells treated in the presence of tetracycline showed the same response as those treated in the absence of tetracycline.) (B) Calu-1 cells infected with pBPSTR1 (C) and three independently isolated clones of Calu-1 cells expressing p16INK4A (clones 6, 14, and 18) were grown in the presence or absence of tetracycline. Cells were treated with ADR or etoposide for 48 h, stained with propidium iodide, and analyzed by FACS. Differences between G1 proportions after chemotherapy treatment in the presence and absence of tetracycline were significant at P ≤ 0.01 (*) or P ≤ 0.005 (**) by Student’s t test. Error bars indicate standard errors of the means. (Data for ADR treatment of the pBPSTR1 population, clone 6, clone 14, and clone 18 were derived from 2, 5, 3, and 3 independent measurements, respectively. Data for etoposide treatments of the pBPSTR1 population, clone 6, clone 14, and clone 18 were derived from 2, 3, 1, and 1 independent measurements, respectively.)
FIG. 4.

Response of Calu-1 cells to DNA damage induced by γ-irradiation, cisplatin, or camptothecin (CPT11). Calu-1 cells infected with pBPSTR1 arrested in G2 18 h after treatment with 10 Gy of irradiation or after 48 h of treatment with cisplatin or camptothecin. Similar treatment of p16INK4A-expressing clones resulted in a significant proportion of cells arresting in G1 (γ-irradiation, clone 18; cisplatin and camptothecin, clone 6 [similar results were observed using other clones]).
TABLE 1.
G1 arrest in p16INK4A-expressing Calu-1 cells in response to γ-irradiation
| h after irradiation | % G1 DNA contenta
|
|
|---|---|---|
| pBPSTR1 | Clone 6 | |
| 0 | 52.4 | 52.4 |
| 4 | 36.2 | 47.6 |
| 8 | 22.0 | 39.6 |
| 12 | 5.1 | 35.3 |
| 16 | 4.6 | 34.1 |
| 18 | 3.1 | 30.9 |
A mass population of Calu-1 cells infected with a control retrovirus (pBSTR1) or a clone (clone 6) of Calu-1 cells infected with a retrovirus expressing p16INK4A were treated with 10 Gy of irradiation. At the indicated times, cells were collected and DNA content was determined by FACS analysis. Similar results were obtained with two additional p16INK4A-expressing clones. The same proportions of G1 DNA were obtained in the presence and absence of nocodazole at 18 h after treatment.
The response to DNA-damaging agents was a true cell cycle arrest since the proportion of cells in S phase decreased from 30% in untreated cells to 5 to 10% after treatment, and the G1 arrest was stable during continued culture in the presence of nocodazole (Fig. 5A). In addition, the G1 arrest was reversible since arrested cells reentered S phase after cessation of DNA-damaging treatments (Fig. 5B). Thus the G1 arrest had the characteristics of a checkpoint response.
FIG. 5.

Stability of G1 arrest in the presence and absence of nocodazole. (A) Calu-1 cells infected with pBPSTR1 alone show a shift from a normal DNA profile for exponentially growing cells to a G2 arrest after 36 h of treatment with nocodazole. p16INK4A-expressing cells (clone 14) show a G1 arrest response after ADR or etoposide treatment (as in A) which does not change after an additional 36 h of nocodazole treatment. Similar results were observed with the other p16INK4A-expressing clones. (B) Calu-1 cells expressing p16INK4A (clone 14) in the absence of tetracycline were induced to arrest in G1 with ADR or γ-irradiation, resulting in less than 10% of these cells having an S-phase DNA content. FACS analysis performed 18 h after withdrawal of the DNA-damaging treatment shows that cells have reentered the cycle, with more than 20% having an S-phase DNA content.
Response of p53−/− fibroblasts to DNA damaging agents.
To ensure that the results observed with Calu-1 cells were not restricted to an engineered cell line, we performed similar experiments on fibroblasts derived from p53−/− mice. Figure 6A shows that early-passage (passage 5) fibroblasts expressed p16INK4A and that levels of expression decreased with continued passage. Because of the absence of p53 in these cells, the expectation was that a G1 arrest checkpoint in response to DNA damage would be absent. Surprisingly, however, Fig. 6B shows that these cells have a persistent G1 arrest response. The proportion of cells arresting in G1 in response to γ-irradiation, ADR, etoposide, or cisplatin correlated with the amount of p16INK4A expressed, so that DNA damage induced a significant proportion of early- but not late-passage cells to arrest in G1. The G1 arrest in p16INK4A-expressing early-passage fibroblasts was stable during continued culture in the presence of nocodazole (Fig. 6B). Intermediate-passage cells expressed intermediate levels of p16INK4A, and the proportion of cells arresting in G1 in response to DNA-damaging agents correlated with these levels of expression (data not shown). In addition, similar to Calu-1 cells, engineered expression of p16INK4A in late-passage cells at levels compatible with cell proliferation restored a G1 arrest response to DNA damage (Fig. 7).
FIG. 6.

Response of p53−/− fibroblasts to DNA-damaging agents. (A) Immunoblot analysis of p16INK4A expression in late- and early-passage primary embryo fibroblasts from p53−/− mice. The blot was stripped and reprobed for cdk4 and proliferating-cell nuclear antigen to demonstrate equal loading. (B) Early- and late-passage fibroblasts were treated with γ-irradiation, ADR, etoposide, and cisplatin and then analyzed for DNA content by propidium iodide staining and FACS analysis. Cells treated with γ-irradiation were analyzed 12 h after treatment, and cells treated with chemotherapeutic agents were analyzed at the end of 24 h of continuous exposure. Early-passage cells were also cultured for an additional 16 h in the presence of nocodazole to demonstrate the stability of the G1 arrest.
FIG. 7.

Effect of ectopic p16INK4A expression in late-passage p53−/− fibroblasts. (A) Immunoblot analysis of p16INK4A expression in a mass population of late-passage p53−/− fibroblasts infected with pBPSTR1 or a single clone of cells infected with pBPSTR1-p16INK4A. The blot was stripped and reprobed for cdk4 to demonstrate equal loading. (B) The control and p16INK4A-expressing cells shown in panel A were analyzed for DNA content before and after treatment with ADR and etoposide. A significant proportion of cells expressing p16INK4A arrested in G1 in response to these agents.
Effect of cdk4 variants and HPV E7 expression on the G1 arrest response.
To confirm that the G1 arrest in Calu-1 cells depended on p16INK4A, we expressed a cdk4 variant in which lysine-35 is replaced by methionine (20) (Fig. 8A). This variant has no kinase activity but binds p16INK4A efficiently (30), and when overexpressed, it sequesters p16INK4A, thereby preventing its interaction with endogenous cdk4. Figure 9 shows that expression of cdk4K35M reversed the G1 arrest response in p16INK4A-expressing Calu-1 cells. A similar result was obtained by expressing cdk4D185N, another kinase-defective variant that may also sequester p16INK4A (39) (Fig. 8A and 9).
FIG. 8.

Expression of cdk4 variants and HPV E7 in p16INK4A-expressing Calu-1 cells. Calu-1 cells expressing p16INK4A (clone 6) were transfected with the indicated expression vectors and controls, and pooled populations of stable transfectants were isolated. Cell lysates were analyzed for expression of transfected proteins by immunoblotting with anti-cdk4 or anti-HA antibodies. All transfected cDNAs encoded HA-tagged proteins except for cdk4K35M (A), and ectopic expression of this transfected cDNA was inferred by the increased amount of total immunoreactive cdk4 compared to pRc/CMV control transfectants. Also shown is the lack of effect on p16INK4A levels by expression of cdk4, cdk4 variants, or E7. (B) The control for E7 expression was the same expression vector into which a mutant E7 (ΔDLYC) unable to bind Rb was cloned (23). However, expression of E7ΔDLYC could not be documented, and this control should be considered a vector control rather than an inactive E7 control.
FIG. 9.

Effect of ectopically expressed cell cycle control proteins on p16INK4A-mediated G1 arrest. Cells transfected with the expression vectors described in the legend to Fig. 8 were analyzed for DNA content by FACS analysis before and after ADR treatment. The FACS patterns are grouped with their appropriate control vectors.
These results suggest that kinase-defective cdk4 variants prevent p16INK4A from inhibiting endogenous cdk4, thereby permitting Rb phosphorylation and reversal of the G1 arrest response to DNA damage. Consistent with this model, overexpression of wild-type cdk4 overcame the G1 arrest response in p16INK4A-expressing cells (Fig. 8A and 9). This was further confirmed by the ability of another cdk4 variant, cdk4R24C to reverse the G1 arrest response (Fig. 8A and 9). This variant has full kinase activity but does not bind and is therefore not inhibited by p16INK4A (41). Finally, the G1 arrest response was abrogated by expressing the HPV E7 protein, which inactivates pocket proteins (Fig. 8B and 9). Thus, the p16INK4A-mediated G1 arrest in response to ADR is dependent on p16INK4A itself and pocket proteins such as Rb and further depends on inhibition of cdk4.
Effect of DNA-damaging agents on cell cycle protein expression and cdk activity.
G1 arrest following DNA damage involves p53-dependent and -independent events. In cells with wild-type p53, DNA damage results in increased levels of p53 (5, 18), which induce increased expression of the non-INK4 cdk inhibitor, p21WAF1 (6), leading to G1 arrest (6, 11, 42). Figure 10A shows that levels of p21WAF1 did not rise following ADR or etoposide treatment, indicating that the G1 arrest response was not due to p53-independent recruitment of the p21WAF1 pathway. Similarly, there was no change in levels of ectopic p16INK4A in Calu-1 cells (Fig. 10A), in levels of endogenous p16INK4A in mouse embryo fibroblasts (Fig. 10B), or in levels of cdk4 in both cell types (Fig. 10A and 11B) following DNA damage. In addition, DNA damage did not increase the amount of p16INK4A complexed to cdk4 and cdk6 (Fig. 10C).
FIG. 11.
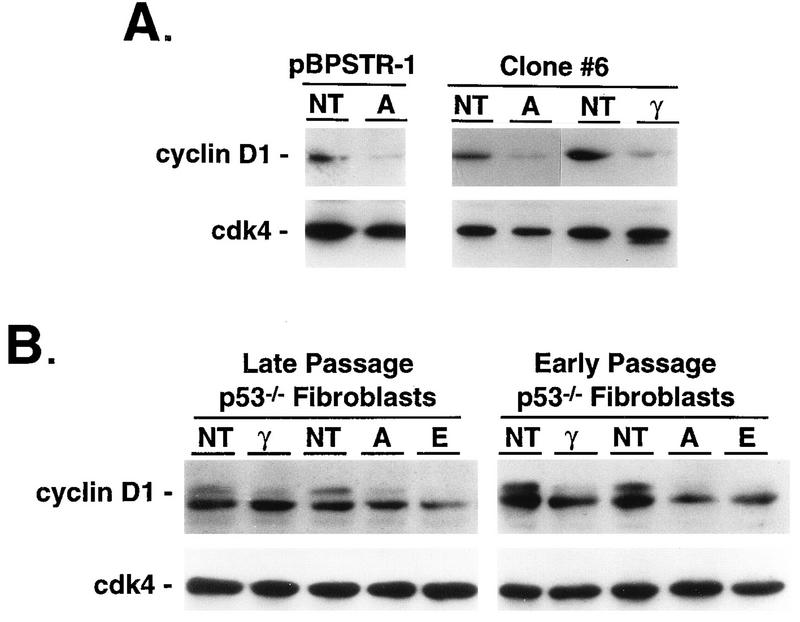
Effect of DNA-damaging agents on expression of cyclin D1. (A) Calu-1 cells infected with pBPSTR1 or with pBPSTR1-p16INK4A (clone 6) were left untreated (NT) or were treated with ADR (A) or γ-irradiation (γ) as described in the legend to Fig. 3, and cell lysates were analyzed by immunoblot for cyclin D1 expression. The blots were stripped and reprobed for cdk4 to demonstrate equal loading. (B) Early- and late-passage fibroblasts from p53−/− mice were untreated or treated with γ-irradiation, ADR, or etoposide (E) as described in the legend to Fig. 6, and cell lysates were examined by immunoblot for cyclin D1 expression. In addition to recognizing the primary cyclin D1 protein, the anti-cyclin D1 antibody used in this analysis also recognizes a less abundant protein of higher apparent molecular weight in murine cells which has been shown by protease mapping to be cyclin D1 as well (2). Similar results were observed with a second anti-cyclin D1 antibody. The blots were stripped and reprobed for cdk4 to demonstrate equal loading.
In contrast, ADR treatment and γ-irradiation routinely produced lower levels of cyclin D1 in engineered Calu-1 cells and in mouse embryo fibroblasts whether or not they expressed p16INK4A (Fig. 11). In the absence of p16INK4A, this decrease was not associated with a measurable reduction in cdk4- or cdk6-mediated Rb kinase activity. However, in the presence of p16INK4A, kinase activity was reduced (Fig. 12).
FIG. 12.
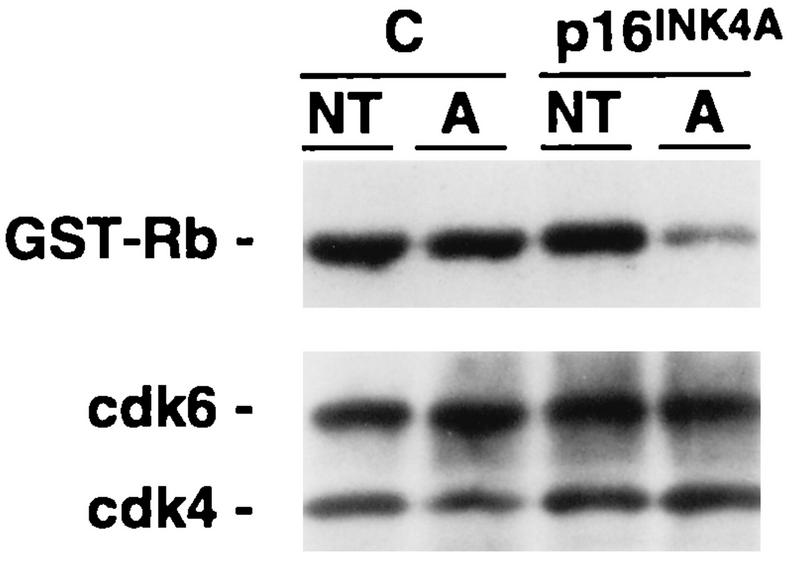
Dependence of reduced Rb kinase activity on p16INK4A expression after DNA damage. Calu-1 cells infected with pBPSTR1 (C) or pBPSTR1-p16INK4A (clone 14) were untreated (NT) or treated with ADR (A) as described in the legend to Fig. 3. Lysates were subjected to immune precipitation using anti-cdk4 and anti-cdk6 antibodies, and the precipitates were used to phosphorylate GST-Rb in vitro. Kinase assays were analyzed by SDS-PAGE followed by electrophoretic transfer to nitrocellulose. Phosphorylated GST-Rb was detected by exposing the nitrocellulose filter to X-ray film. The presence of equal amounts of cdk4 and cdk6 in the precipitates was then determined by staining the nitrocellulose filter with anti-cdk4 and anti-cdk6 antibodies.
DISCUSSION
Our data implicate p16INK4A in a G1 arrest checkpoint in response to DNA damage. We demonstrated this first in Calu-1 NSCLC cells, which express wild-type Rb but do not express p53 or p16INK4A and undergo cell cycle arrest in G2 in response to DNA damage. Although high levels of ectopic p16INK4A expression in these cells produce G1 arrest (33), the levels engineered in the present experiments were compatible with cell proliferation, presumably because cdk4 and cdk6 were in stoichiometric excess compared to p16INK4A, as demonstrated by depletion experiments. While these levels of p16INK4A expression did not inhibit cell proliferation, they did result in G1 arrest in response to a wide variety of DNA-damaging agents that exert their effects in different ways. In addition, the proportion of cells arrested in G1 correlated with the amount of p16INK4A expressed by the cell population.
The G1 arrest response in these cells depended on expression of p16INK4A since it was abrogated by kinase-defective cdk4 variants that sequester p16INK4A. These variants prevent p16INK4A from inhibiting endogenous cdk4, thereby allowing cells to proceed through G1 into S phase. Overexpression of wild-type cdk4 achieved a similar result, again most likely due to p16INK4A sequestration. Furthermore, G1 arrest was reversed by a cdk4 variant incapable of binding p16INK4A and by the HPV E7 protein, indicating that the G1 arrest response was dependent on the ability of p16INK4A to inhibit cdk4 activity in a cell with active pocket proteins, such as Rb.
The G1 arrest response induced by DNA damage was accompanied by decreases in cyclin D1 levels. Although Calu-1 cells engineered to express low levels of p16INK4A proliferated like their parental cells, it is possible that this amount of p16INK4A artificially sensitized them to decreases in cyclin D1 and that p16INK4A is not normally involved in this DNA damage checkpoint. This is unlikely to be the case for several reasons. First, the amount of p16INK4A expressed by engineered Calu-1 cells was similar to that expressed by WI38 diploid fibroblasts and NHBE cells, indicating that the levels of expression approximated physiological levels seen in nontransformed cells in culture.
In addition, our results were not confined to cell lines in which p16INK4A was ectopically expressed but were also observed in primary embryo fibroblasts from p53−/− mice. We found that early-passage cells expressed high levels of p16INK4A and underwent G1 arrest in response to several DNA-damaging agents. This was a surprising result and indicates that DNA damage-induced G1 arrest does not solely depend on the presence of p53 or on events immediately downstream from p53 such as induction of p21WAF1. Late-passage cells expressed much lower levels of p16INK4A and no longer demonstrated a G1 arrest response after DNA damage.
Other reports on studies using human diploid fibroblasts or mouse embryo fibroblasts from wild-type mice have described progressive increases in p16INK4A levels with continued passage (1, 10, 32, 43). The behavior of the p53−/− mouse embryo fibroblasts was different in our experiments, although we did not analyze p16INK4A levels at every passage, so increases in p16INK4A prior to a subsequent decrease may have been missed. Nonetheless, as p16INK4A expression diminished, the proportion of cells arresting in G1 in response to DNA damage progressively decreased. Admittedly, a variety of other genetic (or epigenetic) alterations which might have influenced the DNA damage-induced G1 arrest response could have occurred in these cells during passage. However, there was a clear correlation between the level of endogenous p16INK4A in these p53−/− fibroblasts and their degree of G1 arrest in response to DNA-damaging agents.
The precise mechanism by which p16INK4A functions in a G1 arrest checkpoint remains to be elucidated. We have demonstrated that DNA damage does not induce an increase in p16INK4A levels. Instead, we and others have observed significant decreases in levels of cyclin D1 expression following DNA damage (24, 27). While this response also occurred in transformed cells that do not express p16INK4A, it did not result in a diminution of total cdk4 and cdk6 kinase activities as determined by an in vitro kinase assay. In contrast, when transformed cells were engineered to express p16INK4A, DNA damage-mediated decreases in cyclin D1 levels were associated with a significant decrease in cdk4- and cdk6-mediated Rb kinase activity. Thus, although DNA damage does not produce a change in the levels of p16INK4A in cells that express it, its presence is required for a decrease in Rb kinase activity and resultant G1 arrest following DNA damage.
One simple explanation of these findings would be that lower levels of cyclin D1 after DNA damage permitted increased amounts of p16INK4A to bind to cdk4 and cdk6. However, our data indicate that this is not the case. The depletion experiments (Fig. 10) demonstrated that cdks are in excess and that there are roughly equivalent amounts of p16INK4A-free cdk4 and cdk6 both before and after exposure to DNA damage. Although we cannot exclude the possibility that subtle quantitative changes in the amount of p16INK4A complexed to target kinases could result in a biological effect, it is clear that dramatic accumulation of p16INK4A in cdk4 or cdk6 complexes does not occur following DNA damage.
Rather, it is likely that p16INK4A plays a passive but essential role in the context of other perturbations that occur in the cyclin D-cdk4-Rb axis after DNA damage. For example, tyrosine phosphorylation of cdk4 is required in order for cells to arrest in G1 after UV light treatment (37). Therefore, it is possible that a proportion of cdk4 molecules in the cells we examined becomes tyrosine phosphorylated in response to DNA damage. In the absence of p16INK4A, there must be sufficient amounts of active cyclin D-cdk4 complexes to allow phosphorylation of Rb and bypass of the checkpoint. However, in the presence of p16INK4A, the available cdk4 following DNA damage that is not inhibited by either tyrosine phosphorylation or p16INK4A association may become limiting, leaving insufficient active cyclin D-cdk4 to allow cell cycle progression. In this scenario, steady-state levels of p16INK4A or the amount of p16INK4A complexed to cdk4 need not change following DNA damage, but the presence of p16INK4A is nonetheless essential for G1 arrest to occur.
Another p53-independent contribution to G1 arrest following DNA damage may come from the redistribution of p27Kip1. Unlike INK4 family members, which appear to displace D-type cyclins from cdks, p27Kip1 can exist in complex with the cyclin-cdk holoenzyme (26). In growing cells, most p27Kip1 is associated with cyclin D1-cdk4 (27). UV irradiation has been reported to reduce the levels of both cyclin D1 and p27Kip1, but since the reduction in cyclin D1 is greater, p27Kip1 redistributes to cyclin A-cdk2, causing a reduction in cdk2 kinase activity and G1 arrest (27). p27Kip1 is readily detectable in the cells we examined, and its levels do not change after DNA damage (data not shown). We have not yet investigated the association of p27Kip1 with cyclin-cdk complexes in these cells, but it is tempting to speculate that a more significant redistribution of p27Kip1 to cdk2 complexes might occur in the presence of p16INK4A after DNA damage. If so, this mechanism would again be consistent with a necessary but passive role for p16INK4A in the G1 arrest response.
The ability of p16INK4A to participate in G1 arrest following DNA damage depends on its expression during G1. In this respect, p16INK4A differs from other closely related members of the INK4 family, all of which are potent inhibitors of cdk4 and cdk6. For example, in many cell types, p15INK4B expression depends on transforming growth factor β treatment (9, 29), while the expression of p18INK4C and p19INK4D is cell cycle regulated and restricted to S phase (13). p16INK4A’s expression during G1 in a wider variety of normal cell types than other INK4 family members (34, 36) may explain why it is a more frequent target for inactivation. One would predict that any INK4 family member that is expressed during G1 in a given cell type might be a tumor suppressor in that cell. Consistent with this idea, p15INK4B is expressed during G1 in T lymphocytes and is a tumor suppressor in T-cell leukemias (12, 36).
During tumor progression, loss of the G1 arrest checkpoint in response to DNA damage is essential for the eventual emergence of a malignant clone. Our data indicate that unless p16INK4A activity is lost, the cell types we examined can still arrest in G1 after DNA damage, even in the absence of p53. This may explain why most tumors with wild-type Rb inactivate both p16INK4A and p53 and why the frequency of p16INK4A inactivation in human cancers rivals that of p53. However, while inactivation of p16INK4A in tumors with wild-type Rb is nearly universal, some malignant cells do express wild-type p53. Our data predict that these cells may not have a fully competent G1 arrest checkpoint in response to DNA damage, despite the presence of p53, and that p16INK4A expression may augment this response. We are currently testing this hypothesis in tumor cells that express wild-type p53 and in fibroblasts from p16−/− mice.
ACKNOWLEDGMENTS
We thank David Livingston for helpful comments and suggestions and Steven Reeves for the gift of pBPSTR1. Other plasmids were provided by William Sellers, William Kaelin, David Beach, Charles Sherr, and Ed Harlow. We also thank Michael Reed and Mohamed Ladha for preparation of GST-Rb; Sonya Penfold for maintenance of NHBE cells; Darlene Koestner, Michael Simone, and Maris Handley of the Dana-Farber Cancer Institute Flow Cytometry Facility for technical help, and Laurie Geronimo for administrative assistance.
This work was supported by NIH grant CA72573 to B.J.R. G.I.S. is supported by Aid for Cancer Research. B.J.R. is a Scholar of the Leukemia Society of America. This work was also supported in part by the Novartis/Dana-Farber Drug Discovery Program.
REFERENCES
- 1.Alcorta D A, Xiong Y, Phelps D, Hannon G, Beach D, Barrett J C. Involvement of the cyclin-dependent kinase inhibitor p16 (INK4a) in replicative senescence of normal human fibroblasts. Proc Natl Acad Sci USA. 1996;93:13742–13747. doi: 10.1073/pnas.93.24.13742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bates S, Bonetta L, MacAllan D, Parry D, Holder A, Dickson C, Peters G. CDK6 (PLSTIRE) and CDK4 (PSK-J3) are a distinct subset of the cyclin-dependent kinases that associate with cyclin D1. Oncogene. 1994;9:71–79. [PubMed] [Google Scholar]
- 3.Caamano J, Ruggeri B, Momiki S, Sickler A, Zhang S Y, Klein-Szanto A J. Detection of p53 in primary lung tumors and nonsmall cell lung carcinoma cell lines. Am J Pathol. 1991;139:839–845. [PMC free article] [PubMed] [Google Scholar]
- 4.Chen C, Okayama H. High-efficiency transformation of mammalian cells by plasmid DNA. Mol Cell Biol. 1987;7:2745–2752. doi: 10.1128/mcb.7.8.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Clarke A R, Purdie C A, Harrison D J, Morris R G, Bird C C, Hooper M L, Wyllie A H. Thymocyte apoptosis induced by p53-dependent and independent pathways. Nature. 1993;362:849–852. doi: 10.1038/362849a0. [DOI] [PubMed] [Google Scholar]
- 6.El-Deiry W S, Tokino T, Velculescu V E, Levy D B, Parsons R, Trent J M, Lin D, Mercer W E, Kinzler K W, Vogelstein B. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993;75:817–825. doi: 10.1016/0092-8674(93)90500-p. [DOI] [PubMed] [Google Scholar]
- 7.Ewen M E, Oliver C J, Sluss H K, Miller S J, Peeper D S. p53-dependent repression of CDK4 translation in TGF-β-induced G1 cell-cycle arrest. Genes Dev. 1995;9:204–217. doi: 10.1101/gad.9.2.204. [DOI] [PubMed] [Google Scholar]
- 8.Goldstein A M, Fraser M C, Struewing J P, Hussussian C J, Ranade K, Zametkin D P, Fontaine L S, Organic S M, Dracopoli N C, Clark W H J, Tucker M A. Increased risk of pancreatic cancer in melanoma-prone kindreds with p16INK4 mutations. N Engl J Med. 1995;333:970–974. doi: 10.1056/NEJM199510123331504. [DOI] [PubMed] [Google Scholar]
- 9.Hannon G J, Beach D. p15INK4B is a potential effector of TGF-β-induced cell cycle arrest. Nature. 1994;371:257–261. doi: 10.1038/371257a0. [DOI] [PubMed] [Google Scholar]
- 10.Hara E, Smith R, Parry D, Tahara H, Stone S, Peters G. Regulation of p16CDKN2 expression and its implications for cell immortalization and senescence. Mol Cell Biol. 1996;16:859–867. doi: 10.1128/mcb.16.3.859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Harper J W, Adami G R, Wei N, Keyomarsi K, Elledge S J. The p21 cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell. 1993;75:805–816. doi: 10.1016/0092-8674(93)90499-g. [DOI] [PubMed] [Google Scholar]
- 12.Herman J G, Jen J, Merlo A, Baylin S B. Hypermethylation-associated inactivation indicates a tumor suppressor role for p15INK4B1. Cancer Res. 1996;56:722–727. [PubMed] [Google Scholar]
- 13.Hirai H, Roussel M F, Kato J-Y, Ashmun R A, Sherr C J. Novel INK4 proteins, p19 and p18, are specific inhibitors of cyclin D-dependent kinases CDK4 and CDK6. Mol Cell Biol. 1995;15:2672–2681. doi: 10.1128/mcb.15.5.2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hussussian C J, Struewing J P, Goldstein A M, Higgins P A, Ally D S, Sheahan M D, Clark W J, Tucker M A, Dracopoli N C. Germline p16 mutations in familial melanoma. Nat Genet. 1994;8:15–21. doi: 10.1038/ng0994-15. [DOI] [PubMed] [Google Scholar]
- 15.Kamb A, Gruis N A, Weaver-Feldhaus J, Liu Q, Harshman K, Tavtigian S V, Stocjert E, Day R S I, Johnson B E, Skolnick M H. A cell cycle regulator potentially involved in genesis of many tumor types. Science. 1994;264:436–440. doi: 10.1126/science.8153634. [DOI] [PubMed] [Google Scholar]
- 16.Kamb A, Shattuck-Eidens D, Eeles R, Liu Q, Gruis N A, Ding W, Hussey C, Tran T, Miki Y, Weaver-Feldhaus J, McClure M, Aitken J F, Anderson D E, Bergman W, Frants R, Goldgar D E, Green A, MacLennan R, Martin N G, Meyer L J, Youl P, Zone J J, Skolnick M H, Cannon-Albright L A. Analysis of the p16 gene (CDKN2) as a candidate for the chromosome 9p melanoma susceptibility locus. Nat Genet. 1994;8:22–26. doi: 10.1038/ng0994-22. [DOI] [PubMed] [Google Scholar]
- 17.Koh J, Enders G H, Dynlacht B D, Harlow E. Tumor-derived p16 alleles encoding proteins defective in cell-cycle inhibition. Nature. 1995;375:506–510. doi: 10.1038/375506a0. [DOI] [PubMed] [Google Scholar]
- 18.Lowe S W, Ruley H E, Jacks T, Housman D E. p53-dependent apoptosis modulates the cytotoxicity of anticancer agents. Cell. 1993;74:957–967. doi: 10.1016/0092-8674(93)90719-7. [DOI] [PubMed] [Google Scholar]
- 19.Lukas J, Parry D, Aagaard L, Mann D J, Bartkova J, Strauss M, Peters G, Bartek J. Retinoblastoma-protein-dependent cell cycle inhibition by the tumor suppressor p16. Nature. 1995;375:503–506. doi: 10.1038/375503a0. [DOI] [PubMed] [Google Scholar]
- 20.Matsushime H, Ewen M E, Strom D K, Kato J Y, Hanks S K, Roussel M F, Sherr C J. Identification and properties of an atypical catalytic subunit (p34PSK-J3/cdk4) for mammalian D type G1 cyclins. Cell. 1992;71:323–334. doi: 10.1016/0092-8674(92)90360-o. [DOI] [PubMed] [Google Scholar]
- 21.Matsushime H, Quelle D E, Shurtleff S A, Shibuya M, Sherr C J, Kato J-Y. D-type cyclin-dependent kinase activity in mammalian cells. Mol Cell Biol. 1994;14:2066–2076. doi: 10.1128/mcb.14.3.2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Medema R H, Herrera R E, Lam F, Weinberg R A. Growth suppression by p16ink4 requires functional retinoblastoma protein. Proc Natl Acad Sci USA. 1995;92:6289–6293. doi: 10.1073/pnas.92.14.6289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Munger K, Werness B A, Dyson N, Phelps W C, Harlow E, Howley P M. Complex formation of human papillomavirus E7 proteins with the retinoblastoma tumor suppressor gene product. EMBO J. 1989;8:4099–4105. doi: 10.1002/j.1460-2075.1989.tb08594.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pagano M, Theodoras A M, Tam S W, Draetta G F. Cyclin D1-mediated inhibition of repair and replicative DNA synthesis in human fibroblasts. Genes Dev. 1994;8:1627–1639. doi: 10.1101/gad.8.14.1627. [DOI] [PubMed] [Google Scholar]
- 25.Paulus W, Baur I, Boyce F M, Breakefield X O, Reeves S A. Self-contained, tetracycline-regulated retroviral vector system for gene delivery to mammalian cells. J Virol. 1996;70:62–67. doi: 10.1128/jvi.70.1.62-67.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Polyak K, Kato J, Solomon M J, Sherr C J, Massague J, Roberts J M, Koff A. p27Kip1, a cyclin-Cdk inhibitor, links transforming growth factor-β and contact inhibition to cell cycle arrest. Genes Dev. 1994;8:9–22. doi: 10.1101/gad.8.1.9. [DOI] [PubMed] [Google Scholar]
- 27.Poon R Y, Toyoshima H, Hunter T. Redistribution of the CDK inhibitor p27 between different cyclin-CDK complexes in the mouse fibroblast cell cycle and in cells arrested with lovastatin or ultraviolet irradiation. Mol Biol Cell. 1995;6:1197–1213. doi: 10.1091/mbc.6.9.1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reznikoff C A, Yeager T R, Belair C D, Savelieva E, Puthenveettil J A, Stadler W M. Elevated p16 at senescence and loss of p16 at immortalization in human papillomavirus 16 E6, but not E7, transformed human uroepithelial cells. Cancer Res. 1996;56:2886–2890. [PubMed] [Google Scholar]
- 29.Sandhu C, Garbe J, Bhattacharya N, Daksis J, Pan C-H, Yaswen P, Koh J, Slingerland J M, Stampfer M R. Transforming growth factor β stabilizes p15INK4B protein, increases p15INK4B-cdk4 complexes, and inhibits cyclin D1-cdk4 association in human mammary epithelial cells. Mol Cell Biol. 1997;17:2458–2467. doi: 10.1128/mcb.17.5.2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Serrano M, Gomez-Lahoz E, DePinho R A, Beach D, Bar-Sagi D. Inhibition of ras-induced proliferation and cellular transformation by p16INK4. Science. 1995;267:249–252. doi: 10.1126/science.7809631. [DOI] [PubMed] [Google Scholar]
- 31.Serrano M, Hannon G J, Beach D. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature. 1993;366:704–707. doi: 10.1038/366704a0. [DOI] [PubMed] [Google Scholar]
- 32.Serrano M, Lee H W, Chin L, Cordon-Cardo C, Beach D, DePinho R A. Role of the INK4a locus in tumor suppression and cell mortality. Cell. 1996;85:27–37. doi: 10.1016/s0092-8674(00)81079-x. [DOI] [PubMed] [Google Scholar]
- 33.Shapiro G I, Park J E, Edwards C D, Mao L, Merlo A, Sidransky D, Ewen M E, Rollins B J. Multiple mechanisms of p16INK4A inactivation in non-small cell lung cancer cell lines. Cancer Res. 1995;55:6200–6209. [PubMed] [Google Scholar]
- 34.Sherr C J. Cancer cell cycles. Science. 1996;274:1672–1677. doi: 10.1126/science.274.5293.1672. [DOI] [PubMed] [Google Scholar]
- 35.Stone S, Dayananth P, Kamb A. Reversible, p16-mediated cell cycle arrest as protection from chemotherapy. Cancer Res. 1996;56:3199–3202. [PubMed] [Google Scholar]
- 36.Tam S W, Shay J W, Pagano M. Differential expression and cell cycle regulation of the cyclin-dependent kinase 4 inhibitor p16Ink4. Cancer Res. 1994;54:5816–5820. [PubMed] [Google Scholar]
- 37.Terada Y, Tatsuka M, Jinno S, Okayama H. Requirement for tyrosine phosphorylation of Cdk4 in G1 arrest induced by ultraviolet irradiation. Nature. 1995;376:358–362. doi: 10.1038/376358a0. [DOI] [PubMed] [Google Scholar]
- 38.Vairo G, Livingston D M, Ginsberg D. Functional interaction between E2F-4 and p130: evidence for distinct mechanisms underlying growth suppression by different retinoblastoma protein family members. Genes Dev. 1995;9:869–881. doi: 10.1101/gad.9.7.869. [DOI] [PubMed] [Google Scholar]
- 39.van den Heuvel S, Harlow E. Distinct roles for cyclin-dependent kinases in cell cycle control. Science. 1993;262:2050–2054. doi: 10.1126/science.8266103. [DOI] [PubMed] [Google Scholar]
- 40.Whelan A J, Bartsch D, Goodfellow P J. Brief report: a familial syndrome of pancreatic cancer and melanoma with a mutation in the CDKN2 tumor-suppressor gene. N Engl J Med. 1995;333:975–977. doi: 10.1056/NEJM199510123331505. [DOI] [PubMed] [Google Scholar]
- 41.Wölfel T, Hauer M, Schneider J, Serrano M, Wölfel C, Klehmann-Hieb E, De Plaen E, Hankeln T, Meyer zum Büschenfelde K H, Beach D. A p16INK4a-insensitive CDK4 mutant targeted by cytolytic T lymphocytes in a human melanoma. Science. 1995;269:1281–1284. doi: 10.1126/science.7652577. [DOI] [PubMed] [Google Scholar]
- 42.Xiong Y, Hannon G J, Zhang H, Casso D, Kobayashi R, Beach D. p21 is a universal inhibitor of cyclin kinases. Nature. 1993;366:701–704. doi: 10.1038/366701a0. [DOI] [PubMed] [Google Scholar]
- 43.Zindy F, Quelle D E, Roussel M F, Sherr C J. Expression of the p16INK4a tumor suppressor versus other INK4 family members during mouse development and aging. Oncogene. 1997;15:203–211. doi: 10.1038/sj.onc.1201178. [DOI] [PubMed] [Google Scholar]