

Abstract
To investigate the mechanisms of peroxisome assembly and the molecular basis of peroxisome assembly disorders, we isolated and characterized a peroxisome-deficient CHO cell mutant, ZP139, which was found to belong to human complementation group II, the same group as that of our earlier mutant, ZP105. These mutants had a phenotypic deficiency in the import of peroxisomal targeting signal type 1 (PTS1) proteins. Amino-terminal extension signal (PTS2)-mediated transport, including that of 3-ketoacyl coenzyme A thiolase, was also defective in ZP105 but not in ZP139. PEX5 cDNA, encoding the PTS1 receptor (PTS1R), was isolated from wild-type CHO-K1 cells. PTS1R’s deduced primary sequence comprised 595 amino acids, 7 amino acids less than the human homolog, and contained seven tetratricopeptide repeat (TPR) motifs at the C-terminal region. Chinese hamster PTS1R showed 94, 28, and 24% amino acid identity with PTS1Rs from humans, Pichia pastoris, and Saccharomyces cerevisiae, respectively. A PTS1R isoform (PTS1RL) with 632 amino acid residues was identified in CHO cells; for PTS1R, 37 amino acids were inserted between residues at positions 215 and 216 of a shorter isoform (PTS1RS). Southern blot analysis of CHO cell genomic DNA suggested that these two isoforms are derived from a single gene. Both types of PEX5 complemented impaired import of PTS1 in mutants ZP105 and ZP139. PTS2 import in ZP105 was rescued only by PTS1RL. This finding strongly suggests that PTS1RL is also involved in the transport of PTS2. Mutations in PEX5 were determined by reverse transcription-PCR: a G-to-A transition resulted in one amino acid substitution: Gly298Glu of PTS1RS (G335E of PTS1RL) in ZP105 and Gly485Glu of PTS1RS (G522E of PTS1RL) in ZP139. Both mutations were in the TPR domains (TPR1 and TPR6), suggesting the functional consequence of these domains in protein translocation. The implications of these mutations are discussed.
Compartmentalization of cellular processes into different subcellular compartments is one of the major characteristics of eukaryotic cells. Most proteins of subcellular organelles are synthesized on cytoplasmic polyribosomes and transported to their destined compartments (26). The peroxisome, a single membrane-bounded organelle, contains over 50 different enzymes catalyzing various metabolic pathways, including β-oxidation of very long chain fatty acids and the synthesis of ether-lipids, bile acids, and cholesterol (40). Significant progress has been made in understanding the biogenesis of peroxisomes (7, 8, 12, 30). It is widely accepted that new peroxisomes are formed by growth and division of preexisting peroxisomes (12). Both peroxisomal membrane and matrix proteins are imported posttranslationally into peroxisomes. At least two distinct topogenic signals directing proteins to the peroxisomal matrix have been identified (8, 30). Peroxisomal targeting signal (PTS) type 1 (PTS1) is a C-terminal uncleaved tripeptide (serine-lysine-leucine [SKL] or variants) (11, 14, 16), while type 2 (PTS2) is an N-terminal cleavable peptide comprising 11 to 36 amino acids (23, 31, 35). Both sequences function independently as necessary and sufficient PTSs and are used by evolutionarily diverse organisms, from yeasts to humans.
To investigate the molecular mechanisms involved in peroxisome biogenesis and the genetics-related causes of human peroxisome deficiency disorders, such as Zellweger syndrome and neonatal adrenoleukodystrophy (NALD), we have, to date, isolated seven complementation groups (CGs) of peroxisome-deficient CHO cell mutants by colony autoradiographic screening (47) and the 9-(1′-pyrene)nonanol (P9OH)-UV selection method (17); these mutants include Z24 (39), Z65 (39), ZP92 (27), ZP102 (34), ZP105 (21), ZP104 (21), ZP109 (21), ZP110 (32), and ZP114 (32). All of these mutants resemble fibroblasts from patients with peroxisome deficiency disease with regard to defects in the biogenesis and functions of peroxisomes. The CHO cell mutants Z24, Z65, ZP92, ZP102 or ZP105, and ZP104 or ZP109 are classified into 5 of the 10 human CGs currently described (21, 27, 34); others, such as ZP110 and ZP114, represent 2 CGs distinct from the 10 known human CGs (32). It is plausible that mammalian peroxisome biogenesis requires at least 13 genes or their products, of which only 5, PEX2 (formerly PAF-1) (36), PEX5, coding for the PTS1 receptor (PTS1R) (3, 43), PEX6 (formerly PAF-2) (37, 45), PEX7 (1, 19, 24), and PEX12 (2, 22), are known. We cloned PEX2, PEX6, and PEX12 cDNAs by a genetic phenotype complementation assay of CHO cell mutants Z65, ZP92, and ZP109, respectively (22, 22a, 36, 37). PEX2, PEX5, PEX6, PEX7, and PEX12 are responsible for the genetic events in patients with peroxisome biogenesis disorders (1–3, 9, 19, 22, 24, 28, 43, 45). Thus, peroxisome biogenesis-defective CHO cell mutants are a useful mammalian somatic cell system for the investigation of peroxisome assembly at molecular and cellular levels as well as for the elucidation of the genetic causes of peroxisome biogenesis disorders (8).
We report here the isolation and characterization of CHO cell mutants of CG II with the phenotype of a defect in PTS1 import. We also describe the distinct function of PTS1R in the transport of peroxisomal proteins, including PTS2.
MATERIALS AND METHODS
Isolation of cDNA clones encoding CHO cell PTS1R.
Cloning of human PTS1R cDNA by PCR was described previously (34). About 3 × 105 independent colonies of a cDNA library from wild-type CHO-K1 cells (a generous gift from O. Kuge) were screened with, as a probe, a 32P-labeled 1.8-kb PCR fragment of the human PTS1R cDNA open reading frame (34). One of three positive clones was subcloned into pBluescript II SK(−) (Stratagene, La Jolla, Calif.) at SalI and NotI sites. The nucleotide sequence was determined on both strands by the dideoxy chain termination method with a dye terminator cycle sequencing kit and a 370A DNA sequencer (Applied Biosystems, Foster City, Calif.).
Construction of PTS1R expression plasmids.
A SalI-NotI fragment, a full-length cDNA encoding a short isoform of Chinese hamster PTS1R (termed PTS1RS), was isolated from pBluescript II SK(−) containing PTS1RS cDNA. The NotI-SalI fragment was filled in with the Klenow fragment (Takara, Kyoto, Japan). The fragment was subcloned into the SmaI site of mammalian expression plasmid pUcD2SRαMCSHyg (22a) under the control of the SRα promoter. The resulting plasmid, pUcD2Hyg-PTS1RS, was used in transfection experiments. The DNA fragment with a 37-amino-acid insertion was prepared by amplification from a CHO cell cDNA library with a sense oligonucleotide primer, 264s (residues 264 to 280 of Chinese hamster PTS1RS) (nucleotides are numbered from the first nucleotide of the initiator methionine codon of the PTS1RS open reading frame, unless otherwise indicated) (Table 1), and an antisense primer, 1026r (residues 1010 to 1026). The PCR fragment was subcloned into pUcD2Hyg-PTS1RS with BsmI and AxyI sites. The resulting plasmid was termed pUcD2Hyg-PTS1RL.
TABLE 1.
Synthetic oligonucleotide primers used in this study
| Codea | Sequence | Residuesb |
|---|---|---|
| 61s | 5′-GGTGGTCACCATGGCAA-3′ | 61–77 |
| 264s | 5′-AATTCCTGCAGGACCAG-3′ | 264–280 |
| 724s | 5′-AGAGGCCTGGGTCGATC-3′ | 724–740 |
| 1181s | 5′-AACGAGTCCCTGCAGCG-3′ | 1181–1197 |
| 1926r | 5′-GGAGAGGGAGTCACATC-3′ | 1910–1926 |
| 1236r | 5′-GAGTAGCGCAGCCAGTC-3′ | 1220–1236 |
| 1026r | 5′-TGCACTGCAGCCTCAAA-3′ | 1010–1026 |
s and r, sense and antisense primers, respectively.
Residue positions are from PTS1RS.
Cell lines, culture conditions, and DNA transfection.
Peroxisome-deficient CHO cell mutants were isolated by the P9OH-UV method (17) with TKa cells (wild-type CHO-K1 cells that had been stably transfected with rat PEX2 cDNA) (34). ZP139, one of several CHO cell mutants with the typical phenotype of peroxisome deficiency, i.e., localization of catalase in the cytosol, was found by cell fusion analysis to belong to the same CG as ZP105 (21) (human CG II). CHO cells were cultured in Ham’s F-12 medium supplemented with 10% fetal calf serum under 5% CO2–95% air (39). DNA transfection of cells was done with cationic liposomes [O,O′-ditetradecanolyl-N-(trimethylammonioacetyl)diethanolamine chloride; a gift from A. Ito] prepared by sonication and separately diluted with 300 μl of HEPES-buffered saline containing CaCl2 and MgCl2 (21). Transfection was also done with Lipofectamine (Gibco BRL, Gaithersburg, Md.) as recommended by the manufacturer.
Assays.
Latency of catalase and resistance to P9OH-UV and 12-(1′-pyrene)dodecanoic acid (P12)-UV treatments were determined as described previously (27).
Morphological analysis.
Peroxisomes in CHO cells were visualized by indirect immunofluorescence light microscopy with the monospecific rabbit antibodies described below. Antigen-antibody complexes were detected with fluorescein isothiocyanate-labeled goat anti-rabbit immunoglobulin G antibody (Zymed, South San Francisco, Calif.) under an Axioskop FL microscope (Carl Zeiss, Oberkochen, Germany). Cells were prepared with a fixative containing 4% paraformaldehyde as described previously (27). Differential permeabilization of cells was done by treatments with 25 μg of digitonin per ml and Triton X-100 as described previously (18).
Antibodies.
Anti-PTS1 antibody was raised in rabbits by immunization with a synthetic peptide comprising the C-terminal amino acid sequence (KHLKPLQSKL) (14) of rat acyl coenzyme A (CoA) oxidase (AOx) with N-terminal cysteine, which was linked to keyhole limpet (20). Antibodies to catalase, AOx, 3-ketoacyl-CoA thiolase, and peroxisomal 70-kDa integral membrane protein (PMP70) were used (39).
Cell fusion and labeling of cell protein.
Parent CHO cells and cells to be fused were cocultured for 1 day and then fused with polyethylene glycol as described previously (39). Selection of fused cells was carried out with 1 μM ouabain or 200 U of hygromycin B (Wako, Osaka, Japan) per ml (21, 39). Fusion of CHO cell mutant variants resistant to 6-thioguanine (Tgr) with human fibroblasts was done as described previously (27). Metabolic labeling of cells with 20 μCi of [35S]methionine and [35S]cysteine (New England Nuclear Corp., Boston, Mass.) per ml for 24 h in F-12 medium and immunoprecipitation of peroxisomal proteins from cell lysates were done as described previously (39).
Northern blot analysis.
RNA blots of poly(A)+ RNA from wild-type CHO-K1 cells, ZP105, and ZP139 were hybridized with a 32P-labeled 3.0-kb SalI-NotI fragment of Chinese hamster PTS1RS cDNA under conditions of high stringency. Probe labeling was done with [α-32P]dCTP by use of a Megaprime DNA labeling system (Amersham, Arlington Heights, Ill.). The membrane was also hybridized with a 32P-labeled 1.3-kb cDNA fragment of human glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as a control for load and integrity of the RNA. Washing of the membrane was done twice at room temperature and three times at 65°C with 2× SSPE (1× SSPE is 0.15 M NaCl, 10 mM sodium phosphate, and 1 mM EDTA [pH 7.4])–0.5% sodium dodecyl sulfate (SDS).
Southern blot analysis.
Genomic DNA was prepared from CHO-K1 cells and digested with several restriction enzymes as described previously (38). The digests were separated on an 0.8% agarose gel buffered with 45 mM Tris-borate–1.25 mM EDTA and transferred to a nylon membrane (Biodyne, Port Washington, N.Y.). Prehybridization was done at 37°C for 40 min in prehybridization buffer (5× SSPE, 0.5% SDS, 5× Denhardt’s solution, 50 μg of salmon sperm DNA per ml). Hybridization was done at 37°C for 16 h with an α-32P-labeled SalI-NotI fragment of cDNA encoding Chinese hamster PTS1RS. Washing of the membrane was done sequentially with 2× SSPE–0.5% SDS, once at room temperature and twice at 50°C.
Mutation analysis.
Cloning of PTS1R cDNA from CHO cell mutants ZP139 and ZP105 was performed with reverse transcription (RT)-PCR. Briefly, RT-PCR was done with Superscript RT (Gibco BRL) and 5 μg of total RNA each from mutants ZP139 and ZP105. First-strand cDNA was synthesized with hexaoligonucleotides and amplified with four different sets of primers. For ZP139, first-strand cDNA was amplified with sense primer 61s and antisense primer 1926r by use of Ex Taq polymerase (Takara) in a buffer recommended by the manufacturer. For ZP105, first-strand cDNA was amplified with independent primer sets: sense primer 61s and antisense primer 1026r, sense primer 724s and antisense primer 1236r, and sense primer 1181s and antisense primer 1926r. Amplified DNA fragments were subcloned into the pT7Blue T-vector (Novagen, Madison, Wis.). The DNA sequence was determined on both strands by the dideoxy chain termination method described above.
RESULTS
Isolation and characterization of CHO cell mutants defective in PTS1 import. (i) Anti-PTS1 antibody.
We raised antiserum against PTS1 by immunizing rabbits with PTS1 containing a 10-amino-acid oligopeptide comprising the C-terminal sequence of rat AOx oxidase. The antibody specifically recognized a dozen peroxisomal proteins, including a trifunctional protein (enoyl-CoA isomerase–enoyl-CoA hydratase–3-hydroxyacyl-CoA dehydrogenase) and AOx with the C-terminal tripeptide residues SKL (Fig. 1). AOx, the first enzyme of the peroxisomal β-oxidation system, is a heterodimer comprising 75-kDa A, 53-kDa B, and 22-kDa C polypeptide components (15, 39). B and C are derived from A by proteolytic cleavage within peroxisomes (15, 16). It is noteworthy that the antibody reacted only with the 75-kDa A and 22-kDa C components of AOx, each containing SKL at the C terminus, but not with the 53-kDa B form. It is also apparent that the most intense staining of the AOx A and C components among the peroxisomal proteins, as well as the purified enzyme, may reflect the abundance of antibodies that recognize epitopes comprising a tripeptide and up to 10 amino acid residues of AOx.
FIG. 1.
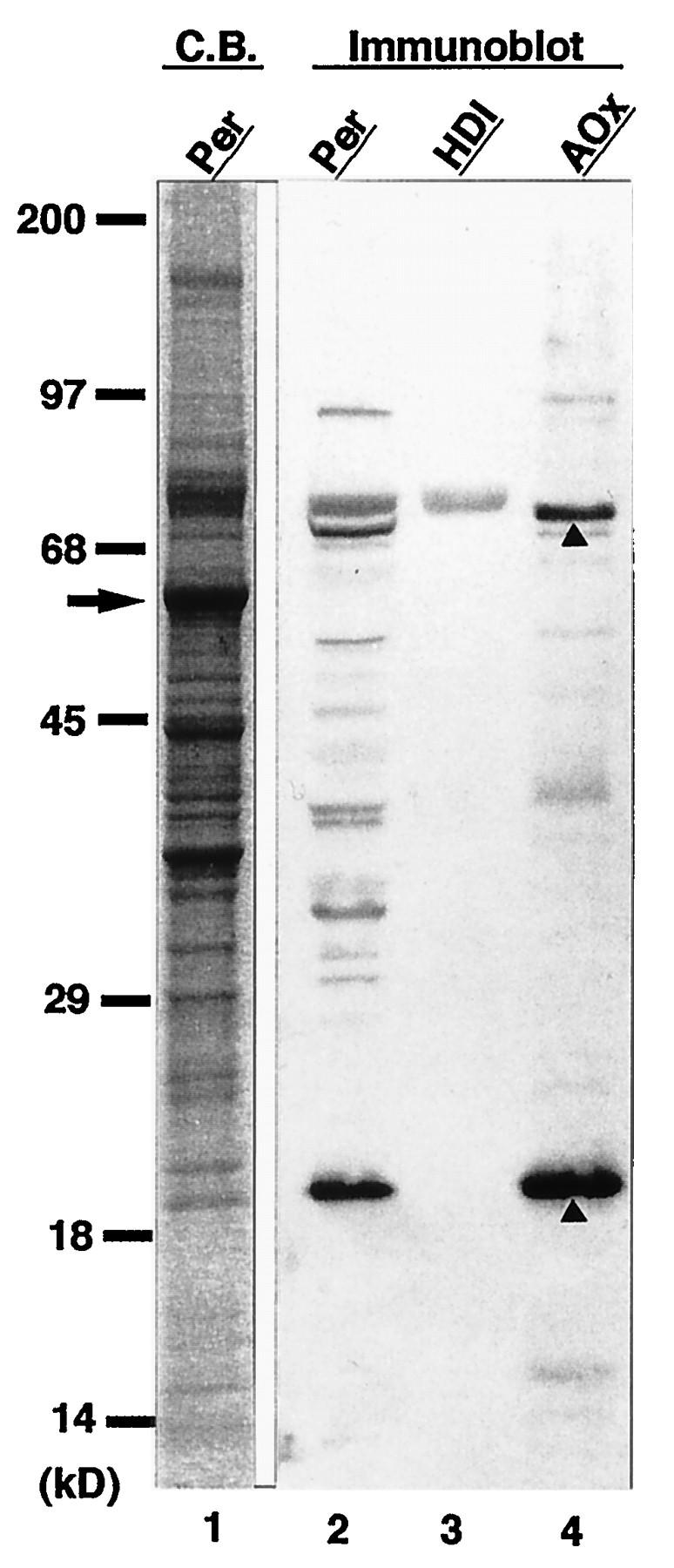
Western blot analysis with rabbit anti-PTS1 peptide antibody. Lanes: 1, Coomassie blue (C.B.)-stained rat liver peroxisomes (20 μg); 2 to 4, immunoblots with anti-PTS1 peptide antibody of peroxisomes (20 μg), rat trifunctional protein (HDI; 1 μg), and rat AOx (1 μg), respectively. Molecular mass markers are on the left. The arrow indicates catalase; arrowheads denote 75-kDa A and 22-kDa C components of AOx.
(ii) Mutant isolation, complementation group analysis, and morphological analysis.
Peroxisome-deficient CHO cell mutants were isolated from PEX2-transformed CHO-K1 cells (TKa cells) by the P9OH-UV selection method. Viable cell colonies were examined for peroxisome morphology with an antibody to catalase, a peroxisomal matrix enzyme (Fig. 2). Mutant colonies showing cytosolic localization of catalase, a phenotype of the peroxisome biogenesis defect, were transfected with human PTS1RS cDNA, which complements abnormalities of fibroblasts from CG II patients. Five mutant cell clones, ZP127, ZP138, ZP139, ZP141, and ZP142, showed peroxisomes as numerous as those present in wild-type CHO-K1 cells (Fig. 2a), suggesting that these mutants belonged to CG II, as concluded by catalase immunocytochemistry (ZP139 is shown in Fig. 3b). ZP105, which had been classified in CG II by cell fusion with ZP102 (21), likewise showed a restoration of peroxisome assembly by human PTS1RS cDNA (Fig. 3a). Thus, it is apparent that ZP105 and the mutants isolated in this study, ZP127, ZP138, ZP139, ZP141, and ZP142, are in the same CG, i.e., CG II. PEX2 and PEX6 cDNAs, complementing genes of group X (the same group as F in Japan) and IV (the same group as C), respectively, did not restore peroxisome biogenesis in these mutants (data not shown). ZP105 and ZP139 showed cytosolic staining with anti-PTS1 peptide antiserum, indicating that the mutants are deficient in PTS1 import (Fig. 2e and f). Punctate staining was noted in wild-type CHO-K1 cells, as was seen with anticatalase antibody (Fig. 2d).
FIG. 2.

Indirect immunofluorescence staining of CHO cell mutants defective in PTS1R. (a to c) CHO-K1, ZP105, and ZP139 stained with anti-rat liver catalase antibody. (d to f) CHO-K1, ZP105, and ZP139 stained with anti-PTS1 peptide antibody. (g to i) CHO-K1, ZP105, and ZP139 stained with anti–rat 3-ketoacyl-CoA thiolase antibody. (j to l) CHO-K1, ZP105, and ZP139 stained with anti-rat PMP70 antibody. Bar, 20 μm.
FIG. 3.

Complementation analysis of CHO cell mutants. (a and b) Mutants ZP105 and ZP139 were transfected with human cDNA encoding PTS1RS 2 days before staining. (c and d) ZP139 fused with ZP105 and Z24, respectively. (e and f) Hybrid cells of wild-type CHO-K1 fused with ZP105 and ZP139, respectively. (g to i) CG II patient fibroblasts fused with ZP105, ZP139, and Z65, respectively. Cells were stained with antibodies to rat catalase (a to f) and human catalase (g to i). Bar, 25 μm.
Six mutants, ZP105, ZP127, ZP138, ZP139, ZP141, and ZP142, as well as wild-type CHO-K1 cells, were stained with rabbit antiserum to rat liver 3-ketoacyl-CoA thiolase, which contains PTS2. CHO-K1 cells showed a punctate staining pattern, presumably peroxisomes (Fig. 2g). Similar particulates, but fewer in number, were detected in mutants ZP138, ZP139 (Fig. 2i), and ZP141, whereas soluble thiolase in the cytoplasm was seen in mutants ZP105 (Fig. 2h), ZP127, and ZP142. Accordingly, the mutants appear to be divided into two distinct groups with respect to PTS2 import, although both are in the same CG. ZP105 and ZP139 were used for further analyses.
Mutants ZP105 and ZP139 were stained with antiserum against PMP70 (Fig. 2k and l). Larger but fewer particles were detected, consistent with earlier findings for other CHO cell mutants (21, 27, 32). Such a particulate appearance is reminiscent of “peroxisomal ghost” vesicles in fibroblasts from Zellweger patients (25, 42, 44). A punctate staining pattern was found in wild-type cells, as was seen with anticatalase antibody (Fig. 2j).
Peroxisomes were not complemented in ZP139 by cell fusion with ZP105, thereby confirming that these mutants are in the same CG, as concluded from the cDNA transfection study (Fig. 3c). By fusion with mutant Z24 of CG I, ZP139 was complemented in peroxisome biogenesis (Fig. 3d), like ZP105 (21), suggesting that ZP105 and ZP139 are distinct from CG I. Fusion of ZP105 and ZP139 with the wild type resulted in normal peroxisomal staining of catalase, indicating that the mutation in both mutants was recessive (Fig. 3e and f).
Peroxisomes were not complemented in cells of fibroblasts obtained from a CG II Zellweger patient and fused with ZP105 and ZP139, whereas numerous punctate catalase-containing structures (peroxisomes) were noted in hybrid cells with PEX2-defective Z65 (36, 38) (Fig. 3g to i). Thus, ZP105 and ZP139 belong to human CG II, consistent with the results from the transfection of human PTS1RS cDNA (Fig. 3a and b).
(iii) Latency of catalase.
The intracellular location of catalase in mutants ZP105 and ZP139 was also examined by assaying catalase latency. At 100 μg of digitonin per ml, full activity of catalase was detected in the mutants, whereas ∼60% of the activity was latent in wild-type CHO-K1 cells (Fig. 4). Catalase of CHO-K1 cells was fully released only when the concentration of digitonin was increased to 300 μg/ml. These results were interpreted to mean that catalase is localized in the cytosol of mutants ZP105 and ZP139, consistent with the morphological results shown in Fig. 2. This finding is also in good agreement with our earlier observations for other mutants (21, 27, 32, 34, 39).
FIG. 4.

Latency of catalase in wild-type and mutant cells. Latency of catalase was determined as described previously (39). Symbols: ○, wild-type CHO-K1; •, ZP105; ▴, ZP139. The results are representative of the average of duplicate assays.
(iv) Properties of CHO cell mutants.
After cell culturing in the presence of P9OH followed by a short exposure to UV, over 90% of mutant ZP105 and ZP139 cells survived, but hardly any of the wild-type cells were viable (Table 2). Both types of mutant cells were highly sensitive to P12-UV treatment, but 80% of CHO-K1 cells were resistant. These cell phenotype properties were also noted in other, previously isolated CHO cell mutants (17, 21, 27, 32, 34, 39, 46).
TABLE 2.
Characteristics of PTS1R-defective CHO cell mutants
| CHO cells | Peroxisomesa | Transport of:
|
Catalase latency (%)b | Survivalc (%) after treatment with:
|
||
|---|---|---|---|---|---|---|
| PTS1 | PTS2 | P9OH-UV | P12-UV | |||
| Wild type | + | + | + | 61 | 0 | 80 |
| ZP105 | − | − | − | 0 | 93d | <0.001d |
| ZP139 | − | − | + | 0 | 94 | <0.01 |
Peroxisomes were assessed by immunostaining with anticatalase antibody. +, presence; −, absence.
Catalase latency represents peroxisomal catalase calculated as described previously (39).
Survival of P9OH-UV- and P12-UV-resistant cells was expressed as a percentage of survival of the unselected control (27).
From Okumoto et al. (21).
Biogenesis of peroxisomal enzymes was investigated in mutants ZP105 and ZP139 labeled with [35S]methionine and [35S]cysteine. 35S-polypeptide components of AOx, 75-kDa A and 53-kDa B, were evident in wild-type CHO-K1 cells, whereas 35S-labeled A polypeptide but not converted form B polypeptide was detected at a reduced level in both ZP105 and ZP139 because of the rapid degradation of AOx component A (Fig. 5A, lanes 1 to 3). A 22-kDa AOx C component was not discerned due to a concomitantly migrating nonspecific protein (Fig. 5A). The third enzyme of the peroxisomal β-oxidation system, 3-ketoacyl-CoA thiolase (which contains PTS2), is synthesized as a larger precursor of 44 kDa and then processed to a 41-kDa mature form (39). The 41-kDa mature 35S-labeled thiolase was apparent in wild-type cells, indicating normal biogenesis of thiolase (Fig. 5A, lane 4). In contrast, only a 44-kDa 35S-labeled precursor of thiolase was detectable in ZP105 and ZP139 (Fig. 5A, lanes 5 and 6), consistent with earlier findings for other CHO cell mutants (21, 27, 32, 39). Thus, peroxisomal proteins are apparently synthesized at a normal level in mutants ZP105 and ZP139, even though precursors such as those for thiolase are not processed.
FIG. 5.

Biogenesis of peroxisomal proteins. (A) Cells were labeled for 24 h with [35S]methionine and [35S]cysteine. Cell types are indicated at the top. Immunoprecipitation was done with rabbit antibodies to rat AOx and 3-ketoacyl-CoA thiolase (Thiolase). Immunoprecipitates were analyzed by SDS–12% polyacrylamide gel electrophoresis. Radioactive polypeptide bands were detected with a FujiX BAS1000 Bio-Imaging analyzer (Fuji Photo Film, Tokyo, Japan). Exposure was for 20 h. Arrows show the positions of AOx components; open and closed arrowheads indicate a larger precursor and a mature version of thiolase, respectively. A faint band migrating nearly at the same position as AOx component B was nonspecific. The AOx C component was indistinguishable from a nonspecific polypeptide (dot). (B) CHO-K1, ZP105, and ZP139 were treated with 25 μg of digitonin per ml under conditions in which the plasma membrane was permeabilized (18) (a to c, respectively) or with 1% Triton X-100 (d to f, respectively). Cells were stained with antibody to 3-ketoacyl-CoA thiolase. Note that thiolase was detected in ZP139 only after Triton X-100 treatment. Bar, 20 μm.
To examine if the thiolase precursor is within the peroxisomal membranes in ZP139, cells were stained with antithiolase antibody after differential permeabilization (Fig. 5B). With 25 μg of digitonin per ml, thiolase was undetectable, while the antibody recognized thiolase with 1% Triton X-100 (Fig. 5B, c and f), as in wild-type CHO-K1 cells (a and d). In contrast, ZP105 showed apparently cytosolic staining of thiolase after either type of treatment (Fig. 5B, b and e). This finding was interpreted to mean that the thiolase precursor is inside the peroxisomal membranes in ZP139. A putative processing protease(s) may be dysfunctional or may not be imported into peroxisomes in this mutant.
Isolation of cDNA for Chinese hamster PTS1R.
We screened a CHO-K1 cell cDNA library with human full-length PTS1R cDNA as a probe and obtained three positive colonies. Plasmids were isolated from each colony, and their sizes were compared. The nucleotide sequence of one of the longer plasmids was determined; the clone contained a 3,032-bp cDNA apparently encoding PTS1RS, which comprised 595 amino acid residues (Fig. 6A). Chinese hamster PTS1RS was shorter by 7 amino acids than human PTS1RS. PCR amplification of a CHO-K1 cell cDNA library with a set of sense and antisense primers, 61s and 1026r or 264s and 1026r, yielded DNA of two different sizes, suggesting that both of the PTS1R isoforms were expressed in CHO cells (data not shown). The larger product, from 264s and 1026r, was sequenced and proved to be identical to PTS1RS cDNA, except for a 111-bp additional sequence which encoded a peptide of 37 amino acids and which was inserted after G at position 720, i.e., between amino acid residues Glu-215 and Gly-216 (Fig. 6A). A restriction fragment from Sse8387I-AxyI digests of the larger PCR product was replaced into Sse8387I-AxyI sites of the Chinese hamster PTS1RS cDNA clone to construct a longer, 3,143-bp form of PTS1R (PTS1RL) cDNA (Fig. 6A). Both PTS1RS and PTS1RL, like Pex5p from Pichia pastoris (13), Saccharomyces cerevisiae (41), and humans (3, 6, 43), contain seven 34-amino-acid tetratricopeptide repeats (TPRs) in their C-terminal halves. Chinese hamster PTS1RS showed 94, 28, and 24% amino acid identity with human PTS1RS, P. pastoris Pex5p, and S. cerevisiae Pex5p, respectively (Fig. 6A). Between CHO cell and human sequences of the 37-amino-acid insert in PTS1RL, 36 amino acids were identical, with a single and acceptable amino acid substitution, suggesting a highly conserved exon in mammals (Fig. 6B).
FIG. 6.

Amino acid sequence of Chinese hamster PTS1RS. (A) The primary sequence deduced from the Chinese hamster (Cricetulus longicaudatus, Cl) PTS1RS cDNA was compared with those deduced from PEX5 from humans (Homo sapiens, Hs), P. pastoris (Pp, formerly PAS8), and S. cerevisiae (Sc, formerly PAS10). Amino acids identical in two or more species, including Chinese hamsters and humans, are shaded. Dashes indicate spaces. TPRs are overlined. The arrow shows the position of the 37-amino-acid insert in PTS1RL. Closed and open arrowheads indicate PTS1R mutation sites in CHO cell mutants ZP105 and ZP139, respectively (see Fig. 10). The DDBJ database accession numbers for Chinese hamster PTS1RS and PTS1RL (PEX5) are AB002564 and AB002565, respectively. (B) Amino acid sequence of the insert comprising 37 residues of Chinese hamster PTS1RL and human PTS1RL. Identical amino acids are shaded.
Fibroblasts obtained from a Zellweger syndrome CG II patient and manifesting a deficiency in the import of both PTS1 and PTS2 (data not shown) were complemented for peroxisomes, as assessed by catalase staining, by transfection of Chinese hamster PTS1RS or PTS1RL cDNA. This result is consistent with the high sequence homology of PTS1R in mammals (Fig. 7).
FIG. 7.

Complementation of CG II fibroblasts by Chinese hamster PTS1R. CG II patient fibroblasts (a) were transfected with cDNA encoding Chinese hamster PTS1RS (b) and PTS1RL (c), respectively. Cells were stained with anti-human catalase antibody. Bar, 25 μm.
Southern blot analysis of genomic DNA from CHO-K1 cells gave a single band in digests with BamHI, EcoRI, XbaI, and XhoI and a major band as well as a minor one in digests with HindIII. This result suggests that PTS1RS and PTS1RL are likely to be derived from a single gene and to be formed by alternative splicing (Fig. 8).
FIG. 8.

Southern blot analysis of CHO cell genomic DNA. Genomic DNA (10 μg) from CHO-K1 cells was digested with the indicated restriction enzymes, separated, transferred to a nylon membrane, and probed with a 32P-labeled PCR product comprising the nucleotide sequence for amino acid residues 63 to 315 of Chinese hamster PTS1RS. DNA size markers are shown on the left. Exposure was for 12 h.
Dysfunction of PTS1R in CHO cell mutants. (i) PTS1R in mutants.
We carried out Northern blot analysis of RNA from mutants ZP105 and ZP139 (Fig. 9). An RNA band of ∼3.0 kb, similar in size to human PTS1R mRNA (3, 43), was detected in both mutants, as it was in the wild type, suggesting that transcription of the PTS1R gene in the mutants was normal. The amounts of PTS1R mRNA were nearly the same, as estimated by taking into account the RNA load from each cell type. PTS1R mRNA from ZP139 appeared to be slightly larger in size than those from ZP105 and wild-type CHO-K1 cells, although CHO-K1 cells showed smeared, apparently double RNA bands (Fig. 9). Blotting of the same RNAs with a control, a human GAPDH cDNA probe, showed a band of apparently the same size but with a difference in intensity, indicative of proper migration in gel electrophoresis and amounts of RNA loads from the three different cell types (Fig. 9, lower panel).
FIG. 9.
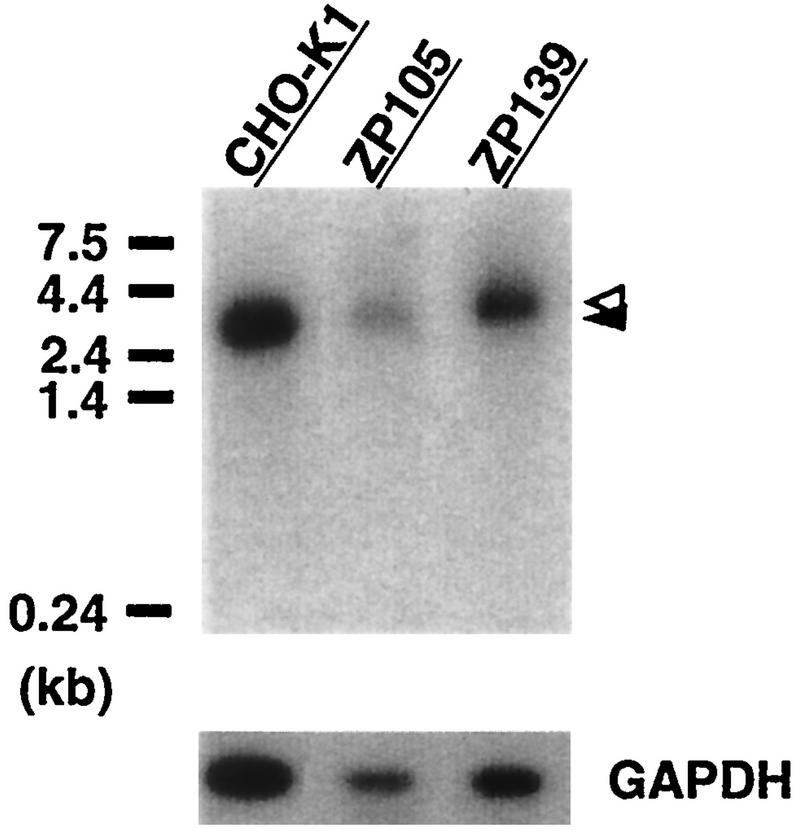
Northern blot analysis of PTS1R. RNA was separated, transferred to a nylon membrane, and hybridized with 32P-labeled cDNA probes for Chinese hamster PTS1RS and human GAPDH. Poly(A)+ RNAs from wild-type CHO-K1 cells (0.6 μg) and mutants ZP105 (0.15 μg) and ZP139 (0.3 μg) were loaded. RNA standards are shown on the left. Open and closed arrowheads indicate a band of PTS1R mRNA from ZP139 and those from CHO-K1 and ZP105, respectively. Exposures were for 12 h (PTS1R) and 1 h (GAPDH).
To investigate the dysfunction of PTS1R in ZP105 and ZP139, we determined the nucleotide sequence of PTS1RS cDNA isolated by RT-PCR from ZP105 and ZP139. In all six cDNA clones isolated from ZP105, nucleotide G at position 893 of a codon for Gly-298 was mutated to A, resulting in a missense mutation, a codon for Glu (Fig. 10). In ZP139, nucleotide G at position 1454 of a codon for Gly-485 was likewise mutated to A, creating a missense codon for Glu. It is noteworthy that mutations in ZP105 and ZP139 were found in TPR1 and TPR6, respectively, implying the significance of the TPR motif in protein translocation.
FIG. 10.

Analysis of PTS1R mutation sites in CHO cell mutants. The nucleotide sequences at residues 886 to 897 of PTS1RS from wild-type and ZP105 cells (upper panel) and residues 1450 to 1461 from wild-type and ZP139 cells (lower panel) are shown.
The apparent difference in the levels of PTS1RS and PTS1RL mRNAs between ZP105 and ZP139 could be due to the instability of PTS1RL mRNA caused by one missense mutation and that of PTS1RS mRNA caused by the other missense mutation. Alternatively, transcription of the PTS1R gene might be altered by, e.g., alternative splicing.
(ii) Complementation of protein transport by PTS1R cDNA.
When ZP105 was transfected with cDNA encoding PTS1RS or PTS1RL from wild-type CHO-K1 cells, catalase and PTS1 were found by punctate staining, indicating complementation of peroxisomal protein import (Fig. 11A, a to d). Thiolase-positive particles were detected after transfection of cDNA for PTS1RL but not of that for PTS1RS (Fig. 11A, e and f). Catalase and PTS1 in ZP139 were observed in particulates, as in ZP105, after the introduction of cDNA coding for either PTS1RS or PTS1RL, whereas thiolase-containing particles in both types of transfectants were like those in untransfected ZP139 (Fig. 11B). Transfection of a mock vector did not alter the intracellular location of these proteins in both mutants (data not shown). Taken together, these results indicate that PTS1R is involved in the import of not only PTS1 but also PTS2. Dysfunction of PTS1R, such as that caused by a point mutation, is most likely to be the primary defect in the mutants.
FIG. 11.

Transfection of PTS1R cDNA to CHO cell mutants. (A) CHO cell mutant ZP105 was transfected with Chinese hamster PTS1R cDNA. Transfection was done with a plasmid expressing PTS1RS (a, c, and e) or PTS1RL (b, d, and f). Cells were stained with rabbit antibodies to rat catalase (a and b), PTS1 peptide (c and d), and rat 3-ketoacyl-CoA thiolase (e and f). Bar, 20 μm. (B) Transfection of mutant ZP139 and staining were done as described in panel A. Bar, 20 μm.
To assess the impaired function of PTS1R in mutants ZP105 and ZP139, ZP105- and ZP139-derived cDNAs encoding PTS1RL and having mutations G335E and G522E, respectively, were transfected back to the mutant cells. Catalase was present in the cytosol in a diffuse pattern in ZP105 transfected with ZP105- or ZP139-derived cDNA, implying the dysfunction of both mutated forms of PTS1RL (Fig. 12A, a and c). Likewise, ZP139 showed cytosolic staining of catalase in mutant PTS1RL transfectants (Fig. 12A, b and d). Nearly the same results were obtained when these transfected cells were stained with anti-PTS1 antibody (data not shown; see below). Transfection of ZP105- and ZP139-derived mutant PTS1RS cDNA gave results similar to those obtained with PTS1RL (data not shown). In contrast, import of PTS2 but not PTS1 was apparently restored in ZP105 after transfection of ZP139-derived PTS1RL cDNA, implying that ZP139-type PTS1RL is functional in mediating PTS2 transport (Fig. 12B). This result is consistent with the phenotype of ZP139 showing peroxisomal import of PTS2.
FIG. 12.

Transfection of mutant PTS1R cDNA to ZP105 and ZP139. (A) Mutants ZP105 and ZP139 were transfected with mutant PTS1RL cDNAs derived from ZP105 (a and b) and ZP139 (c and d). Cells were stained with anticatalase antibody. Bar, 20 μm. (B) ZP105 cells transfected with ZP139-derived PTS1RL cDNA were stained with antisera to PTS1 peptide (a) and thiolase (a PTS2 peptide) (b), respectively. Note that only thiolase import was restored. Bar, 20 μm.
We conclude from these results that site mutations G298E and G485E in PTS1RS (G335E and G522E in PTS1RL) are the primary defects in impaired peroxisome biogenesis in the CHO cell mutants ZP105 and ZP139, respectively.
DISCUSSION
The investigation of molecular mechanisms involved in peroxisome biogenesis has been one of the major foci of research on peroxisomes and is directly linked to elucidation of the primary defects of peroxisome biogenesis disorders, including Zellweger syndrome and NALD. Human cDNA encoding PTS1R was independently isolated from PEX5 by an expressed sequence-tagged homology search with P. pastoris PAS8 (3), immunoscreening of a cDNA expression library (43), and a yeast two-hybrid system (6). Human PEX5 was shown to be responsible for primary defects in two patients with CG II disorders (3, 43; this study). Human PTS1RS restored PTS1 import in fibroblasts from patients with Zellweger syndrome and NALD of CG II.
To search for a clue as to the mechanism of PTS1 import in vivo, in the present work we isolated peroxisome assembly-defective CHO cell mutants of CG II using TKa cells, wild-type CHO-K1 cells transformed with rat PEX2 (34). After several cycles of the mutant isolation procedure, i.e., mutagenesis, P9OH-UV treatment, and transfection of human PTS1RS cDNA followed by indirect immunofluorescence microscopy, five mutant cell clones were isolated. In the present work, no PEX2-defective Z65-type mutants were isolated, as in our recent studies (21, 32, 34), confirming the efficacy of TKa cells in isolating non-PEX2-deficient mutants. All five mutants were determined to belong to CG II by cell fusion with ZP105, which was previously shown to be in CG II, likewise by cell fusion with ZP102 (21). Cell fusion of ZP105 and ZP139 with fibroblasts from CG II patients confirmed that these mutants are indeed in CG II. Mutants ZP127, ZP142, and ZP105 showed typical and common properties characterized for the earlier mutants Z24, Z65, and ZP92, including a recessive lesion(s), absence of morphologically recognizable peroxisomes, no latency of catalase, and high sensitivity to P12-UV treatment, despite normal synthesis of peroxisomal proteins. The mutants also contained the peroxisomal ghost-like vesicular structures present in all previously examined CHO cell mutants (21, 27, 32) and fibroblasts from patients with peroxisome deficiency diseases (25, 42, 44). The physiological significance of peroxisomal ghosts is unknown. These mutants showed the phenotype of a defect in both PTS1 and PTS2 import, whereas another group of mutants, ZP138, ZP139, ZP140, and ZP141, were defective in the transport of PTS1 but not PTS2. Accordingly, the mutants isolated in the present work represent typical somatic mammalian cell mutants of CG II with apparently two distinct phenotypes. Fibroblasts with such distinct phenotypes from CG II patients were recently identified (18, 29, 43).
Chinese hamster PTS1RS comprises 595 amino acids and is 7 residues shorter than human PTS1RS, where 1 amino acid insertion and the deletion of 8 residues occur at positions 195 and 271 through 278, respectively (Fig. 6A), suggesting that these residues are not essential for the function of PTS1R. This inference is consistent with findings of complementation of peroxisome biogenesis in ZP105 and ZP139 by human PTS1RS cDNA as well as in CG II fibroblasts by Chinese hamster cDNAs for PTS1RS and PTS1RL (Fig. 3 and 7). Homology of over 90% was noted between Chinese hamster PTS1R and human PTS1R, including the TPR domain in the C-terminal part, which is more conserved than the N-terminal region (97% versus 91%). PTS1R from yeasts and mammals contains seven TPRs, suggesting the importance of these regions for the function of the PTS1R, such as interactions with other protein molecules. TPR1 to TPR3 of seven TPR domains appear to be required for binding to PTS1 (33).
We delineated the mutation sites of PTS1R in ZP105 and ZP139: Gly298Glu in TPR1 of PTS1RS in ZP105 and Gly485Glu in TPR6 in ZP139. Similar permutations noted in two CG II patients were the nonsense transition Arg390Ter in TPR3, apparently the genetic cause in a Zellweger syndrome patient, and the missense transversion Asn489Lys in TPR6, resulting in a defect of PTS1 import but not of PTS2 import in an NALD patient (3). Loss of PTS1R function in protein import by a point mutation in the TPR implies the functional consequence of TPRs, such as the recognition of PTS1 and PTS2. PTS1R with a mutation in TPR1 or TPR6 was incompetent in restoring peroxisome biogenesis, indicative of dysfunction of both mutant PTS1Rs.
These findings obtained with two phenotypically distinct CHO cell mutants can be reconciled by a working model of PTS1R mediating the translocation of PTS1 and PTS2 polypeptides (Fig. 13). PTS1 peptides are transported to peroxisomes by either PTS1RS or PTS1RL in the cytosol and are imported into peroxisomes by machinery comprising components including the PTS1R-docking protein encoded by PEX13 (4, 5, 10). A mutation in the TPR domain abolishes PTS1 import. Given the fact that PTS2 import in ZP105 was restored by wild-type PTS1RL but not PTS1RS, PTS1RL plays an important role in PTS2 import. Moreover, PTS2 import in ZP105 was complemented by transfection of ZP139-derived PTS1RL cDNA, confirming that ZP139-type PTS1RL is functional in mediating PTS2 transport. One possible model for PTS2 transport by PTS1R is as follows. Polypeptides with PTS2, such as 3-ketoacyl-CoA thiolase, can be translocated by PTS1RL in which the insert of 37 amino acids is evidently required. PTS1RL may recognize PTS2 through PTS2R (Pex7p) (1, 19, 24), possibly by interacting with a Trp-Asp (WD) motif domain in Pex7p. PTS1RL may not directly bind PTS2 because fibroblasts obtained from patients with rhizomelic chondrodysplasia punctata and manifesting only the defect in PTS2 import were complemented only by PEX7 (1, 19, 24). We cannot exclude the possibility that PTS2 may be imported with the aid of a heteromeric dimer or oligomer comprising PTS1RS and PTS1RL.
FIG. 13.

Hypothetical model of protein transport by PTS1R. PTS1 is translocated by either PTS1RS or PTS1RL. PTS2, such as 3-ketoacyl-CoA thiolase, is transported by PTS1RL, presumably in concert with Pex7p (PTS2R) (for details, see the text).
ACKNOWLEDGMENTS
We thank O. Kuge for the CHO-K1 cDNA library and T. Harano, S. Tamura, K. Mizuno, and M. Ohara for helpful comments. We also thank T. Sakaguchi and N. Matsumoto for technical assistance.
This work was supported in part by grants-in-aid for scientific research (07408016, 08249232, and 08557011 to Y.F.) from the Ministry of Education, Science, Sports and Culture, by a CREST grant (to Y.F.) from the Japan Science and Technology Corporation, and by grants (to Y.F.) from the Mitsubishi Foundation, Terumo Life Science Foundation, Naito Foundation, Shorai Foundation for Science and Technology, and Ciba-Geigy Foundation (Japan) for the Promotion of Science.
REFERENCES
- 1.Braverman N, Steel G, Obie C, Moser A, Moser H, Gould S J, Valle D. Human PEX7 encodes the peroxisomal PTS2 receptor and is responsible for rhizomelic chondrodysplasia punctata. Nat Genet. 1997;15:369–376. doi: 10.1038/ng0497-369. [DOI] [PubMed] [Google Scholar]
- 2.Chang C-C, Lee W-H, Moser H, Valle D, Gould S J. Isolation of the human PEX12 gene, mutated in group 3 of the peroxisome biogenesis disorders. Nat Genet. 1997;15:385–388. doi: 10.1038/ng0497-385. [DOI] [PubMed] [Google Scholar]
- 3.Dodt G, Braverman N, Wong C S, Moser A, Moser H W, Watkins P, Valle D, Gould S J. Mutations in the PTS1 receptor gene, PXR1, define complementation group 2 of the peroxisome biogenesis disorders. Nat Genet. 1995;9:115–125. doi: 10.1038/ng0295-115. [DOI] [PubMed] [Google Scholar]
- 4.Elgersma Y, Kwast L, Klein A, Voorn-Brouwer T, van den Berg M, Metzig B, America T, Tabak H F, Distel B. The SH3 domain of the Saccharomyces cerevisiae peroxisomal membrane protein Pex13p functions as a docking site for Pex5p, a mobile receptor for the import of PTS1-containing proteins. J Cell Biol. 1996;135:97–109. doi: 10.1083/jcb.135.1.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Erdmann R, Blobel G. Identification of Pex13p, a peroxisomal membrane receptor for the PTS1 recognition factor. J Cell Biol. 1996;135:111–121. doi: 10.1083/jcb.135.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fransen M, Brees C, Baumgart E, Vanhooren J C, Baes M, Mannaerts G P, Veldhoven P P V. Identification and characterization of the putative human peroxisomal C-terminal targeting signal import receptor. J Biol Chem. 1995;270:7731–7736. doi: 10.1074/jbc.270.13.7731. [DOI] [PubMed] [Google Scholar]
- 7.Fujiki Y. Approaches to studies on peroxisome biogenesis and human peroxisome-deficient disorders. Ann N Y Acad Sci. 1996;804:491–501. doi: 10.1111/j.1749-6632.1996.tb18639.x. [DOI] [PubMed] [Google Scholar]
- 8.Fujiki Y. Molecular defects in genetic diseases of peroxisomes. Biochim Biophys Acta. 1997;1361:235–250. doi: 10.1016/s0925-4439(97)00051-3. [DOI] [PubMed] [Google Scholar]
- 9.Fukuda S, Shimozawa N, Suzuki Y, Tomatsu S, Tsukamoto T, Hashiguchi N, Osumi T, Masuno M, Imaizumi K, Kuroki Y, Fujiki Y, Orii T, Kondo N. Human peroxisome assembly factor-2 (human PAF-2): a gene responsible for group C peroxisome biogenesis disorder in humans. Am J Hum Genet. 1996;59:1210–1220. [PMC free article] [PubMed] [Google Scholar]
- 10.Gould S J, Kalish J E, Morrell J C, Bjorkman J, Urquhart A J, Crane D I. Pex13p is an SH3 protein of the peroxisome membrane and a docking factor for the predominantly cytoplasmic PTS1 receptor. J Cell Biol. 1996;135:85–95. doi: 10.1083/jcb.135.1.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gould S J, Keller G-A, Hosken N, Wilkinson J, Subramani S. A conserved tripeptide sorts proteins to peroxisomes. J Cell Biol. 1989;108:1657–1664. doi: 10.1083/jcb.108.5.1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lazarow P B, Fujiki Y. Biogenesis of peroxisomes. Annu Rev Cell Biol. 1985;1:489–530. doi: 10.1146/annurev.cb.01.110185.002421. [DOI] [PubMed] [Google Scholar]
- 13.McCollum D, Monosov E, Subramani S. The pas8 mutant of Pichia pastoris exhibits the peroxisomal protein import deficiencies of Zellweger syndrome cells—the PAS8 protein binds to the COOH-terminal tripeptide peroxisomal targeting signal, and is a member of the TPR protein family. J Cell Biol. 1993;121:761–774. doi: 10.1083/jcb.121.4.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miura S, Kasuya-Arai I, Mori H, Miyazawa S, Osumi T, Hashimoto T, Fujiki Y. Carboxyl-terminal consensus Ser-Lys-Leu-related tripeptide of peroxisomal proteins functions in vitro as a minimal peroxisome-targeting signal. J Biol Chem. 1992;267:14405–14411. [PubMed] [Google Scholar]
- 15.Miyazawa S, Hayashi H, Hijikata M, Ishii N, Furuta S, Kagamiyama H, Osumi T, Hashimoto T. Complete nucleotide sequence of cDNA and predicted amino acid sequence of rat acyl-CoA oxidase. J Biol Chem. 1987;262:8131–8137. [PubMed] [Google Scholar]
- 16.Miyazawa S, Osumi T, Hashimoto T, Ohno K, Miura S, Fujiki Y. Peroxisome targeting signal of rat liver acyl-coenzyme A oxidase resides at the carboxy terminus. Mol Cell Biol. 1989;9:83–91. doi: 10.1128/mcb.9.1.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morand O H, Allen L-A H, Zoeller R A, Raetz C R H. A rapid selection for animal cell mutants with defective peroxisomes. Biochim Biophys Acta. 1990;1034:132–141. doi: 10.1016/0304-4165(90)90066-6. [DOI] [PubMed] [Google Scholar]
- 18.Motley A, Hettema E, Distel B, Tabak H. Differential protein import deficiencies in human peroxisome assembly disorders. J Cell Biol. 1994;125:755–767. doi: 10.1083/jcb.125.4.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Motley A M, Hettema E H, Hogenhout E M, Brites P, ten Asbroek A L M A, Wijburg F A, Baas F, Heijmans H S, Tabak H F, Wanders R J A, Distel B. Rhizomelic chondrodysplasia punctata is a peroxisomal protein targeting disease caused by a non-functional PTS2 receptor. Nat Genet. 1997;15:377–380. doi: 10.1038/ng0497-377. [DOI] [PubMed] [Google Scholar]
- 20.Niman H L, Houghten R A, Walker L E, Reisfeld R A, Wilson I A, Hogle J M, Lerner R A. Generation of protein-reactive antibodies by short peptides is an event of high frequency: implications for the structural basis of immune recognition. Proc Natl Acad Sci USA. 1983;80:4949–4953. doi: 10.1073/pnas.80.16.4949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Okumoto K, Bogaki A, Tateishi K, Tsukamoto T, Osumi T, Shimozawa N, Suzuki Y, Orii T, Fujiki Y. Isolation and characterization of peroxisome-deficient Chinese hamster ovary cell mutants representing human complementation group III. Exp Cell Res. 1997;233:11–20. doi: 10.1006/excr.1997.3552. [DOI] [PubMed] [Google Scholar]
- 22.Okumoto K, Fujiki Y. PEX12 encodes an integral membrane protein of peroxisomes. Nat Genet. 1997;17:265–266. doi: 10.1038/ng1197-265. [DOI] [PubMed] [Google Scholar]
- 22a.Okumoto, K., et al. Submitted for publication.
- 23.Osumi T, Tsukamoto T, Hata S, Yokota S, Miura S, Fujiki Y, Hijikata M, Miyazawa S, Hashimoto T. Amino-terminal presequence of the precursor of peroxisomal 3-ketoacyl-CoA thiolase is a cleavable signal peptide for peroxisomal targeting. Biochem Biophys Res Commun. 1991;181:947–954. doi: 10.1016/0006-291x(91)92028-i. [DOI] [PubMed] [Google Scholar]
- 24.Purdue P E, Zhang J W, Skoneczny M, Lazarow P B. Rhizomelic chondrodysplasia punctata is caused by deficiency of human PEX7, a homologue of the yeast PTS2 receptor. Nat Genet. 1997;15:381–384. doi: 10.1038/ng0497-381. [DOI] [PubMed] [Google Scholar]
- 25.Santos M J, Hoefler S, Moser A B, Moser H W, Lazarow P B. Peroxisome assembly mutations in humans: structural heterogeneity in Zellweger syndrome. J Cell Physiol. 1992;151:103–112. doi: 10.1002/jcp.1041510115. [DOI] [PubMed] [Google Scholar]
- 26.Schatz G, Dobberstein B. Common principles of protein translocation across membranes. Science (Washington, DC) 1996;271:1519–1526. doi: 10.1126/science.271.5255.1519. [DOI] [PubMed] [Google Scholar]
- 27.Shimozawa N, Tsukamoto T, Suzuki Y, Orii T, Fujiki Y. Animal cell mutants represent two complementation groups of peroxisome-defective Zellweger syndrome. J Clin Invest. 1992;90:1864–1870. doi: 10.1172/JCI116063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shimozawa N, Tsukamoto T, Suzuki Y, Orii T, Shirayoshi Y, Mori T, Fujiki Y. A human gene responsible for Zellweger syndrome that affects peroxisome assembly. Science (Washington, DC) 1992;255:1132–1134. doi: 10.1126/science.1546315. [DOI] [PubMed] [Google Scholar]
- 29.Slawecki M L, Dodt G, Steinberg S, Moser A B, Moser H W, Gould S J. Identification of three distinct peroxisomal protein import defects in patients with peroxisome biogenesis disorders. J Cell Sci. 1995;108:1817–1829. doi: 10.1242/jcs.108.5.1817. [DOI] [PubMed] [Google Scholar]
- 30.Subramani S. Protein import into peroxisomes and biogenesis of the organelle. Annu Rev Cell Biol. 1993;9:445–478. doi: 10.1146/annurev.cb.09.110193.002305. [DOI] [PubMed] [Google Scholar]
- 31.Swinkels B W, Gould S J, Bodnar A G, Rachubinski R A, Subramani S. A novel, cleavable peroxisomal targeting signal at the amino-terminus of the rat 3-ketoacyl-CoA thiolase. EMBO J. 1991;10:3255–3262. doi: 10.1002/j.1460-2075.1991.tb04889.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tateishi K, Okumoto K, Shimozawa N, Tsukamoto T, Osumi T, Suzuki Y, Kondo N, Okano I, Fujiki Y. Newly identified Chinese hamster ovary cell mutants defective in peroxisome biogenesis represent two novel complementation groups in mammals. Eur J Cell Biol. 1997;73:352–359. [PubMed] [Google Scholar]
- 33.Terlecky S R, Nuttley W M, McCollum D, Sock E, Subramani S. The Pichia pastoris peroxisomal protein PAS8p is the receptor for the C-terminal tripeptide peroxisome targeting signal. EMBO J. 1995;14:3627–3634. doi: 10.1002/j.1460-2075.1995.tb00032.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tsukamoto T, Bogaki A, Okumoto K, Tateishil K, Fujiki Y, Shimozawa N, Suzuki Y, Kondo N, Osumi T. Isolation of a new peroxisome deficient CHO cell mutant defective in peroxisome targeting signal-1 receptor. Biochem Biophys Res Commun. 1997;230:402–406. doi: 10.1006/bbrc.1996.5971. [DOI] [PubMed] [Google Scholar]
- 35.Tsukamoto T, Hata S, Yokota S, Miura S, Fujiki Y, Hijikata M, Miyazawa S, Hashimoto T, Osumi T. Characterization of the signal peptide at the amino terminus of the rat peroxisomal 3-ketoacyl-CoA thiolase precursor. J Biol Chem. 1994;269:6001–6010. [PubMed] [Google Scholar]
- 36.Tsukamoto T, Miura S, Fujiki Y. Restoration by a 35K membrane protein of peroxisome assembly in a peroxisome-deficient mammalian cell mutant. Nature (London) 1991;350:77–81. doi: 10.1038/350077a0. [DOI] [PubMed] [Google Scholar]
- 37.Tsukamoto T, Miura S, Nakai T, Yokota S, Shimozawa N, Suzuki Y, Orii T, Fujiki Y, Sakai F, Bogaki A, Yasumo H, Osumi T. Peroxisome assembly factor-2, a putative ATPase cloned by functional complementation on a peroxisome-deficient mammalian cell mutant. Nat Genet. 1995;11:395–401. doi: 10.1038/ng1295-395. [DOI] [PubMed] [Google Scholar]
- 38.Tsukamoto T, Shimozawa N, Fujiki Y. Peroxisome assembly factor-1: nonsense mutation in a peroxisome-deficient Chinese hamster ovary cell mutant and deletion analysis. Mol Cell Biol. 1994;14:5458–5465. doi: 10.1128/mcb.14.8.5458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tsukamoto T, Yokota S, Fujiki Y. Isolation and characterization of Chinese hamster ovary cell mutants defective in assembly of peroxisomes. J Cell Biol. 1990;110:651–660. doi: 10.1083/jcb.110.3.651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.van den Bosch H, Schutgens R B H, Wanders R J A, Tager J M. Biochemistry of peroxisomes. Annu Rev Biochem. 1992;61:157–197. doi: 10.1146/annurev.bi.61.070192.001105. [DOI] [PubMed] [Google Scholar]
- 41.Van der Leij I, Franse M M, Elgersma Y, Distel B, Tabak H F. PAS10 is a tetratricopeptide-repeat protein that is essential for the import of most matrix proteins into peroxisomes of Saccharomyces cerevisiae. Proc Natl Acad Sci USA. 1993;90:11782–11786. doi: 10.1073/pnas.90.24.11782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wendland M, Subramani S. Presence of cytoplasmic factors functional in peroxisomal protein import implicates organelle-associated defects in several human peroxisomal disorders. J Clin Invest. 1993;92:2462–2468. doi: 10.1172/JCI116854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wiemer E A, Nuttley W M, Bertolaet B L, Li X, Francke U, Wheelock M J, Anne U K, Johnson K R, Subramani S. Human peroxisomal targeting signal-1 receptor restores peroxisomal protein import in cells from patients with fatal peroxisomal disorders. J Cell Biol. 1995;130:51–65. doi: 10.1083/jcb.130.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wiemer E A C, Brul S, Just W W, van Driel R, Brouwer-Kelder E, van den Berg M, Weijers P J, Schutgens R B H, van den Bosch H, Schram A, Wanders R J A, Tager J M. Presence of peroxisomal membrane proteins in liver and fibroblasts from patients with the Zellweger syndrome and related disorders: evidence for the existence of peroxisomal ghosts. Eur J Cell Biol. 1989;50:407–417. [PubMed] [Google Scholar]
- 45.Yahraus T, Braverman N, Dodt G, Kalish J E, Morrell J C, Moser H W, Valle D, Gould S J. The peroxisome biogenesis disorder group 4 gene, PXAAA1, encodes a cytoplasmic ATPase required for stability of the PTS1 receptor. EMBO J. 1996;15:2914–2923. [PMC free article] [PubMed] [Google Scholar]
- 46.Zoeller R A, Morand O H, Raetz C R H. A possible role for plasmalogens in protecting animal cells against photosensitized killing. J Biol Chem. 1988;263:11590–11596. [PubMed] [Google Scholar]
- 47.Zoeller R A, Raetz C R H. Isolation of animal cell mutants deficient in plasmalogen biosynthesis and peroxisome assembly. Proc Natl Acad Sci USA. 1986;83:5170–5174. doi: 10.1073/pnas.83.14.5170. [DOI] [PMC free article] [PubMed] [Google Scholar]