

Abstract
Members of the kexin family of processing enzymes are responsible for the cleavage of many proproteins during their transport through the secretory pathway. The enzymes themselves are made as inactive precursors, and we investigated the activation process by studying the maturation of Krp1, a kexin from the fission yeast Schizosaccharomyces pombe. Using a cell-free translation-translocation system prepared from Xenopus eggs, we found that Krp1 is made as a preproprotein that loses the presequence during translocation into the endoplasmic reticulum. The prosequence is also rapidly cleaved in a reaction that is autocatalytic and probably intramolecular and is inhibited by disruption of the P domain. Prosequence cleavage normally occurs at Arg-Tyr-Lys-Arg102↓ (primary cleavage site) but can occur at Lys-Arg82 (internal cleavage site) and/or Trp-Arg99 when the basic residues are removed from the primary site. Cleavage of the prosequence is necessary but not sufficient for activation, and Krp1 is initially unable to process substrates presented in trans. Full activation is achieved after further incubation in the extract and is coincident with the addition of O-linked sugars. O glycosylation is not, however, essential for activity, and the crucial event appears to be cleavage of the initially cleaved prosequence at the internal site. Our results are consistent with a model in which the cleaved prosequence remains noncovalently associated with the catalytic domain and acts as an autoinhibitor of the enzyme. Inhibition is then relieved by a second (internal) cleavage of the inhibitory prosequence. Further support for this model is provided by our finding that overexpression of a Krp1 prosequence lacking a cleavable internal site dramatically reduced the growth rate of otherwise wild-type S. pombe cells, an effect that was not seen after overexpression of the normal, internally cleavable, prosequence or prosequences that lack the Lys-Arg102 residues.
The maturation of prohormones and propolypeptides often involves cleavage at pairs of basic residues as the precursor is transported through the secretory pathway. In many cases, cleavage is performed by a member of the prohormone convertase or kexin family of endopeptidases (the Kex2 protease from Saccharomyces cerevisiae was the first member of the family to be characterized) (for a review, see reference 45). The kexins themselves are also made as inactive precursors, and there has been considerable interest in defining the mechanism by which they are activated. The process has been studied in systems that include mammalian cells, yeast, Xenopus oocytes, and cell extracts, and the activation mechanism is thought to be broadly similar for each member of the family. All are synthesized with an amino-terminal presequence, a prosequence, a subtilisin-like catalytic domain, a P domain (12, 15), and a carboxy-terminal region that may (e.g., Krp1, Kex2, and furin) or may not contain a hydrophobic transmembrane domain (Fig. 1). The presequence directs the precursor to the secretory pathway and is removed during segregation into the endoplasmic reticulum (ER). The prosequence is presumed to play a role in the correct folding of the catalytic domain and is usually removed in the ER (7, 31, 49) in a reaction that is believed to be autocatalytic and probably intramolecular (6, 16, 29), although intermolecular cleavage can occur (29). The mature enzyme is then transported along the secretory pathway to its site of action.
FIG. 1.

Schematic of Krp1. Krp1 is a type I membrane protein with an N-terminal presequence (diagonal lines), a single transmembrane domain (wavy lines), and a short cytoplasmic domain (residues 696 to 709). The residues at the end of the prosequence are shown to highlight the location of the primary (Lys-Arg102) and internal (Lys-Arg82) cleavage sites. The locations of residues used to truncate Krp1 are also indicated (Y611, R667, and K696). There are five potential sites for N glycosylation (lollipops) and a Ser/Thr-rich region (light shading just N terminal of the transmembrane domain) with several potential sites for O glycosylation. The catalytic domain contains the active-site residues (Asp, His, Asn, and Ser) and is followed by the P domain (dark shading), a region of high sequence homology between members of the kexin family (15); residues near the predicted C terminus of the P domain are shown for comparison. S. pombe (Sp) Krp1 (8), Saccharomyces cerevisiae (Sc) Kex2 (10), Kluyveromyces lactis (Kl) Kex1 (46), Mus musculus (Mm) PC6 (33), Homo sapiens (Hs) PACE4 (20), Drosophila melanogaster (Dm) fur1 (36), Dm fur2 (37), Hs PC2 (43), Hs PC3 (= PC1) (44), Hs PC7 (= PC8) (3), Hs fur1 (48), X. laevis (Xl) fur1 (21), and Mm PC4 (34) sequences are shown. AAs, amino acids.
Prosequence cleavage at the primary cleavage site is necessary for the kexin to become active but is not sufficient for activation, and additional steps are required before the enzyme is able to cleave substrates presented in trans (7, 31). The first insights into the nature of these additional steps came not from kexins but from studies of the bacterial serine proteinases subtilisin and α-lytic protease. These enzymes are evolutionarily related to the eukaryotic kexins and are also synthesized with an N-terminal prosequence that facilitates correct folding of the protein before being removed by intramolecular cleavage (18, 35, 42, 52). The cleaved prosequence is not released from the enzyme but remains noncovalently associated with the catalytic domain and acts as an autoinhibitor (4, 13, 25, 26). The enzyme is able to become fully active only when the prosequence is degraded (18). A recent study of furin maturation suggests that the prosequence plays a similar inhibitory role in the activation of kexins (1). To investigate whether the cleaved prosequence plays a role in the activation of other kexins, we studied the maturation of Krp1, a kexin from the fission yeast Schizosaccharomyces pombe (8). Because Krp1 is an essential enzyme and its overexpression is also lethal (8), it has not been possible to perform these studies in its normal environment within the cell. Maturation has therefore been studied in vitro by using a coupled translation-translocation system prepared from Xenopus laevis eggs (28, 29), a system that has proved invaluable for the analysis of other members of the kexin family (29, 39). We found that prosequence cleavage is autocatalytic and probably intramolecular and that the efficiency of this reaction is influenced by both the sequence at the cleavage site and by the integrity of other parts of the protein. The cleaved prosequence seems to inhibit the activation of Krp1, and the enzyme is able to become fully active only after an additional, internal cleavage of the prosequence. Further support for this model comes from in vivo analysis, showing that overexpression of a mutant prosequence lacking the internal cleavage site inhibited the growth of otherwise wild-type yeast cells, presumably via inhibition of Krp1. Our results are similar to those reported for furin, although there are some differences between the two enzymes and the significance of these is discussed.
MATERIALS AND METHODS
Krp1 constructs.
The starting template for constructing various mutants was a 2,320-bp EcoRV-SnaBI fragment containing the entire coding region of the krp1 gene (8). The SnaBI restriction site lies just downstream of the stop codon, whereas the EcoRV site was introduced immediately upstream of the initiator codon by PCR with primer JO95 (5′-GGGATATCCCAGCACCATGCATCC-3′ [the EcoRV site is underlined, and the initiator ATG is shown in boldface]). All PCRs used Pwo DNA polymerase (from Pyrococcus woesei; supplied by Boehringer Mannheim, Lewes, East Sussex, United Kingdom), as this has a 3′-5′ exonuclease, or proofreading, activity and greatly reduces the introduction of errors during amplification. All constructs were sequenced by the dideoxynucleotide method with double-stranded DNA to confirm that only the appropriate changes had been introduced.
Truncated forms of Krp1 were generated by PCR with JO95 as the sense primer and an appropriate antisense primer that included two stop anticodons (TCATCA [shown in boldface]) and an EcoRV restriction site (underlined); JO617(5′-GGGATATCATCACCAATTTTCAAACGTACC-3′) for Krp1[W578*],JO616 (5′-GGGATATCATCACAACTGCCAATTTTCAAACG-3′) for Krp1[L580*], JO618 (5′-GGGATATCATCACAAAGCCAACTGCCAATTTTC-3′) for Krp1[L582*], JO615 (5′-GGGATATCATCACCACAAAGCCAACTGCCAA-3′) for Krp1[W583*], JO516 (5′-GGGATATCATCATCCCCACAAAGCCAACTG-3′) for Krp1[G584*], JO517 (5′-GGGATATCATCATTCTCCCCACAAAGCCAAC-3′) for Krp1[E585*], JO613 (5′-GGGATATCATCAAGAAGGGTTTTCAGATTCTCCC-3′) for Krp1[S590*], JO518 (5′-GGGATATCATCAGTATATTCCCAAGACCATC-3′) for Krp1[Y611*], JO519 (5′-GGGATATCATCATCTATAAGAGGGTTCCAATAC-3′) for Krp1[Y667*], JO520 (5′-GGGATATCATCATTTCCAAAATGCGGAAATCC-3′) for Krp1[K696*], JO456 (5′-GGGATATCATCATCGTTTTCGAATTGAACTTTG-3′) for Krp1[R82*],JO457 (5′-GGGA TATCATCAGCGCCATCTGGGCGTCTGGGC-3′) forKrp1[R99*], and JO458 (5′-GGGATATCATCACCGCTTGTAGCGCCATCTGGG-3′) for Krp1[R102*]. The PCR products were digested with EcoRV and cloned into pBluescript-KS (Stratagene Ltd., Cambridge, United Kingdom).
To change Ser371 to Ala, we took advantage of a KpnI restriction site immediately upstream of the appropriate codon in the krp1 gene and designed a sense primer that incorporated this restriction site and the necessary TCA-to-GCA change (JO459 is 5′-GGTGGTACCGCAGCGGCTGC-3′, where the KpnI site is underlined and the T-to-G mutation is shown in boldface). JO459 and JO66 (an antisense primer that lies downstream of the SnaBI site) were used to amplify the C terminus of krp1, and this was then digested with KpnI and SnaBI. The mutated fragment was exchanged for the equivalent region from the wild-type krp1 gene to produce krp1S371A.
Changes to the basic residues at the primary and/or internal cleavage sites were achieved by exploiting the EcoRV site that we had previously introduced immediately upstream of the initiator ATG (with JO95) and a unique BamHI site that corresponds to codons 122 to 124 of the krp1 open reading frame. This region can be amplified with JO95 and JO58 (antisense to a region just downstream of the BamHI site). The mutation of Arg102 to Ala was therefore achieved by first amplifying upstream regions with JO95 and JO420 (5′-ACTCGCATCCGCCTTGTAGCG-3′; antisense primer that changes the Arg102 anticodon from CCG to CGC [shown in bold] for Ala) and then amplifying C-terminal regions with JO419 (5′-CGCTACAAGGCGGATGCGAGT-3′; sense primer that changes the Arg102 codon from CGG to GCG [shown in bold] for Ala) and JO58. The products from these individual reactions were mixed and amplified with JO95 and JO58 to generate the full-length EcoRV-BamHI fragment containing the Arg102-to-Ala mutation. This was then exchanged for the equivalent fragment from the wild-type krp1 gene to give krp1[KR82][KA102]. The Arg102-to-Lys change was made in the same way by using JO349 (5′-CGCTACAAGAAGGATGCGAGT-3′; changes the CGG codon to AAG [shown in boldface]) as the sense primer and JO350 (5′-ACTCGCATCCTTCTTGTAGCG-3′; changes the CCG anticodon to CTT [shown in boldface]) as the antisense primer.
Mutations at the internal site took advantage of a unique BstBI site immediately upstream of the appropriate region in the krp1 gene. We therefore designed sense primers that incorporated the BstBI site and made the appropriate change at the internal cleavage site. A PCR with the relevant primer and JO58 (see above) therefore amplifies a mutated BstBI-BamHI fragment that can then be exchanged for the equivalent fragment of the krp1 gene. By using constructs already containing appropriate mutations at the primary cleavage site, we were able to prepare constructs mutated at both sites. The following two sense primers were used to make the changes at the internal site (the changed codons [Lys81 is normally AAA, and Arg82 is normally CGA] are shown in boldface, and the BstBI site is underlined): JO426 (5′-GTTCAATTCGAAAAGCCGGCATTGATGCC-3′) for Krp1[KA82] and JO427 (5′-GTTCAATTCGAATTGCCGGCATTGATGCC-3′) for Krp1[IA82].
All oligonucleotides were synthesized on an automatic synthesizer (model BT8510; Biotech Instruments) by using the materials and conditions recommended by the manufacturer. (This work was performed by Alta Bioscience at The University of Birmingham, Birmingham, United Kingdom.)
In vitro transcription.
mRNA was synthesized in an in vitro transcription reaction with SP6 RNA polymerase and templates produced by subcloning the required Krp1 constructs into vector pSP64T (22). Transcription reactions were performed as described previously (28). The construction of pSP64Tmap2 has already been described (8); the map2 gene encodes the precursor of the S. pombe P-factor mating pheromone (19).
Xenopus egg extract.
Xenopus egg extract was prepared as described previously (28). Translations were initiated by adding mRNA (final concentration, 100 μg/ml) to an aliquot of extract containing 10% (vol/vol) nuclease-treated rabbit reticulocyte lysate (Promega, Southampton, United Kingdom), 10 μM creatine phosphate, 0.2 μM spermidine, and 2 mCi of [3H]leucine (130 Ci/mmol) (Amersham International, Little Chalfont, Buckinghamshire, United Kingdom) per ml. All samples were incubated at 21°C for 1 h. When required, cycloheximide was added to a final concentration of 2 mM to inhibit further translation, and the incubation was continued at 21°C. The analysis of catalytic fragments by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was performed as described previously (28, 29), and cleaved prosequences were separated on a 15% peptide gel by using an SDS-Tris-tricine buffer system (38). Quantitative analysis of the amount of Krp1-related material was made by using a PhosphorImager (Molecular Dynamics) with ImageQuant software. N-terminal radiosequencing was performed as described previously (8). Briefly, proteins were transferred to a polyvinylidene difluoride (PVDF) membrane with a semidry blotter and the appropriate section of the membrane (identified by autoradiography) was excised and subjected to cycle sequencing in an ABI 473A protein sequencer (Applied Biosystems) (this was performed by Alta Bioscience, University of Birmingham). The injection of Xenopus laevis oocytes with Krp1 mRNA or a control mRNA (Map2 mRNA) and the preparation of membrane extracts for incubation with Krp1S371A were performed as described previously (8).
Krp1 activity assays.
Samples of extract containing translated Krp1 were diluted by the addition of ice-cold 100 mM HEPES (pH 6.5)–2 mM CaCl2–1 mM phenylmethylsulfonyl fluoride–1 mM tosyl-l-phenylalanine chloromethyl ketone to osmotically lyse membrane vesicles. These were collected by centrifugation at full speed in a microcentrifuge at 4°C, and the pellets were resuspended in assay buffer (100 mM HEPES [pH 6.5], 2 mM CaCl2, 0.5% Triton X-100, 1 mM phenylmethylsulfonyl fluoride, and 1 mM tosyl-l-phenylalanine chloromethyl ketone). Reaction mixtures were assembled in two halves, with one half containing membranes from the extract and the other containing the fluorogenic peptide Boc.Arg-Thr-Lys-Arg–4-methyl-comaryl-7-amide (MCA) (Peptides Institute Inc., Louisville, Ky.) at 100 μM. All reactions were performed at 29°C, and both halves were preincubated at this temperature for 5 min before mixing. The increase in fluorescence was monitored with a luminescence spectrometer (model LS-5; Perkin-Elmer) at an excitation wavelength of 365 nm and an emission wavelength of 460 nm.
Expressing prosequence constructs in S. pombe.
The prosequence regions of relevant Krp1 constructs were amplified by a PCR approach similar to that described above for generating truncated forms of Krp1. Briefly, sense primer JO638 introduced a BamHI restriction site just upstream of the initiator ATG (5′-GGGGGATCCAGCACCATGCATCCTGCTTTGC-3′ [the BamHI site is underlined, and the initiator ATG is shown in bold]) and a stop anticodon (TCA [shown in bold]) and a BamHI restriction site (underlined) were introduced at the required positions with the appropriate antisense primer, JO458 (5′-GGGGATCCTCACCGCTTGTAGCGCCATCTG-3′) for prosequences that terminate immediately after Arg102 (Krp1[R102*]) and JO451 (5′-CCGGATCCTCAGTAGCGCCATCTGGGCG-3′) for prosequences that terminate after Tyr100 (Krp1[Y100*]). By using different Krp1 constructs as templates for PCR, we were able to generate prosequences with different motifs at the internal cleavage site ([KR82] and [KA82]). The PCR products were cloned into the BamHI site of expression vector pREP3X (30), sequenced to confirm that only the appropriate changes had been introduced, and transformed into S. pombe JY330 (a heterothallic haploid strain of the Plus mating type). Transformants were cultured in either minimal medium or minimal medium lacking thiamine to induce expression from the nmt1 promoter (30). All yeast procedures were performed by standard methods (32).
RESULTS
Primary cleavage of the prosequence is autocatalytic.
A 1-h translation of Krp1 mRNA in Xenopus egg extract generated two products (Fig. 2, lane 1). We had previously used radiosequencing to show that the smaller polypeptide (apparent Mr of ∼76,000) is the C-terminal portion of Krp1 generated by cleavage after Lys-Arg102 (8), and we believe that the other product (apparent Mr of ∼90,000) is proKrp1 (i.e., Krp1 that lacks the presequence but still contains its prosequence). To investigate whether cleavage of the prosequence is autocatalytic, we used site-directed mutagenesis to change Ser371 to Ala (a product referred to as Krp1S371A). A sequence comparison with other related enzymes indicated that Ser371 of Krp1 is likely to form an essential part of the active site, and if so, Krp1S371A would be expected to be catalytically inactive (14, 15, 40). Therefore, the fact that translation of Krp1S371A mRNA in Xenopus egg extract produced only proKrp1 (Fig. 2, lane 2) suggests that prosequence cleavage at the primary site is an autocatalytic event. To confirm that Krp1 can cleave the prosequence from proKrp1, we mixed the product from Krp1S371A with membranes prepared from Xenopus oocytes that had been injected with Krp1 mRNA (Fig. 2, lane 3). These membranes provide a source of active Krp1 that is relatively free from related proteases (8), and the conversion of proKrp1 to Krp1 demonstrates that Krp1 can cleave its own prosequence through an intermolecular interaction.
FIG. 2.

Prosequence cleavage is autocatalytic. Krp1S371A or Krp1[IA82][KK102]S371A mRNA was translated in Xenopus egg extract for 1 h in the presence of [3H]leucine and was then mixed in the presence of 1% Triton X-100 with membrane extracts prepared from Xenopus oocytes that had been injected with either a control mRNA (lanes 2 and 5) or Krp1 mRNA (lanes 3 and 6). The digestion products were separated by SDS-PAGE. Lane 1 is included to aid interpretation. It contained the products obtained from a 1-h translation of Krp1 mRNA in egg extract; the products are proKrp1 (upper band) and Krp1 (lower band). Lane 4 was from a 1-h translation of Krp1[IA82][KK102] mRNA and demonstrates that the Lys-Lys102 motif at the primary cleavage site is cleaved in an active form of Krp1. The positions of molecular weight markers (in thousands) are shown on the left.
We took advantage of the inability of Krp1S371A to remove its own prosequence to identify the site of presequence cleavage. Krp1S371A mRNA was translated in egg extract, and proKrp1S371A was transferred to a PVDF membrane before being subjected to amino acid sequencing (8). Peaks of [3H]leucine were observed in cycles 4, 9, and 11 (not shown), which suggests a presequence that is cleaved after Ser23 in the sequence Val-Ser-Ser23-Cys-Ser-Pro.
N-terminal requirements for prosequence cleavage.
A striking feature of the prosequences of most kexins, including Krp1, is the presence of two distinct clusters of basic amino acids (41). One cluster (Arg-XXX-Lys-Arg) is the position that we have identified as the site of initial cleavage of the Krp1 prosequence (8) and others have shown to be the site of prosequence cleavage in furin (7, 24, 31, 49), Kex2 (2, 51), PC2 (29), and PC3 (16, 27, 50). This is often referred to as the primary cleavage site. The second cluster of basic residues has been studied less extensively, and until recently (see below), there has been no convincing evidence of cleavage at this internal site. We therefore decided to investigate cleavage in more detail by generating mutant forms of Krp1 that lack the normal basic motifs at either or both of these sites and analyzing the effects in Xenopus egg extract (Fig. 3).
FIG. 3.

N-terminal requirements for prosequence cleavage. (A) Krp1 mRNA and mRNAs from various Krp1 mutants that lack the normal basic motifs at either the primary or internal site were translated in Xenopus egg extract for 1 h in the presence of [3H]leucine before being analyzed by SDS-PAGE on a low-percentage gel (lanes 1 to 9). Krp1[IA82][KA102] mRNA was also translated for 6 h before analysis in order to investigate the slow processing of this mutant (lane 10). The positions of molecular weight markers (in thousands) are shown on the left. (B) Selected samples were also analyzed on a high-percentage acrylamide gel (38). To aid in the interpretation of prosequence fragments, we prepared versions of Krp1 with stop codons introduced immediately after Arg102 (Krp1[R102*]), Arg99 (Krp1[R99*]), and Arg82 (Krp1[R82*]). Translation of these truncated proteins in extract resulted in translocation into microsomes and presequence cleavage to generate markers that are appropriate for different prosequences.
Mutations that disrupted the internal cluster of basic residues (without affecting the primary cleavage site) did not prevent cleavage of the prosequence at the primary site (Fig. 3A, lanes 2 and 3). This is not unexpected, but it indicates that cleavage of the prosequence at Lys-Arg102 does not require prior cleavage at the internal site. We confirmed this by using a high-percentage acrylamide gel to demonstrate that the cleaved prosequence was indeed full-length and comigrated with the product obtained by translating Krp1[R102*] in extract (Fig. 3B, lane 1). Krp1[R102*] is a shortened form of Krp1 that is truncated immediately after Arg102 to give a product that contains residues 24 to 102 (inclusive) of full-length Krp1 (the first 23 residues are removed as the presequence).
Changing the primary cleavage site from the normal Lys-Arg motif to Lys-Lys also had little effect on processing at this position (Fig. 3A, lanes 4 through 6) (see below), but converting the dibasic motif at the primary site to Lys-Ala inhibited cleavage at this position and allowed us to investigate processing at other sites within the prosequence (Fig. 3A, lanes 7 through 9). A mutant possessing the normal Lys-Arg82 motif at the internal site was cleaved at this position to generate a catalytic fragment that starts at residue 83 (Fig. 3A, lane 7; the identity of this product was confirmed by radiosequencing [not shown]) and a prosequence that corresponds to residues 24 to 82 of Krp1 (Fig. 3B, lane 2). The catalytic fragment underwent a second cleavage to generate a product with an apparent Mr that is between those of products cleaved at the primary site and products cleaved at the internal site. This cleavage must occur after the cleavage at Lys-Arg82, as we see only one product on the high-percentage gel (Fig. 3B, lane 2); we would not expect to detect the small peptide produced from the catalytic fragment by this second cleavage. The identity of this second product is discussed below.
Removing the dibasic motifs at both the primary and internal sites greatly reduced processing of the proprotein (Fig. 3A, lanes 8 and 9), and only limited cleavage was observed after prolonged incubation in extract (Fig. 3A, lane 10). The catalytic fragment produced by this cleavage comigrated with the product generated by the second cleavage of Krp1[KA102] (Fig. 3A, lane 7), whereas the prosequence migrated between those produced after digestion at the primary and internal sites (Fig. 3B, lane 3). Krp1 contains three Arg residues between the internal and primary cleavage sites (Fig. 1), and we suspected that this product was generated by cleavage at one of these residues. Given that Kex2 can cleave substrates at Pro-Arg motifs (53), we expected this cleavage to occur after Pro-Arg97 and were surprised that sequencing showed that cleavage occurs after Trp-Arg99 (Fig. 4). This site is clearly less accessible in full-length proKrp1 than in the protein that has already been cleaved at Lys-Arg102.
FIG. 4.

Prosequence cleavage between the primary and internal sites. Krp1 (shown as Krp1[KR82][KR102] to emphasize the sequences at the primary and internal sites) and Krp1[IA82][KA102] mRNAs were translated in Xenopus egg extract for 1 h in the presence of [3H]leucine, and the products were separated by SDS-PAGE before being transferred to a PVDF membrane. The positions of proteins were identified by autoradiography, and the sections of the membrane corresponding to the proteins were excised and subjected to cycle sequencing. The amount of [3H]leucine in each cycle was determined by scintillation counting and compared to the protein sequences. The cleavage sites are indicated by black arrows, and shaded arrows indicate the positions of mutated sites in Krp1[IA82][KA102].
Primary cleavage of the prosequence is intramolecular.
As Krp1 does not normally cleave a substrate that contains a Lys-Lys motif (8), the apparently efficient cleavage of mutant Krp1 proteins containing Lys-Lys102 (Fig. 3A, lanes 4 through 6) appeared to contradict our earlier observation that the primary cleavage of the prosequence is autocatalytic. We addressed this issue by combining the Lys-Lys102 change with the Ser371-to-Ala mutation that inactivates the enzyme. To avoid potential problems with cleavage at the internal site, we also introduced the Ile-Ala82 change that completely inhibits cleavage at this site (Fig. 3, lanes 9 and 10). As expected, translation of Krp1[IA82][KK102]S371A mRNA in Xenopus egg extract generated a single product corresponding to the proenzyme (Fig. 2, lane 5). However, in contrast to the result with proKrp1S371A (Fig. 2, lanes 2 and 3), active Krp1 was unable to remove the prosequence from proKrp1[IA82][KK102]S371A when it was presented in trans (Fig. 2, lane 6). This difference is consistent with our previous work showing that Krp1 has a strong preference for substrates containing Lys-Arg motifs rather than Lys-Lys motifs (8). Therefore, the efficient cleavage of the Lys-Lys102 site when it was present as part of an enzymatically active form of Krp1 (Fig. 2, lane 4, and 3, lanes 4 through 6) suggests that initial cleavage of the prosequence is not an intermolecular event but, rather, an intramolecular event normally.
C-terminal requirements for prosequence cleavage.
There have been several studies to investigate the role of C-terminal sequences in the maturation of kexins (15, 17, 47). Much of this work has focused on the role of the P domain, a conserved region of about 150 residues that is found just C terminal of the catalytic domain and is present only in subtilisin-like enzymes implicated in proteolytic processing rather than degradation (12, 15). Disruption of the P domain blocks the production of active enzyme, and although the nature of the defect has not always been determined, it appears that this region is required for efficient cleavage of the prosequence (15). We therefore investigated the role of the C terminus in the maturation of Krp1.
Stop codons were introduced at various positions along the krp1 open reading frame, and mRNAs corresponding to the various truncated enzymes were translated in egg extract (Fig. 5). Truncations that successively removed the cytoplasmic tail (Krp1[K696*]), the transmembrane domain (Krp1[R667*]), and the Ser/Thr-rich domain (Krp1[Y611*]) had no effect on cleavage of the prosequence (Fig. 5, lanes 4 through 6), which appeared to occur as efficiently as it did in the full-length protein (Fig. 5, lane 1) (full-length Krp1 contains 709 residues). In contrast, truncating Krp1 at or N terminal of Ser590 dramatically inhibited cleavage of the prosequence (Fig. 5, lanes 2 and 3); only partial processing was observed even after prolonged incubation (Fig. 5, lanes 7 and 8). It should be noted that for brevity, Fig. 5 contains only the results for Krp1[W578*] and Krp1[S590*] and that we obtained very similar results with a series of mutants truncated between these two points (Krp1[L580*], Krp1[L582*], Krp1[W583*], Krp1[G584*], and Krp1[E585*]). Further truncations between Ser590 and Tyr611 are needed to more closely define the region required for efficient cleavage of the prosequence.
FIG. 5.

C-terminal requirements for prosequence cleavage. Krp1 mRNA and mRNAs from truncated forms of Krp1 were translated in Xenopus egg extract for 1 h in the presence of [3H]leucine before being analyzed by SDS-PAGE (lanes 1 to 6). mRNAs from Krp1[W578*], Krp1[S590*], and Krp1[Y611*] (included to provide a convenient reference point between processed and unprocessed proteins) were also translated for 24 h before analysis (lanes 7 to 9). The positions of molecular weight markers (in thousands) are shown on the left.
Activation of Krp1 in Xenopus egg extract.
Autoproteolytic cleavage of the prosequence indicates that Krp1 becomes active soon after synthesis in Xenopus egg extract. However, we were unable to detect any proteolytic activity in such samples when fluorogenic substrates were used under conditions that supported cleavage by Krp1 prepared from yeast membranes (not shown). The inability of these samples to process substrates presented in trans is reminiscent of earlier studies in which furin activation was shown to require events that occurred after the cleavage of the prosequence at the primary site (31, 49). We therefore investigated whether egg extract would support the additional steps required to activate Krp1. To simplify interpretation of the results, we translated Krp1 mRNA for 1 h in the presence of [3H]leucine before adding cycloheximide to inhibit any further translation. Incubation was continued for various periods, and samples were analyzed by SDS-PAGE and assayed for the ability to cleave an appropriate substrate (Fig. 6). Little or no Krp1 activity was detected in early samples, but considerable activity was detected in samples incubated for 15 and 23 h. As translation had been inhibited by the addition of cycloheximide, this activity was not the result of increased production of Krp1; it was due to activation of the enzyme.
FIG. 6.

Activation of Krp1 in Xenopus egg extract. Krp1 mRNA was translated in Xenopus egg extract for 1 h in the presence of [3H]leucine. Cycloheximide was added (final concentration, 2 mM) to inhibit any further translation, and incubation was continued for various times (as indicated) before samples were analyzed by SDS-PAGE. The amount of Krp1 in each sample was determined by using a PhosphorImager (Molecular Dynamics) with ImageQuant software, and equivalent amounts of protein were assayed for the ability to cleave the fluorogenic substrate Boc-Arg-Thr-Lys-Arg-MCA. Activity was determined under initial rate conditions and is expressed relative to the maximum activity observed in the experiment. The positions of molecular weight markers (in thousands) are shown to the left of the gel.
Activation of Krp1 appears to correlate with a significant decrease in the mobility of the protein. Such heterogeneous changes often accompany the addition of carbohydrates to proteins. We investigated whether Krp1 underwent O glycosylation or whether there was further elaboration of the N-linked sugars that are added soon after translocation into membranes (Fig. 7). The tripeptide (acetyl)-Asn-Tyr-Thr-(amide) competes with proteins for the transfer of sugar chains from their dolichol lipid carrier (23) and has previously been shown to completely inhibit N glycosylation of Krp1 in egg extract (8). It did not, however, prevent the decrease in the mobility of Krp1 (Fig. 7A), which suggests that this modification is not due to the elaboration of N-linked sugars. As no equivalent inhibitor of O glycosylation was available, we sought to investigate this indirectly by exploiting the ability of glycoproteins to bind to concanavalin A (ConA)-Sepharose (Fig. 7B). ConA preferentially binds to α-mannopyranosyl and α-glycopyranosyl residues but recognizes a somewhat wider range of both N- and O-linked sugars; therefore, the binding of Krp1 to ConA-Sepharose in the absence of N glycosylation is reasonable evidence that this protein is O glycosylated. In contrast, the Map2 protein does not become O glycosylated and fails to bind ConA when N glycosylation is inhibited. The most likely location for O glycosylation of Krp1 is the Ser/Thr-rich domain located just proximal to the transmembrane domain (19 Ser or Thr residues between positions 619 and 647) (9). Such domains become O glycosylated in other transmembrane proteins, including Kex2 (10). The suggestion that the heterogeneity we observed was due to O glycosylation of this region was provided by the Krp1[Y611*] mutant described above. This mutant is truncated just N terminal of the Ser/Thr-rich domain. Although it became N glycosylated soon after translation, it did not undergo any further modifications during extended incubation in extract (Fig. 5, lane 9). The Krp1[Y611*] mutant also demonstrated that the correlation between O glycosylation and activation is not obligatory and that like Kex2 (11, 15), Krp1 can become active without being O glycosylated (Table 1). It appears therefore that O glycosylation merely provides a convenient marker for some other event that is essential for the activation of Krp1.
FIG. 7.

Krp1 undergoes both N glycosylation and O glycosylation in Xenopus egg extract. Krp1 mRNA was translated in egg extract for 1 h in the presence of [3H]leucine before a 23-h chase (cycloheximide was added after 1 h to inhibit further translation). Translations were also performed in the presence of the tripeptide (acetyl)-Asn-Tyr-Thr-(amide) (NYT; final concentration, 10 mM) to inhibit N glycosylation (8, 23). (A) Samples were analyzed by SDS-PAGE. (B) Samples from extract (prepared in the presence and absence of NYT) were diluted in binding buffer (20 mM Tris [pH 7.5], 0.5 M NaCl, 0.5% Triton X-100) and allowed to bind to ConA-Sepharose in the same buffer. Unbound materials were removed by extensive washing, and beads were resuspended in electrophoresis sample buffer and analyzed by SDS-PAGE. T, total sample prior to incubation with ConA-Sepharose; B, material remaining bound to beads after being washed. Map2, a protein that undergoes N glycosylation but not O glycosylation (8, 19), was included as a control for the inhibition of N glycosylation by NYT and for the specificity of beads. The positions of molecular weight markers (in thousands) are shown on the left.
TABLE 1.
Relative activities of various Krp1 mutantsa
| Construct | Internal site | Primary site | Activity |
|---|---|---|---|
| Krp1 | Lys-Arg | Lys-Arg | 100 |
| Krp1S371A | Lys-Arg | Lys-Arg | <1 |
| Krp1[IA82][KK102]S371A | Ile-Ala | Lys-Lys | <1 |
| Krp1[KA82][KR102] | Lys-Ala | Lys-Arg | 1–10 |
| Krp1[IA82][KR102] | Ile-Ala | Lys-Arg | 1–10 |
| Krp1[KR82][KK102] | Lys-Arg | Lys-Lys | >120 |
| Krp1[KA82][KK102] | Lys-Ala | Lys-Lys | 1–10 |
| Krp1[IA82][KK102] | Ile-Ala | Lys-Lys | 1–10 |
| Krp1[KR82][KA102] | Lys-Arg | Lys-Ala | 10–20 |
| Krp1[KA82][KA102] | Lys-Ala | Lys-Ala | 10–20 |
| Krp1[IA82][KA102] | Ile-Ala | Lys-Ala | 10–20 |
| Krp1[W578*] | Lys-Arg | Lys-Arg | 1–10 |
| Krp1[S590*] | Lys-Arg | Lys-Arg | 1–10 |
| Krp1[Y611*] | Lys-Arg | Lys-Arg | 80–90 |
Various mRNAs were translated in Xenopus egg extract for 1 h in the presence of [3H]leucine. Cycloheximide was added (final concentration, 2 mM) to inhibit any further translation, and incubation was continued for 23 h to allow maturation to occur. Samples were analyzed by SDS-PAGE, and the amount of Krp1-related material in each sample was determined by using a PhosphorImager (Molecular Dynamics) with ImageQuant software (an allowance was made for the number of leucine residues in each construct). Equivalent amounts of protein were assayed for the ability to cleave the fluorogenic substrate Boc-Arg-Thr-Lys-Arg-MCA under initial rate conditions. The results are expressed relative to the activity of Krp1. Each sample was translated and assayed on at least three occasions, and each result was within the range indicated.
To discover which mutants could become active in the extract, we translated the appropriate mRNAs for 1 h before adding cycloheximide to inhibit further translation and allowing the incubation to continue for a further 23 h. Equivalent amounts of Krp1 and various mutant enzymes were assayed for activity (Table 1). As expected, the active-site mutants Krp1S371A and Krp1[IA82][KK102]S371A were inactive, but all of the other mutants developed at least some activity. Removing the cytoplasmic tail, transmembrane domain, and Ser/Thr-rich region (e.g., Krp1[Y611*]) had only a small effect on the activation process, but truncating Krp1 at or near the C-terminal end of the P domain (e.g., Krp1[S590*]) had a more dramatic effect. These mutants attained less than 10% of the activity associated with the full-length protein. Mutating the primary cleavage site from Lys-Arg102 to Lys-Ala102 prevented proper cleavage of the prosequence and caused at least a fivefold reduction in activity. These mutants were cleaved at Trp-Arg99 (Fig. 4), and there appeared to be no difference in activation between constructs which were cleaved only at this site (e.g., Krp1[KA82][KA102]) and those which were initially cleaved at Lys-Arg82 (e.g., Krp1[KR82][KA102]). It was perhaps not surprising to find that incorrect cleavage of the prosequence affected activity, but we were more surprised to discover that the nature of the cleaved prosequence appears to influence subsequent activation of the enzyme. Indeed, only one mutant (Krp1[KR82][KK102]) developed activity that was similar to that of Krp1. We considered it to be perhaps significant that this was the only construct in which the internal cleavage site was the same as that in the wild-type enzyme, and we wondered whether cleavage at this site was required for activation of the enzyme. To investigate this, we examined the prosequences produced from a selection of constructs and found that activation did appear to correlate with internal cleavage of the prosequence (Fig. 8). Internal cleavage of the Krp1 prosequence appeared to be coincident with its activation (Fig. 8, lane 1), whereas the prosequence initially cleaved from the Krp1[IA82] mutant did not undergo secondary cleavage at the internal site (Fig. 8, lane 2) and the enzyme did not become active (Table 1). As there is no apparent difference in the initial cleavage of the prosequence from these two enzymes (both are cleaved at the primary site of Lys-Arg102) and they contain identical catalytic domains, we suggest that the inability of the mutant to become active is due to its failure to cleave the internal site of the initially cleaved prosequence. In the other two mutants whose results are shown in Fig. 8, mutation of the primary cleavage site leads to the production of shortened prosequences that do not undergo subsequent cleavage but are also unable to prevent the enzymes from becoming at least partially active.
FIG. 8.
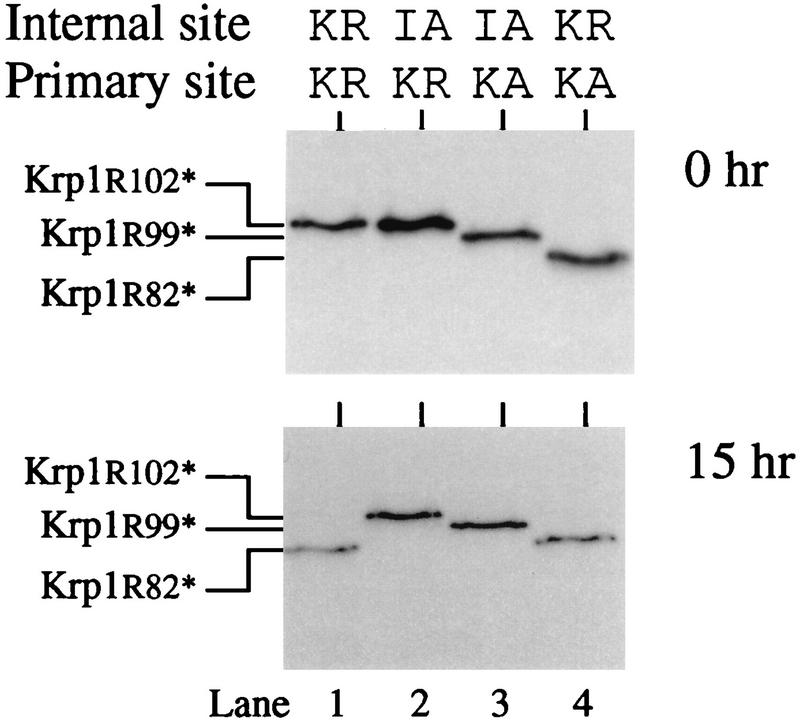
Internal cleavage of the initially cleaved prosequence is required for activation of Krp1. Krp1 mRNA and mRNAs from various Krp1 mutants that lack the normal basic motifs at either the primary or internal site were translated in Xenopus egg extract for 1 h in the presence of [3H]leucine. (Top) Samples were analyzed by SDS-PAGE on a high-percentage gel. (Bottom) Duplicate samples were treated in the same way except that cycloheximide was added after 1 h (to inhibit translation) and incubation was continued for a further 15 h before samples were analyzed. Mutant forms of Krp1 with stop codons introduced immediately after Arg102 (Krp1[R102*]), Arg99 (Krp1[R99*]), and Arg82 (Krp1[R82*]) were translated in extract to provide appropriate markers and aid in the interpretation of results.
The prosequence interacts with Krp1 in vivo.
Our studies with Xenopus egg extract helped us to define events in the maturation of Krp1, and although we believe that our findings reflect events in this fission yeast, we sought to confirm this in a more physiological environment. Unfortunately, such studies are complicated by the fact that S. pombe appears to require a tightly controlled level of Krp1 activity and that both the loss and overexpression of the enzyme are deleterious to cell growth (8). It was not possible therefore to simply repeat the in vitro analysis in vivo. We took the alternative approach of expressing various Krp1 prosequence constructs in otherwise wild-type yeast cells (Table 2). These cells contained the normal chromosomal copy of the krp1 gene, and cells transformed with pREP3X vector alone had a doubling time of about 4 h. The expression of Krp1[KR82][KR102][R102*] (which has the Lys-Arg motif at both the primary and internal cleavage sites and terminates immediately after Arg102 to generate a product that is equivalent to the prosequence expected from the initial cleavage of wild-type Krp1) had little effect on cell growth, but the expression of Krp1[KA82][KR102][R102*] (same as Krp1[KR82][KR102][R102*] but with a noncleavable internal site) caused a dramatic decrease in the rate of growth. Such a decrease in the growth rate would be expected if Krp1[KA82][KR102][R102*] inhibited Krp1. The specificity of this interaction was supported by our finding that Krp1[KA82][Y100*] did not affect cell growth. This construct generates a prosequence that lacks the C-terminal Lys-Arg motif of the normal prosequence, which suggests that these residues are important for the apparent inhibition of Krp1 by Krp1[KA82][KR102][R102*].
TABLE 2.
Inhibitory properties of different prosequences in vivoa
| Prosequence | Internal site | Primary site | Doubling time
|
|
|---|---|---|---|---|
| Actual (h)b | Relative | |||
| Krp1[KR82][KR102][R102*] | Lys-Arg | Lys-Arg | 4.12 | 1.00 |
| Krp1[KA82][KR102][R102*] | Lys-Ala | Lys-Arg | 5.55 | 1.35 |
| Krp1[KR82][Y100*] | Lys-Arg | Absent | 4.21 | 1.02 |
| Krp1[KA82][Y100*] | Lys-Ala | Absent | 4.13 | 1.00 |
Plasmids containing the indicated Krp1 prosequence constructs under the transcriptional control of the nmt1 promoter were transformed into S. pombe cells expressing wild-type Krp1 from its chromosomal allele. Transformants were cultured at 29°C in the absence of thiamine to induce expression from the nmt1 promoter.
Data are averages of five separate determinations.
DISCUSSION
Using an in vitro system prepared from Xenopus eggs, we investigated events in the activation of Krp1, the only member of the kexin family of proprotein convertases present in the fission yeast S. pombe. Krp1 is a type I membrane protein that is synthesized as a preproenzyme. The presequence is removed during translocation into the ER, where the proKrp1 becomes N glycosylated. Cleavage of the prosequence at Lys-Arg102 is autocatalytic and can occur intermolecularly, although we suspect it is normally intramolecular because mutants containing Lys-Lys102 are cleaved much more efficiently than would be expected from the relative inability of Krp1 to cleave Lys-Lys-containing substrates in trans (8). Indeed, an inactive form of Krp1 that contains the S371A mutation as well as the Lys-Lys102 motif was not cleaved by incubation with Krp1. A closer analysis of the kinetics of this processing event will be required to prove that prosequence cleavage is intramolecular. Prosequence cleavage was unaffected in mutants lacking the cytoplasmic tail, the transmembrane domain, and even the Ser/Thr-rich region near the C terminus of the protein but was inhibited by deletions that affect the P domain. Even though it was initially identified as a homologous region C terminal to the catalytic domain, mutational analyses have demonstrated a functional role for the P domain in kexin maturation (6, 15, 17). Although our results are similar to those obtained with the P domain of Kex2 (15), they do not allow us to identify the end of the P domain as accurately as was possible in the earlier study. The high level of sequence similarity between the two enzymes in this region suggests that the different results are probably due to differences in the assays used to monitor the effects of these mutations rather than to any significant difference in the precise role of the P domain.
Cleavage of the prosequence at the primary site was necessary but not sufficient for activation of Krp1, and despite the ability of the enzyme to cleave its own prosequence, it was initially unable to process substrates presented in trans. The enzyme became fully active only upon further incubation in extract. The relationship between activation and O glycosylation appeared to be fortuitous, however, as a truncated form of Krp1 that failed to become O glycosylated did become active. It seems more likely that this is simply a convenient marker for another event that is necessary for activation. Our results do not precisely define this event but show that activation is accompanied by internal cleavage of the initially cleaved prosequence and, furthermore, that mutant enzymes in which this cleavage is prevented fail to become activated. These results are consistent with a model in which the cleaved prosequence remains noncovalently associated with the catalytic domain of Krp1 and acts as an autoinhibitor. Inhibition is relieved by cleavage of the prosequence by Krp1 at the internal site, and the enzyme is then able to process other substrates. This model is further supported by the finding that overexpression of a noncleavable prosequence reduced the growth rate of yeast cells, presumably through inhibition of Krp1. The reduction in growth rate and presumably therefore the interaction with Krp1 require the presence of the two basic residues (Lys-Arg102) at the C terminus of the prosequence.
Our results are similar to those recently reported for the maturation of furin (1). The furin prosequence is removed autoproteolytically but remains associated with the mature part of the enzyme and acts as a potent inhibitor. Inhibition is relieved by cleavage of the prosequence at the internal site. Indeed, the only significant difference between furin and Krp1 appears to be the trigger that allows cleavage of the inhibitory prosequence. The activation of furin requires its movement from the ER to the Golgi complex and appears to be triggered by the decreased pH and increased Ca2+ associated with this transport. These changes are thought to reduce the interaction between furin and its prosequence and allow the enzyme to cleave the inhibitory peptide at the internal site. Such compartment-specific activation of furin may prevent inappropriate cleavage of substrates in the ER or may be required to allow the enzyme sufficient time to adopt its correct conformation. As predicted by this model, inhibiting the transport of furin to the Golgi complex by adding an ER retrieval signal prevents the enzyme from becoming active (31). This ER-retained form of furin can be activated in vitro by mimicking the changes normally associated with transport to the Golgi complex (1). In contrast, despite using conditions similar to those used for furin (1), we were unable to promote the activation of Krp1 by changing the pH and/or Ca2+ concentration (not shown). This may suggest that the activation of the yeast enzyme does not require its export from the ER. Indeed, we sometimes observed activation of Krp1 in Xenopus egg extract in the absence of O glycosylation (not shown), but we cannot yet discriminate between Krp1 molecules that remain in the ER and those that are transported to the Golgi complex but are not O glycosylated. It is perhaps significant that the addition of an ER retrieval signal to Kex2 does not prevent its activation (5).
A possible molecular explanation for the pH-dependent activation of furin is ionization of the histidine residues near the C terminus of the prosequence (1). These may influence the interaction between the prosequence and the catalytic domain and may be affected by the transport of the enzyme from the ER to the Golgi complex. Changes in the ionization state of these residues may reduce the stability of the prosequence-enzyme complex and trigger activation of the enzyme. The positions of these histidine residues are conserved in almost all of the kexins identified in higher eukaryotes but are absent from both Krp1 and Kex2, which is consistent with evidence that activation of the yeast enzymes, like that of the bacterial enzymes, is not pH dependent.
Many members of the kexin family contain a cluster of basic residues that can be aligned with the internal site of furin and Krp1 (41), and cleavage at this site may be a common feature in the activation of these other enzymes. However, not all kexins contain this second site. If the inhibitory prosequence is therefore a common theme in this family, it will be necessary to invoke other mechanisms of degradation; it is perhaps significant that furin can be activated by digestion with trypsin (1).
Our results have gone some way to defining the events required for the activation of Krp1, but they have revealed little about how these events are controlled at the molecular level. For example, how does the prosequence remain associated with the enzyme and what events are required to trigger dissociation? It will also be interesting to discover whether the prosequence physically dissociates from the Krp1 before being inactivated by cleavage at the internal site. We are using Xenopus egg extract to address these and other questions and are complementing our in vitro work with in vivo analysis.
ACKNOWLEDGMENTS
We thank Kevin Davis and Glenn Matthews for expert technical assistance during this work.
This work was supported by a studentship from the Biotechnology and Biological Sciences Research Council (ref. 94305555) (D.P.) and by grants from the Cancer Research Campaign (ref. Sp1972). J.D. is a Lister Institute Research Fellow.
REFERENCES
- 1.Anderson E D, Van Slyke J K, Thulin C D, Jean F, Thomas G. Activation of furin endoprotease is a multiple-step process: requirements for acidification and internal propeptide cleavage. EMBO J. 1997;16:1508–1518. doi: 10.1093/emboj/16.7.1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brenner C, Fuller R S. Structural and enzymatic characterisation of a purified prohormone processing enzyme: secreted, soluble KEX2 protease. Proc Natl Acad Sci USA. 1992;89:922–926. doi: 10.1073/pnas.89.3.922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bruzzaniti A, Goodge K, Jay P, Taviaux S A, Lam M H C, Berta P, Martin T J, Moseley J M, Gillespie M T. C8, a new member of the convertase family. Biochem J. 1996;314:727–731. doi: 10.1042/bj3140727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bryan P, Wang L, Hoskins J, Ruvinov S, Strausberg S, Alexander P, Almog O, Gilliland G, Gallagher T. Catalysis of a protein folding reaction: mechanistic implications of the 2.0 Å structure of the subtilisin-prodomain complex. Biochemistry. 1995;34:10310–10318. doi: 10.1021/bi00032a026. [DOI] [PubMed] [Google Scholar]
- 5.Chaudhuri B, Latham S E, Stephan C. A mutant Kex2 enzyme with a C-terminal HDEL sequence releases correctly folded human insulin-like growth factor-1 from a precursor accumulated in the yeast endoplasmic reticulum. Eur J Biochem. 1992;210:811–822. doi: 10.1111/j.1432-1033.1992.tb17484.x. [DOI] [PubMed] [Google Scholar]
- 6.Creemers J W M, Siezen R J, Roebroek A J M, Ayoubi T A Y, Huylebroeck D, Van de Ven W J. Modulation of furin-mediated proprotein processing activity by site-directed mutagenesis. J Biol Chem. 1993;268:21826–21834. [PubMed] [Google Scholar]
- 7.Creemers J W M, Vey M, Schäfer W, Ayoubi T A Y, Roebroek A J M, Klenk H-D, Garten W, Van de Ven W J M. Endoproteolytic cleavage of its propeptide is a prerequisite for efficient transport of furin out of the endoplasmic reticulum. J Biol Chem. 1995;270:2695–2702. doi: 10.1074/jbc.270.6.2695. [DOI] [PubMed] [Google Scholar]
- 8.Davey J, Davis K, Imai Y, Yamamoto M, Matthews G. Isolation and characterisation of krp, a dibasic endopeptidase required for cell viability in the fission yeast Schizosaccharomyces pombe. EMBO J. 1994;13:5910–5921. doi: 10.1002/j.1460-2075.1994.tb06936.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Elhammer A P, Poorman R A, Brown E, Maggiora L L, Hoogerheide J H, Kezdy F J. The specificity of UDP-GalNAc:polypeptide N-acetylgalactosaminyltransferase as inferred from a database of in vivo substrates and from the in vitro glycosylation of proteins and peptides. J Biol Chem. 1993;268:10029–10038. [PubMed] [Google Scholar]
- 10.Fuller R S, Brake A J, Thorner J. Intracellular targeting and structural conservation of a prohormone-processing endoprotease. Science. 1989;246:482–485. doi: 10.1126/science.2683070. [DOI] [PubMed] [Google Scholar]
- 11.Fuller R S, Brake A, Thorner J. Yeast prohormone processing enzyme (KEX2 gene product) is a calcium-dependent serine protease. Proc Natl Acad Sci USA. 1989;86:1434–1438. doi: 10.1073/pnas.86.5.1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fuller R S, Brenner C, Gluschankof P, Wilcox C A. The yeast prohormone-processing Kex2 protease, an enzyme with specificity for paired basic residues. In: Jörnvall H, Höög J-O, Gustavsson A-M, editors. Methods in protein sequence analysis. Berlin, Germany: Birkhäuser; 1991. pp. 205–214. [Google Scholar]
- 13.Gallagher T, Gilliland G, Wang L, Bryan P. The prosegment subtilisin BPN′ complex: crystal structure of a specific ‘foldase.’. Structure. 1995;3:907–914. doi: 10.1016/S0969-2126(01)00225-8. [DOI] [PubMed] [Google Scholar]
- 14.Germain D, Dumas F, Vernet T, Bourbonnais Y, Thomas D Y, Boileau G. The pro-region of the Kex2 endoprotease of Saccharomyces cerevisiae is removed by self-processing. FEBS Lett. 1992;299:283–286. doi: 10.1016/0014-5793(92)80132-z. [DOI] [PubMed] [Google Scholar]
- 15.Gluschankof P, Fuller R S. A C-terminal domain conserved in precursor processing proteases is required for intramolecular N-terminal maturation of pro-Kex2 protease. EMBO J. 1994;13:2280–2288. doi: 10.1002/j.1460-2075.1994.tb06510.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goodman L J, Gorman C M. Autoproteolytic activation of the mouse prohormone convertase mPC1. Biochem Biophys Res Commun. 1994;201:795–804. doi: 10.1006/bbrc.1994.1771. [DOI] [PubMed] [Google Scholar]
- 17.Hatsuzawa K, Nagahama M, Takahashi S, Takada K, Murakami K, Nakayama K. Purification and characterisation of furin, a Kex2-like processing endoprotease, produced in Chinese hamster ovary cells. J Biol Chem. 1992;267:16094–16099. [PubMed] [Google Scholar]
- 18.Ikemura H, Inouye M. In vitro processing of pro-subtilisin produced in Escherichia coli. J Biol Chem. 1988;263:12959–12693. [PubMed] [Google Scholar]
- 19.Imai Y, Yamamoto M. The fission yeast mating pheromone P-factor: its molecular structure, gene structure and physiological activities to induce gene expression and G1 arrest in the mating partner. Genes Dev. 1994;8:328–338. doi: 10.1101/gad.8.3.328. [DOI] [PubMed] [Google Scholar]
- 20.Kiefer M C, Tucker J E, Joh R, Landsberg K E, Saltman D, Barr P J. Identification of a second subtilisin-like protease gene in the fes/fps region of chromosome 15. DNA Cell Biol. 1991;10:757–769. doi: 10.1089/dna.1991.10.757. [DOI] [PubMed] [Google Scholar]
- 21.Korner J, Chun J, Harter D, Axel R. Isolation and functional expression of a mammalian prohormone processing enzyme, murine prohormone convertase-1. Proc Natl Acad Sci USA. 1991;88:6834–6838. doi: 10.1073/pnas.88.15.6834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Krieg P A, Melton D A. Functional messenger RNAs are produced by SP6 in vitro transcription of cloned cDNAs. Nucleic Acids Res. 1984;12:7057–7070. doi: 10.1093/nar/12.18.7057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lau J T, Welply J K, Shenbagamartin P, Naider F, Lennarz W J. Substrate recognition by oligosaccharyl transferase. J Biol Chem. 1983;258:15255–15260. [PubMed] [Google Scholar]
- 24.Leduc R, Molloy S S, Thorne B A, Thomas G. Activation of human furin precursor processing endoprotease occurs by an intramolecular autoproteolytic cleavage. J Biol Chem. 1992;267:14304–14308. [PubMed] [Google Scholar]
- 25.Li Y, Inouye M. Autoprocessing of prothiolsubtilisin E in which active-site serine 211 is altered to cysteine. J Biol Chem. 1994;269:4169–4174. [PubMed] [Google Scholar]
- 26.Li Y, Hu Z, Jordan F, Inouye M. Functional analysis of the propeptide of subtilisin E as an intramolecular chaperone for protein folding. J Biol Chem. 1995;270:25127–25132. doi: 10.1074/jbc.270.42.25127. [DOI] [PubMed] [Google Scholar]
- 27.Lindberg I. Evidence for cleavage of the PC1/PC3 pro-segment in the endoplasmic reticulum. Mol Cell Neurosci. 1994;5:263–268. doi: 10.1006/mcne.1994.1030. [DOI] [PubMed] [Google Scholar]
- 28.Matthews G, Colman A. A highly efficient, cell-free translation/translocation system prepared from Xenopus eggs. Nucleic Acids Res. 1991;19:6405–6412. doi: 10.1093/nar/19.23.6405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Matthews G, Shennan K I J, Seal A J, Taylor N A, Colman A, Docherty K. Autocatalytic maturation of the prohormone convertase PC2. J Biol Chem. 1994;269:588–592. [PubMed] [Google Scholar]
- 30.Maundrell K. Thiamine-repressible expression vectors pREP and pRIP for fission yeast. Gene. 1993;123:127–130. doi: 10.1016/0378-1119(93)90551-d. [DOI] [PubMed] [Google Scholar]
- 31.Molloy S S, Thomas L, Van Slyke J K, Stenberg P E, Thomas G. Intracellular trafficking and activation of the furin proprotein convertase: localisation to the TGN and recycling from the cell surface. EMBO J. 1994;13:18–33. doi: 10.1002/j.1460-2075.1994.tb06231.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moreno S, Klar A, Nurse P. An introduction to molecular genetic analysis of the fission yeast Schizosaccharomyces pombe. Methods Enzymol. 1991;194:795–823. doi: 10.1016/0076-6879(91)94059-l. [DOI] [PubMed] [Google Scholar]
- 33.Nakagawa T, Hosaka M, Torii S, Watanabe T, Murakami K, Nakayama K. Identification and functional expression of a new member of the mammalian Kex2-like processing endoprotease family—its striking structural similarity to PACE4. J Biochem. 1993;113:132–135. doi: 10.1093/oxfordjournals.jbchem.a124015. [DOI] [PubMed] [Google Scholar]
- 34.Nakayama K, Kim W-S, Torii S, Hosaka M, Nakagawa T, Ikemizu J, Baba T, Murakami K. Identification of the fourth member of the mammalian endoprotease family homologous to the yeast Kex2 protease. J Biol Chem. 1992;267:5897–5900. [PubMed] [Google Scholar]
- 35.Power S D, Adams R M, Wells J A. Secretion and autoproteolytic maturation of subtilisin. Proc Natl Acad Sci USA. 1986;83:3096–3100. doi: 10.1073/pnas.83.10.3096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Roebroek A J M, Pauli I G L, Zhang Y, Van de Ven W J M. cDNA sequence of a Drosophila melanogaster gene, dfur1, encoding a protein structurally related to the subtilisin-like proprotein processing enzyme furin. FEBS Lett. 1991;289:133–137. doi: 10.1016/0014-5793(91)81052-a. [DOI] [PubMed] [Google Scholar]
- 37.Roebroek A J M, Creemers J W M, Pauli I G L, Kurzikdumke U, Rentrop M, Gateff E A F, Leunissen J A M, Van de Ven W J M. Cloning and functional expression of dfurin2, a subtilisin-like proprotein processing enzyme of Drosophila melanogaster with multiple repeats of a cysteine motif. J Biol Chem. 1992;267:17208–17215. [PubMed] [Google Scholar]
- 38.Schägger H, von Jagow G. Tricine-sodium dodecyl sulfate polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa. Anal Biochem. 1987;166:369–379. doi: 10.1016/0003-2697(87)90587-2. [DOI] [PubMed] [Google Scholar]
- 39.Shennan K I J, Taylor N A, Jermany J L, Matthews G, Docherty K. Differences in pH optima and calcium requirements for maturation of the prohormone convertases PC2 and PC3 indicate different intracellular locations for these events. J Biol Chem. 1995;270:1402–1407. doi: 10.1074/jbc.270.3.1402. [DOI] [PubMed] [Google Scholar]
- 40.Siezen R J, Leunissen J A M. Subtilases: the superfamily of subtilisin-like serine proteases. Protein Sci. 1997;6:501–523. doi: 10.1002/pro.5560060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Siezen R J, Leunissen J A M, Shinde U. Homology analysis of the propeptides of subtilisin-like serine proteases (subtilases) In: Shinde U, Inouye M, editors. Intramolecular chaperones and protein folding. R. G. Austin, Tex: Landes Company; 1995. pp. 233–255. [Google Scholar]
- 42.Silen J L, Frank D, Fujishige A, Bone R, Agard D A. Analysis of prepro-α-lytic protease expression in Escherichia coli reveals that the pro region is required for activity. J Bacteriol. 1989;171:1320–1325. doi: 10.1128/jb.171.3.1320-1325.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Smeekens S P, Steiner D F. Identification of a human insulinoma cDNA encoding a novel mammalian protein structurally related to the yeast dibasic processing protease Kex2. J Biol Chem. 1990;265:2997–3000. [PubMed] [Google Scholar]
- 44.Smeekens S P, Avruch A S, Lamendola J, Chan S J, Steiner D F. Identification of a cDNA encoding a second putative prohormone convertase related to PC2 in AtT20 cells and islets of Langerhans. Proc Natl Acad Sci USA. 1991;88:340–344. doi: 10.1073/pnas.88.2.340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Steiner D F, Smeekens S P, Ohagi S, Chan S J. The new enzymology of precursor processing endoproteases. J Biol Chem. 1992;267:23435–23438. [PubMed] [Google Scholar]
- 46.Tanguy-Rougeaua C, Weslowski-Louvel M, Fukuhara H. The Kluyveromyces lactis KEX1 gene encodes a subtilisin-like serine proteinase. FEBS Lett. 1988;234:464–470. doi: 10.1016/0014-5793(88)80139-x. [DOI] [PubMed] [Google Scholar]
- 47.Taylor N A, Shennan K I J, Cutler D F, Docherty K. Mutations within the propeptide, the primary cleavage site or the catalytic site, or deletion of C-terminal sequences, prevents secretion of proPC2 from transfected COS-7 cells. Biochem J. 1997;321:367–373. doi: 10.1042/bj3210367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Van den Ouweland A M V, Duijnhoven H L P, Keizer G D, Dorsser L C J, Van de Ven W J M. Structural homology between the human Fur gene product and the subtilisin-like protease encoded by yeast KEX-2. Nucleic Acids Res. 1990;18:664. doi: 10.1093/nar/18.3.664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vey M, Schäfer W, Berghöfer S, Klenk H-D, Garten W. Maturation of the trans-Golgi network protease furin: compartmentalisation of propeptide removal, substrate cleavage and COOH-terminal truncation. J Cell Biol. 1994;127:1829–1842. doi: 10.1083/jcb.127.6.1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vindrola O, Lindberg I. Biosynthesis of the prohormone convertase-mPC1 in AtT-20 cells. Mol Endocrinol. 1992;6:1088–1094. doi: 10.1210/mend.6.7.1508222. [DOI] [PubMed] [Google Scholar]
- 51.Wilcox C A, Fuller R S. Posttranslational processing of the prohormone-cleaving Kex2 protease in the Saccharomyces cerevisiae secretory pathway. J Cell Biol. 1991;115:297–307. doi: 10.1083/jcb.115.2.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhu X, Ohta Y, Jordan F, Inouye M. Pro-sequence of subtilisin can guide the refolding of denatured subtilisin in an intermolecular process. Nature. 1989;339:483–484. doi: 10.1038/339483a0. [DOI] [PubMed] [Google Scholar]
- 53.Zhu Y S, Zhang X Y, Cartwright C P, Tipper D J. Kex2-dependent processing of yeast K1 killer preprotoxin includes cleavage at ProArg-44. Mol Microbiol. 1992;6:511–520. doi: 10.1111/j.1365-2958.1992.tb01496.x. [DOI] [PubMed] [Google Scholar]