

Abstract
The Saccharomyces cerevisiae cell cycle is arrested in G1 phase by the mating factor pathway. Genetic evidence has suggested that the G1 cyclins Cln1, Cln2, and Cln3 are targets of this pathway whose inhibition results in G1 arrest. Inhibition of Cln1- and Cln2-associated kinase activity by the mating factor pathway acting through Far1 has been described. Here we report that Cln3-associated kinase activity is inhibited by mating factor treatment, with dose response and timing consistent with involvement in cell cycle arrest. No regulation of Cln3-associated kinase was observed in a fus3 kss1 strain deficient in mating factor pathway mitogen-activated protein (MAP) kinases. Inhibition occurs mainly at the level of specific activity of Cln3-Cdc28 complexes. Inhibition of the C-terminally truncated Cln3-1-associated kinase is not observed; such truncations were previously identified genetically as causing resistance to mating factor-induced cell cycle arrest. Regulation of Cln3-associated kinase specific activity by mating factor treatment requires Far1. Overexpression of Far1 restores inhibition of C-terminally truncated Cln3-1-associated kinase activity. G2/M-arrested cells are unable to regulate Cln3-associated kinase, possibly because of cell cycle regulation of Far1 abundance. Inhibition of Cln3-associated kinase activity by the mating factor pathway may allow this pathway to block the earliest step in normal cell cycle initiation, since Cln3 functions as the most upstream G1-acting cyclin, activating transcription of the G1 cyclins CLN1 and CLN2 as well as of the S-phase cyclins CLB5 and CLB6.
The G1 cyclins of Saccharomyces cerevisiae, encoded by CLN1, CLN2, and CLN3, cause postmitotic G1-phase cells to enter the cell division cycle (8, 37, 38, 45). Their direct role in this cell cycle transition, called Start, likely includes the activation of transcription of numerous genes (including CLN1 and CLN2 themselves, other cyclin genes, and genes required for DNA replication) (8, 26), the induction of cell polarization, and bud emergence (8). They also carry out several steps essential for activation of B-type cyclin (Clb)-dependent kinases: inducing proteolysis of the B-type cyclin-dependent kinase inhibitor Sic1 and blocking Clb proteolysis (1, 12, 37, 38). Cln3 is likely specialized for transcriptional activation (12, 29, 51, 55), whereas Cln1 and Cln2 are probably directly involved in bud emergence and other ‘Start-specific’ processes. Therefore, inactivation of Cln cyclins effectively blocks all postmitotic cell cycle events.
The mating factor signal transduction pathway in budding yeast is well characterized genetically and biochemically (27, 28). Binding of mating factor to a seven-transmembrane receptor activates a heterotrimeric G protein, the beta and gamma subunits of which carry signal to a mitogen-activated protein (MAP) kinase cascade. The MAP kinases, Fus3 and Kss1 (5, 15), are presumed to phosphorylate targets resulting in various aspects of the mating factor response. Far1 has been proposed as a target of the MAP kinases required for cell cycle arrest (16, 43, 44, 54). Far1 is required for cell cycle arrest only in the presence of Cln2 (3). This correlates with Far1-dependent inhibition of Cln2-associated kinase activity in vivo and in vitro (44). Far1 forms a complex with each of the three Cln-Cdc28 complexes (54). In addition, CLN1 and CLN2 transcription is strongly reduced in the presence of mating factor (57, 58). C-terminal truncations in Cln3 result in mating factor resistance (6, 36). Resistance due to truncated Cln3 is weakened but not abolished by deletion of CLN1 and CLN2 (11, 13).
These observations lead to a simple model in which cell cycle arrest by mating factor is mediated by inhibition of CLN function (8). While the mating factor pathway can inactivate Cln1 and Cln2 function both by transcriptional turnoff and by Far1-dependent inhibition of kinase activity, there is no information on how Cln3 function might be blocked by the mating factor pathway. It is logically required that Cln3 function be blocked in some way. Even the complete genetic absence of CLN1 and CLN2 has no effect on viability or growth rate provided CLN3 is present (7, 46), so that even complete inhibition of CLN1 and CLN2 cannot account for mating factor-induced cell cycle arrest. CLN3 transcription continues in the presence of mating factor (36). While previous data indicated no inhibitory effect of the mating factor pathway on Cln3-associated kinase activity (55), these experiments were performed in a cdc34 mutant strain background. Cdc34 is a ubiquitin-conjugating enzyme (21) whose mutation has pleiotropic phenotypes, including the strong elevation of Cln3-associated kinase activity for unknown reasons (56). We therefore reexamined the regulation of Cln3-associated kinase activity by the mating factor pathway without the use of the cdc34 mutation and detected significant regulation of Cln3-Cdc28 kinase activity.
MATERIALS AND METHODS
Yeast strains and plasmids.
Strains used in this study are listed in Table 1. Standard genetic methods were used for all strain constructions. All strains were congenic with BF264-15D. Most alleles used were described previously: cln deletion alleles (11), leu2::LEU2::GAL1::CLN3 (31), far1::URA3 and GAL1::FAR1 (34), cln3::URA3::GAL1::CLN3C or cln3::URA3::GAL1::CLN3-1C (three-hemagglutinin [HA] epitope-tagged version of CLN3 or CLN3-1 coding sequence) (56) (for GAL1::CLN3C, we used marker-swapped [9] versions in which URA3 was disrupted with LEU2 or TRP1); fus3::LEU2 (15), and kss1::URA3 (5). The trp1::TRP1::GAL1::CLN3C strain was made by transferring the insert from KL002 (29) to the integrating RS404 vector (50); the resulting plasmid, KL036, was digested with EcoRV to target integration to trp1 and used to transform appropriate yeast strains. In most experiments using the latter allele, spontaneous Trp+ derivatives (lacking GAL1::CLN3C) of the transformed strain were used as untagged controls.
TABLE 1.
S. cerevisiae strains used
| Strain | Genotype |
|---|---|
| 1255-5C | CLN1 CLN2 CLN3 |
| 1729-20D | CLN1 CLN2 CLN3 |
| 2928-2B | CLN1 CLN2 cln3::LEU2::GAL1::CLN3C |
| FCMT34-1 | CLN1 CLN2 cln3::URA3::GAL1::CLN3-1C |
| FCMT34-2 | CLN1 CLN2 cln3::URA3::GAL1::CLN3-1C |
| 2202-16A | cln1-del cln2-del cln3::LEU2::GAL1::CLN3C |
| 1807-31B | cln1-del cln2-del cln3::URA3::GAL1::CLN3-1C |
| BOY521-3a | CLN1 CLN2 CLN3 kss1::URA3 fus3::LEU2 TRP1 |
| BOY521-3b | CLN1 CLN2 CLN3 trp1::TRP1::GAL1::CLN3C kss1::URA3 fus3::LEU2 |
| BOY521-3c | CLN1 CLN2 CLN3 trp1::TRP1::GAL1::CLN3C kss1::URA3 fus3::LEU2 |
| 1255-5C-4a | CLN1 CLN2 CLN3 TRP1 |
| 1255-5C-4b | CLN1 CLN2 CLN3 trp1::TRP1::GAL1::CLN3C |
| 1729-5C | CLN1 CLN2 cln3::LEU2::GAL1::CLN3C |
| 1729-19A | CLN1 CLN2 cln3::LEU2::GAL1::CLN3C far1::URA3 |
| 1729-23D | cln1-del cln2-del cln3::LEU2::GAL1::CLN3C |
| 1729-11B | cln1-del cln2-del cln3::LEU2::GAL1::CLN3C far1::URA3 |
| 1723-6C | CLN1 CLN2 cln3::TRP1::GAL1::CLN3C leu2::LEU2::GAL1::FAR1 |
| BOY747 | cln1-del cln2-del cln3-del leu2::LEU2::GAL1::CLN3 |
| BOY901 | cln1-del cln2-del cln3-del leu2::LEU2::GAL1::CLN3 far1::URA3 |
| 1808-3 | CLN1 CLN2 CLN3 leu2::LEU2::GAL1::FAR1(Δ30) far1::URA3 |
| 1808-2 | CLN1 CLN2 cln3::TRP1::GAL1::CLN3C leu2::LEU2::GAL1::FAR1(Δ30) far1::URA3 |
All strains were congenic with BF264-15Daub (MATa bar1 trp1 leu2 ura3 ade1 his2) (46). See Materials and Methods for source of alleles.
Assay for mating factor sensitivity.
For the halo assay (15), exponentially growing cells were spread on YEPD or YEP-galactose plates, and paper discs containing 15 μl of 0.2, 0.1, and 0.05 mM α-factor were placed on the surface. Plates were incubated at 30°C for 2 days. For the budding assay, different doses of mating factor were added to exponentially growing cultures in liquid medium (usually YEP-galactose). At intervals, aliquots were removed, fixed, and sonicated, and the proportion of unbudded cells was determined microscopically.
Immunoprecipitation, histone H1 kinase activity assay, and immunoblot analysis.
Cells from 100 ml of culture were collected by filtration on a Millipore filter, rinsed off the filter in LSHN buffer (10 mM HEPES [pH 7.5], 50 mM NaCl, 10% glycerol), and centrifuged at 1,000 rpm. Before centrifugation, 1 ml from each 100 ml of culture was saved for fixing. Fixing solution contains 0.037% formaldehyde in 1× phosphated-buffered saline. Cell pellets were resuspended in 1 ml of LSHN, transferred to a microcentrifuge tube, and pelleted. Washed cell pellets were resuspended in buffer N (44) (50 mM Tris-HCl [pH 7.5], 100 mM NaCl, 0.1 mM EDTA, 10 mM NaF, 60 mM β-glycerophosphate, 0.1% Nonidet P-40 [NP-40]) with protease inhibitors (aprotinin [10% by volume of Sigma A2679], 10 μg of pepstatin per ml, 0.5 mM phenylmethylsulfonyl fluoride). Glass beads (425 to 600 μm) were added, and samples were vortexed in a Vortex Genie Sleeve. Vortexing was done twice for 3 min at 4°C. Cell lysates were centrifuged at 15,000 rpm for 2 min to remove cell debris. Cell lysates were immunoprecipitated with anti-HA monoclonal antibody 12CA5 (BabCo) (5 μg in each reaction) for 1 h on ice. After centrifugation at 15,000 rpm for 2 min, immune complexes were adsorbed onto protein A-agarose (Repligen) by addition of 35 μl of slurry followed by a 1-h rotation at 4°C. Immune complexes were washed with LSHNN buffer (10 mM HEPES [pH 7.5], 50 mM NaCl, 10% glycerol, 0.1% NP-40) three times, washed with kinase buffer twice, and resuspended in kinase buffer (10 mM HEPES [pH 7.5], 10 mM MgCl2, 1 mM dithiothreitol). Immune complexes were incubated with 50 μM ATP, 2 μg of histone H1, (Boerhinger), and [γ-32P]ATP (2 μCi). Incubation was carried out at 30°C for 10 to 30 min (the reaction was approximately linear for the first 10 min and then slowed; the different incubation times did not make a significant difference for the final results). The reaction was stopped by adding sample buffer (62.5 mM Tris-HCl [pH 6.8], 10% glycerol, 2% sodium dodecyl sulfate [SDS], 0.0025% bromophenol blue, 2% β-mercaptoethanol). Samples were denatured at 95°C for 5 min. Denatured samples were electrophoresed on SDS–12% polyacrylamide gels. Gels were transferred to an Immobilon-P membrane with semidry blot transfer (Hoefer), and the membrane was exposed to film for 30 to 60 min to detect kinase activity. Kinase assays were quantitated with a PhosphorImager.
For immunoblot analysis, immunoprecipitates were denatured in sample buffer for 5 min at 95°C. Denatured samples were loaded for SDS-polyacrylamide gel electrophoresis. Gels were transferred by electroblotting to an Immobilon-P membrane for 1 h at 4°C. After transfer, the membrane was incubated with blocking buffer (1× phosphate-buffered saline, 0.1% NP-40, 0.5% Tween 20, 0.5% [wt/vol] bovine serum albumin, 1% nonfat dried milk, 0.02% NaN3) and rinsed in the same buffer without NaN3. Polyclonal anti-HA antibody (BabCo) was used at a 1:5,000 dilution, and polyclonal anti-CDC28 antibody (a gift from R. Deshaies) was used at a 1:7,500 dilution. Polyclonal anti-rabbit horseradish peroxidase was used at a 1:1,000 dilution. Enhanced chemiluminescence reagents (Pierce) were added to each blot for 10 to 15 min.
Other methods.
Cell cycle synchronization and RNA hybridization analysis were carried out as described previously (40). DNA flow cytometry was carried out as described elsewhere (17).
RESULTS
Cln3-Cdc28 kinase activity is inhibited by mating factor treatment; effective regulation is dependent on the Cln3 C terminus.
When we measured Cln3-associated histone H1 kinase activity in asynchronously cycling cultures expressing HA epitope-tagged Cln3 from the GAL1 promoter, compared to cultures treated with mating factor for one generation time, we found a striking decline in activity (Fig. 1). The exact degree of regulation is somewhat difficult to quantitate because in many experiments the Cln3-associated activity is reduced to near the background signal observed with cells not expressing an epitope-tagged protein, but we observe at least 5- to 10-fold inhibition across various experiments. Similar inhibition of Cln3-associated kinase activity was observed at a range of ATP concentrations from 5 to 100 μM (data not shown), suggesting that the drop in kinase specific activity was not related to a drop in Km for ATP.
FIG. 1.
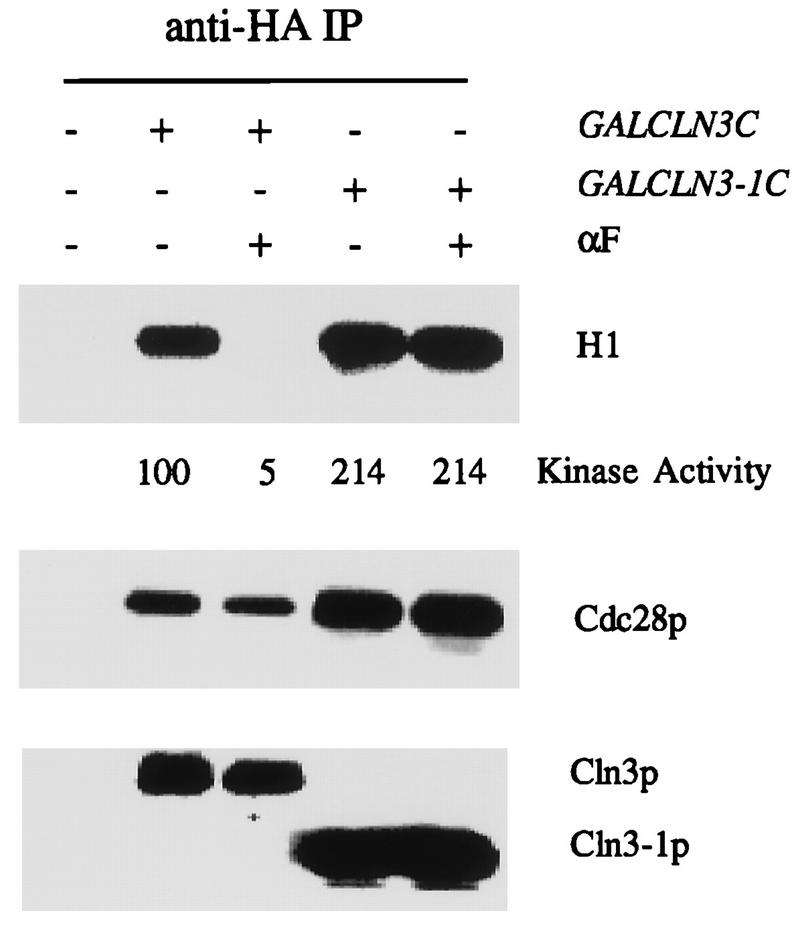
Regulation of Cln3-associated kinase activity by the mating factor pathway dependent on the Cln3 C terminus. Cells of genotype MATa bar1 GAL1::CLN3C (2928-2B) or GAL1::CLN3-1C (FCMT34-1) (three-HA epitope-tagged version of CLN3 or CLN3-1 coding sequence [56]) and a congenic CLN3 (untagged) control (1255-5C) were grown in YEP-Gal medium overnight at 30°C to log phase (optical density at 660 nm [OD660] of ∼0.5). α-Factor (αF) was added to 0.5 μM, and incubation continued for 2.5 h. Cells were extracted, and extracts were immunoprecipitated (IP) with antibody against the epitope tag. Immunoprecipitates were immunoblotted with anti-HA antibody or anti-Cdc28 antibody. Cln3-associated histone H1 kinase activity was determined and quantitated with a PhosphorImager. The background H1 phosphorylation signal from the untagged control was subtracted from all values before standardizing to the signal from sample 2 (tagged Cln3, no mating factor, set at 100 arbitrary units).
In many experiments, we noted some reduction in Cln3 protein levels following α-factor treatment. We quantitated the decrease in Cln3 levels that we observed by a combination of densitometry and serial dilution of standards on immunoblots and found that the average reduction was approximately twofold (data not shown). In no experiment was the quantitated reduction in Cln3 abundance sufficient to explain the reduction in Cln3-associated kinase activity.
Cln3 is likely to function by activating protein kinase activity of the cyclin-dependent kinase Cdc28 (10, 56). We detected approximately similar levels of Cdc28 coimmunoprecipitated with Cln3 from extracts of cycling or arrested cultures (Fig. 1), indicating that the regulation of Cln3-associated kinase activity was not at the level of Cdc28 binding. Therefore, we conclude that the specific activity of the Cln3-Cdc28 complex was reduced by mating factor treatment.
C-terminal truncation of Cln3 results in mating factor resistance (6, 36). We therefore examined whether kinase activity associated with truncated Cln3 was sensitive to mating factor treatment, using strains containing GAL1::CLN3-1C (Fig. 1 and 2). The mating factor resistance of cells expressing truncated Cln3 is significantly although not completely dependent on CLN1 and CLN2 (13). Therefore, we tested regulation of Cln3-1-associated kinase in a cln1 cln2 background (Fig. 2). We observed little regulation of Cln3-1 kinase activity in these strains, although they were significantly less resistant to division arrest by mating factor than isogenic CLN1 CLN2 GAL1::CLN3-1 controls by halo assay (data not shown).
FIG. 2.

C-terminus-dependent regulation of Cln3-associated kinase activity by the mating factor pathway is independent of CLN1 and CLN2. Strains of genotype MATa bar1 GAL1::CLN3C or GAL1::CLN3-1C and a congenic CLN3 (untagged [U]) control (1255-5C) were grown in YEP-Gal medium overnight at 30°C to log phase (OD660 of ∼0.5). For GAL1::CLN3C and GAL1::CLN3-1C strains, CLN1 CLN2 strains (2928-2B and FCMT34-1) and congenic cln1 cln2 strains (2202-16A and 1807-31B) were tested. α-Factor (αF) was added to 0.5 μM, and incubation continued for 2.5 h. Cells were extracted, and extracts were immunoprecipitated (IP) with antibody against the epitope tag. Immunoprecipitates were immunoblotted with anti-HA antibody. Cln3-associated histone H1 kinase activity was determined and quantitated with a PhosphorImager. The background H1 phosphorylation signal from the untagged control was subtracted from all values before standardizing to the signal from sample 3 (tagged Cln3, no mating factor, set at 100 arbitrary units). The amount of Cln3 in the immunoprecipitates was estimated by densitometry of the Western blot signal. The specific activity (S.A.) of the kinase is the kinase recovered divided by the Cln3 Western blot signal, in arbitrary units.
We have used Cln3-overexpressing strains for the experiments reported here, since Cln3-associated kinase activity was relatively low even in these overexpressors. This should not strongly limit interpretation of our results, and preliminary results with Cln3 expressed from its own promoter confirm inhibition of activity by the mating factor pathway; these experiments are difficult, however, because the Cln3-associated kinase detected is near the background signal from cells not expressing epitope-tagged protein (data not shown).
Dose response and kinetics of inhibition of Cln3-associated kinase compared to cell cycle inhibition.
If the observed inhibition of Cln3-associated kinase activity is biologically significant for cell cycle arrest induced by mating factor, then it should occur with timing and dose response consistent with cell cycle arrest. Indeed, we observed significant inhibition of Cln3-associated kinase at doses of mating factor lower than those required for effective cell cycle arrest (monitored by accumulation of unbudded cells), and maximal inhibition of kinase activity occurred at the minimum dose for maximal cell cycle arrest (Fig. 3). The decrease in Cln3-associated kinase activity following addition of mating factor preceded the accumulation of unbudded G1 cells. These correlational results are consistent with the idea that inhibition of Cln3-associated kinase activity is required for cell cycle arrest.
FIG. 3.

Time course and dose response of inhibition of Cln3-associated kinase activity compared to cell cycle arrest. Strains of genotype MATa bar1 GAL1::CLN3C (2928-2B) and a CLN3 (untagged [U]) control (1255-5C) were grown in YEP-Gal medium overnight at 30°C to log phase (OD660 of ∼0.5). (A) α-Factor (αF) was added to 0.5 μM. At the indicated times, cells were harvested. (B) The indicated concentrations of α-factor were added, and incubation continued for 2.5 h, when cells were harvested. (A and B) The percentage of unbudded cells was determined microscopically. Cells were extracted, and extracts immunoprecipitated (IP) with antibody against the epitope tag. Immunoprecipitates were immunoblotted with anti-HA antibody. Cln3-associated histone H1 kinase activity was determined and quantitated with a PhosphorImager. The background H1 phosphorylation signal from the untagged control was subtracted from all values before standardizing to the signal from the time zero sample (tagged Cln3, no mating factor, set at 100 arbitrary units).
Regulation of Cln3-associated kinase activity depends on MAP kinase function and on Ste12.
The mating factor pathway has as downstream effectors a redundant pair of MAP kinases, Fus3 and Kss1 (5, 15, 16, 27, 28). We examined the effect of disruption of either or both of these genes on regulation of Cln3-associated kinase activity. We found that while neither gene was essential for regulation of Cln3-associated kinase (data not shown), at least one of the pair was required (Fig. 4).
FIG. 4.

Regulation of Cln3-associated kinase activity requires a mating factor pathway-regulated MAP kinase. Cells of genotype MATa bar1 GAL1::CLN3C and a CLN3 (untagged) control were grown in YEP-Gal medium overnight at 30°C to log phase (OD660 of ∼0.5). For both tagged and untagged strains, both FUS3 KSS1 (1255-5C-4b and 1255-5C-4a) and fus3 kss1 (BOY521-3b, -3c, and -3a) strains were tested. α-Factor (αF) was added to 0.5 μM, and incubation continued for 2.5 h. Cells were extracted, and extracts were immunoprecipitated (IP) with antibody against the epitope tag. Immunoprecipitates were immunoblotted with anti-HA antibody. Cln3-associated histone H1 kinase specific activity (S.A.) was determined and quantitated with a PhosphorImager. The background H1 phosphorylation signals from the untagged controls were subtracted from all values before standardizing to the signal from sample 9 (tagged Cln3, FUS3 KSS1 wild type, no mating factor, set at 100 arbitrary units).
Deletion of STE5, which encodes a potential scaffold protein required for MAP kinase activation (4), also completely blocks regulation of Cln3-associated kinase by mating factor treatment (data not shown). The Ste12 transcription factor is essential for transcriptional induction and cell cycle arrest in response to mating factor (14, 18). Unlike Ste5, Ste12 is not required for activation of the Fus3 MAP kinase in response to mating factor (27, 28, 60). Nevertheless, Ste12 is required for regulation of Cln3-associated kinase activity by the mating factor pathway (data not shown).
Far1 is required for regulation of Cln3-associated kinase specific activity.
When α-factor was added to cultures of far1 or FAR1 strains expressing GAL1::CLN3-HA, the far1 strain exhibited little or no regulation of Cln3-associated kinase activity by mating factor (Fig. 5). Previous genetic and biochemical results (3, 43, 44, 54) suggested that Far1 functioned as an inhibitor of Cln1 and Cln2 function. Therefore, we asked whether regulation of Cln3-associated kinase would remain Far1 dependent in the absence of Cln1 and Cln2. We found that deletion of CLN1 and CLN2 largely restored the apparent sensitivity of recovered Cln3-associated kinase to mating factor in the absence of Far1. However, in this case the inhibition of Cln3-associated kinase appeared to be largely at the level of Cln3 protein abundance. In seven experiments, we found an average decrease in Cln3 abundance in far1 cln1 cln2 strains treated with mating factor of 2.9-fold (standard error of mean [SEM] = 0.6), with a decrease in total recovered kinase activity of 4.7-fold (SEM = 0.6); the decrease in Cln3 abundance in FAR1 cln1 cln2 strains treated in parallel was 1.4-fold (SEM = 0.2) with a decrease in total recovered kinase activity of 10.2-fold (SEM = 2.2) (Fig. 5 and data not shown; in these experiments, recovered kinase activity was quantitated by PhosphorImager analysis, and Cln3 protein abundance was quantitated by densitometric scanning combined in some experiments with serial dilution to be sure that the scanner was in a linear range). Thus, most or all of the regulation by the mating factor pathway of the specific activity of Cln3-associated kinase activity (defined as kinase activity recovered per Cln3 protein immunoprecipitated) requires Far1, even in the absence of CLN1 and CLN2.
FIG. 5.

Far1 is required for regulation of Cln3-associated kinase specific activity. (A) Cells of genotype MATa bar1 GAL1::CLN3C and a CLN3 (untagged) control (1255-5C) were grown in YEP-Gal medium overnight at 30°C to log phase (OD660 of ∼0.5). Strains tested were FAR1 (2928-2B) or far1::URA3 (1729-19A). α-Factor (αF) was added to 0.5 μM, and incubation continued for 2.5 h. Cells were extracted, and extracts were immunoprecipitated (IP) with antibody against the epitope tag. Immunoprecipitates were immunoblotted with anti-HA antibody. Cln3-associated histone H1 kinase specific activity (S.A.) was determined and quantitated with a PhosphorImager. The background H1 phosphorylation signals from the untagged controls were subtracted from all values before standardizing to the signal from sample 2 (tagged Cln3, no mating factor, set at 100 arbitrary units). (B) Cells of genotype MATa bar1 GAL1::CLN3C and a CLN3 (untagged [UT]) control (1729-20D) were grown in YEP-Gal medium overnight at 30°C to log phase (OD660 of ∼0.5). Strains tested were CLN1 CLN2 FAR1 (1729-5C), CLN1 CLN2 far1::URA3 (1729-19A), cln1 cln2 FAR1 (1729-23D), and cln1 cln2 far1::URA3 (1729-11B). α-Factor was added to 0.5 μM, and incubation continued for 2.5 h. Cells were extracted and extracts immunoprecipitated with antibody against the epitope tag. Immunoprecipitates were immunoblotted with anti-HA antibody. Cln3-associated histone H1 kinase activity was determined and quantitated with a PhosphorImager. The background H1 phosphorylation signals from the untagged controls were subtracted from all values before standardizing to the signal from sample 2 (tagged Cln3, FAR1 CLN1 CLN2 strain, no mating factor, set at 100 arbitrary units). (C) Cells of genotype MATa bar1 GAL1::CLN3C and a CLN3 (untagged) control (1255-5C) were grown in YEP-Gal medium overnight at 30°C to log phase (OD660 of ∼0.5). Strains tested were cln1 cln2 FAR1 (1729-23D) and cln1 cln2 far1::URA3 (1729-11B). α-Factor was added to various concentrations (0, 0.001, 0.01, 0.1, and 0.5 μM), and incubation continued for 2.5 h. Cells were extracted, and extracts were immunoprecipitated with antibody against the epitope tag. Immunoprecipitates were immunoblotted with anti-HA antibody. Cln3-associated histone H1 kinase activity was determined and quantitated with a PhosphorImager. The background H1 phosphorylation signal from the untagged control was subtracted from all values before standardizing to the signal from sample 3 (tagged Cln3, no mating factor, set at 100 arbitrary units). In panels B and C, the amount of Cln3 in the immunoprecipitates was estimated by densitometry of the Western blot signal. The specific activity (S.A.) of the kinase is the kinase recovered divided by the Cln3 Western blot signal, in arbitrary units.
Cln3 protein abundance is controlled by C-terminal sequences and C-terminal phosphorylation destabilizing the protein (10, 56, 59). Therefore, we wished to examine more closely whether Far1 could regulate kinase specific activity associated with truncated Cln3, which might escape regulation of Cln3 protein abundance. As shown above, cells expressing GAL1::CLN3-1–HA do not exhibit mating factor-induced inhibition of Cln3-1-associated kinase. In contrast, when we simultaneously overexpress FAR1 by using a GAL1::FAR1 construct, we find significant regulation of Cln3-1-associated kinase by mating factor treatment (Fig. 6). This is observed in the presence or absence of CLN1 and CLN2 (data not shown), suggesting that the effect may be direct. In addition, we observe strong mating factor-dependent coimmunoprecipitation of Far1 with Cln3-1–HA, consistent with a direct Far1-dependent inhibition of Cln3-1-associated kinase (Fig. 6). This mating factor-dependent increase in binding is not due to an increase in overall Far1 levels (data not shown). These results demonstrate a Far1-dependent and possibly direct mechanism for inhibition of Cln3-associated kinase specific activity that is active against both full-length and truncated Cln3. The failure of regulation of truncated Cln3 without FAR1 overexpression may be due to acceleration of cell cycle Start by Cln3 truncation (6, 36), since this acceleration strongly reduces Far1 protein abundance (31).
FIG. 6.

FAR1 overexpression restores mating factor regulation of Cln3-1-associated kinase activity. (A) Cells of genotype MATa bar1 GAL1::CLN3C (2928-2B) or GAL1::CLN3-1C (three-HA epitope-tagged version CLN3-1 coding sequence [56]) (FCMT34-1, FCMT34-2, 1723-6C) and a CLN3 (untagged [U]) control (1255-5C) were grown in YEP-Gal medium overnight at 30°C to log phase (OD660 of ∼0.5). One GAL1::CLN3-1C strain (1723-6C) also expressed FAR1 from the GAL1 promoter. α-Factor (αF) was added to 0.5 μM, and incubation continued for 2.5 h. Cells were extracted, and extracts were immunoprecipitated (IP) with antibody against the epitope tag. Immunoprecipitates were immunoblotted with anti-HA antibody or anti-Far1 antibody. Cln3-associated histone H1 kinase activity was determined and quantitated with a PhosphorImager. The background H1 phosphorylation signals from the untagged controls were subtracted from all values before standardizing to the signal from sample 3 (tagged Cln3, no mating factor, set at 100 arbitrary units). (B) Strain 1723-6C (MATa bar1 GAL1::CLN3-1C GAL1::FAR1) was incubated for 2.5 h in 0, 0.001, 0.005, 0.01, 0.05, 0.1, 0.5, or 1 μM α-factor. Cln3-1 was immunoprecipitated, and H1 kinase activity and coimmunoprecipitated Far1 were assayed as for panel A.
Although Far1 binding to full-length Cln3 is not readily detected even under conditions where Cln3 kinase is inhibited (Fig. 6), binding can be detected when Far1 is overexpressed from the GAL1 promoter (data not shown). Tyers and Futcher (54) demonstrated binding of Far1 to Cln3 without overexpression. We do not know the stoichiometry of Cln3 and Far1 in these complexes.
To further characterize the mating factor response in the simultaneous absence of FAR1, CLN1, and CLN2, we blocked cln1 cln2 GAL1::CLN3 FAR1 and cln1 cln2 GAL1::CLN3 far1::URA3 strains in G1 by switch from galactose to raffinose medium and then added back galactose medium with or without mating factor addition (Fig. 7). In the absence of mating factor, the FAR1 and the far1 cultures synchronously activated transcription of PCL1, cln2, and histone H2A, budded, and entered S phase, with identical kinetics. Strikingly, while the FAR1 culture incubated in the presence of mating factor was blocked for all of these events, the far1::URA3 culture carried out all of these events (with some delay) (Fig. 7). (Ultimate inhibition of proliferation by mating factor in cln1 cln2 far1 GAL1::CLN3 strains occurs, but at an aberrant postreplication cell cycle position [42].) This result extends the observation that Cln3 kinase specific activity is not inhibited by mating factor in the absence of Far1 to a biological readout for Cln3 function (induction of new gene transcription and ultimately budding and DNA replication). There is clearly a significant delay in many early cell cycle events in this protocol caused by mating factor in the far1 strain, for unknown reasons.
FIG. 7.

FAR1 is required for cell cycle arrest in G1 in cln1 cln2 GAL1::CLN3 strains. Cells of genotype cln1 cln2 cln3 GAL1::CLN3 that were either FAR1 (BOY747) or far1::URA3 (BOY901) were grown in YEP-Gal medium overnight at 30°C to log phase (OD660 of ∼0.5). Cells were collected by filtration, resuspended in YEP-raffinose medium, incubated for 2.5 h at 30°C, and then released from the G1 block by addition of galactose to 3% as described previously (31). Ten minutes before galactose addition, 0.5 μM α-factor (αF) was added to one half of each culture. DNA content was analyzed by flow cytometry, and Northern blot analysis was performed, probing for the indicated transcripts. Open circles, FAR1 culture; closed circles, far1::URA3 culture.
The ability to regulate Cln3-Cdc28 kinase may not be uniform across the cell cycle.
Inhibition of Cln3-associated kinase is slow relative to the very rapid induction of transcription of mating factor-induced genes such as FUS1 (30, 53), which is nearly maximal within 15 min. One possibility to explain this would be if the ability to inhibit Cln3-associated kinase were restricted to only a portion of the cell cycle, meaning that cells would have to accumulate in a specific window of the cell cycle before kinase regulation was detectable. To test this, we blocked cells in G2/M with nocodazole. We observed that under these conditions, mating factor had no effect on Cln3-associated kinase (Fig. 8), although we know that mating factor signalling (based on induction of transcription of FUS1) is fully functional in nocodazole-blocked cells (41). This result supports the idea that slow kinetics of inhibition of Cln3-associated kinase might reflect transit of cells out of compartments of the cell cycle (for example, G2/M) in which Cln3-associated kinase is refractory to inhibition. We also noted that levels of both Cln3 protein and Cln3-associated kinase were elevated approximately twofold in the nocodazole-blocked cultures. Thus, some of the moderate decline in Cln3 protein levels that we observe in mating factor-treated cultures may be due to an indirect cell cycle position effect on Cln3 protein stability.
FIG. 8.

G2/M-arrested cells are unable to regulate Cln3-associated kinase activity due to a deficit in Far1 protein abundance. (A) Cells of genotype MATa bar1 GAL1::CLN3C (2928-2B) and a CLN3 (untagged) control (1255-5C) were grown in YEP-Gal medium overnight at 30°C to log phase (OD660 of ∼0.5). Nocodazole (NZ; 15 μg/ml) was added to half of each culture, and incubation continued for 2.5 h to allow mitotic arrest. α-Factor (αF) was added to 0.5 μM, and incubation continued. Cells were extracted, and extracts were immunoprecipitated (IP) with antibody against the epitope tag. Immunoprecipitates were immunoblotted with anti-HA antibody. Cln3-associated histone H1 kinase activity was determined and quantitated with a PhosphorImager. The background H1 phosphorylation signals from the untagged controls were subtracted from all values before standardizing to the signal from sample 2 (tagged Cln3, no mating factor, set at 100 arbitrary units). Lanes 1, 2, 7, and 8 are from untagged cells at 0 and 90 min of α-factor treatment. Other lanes are from tagged cells at 0 (lanes 3 and 9), 30 (lanes 4 and 10), 60 (lanes 5 and 11), and 90 min of α-factor treatment. (B) The experiment was identical to that in panel A except that tagged and untagged strains (1808-2 and 1808-3) contained GAL1::FAR1Δ30 (32) and were incubated in α-factor for 30 min after 2.5 h of nocodazole treatment.
A deficit in Far1 protein contributes to lack of regulation of Cln3-associated kinase activity in G2/M-blocked cells.
FAR1 transcription and Far1 protein stability are strongly cell cycle regulated (31, 32), and very low levels of Far1 protein accumulate in nocodazole-blocked cells even when the cells are treated with mating factor (data not shown). Thus, a requirement for Far1 could explain the inability of mating factor to inhibit Cln3-associated kinase in nocodazole-blocked cells. To test this idea, we overexpressed Far1 in cells that were either cycling or blocked in nocodazole. We used Far1 with a deletion of amino acids 2 to 30 to circumvent proteolytic control that strongly reduces Far1 protein levels in G2/M-blocked cells (31, 32). We found significant rescue of mating factor-dependent inhibition of Cln3-associated kinase in G2/M-arrested cultures by ectopic expression of Far1 (Fig. 8), suggesting that Far1 regulation accounts for at least some of the inability to regulate Cln3-associated kinase under these conditions.
DISCUSSION
Cln3-associated kinase is a logical target for the mating factor pathway.
As discussed in the introduction, Cln3 function needs to be blocked by the mating factor pathway in some way to account for cell cycle arrest. We have now demonstrated inhibition of Cln3-associated kinase activity by the mating factor pathway. A previous study that failed to detect such inhibition (55) either used cdc34 mutant strains or else examined Cln3 kinase expressed at such low abundance that reliable quantitation was not possible. cdc34 mutants have a significant defect in their mating factor signal transduction pathway, at least at a restrictive temperature (42); since Cdc34 is a ubiquitin-conjugating enzyme with multiple targets probably including Far1 (31) and the B-type cyclin-Cdc28 inhibitor Sic1 (48), the use of these mutant cells is problematical. For these reasons, we have used Cln3 overexpressors in these experiments to raise the kinase activity to an easily detectable level, thus allowing us to avoid the use of the cdc34 mutant strain. (We have not tested cdc34 mutant strains under our conditions, and so this ad hoc explanation for the discrepancy in results is untested.)
The three CLN genes are functionally redundant (i.e., any one is sufficient for viability, and loss of all three results in cell cycle arrest) (7, 46). Nevertheless, this apparent redundancy may be misleading, since these genes are arranged in a regulatory hierarchy, with Cln3 acting as a transcriptional activator of CLN1 and CLN2 as well as of other cyclin genes, such as CLB5,6 and PCL1,2, and of other genes (12, 17, 20, 26, 29, 33, 49, 51, 55). CLN1 and CLN2, as well as these other cyclin genes, then may carry out activation of downstream events resulting in cell cycle Start (8, 12). According to this hierarchical view, Cln3-associated kinase is a logical target for the mating factor pathway since its inactivation could contribute to efficient cell cycle arrest at the correct cell cycle position. In contrast, once CLN1 and CLN2 transcription has been activated, the mating factor pathway overall is inactivated (40); in addition, Far1, which is essential for regulation of CLN function by the mating factor pathway (3, 44) (see above), is rapidly degraded (31, 32). Nevertheless, inhibition of Cln3-associated kinase activity and of CLN1 and CLN2 transcription is not sufficient to account for mating factor-induced cell cycle arrest, since placing CLN1 or CLN2 under control of constitutive promoters does not result in full resistance to mating factor arrest unless the promoter is very strong (40, 44, 57). It is not known whether placing a sufficient number of downstream transcriptional targets of Cln3 (such as CLB5, PCL1, and others [12, 17, 20, 26, 29, 33, 49, 51, 55]) under constitutive promoters in addition to CLN1 and CLN2 would result in mating factor resistance. The mating factor pathway appears likely to have direct effects on the transcription of some of these genes independent of its effect on Cln-Cdc28 activity (23, 57).
Possible mechanism of inhibition of Cln3-associated kinase activity.
Our results suggest that Far1 is required for significant reduction of Cln3-associated kinase specific activity, for the following reasons: first, significant inhibition of Cln3-associated kinase specific activity is observed only in FAR1 strains; second, conditions in which Cln3-associated kinase inhibition is not observed (in CLN3-1-overexpressing cells and in nocodazole-treated cells) are conditions in which Far1 does not accumulate (31, 32), and overexpression of FAR1 restores significant inhibition in these circumstances. Far1 may act directly to regulate Cln3-associated kinase activity, as shown previously for Cln1- and Cln2-associated kinase activity (44). Preliminary results suggest that an in vitro inhibitor of Cln3-associated kinase activity may accumulate in mating factor-treated wild-type cells but not in mating factor-treated far1 cells (24). Inhibition of Cln3-associated kinase is not associated with loss of binding to Cdc28 (Fig. 1). We have attempted to remove inhibitors from Cln3 immunoprecipitates by washing in high-salt buffers, as described for Far1-Cln2 complexes by Peter and Herskowitz (44), but we have not found washing conditions that restore Cln3-associated kinase activity in immunoprecipitates from mating factor-treated cells, perhaps because Cln3-associated kinase activity is itself relatively sensitive to incubation in high salt (22).
In addition to Far1, the only characterized cyclin-dependent kinase inhibitor in budding yeast is Sic1 (34, 39, 48); we have found only minor effects of deletion of SIC1 on Cln3-associated kinase by mating factor treatment (data not shown).
Tyers et al. (56) showed that phosphorylation of Cln3-Cdc28 complexes was required for kinase activity. It is unclear whether the activating phosphorylations are on Cln3 or on Cdc28. The only known activating site of phosphorylation on Cdc28 is at Thr169 (19, 25, 52). The low level of Cln3-Cdc28 complex that we can purify is discouraging with respect to direct assay for phosphorylation of this residue in these complexes dependent on mating factor pathway activity. Known sites of phosphorylation on Cln3 are thought to be associated with its degradation, rather than with associated kinase activity (59).
Another mode of regulation of cyclin-dependent kinases is via inhibitory phosphorylation of the kinase catalytic subunit at Thr18 and Tyr19 (2, 35). We have assayed regulation of Cln3-associated kinase activity in a strain with a disrupted SWE1 gene. Swe1 is the only identified Cdc28-inhibitory kinase in budding yeast (2). We find no difference in regulation of Cln3-associated kinase activity as a consequence of SWE1 disruption (data not shown). We have also found normal mating factor-induced inhibition of Cln3-associated kinase in a strain expressing Cdc28 mutant at both Thr18 and Tyr19 (CDC28-T18V,Y19F) (data not shown).
Thus, at present our data suggest that Far1 is the most likely candidate for inhibiting Cln3-associated kinase specific activity. Why, then, are far1 cln1 cln2 strains mating factor sensitive (3)? Our data do not exclude the possibility of Far1-independent mechanisms that can regulate Cln3-associated kinase, especially at low levels of expression; alternatively, Cln3-associated kinase at endogenous levels of expression may simply be insufficient for efficient execution of Start due to other responses to mating factor that could be Far1 independent (for example, transcription of the SCB- and MCB-regulated genes, that come on at Start, has been suggested to be under direct control of the mating factor pathway [23, 57].) Also, far1 cln1 cln2 strains were reported to require more mating factor for efficient arrest than FAR1 cln1 cln2 strains (3).
Far1 could act by alteration of the folded structure of Cln3-associated Cdc28 to block catalytic activity, as was shown for the p27 inhibitor of Cdk2-cyclin A (47). Alternatively, it could inhibit by binding and sequestration of Cln3-Cdc28 complexes without direct inactivation of the catalytic machinery.
In conclusion, we have demonstrated that the kinase activity associated with Cln3, the most upstream G1 cyclin, is inhibited by the mating factor pathway. This inhibition is likely to require Far1 after its activation by the MAP kinases Fus3 and Kss1. Further work is required to understand the mechanism of inhibition.
ACKNOWLEDGMENTS
Thanks go to A. Gartner, M. Hoek, and M. Miller for communicating unpublished data, to M. Tyers for providing strains and plasmids, and to M. Tyers, J. Roberts, and A. Gartner for useful discussions.
This work was supported by PHS grant GM49716.
REFERENCES
- 1.Amon A, Irniger S, Nasmyth K. Closing the cell cycle circle in yeast: G2 cyclin proteolysis initiated at mitosis persists until the activation of G1 cyclins in the next cycle. Cell. 1994;77:1037–1050. doi: 10.1016/0092-8674(94)90443-x. [DOI] [PubMed] [Google Scholar]
- 2.Booher R N, Deshaies R J, Kirschner M W. Properties of Saccharomyces cerevisiae weel and its differential regulation of p34CDC28 in response to G1 and G2 cyclins. EMBO J. 1993;12:3417–3426. doi: 10.1002/j.1460-2075.1993.tb06016.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chang F, Herskowitz I. Identification of a gene necessary for cell cycle arrest by a negative growth factor of yeast: FAR1 is an inhibitor of a G1 cyclin, CLN2. Cell. 1990;63:999–1011. doi: 10.1016/0092-8674(90)90503-7. [DOI] [PubMed] [Google Scholar]
- 4.Choi K-Y, Satterberg B, Lyons D M, Elion E A. Ste5 tethers multiple protein kinases in the MAP kinase cascade required for mating in S. cerevisiae. Cell. 1994;78:499–512. doi: 10.1016/0092-8674(94)90427-8. [DOI] [PubMed] [Google Scholar]
- 5.Courchesne W E, Kunisawa R, Thorner J. A putative protein kinase overcomes pheromone-induced arrest of cell cycling in S. cerevisiae. Cell. 1989;58:1107–1119. doi: 10.1016/0092-8674(89)90509-6. [DOI] [PubMed] [Google Scholar]
- 6.Cross F R. DAF1, a mutant gene affecting size control, pheromone arrest, and cell cycle kinetics of Saccharomyces cerevisiae. Mol Cell Biol. 1988;8:4675–4684. doi: 10.1128/mcb.8.11.4675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cross F R. Cell cycle arrest caused by CLN gene deficiency in Saccharomyces cerevisiae resembles START-I arrest and is independent of the mating-pheromone signalling pathway. Mol Cell Biol. 1990;10:6482–6490. doi: 10.1128/mcb.10.12.6482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cross F R. Starting the cell cycle: what’s the point? Curr Opin Cell Biol. 1995;7:790–797. doi: 10.1016/0955-0674(95)80062-x. [DOI] [PubMed] [Google Scholar]
- 9.Cross F R. ‘Marker swap’ plasmids: convenient tools for budding yeast molecular genetics. Yeast. 1997;13:647–653. doi: 10.1002/(SICI)1097-0061(19970615)13:7<647::AID-YEA115>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 10.Cross F R, Blake C M. The yeast Cln3 protein is an unstable activator of Cdc28. Mol Cell Biol. 1993;13:3266–3271. doi: 10.1128/mcb.13.6.3266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cross F R, Tinkelenberg A H. A potential positive feedback loop controlling CLN1 and CLN2 gene expression at the start of the yeast cell cycle. Cell. 1991;65:875–883. doi: 10.1016/0092-8674(91)90394-e. [DOI] [PubMed] [Google Scholar]
- 12.Dirick L, Bohm T, Nasmyth K. Roles and regulation of Cln-Cdc28 kinases at the start of the cell cycle of Saccharomyces cerevisiae. EMBO J. 1995;14:4803–4813. doi: 10.1002/j.1460-2075.1995.tb00162.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dirick L, Nasmyth K. Positive feedback in the activation of G1 cyclins in yeast. Nature. 1991;351:754–757. doi: 10.1038/351754a0. [DOI] [PubMed] [Google Scholar]
- 14.Dolan J W, Kirkman C, Fields S. The yeast STE12 protein binds to the DNA sequence mediating pheromone induction. Proc Natl Acad Sci USA. 1989;86:5703–5707. doi: 10.1073/pnas.86.15.5703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Elion E A, Grisafi P L, Fink G R. FUS3 encodes a cdc2+/CDC28-related kinase required for the transition from mitosis into conjugation. Cell. 1990;60:649–664. doi: 10.1016/0092-8674(90)90668-5. [DOI] [PubMed] [Google Scholar]
- 16.Elion E A, Satterberg B, Kranz J E. FUS3 phosphorylates multiple components of the mating signal transduction cascade: evidence for STE12 and FAR1. Mol Biol Cell. 1993;4:495–510. doi: 10.1091/mbc.4.5.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Epstein C B, Cross F R. CLB5: a novel B cyclin from budding yeast with a role in S phase. Genes Dev. 1992;6:1695–1706. doi: 10.1101/gad.6.9.1695. [DOI] [PubMed] [Google Scholar]
- 18.Errede B, Ammerer G. STE12, a protein involved in cell-type-specific transcription and signal transduction in yeast, is part of protein-DNA complexes. Genes Dev. 1989;3:1349–1361. doi: 10.1101/gad.3.9.1349. [DOI] [PubMed] [Google Scholar]
- 19.Espinoza F H, Farrell A, Erdjument-Bromage H, Tempst P, Morgan D O. A cyclin-dependent kinase-activating kinase (CAK) in budding yeast unrelated to vertebrate CAK. Science. 1996;273:1714–1717. doi: 10.1126/science.273.5282.1714. [DOI] [PubMed] [Google Scholar]
- 20.Espinoza F H, Ogas J, Herskowitz I, Morgan D O. Cell cycle control by a complex of the cyclin HCS26 (PCL1) and the kinase PHO85. Science. 1994;266:1388–1391. doi: 10.1126/science.7973730. [DOI] [PubMed] [Google Scholar]
- 21.Goebl M G, Yochem J, Jentsch S, McGrath J P, Varshavsky A, et al. The yeast cell cycle gene CDC34 encodes a ubiquitin-conjugating enzyme. Science. 1988;241:1331–1335. doi: 10.1126/science.2842867. [DOI] [PubMed] [Google Scholar]
- 22.Hoek, M., and F. R. Cross. 1997. Unpublished data.
- 23.Horecka J, Sprague G J., Jr Identification and characterization of FAR3, a gene required for pheromone-mediated G1 arrest in Saccharomyces cerevisiae. Genetics. 1996;144:905–921. doi: 10.1093/genetics/144.3.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jeoung, D.-I., and F. R. Cross. 1997. Unpublished data.
- 25.Kaldis P, Sutton A, Solomon M J. The Cdk-activating kinase (CAK) from budding yeast. Cell. 1996;86:553–564. doi: 10.1016/s0092-8674(00)80129-4. [DOI] [PubMed] [Google Scholar]
- 26.Koch C, Nasmyth K. Cell cycle regulated transcription in yeast. Curr Opin Cell Biol. 1994;6:451–459. doi: 10.1016/0955-0674(94)90039-6. [DOI] [PubMed] [Google Scholar]
- 27.Kurjan J. The pheromone response pathway in Saccharomyces cerevisiae. Annu Rev Genet. 1993;27:147–179. doi: 10.1146/annurev.ge.27.120193.001051. [DOI] [PubMed] [Google Scholar]
- 28.Leberer E, Thomas D Y, Whiteway M. Pheromone signalling and polarized morphogenesis in yeast. Curr Opin Genet Dev. 1997;7:59–66. doi: 10.1016/s0959-437x(97)80110-4. [DOI] [PubMed] [Google Scholar]
- 29.Levine K, Huang K, Cross F R. Saccharomyces cerevisiae G1 cyclins differ in their intrinsic functional specificities. Mol Cell Biol. 1996;16:6794–6803. doi: 10.1128/mcb.16.12.6794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McCaffrey G, Clay F J, Kelsey K, Sprague G F. Identification and regulation of a gene required for cell fusion during mating of the yeast Saccharomyces cerevisiae. Mol Cell Biol. 1987;7:2680–2690. doi: 10.1128/mcb.7.8.2680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McKinney J D, Chang F, Heintz N, Cross F R. Negative regulation of FAR1 at the Start of the yeast cell cycle. Genes Dev. 1993;7:833–843. doi: 10.1101/gad.7.5.833. [DOI] [PubMed] [Google Scholar]
- 32.McKinney J D, Cross F R. FAR1 and the G1 phase specificity of cell cycle arrest by mating factor in Saccharomyces cerevisiae. Mol Cell Biol. 1995;15:2509–2516. doi: 10.1128/mcb.15.5.2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Measday V, Moore L, Ogas J, Tyers M, Andrews B. The PCL2 (ORFD)-PHO85 cyclin-dependent kinase complex: a cell cycle regulator in yeast. Science. 1994;266:1391–1395. doi: 10.1126/science.7973731. [DOI] [PubMed] [Google Scholar]
- 34.Mendenhall M D. An inhibitor of p34CDC28 protein kinase activity from Saccharomyces cerevisiae. Science. 1993;259:216–219. doi: 10.1126/science.8421781. [DOI] [PubMed] [Google Scholar]
- 35.Morgan D O. Principles of CDK regulation. Nature. 1995;374:131–134. doi: 10.1038/374131a0. [DOI] [PubMed] [Google Scholar]
- 36.Nash R, Tokiwa G, Anand S, Erickson K, Futcher A B. The WHI1+ gene of Saccharomyces cerevisiae tethers cell division to cell size and is a cyclin homolog. EMBO J. 1988;7:4335–4346. doi: 10.1002/j.1460-2075.1988.tb03332.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nasmyth K. Control of the yeast cell cycle by the Cdc28 protein kinase. Curr Opin Cell Biol. 1993;5:166–179. doi: 10.1016/0955-0674(93)90099-c. [DOI] [PubMed] [Google Scholar]
- 38.Nasmyth K. At the heart of the budding yeast cell cycle. Trends Genet. 1996;12:405–412. doi: 10.1016/0168-9525(96)10041-x. [DOI] [PubMed] [Google Scholar]
- 39.Nugroho T T, Mendenhall M D. An inhibitor of yeast cyclin-dependent protein kinase plays an important role in ensuring the genomic integrity of daughter cells. Mol Cell Biol. 1994;14:3320–3328. doi: 10.1128/mcb.14.5.3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Oehlen L J W M, Cross F R. G1 cyclins CLN1 and CLN2 repress the mating factor response pathway at Start in the yeast cell cycle. Genes Dev. 1994;8:1058–1070. doi: 10.1101/gad.8.9.1058. [DOI] [PubMed] [Google Scholar]
- 41.Oehlen, L. J. W. M., and F. R. Cross. 1997. Unpublished data.
- 42.Oehlen, L. J. W. M., D.-I. Jeoung, and F. R. Cross. Cyclin specificity and G1 arrest specificity of the mating factor pathway. Submitted for publication.
- 43.Peter M, Gartner A, Horecka J, Ammerer G, Herskowitz I. FAR1 links the signal transduction pathway to the cell cycle machinery in yeast. Cell. 1993;73:747–760. doi: 10.1016/0092-8674(93)90254-n. [DOI] [PubMed] [Google Scholar]
- 44.Peter M, Herskowitz I. Direct inhibition of the yeast cyclin-dependent kinase Cdc28-Cln by Far1. Science. 1994;265:1228–1231. doi: 10.1126/science.8066461. [DOI] [PubMed] [Google Scholar]
- 45.Pringle J R, Hartwell L H. The Saccharomyces cerevisiae cell cycle. In: Strathern J N, Jones E W, Broach J R, editors. The molecular biology of the yeast Saccharomyces: life cycle and inheritance. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1981. pp. 97–142. [Google Scholar]
- 46.Richardson H E, Wittenberg C, Cross F, Reed S I. An essential G1 function for cyclin-like proteins in yeast. Cell. 1989;59:1127–1133. doi: 10.1016/0092-8674(89)90768-x. [DOI] [PubMed] [Google Scholar]
- 47.Russo A A, Jeffrey P D, Patten A K, Massagu J, Pavletich N P. Crystal structure of the p27Kip1 cyclin-dependent-kinase inhibitor bound to the cyclin A Cdk2 complex. Nature. 1996;382:325–331. doi: 10.1038/382325a0. [DOI] [PubMed] [Google Scholar]
- 48.Schwob E, Böhm T, Mendenhall M D, Nasmyth K. The B-type cyclin kinase inhibitor p40SIC1 controls the G1 to S transition in S. cerevisiae. Cell. 1994;79:233–244. doi: 10.1016/0092-8674(94)90193-7. [DOI] [PubMed] [Google Scholar]
- 49.Schwob E, Nasmyth K. CLB5 and CLB6, a new pair of B cyclins involved in DNA replication in Saccharomyces cerevisiae. Genes Dev. 1993;7:1160–1175. doi: 10.1101/gad.7.7a.1160. [DOI] [PubMed] [Google Scholar]
- 50.Sikorski R S, Hieter P. A system of shuttle vectors and yeast strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stuart D, Wittenberg C. CLN3, not positive feedback, determines the timing of CLN2 transcription in cycling cells. Genes Dev. 1995;9:2780–2794. doi: 10.1101/gad.9.22.2780. [DOI] [PubMed] [Google Scholar]
- 52.Thuret J Y, Valay J G, Faye G, Mann C. Civ1 (CAK in vivo), a novel Cdk-activating kinase. Cell. 1996;86:565–576. doi: 10.1016/s0092-8674(00)80130-0. [DOI] [PubMed] [Google Scholar]
- 53.Trueheart J, Boeke J D, Fink G R. Two genes required for cell fusion during yeast conjugation: evidence for a pheromone-induced surface protein. Mol Cell Biol. 1987;7:2316–2328. doi: 10.1128/mcb.7.7.2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tyers M, Futcher B. Far1 and Fus3 link the mating pheromone signal transduction pathway to three G1-phase Cdc28 kinase complexes. Mol Cell Biol. 1993;13:5659–5669. doi: 10.1128/mcb.13.9.5659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tyers M, Tokiwa G, Futcher B. Comparison of the Saccharomyces cerevisiae G1 cyclins: Cln3 may be an upstream activator of Cln1, Cln2 and other cyclins. EMBO J. 1993;12:1955–1968. doi: 10.1002/j.1460-2075.1993.tb05845.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tyers M, Tokiwa G, Nash R, Futcher B. The Cln3-Cdc28 kinase complex of S. cerevisiae is regulated by proteolysis and phosphorylation. EMBO J. 1992;11:1773–1784. doi: 10.1002/j.1460-2075.1992.tb05229.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Valdivieso M H, Sugimoto K, Jahng K-Y, Fernandes P M B, Wittenberg C. FAR1 is required for posttranscriptional regulation of CLN2 gene expression in response to mating pheromone. Mol Cell Biol. 1993;13:1013–1022. doi: 10.1128/mcb.13.2.1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wittenberg C, Sugimoto K, Reed S I. G1-specific cyclins of S. cerevisiae: Cell cycle periodicity, regulation by mating pheromone, and association with the p34CDC28 protein kinase. Cell. 1990;62:225–237. doi: 10.1016/0092-8674(90)90361-h. [DOI] [PubMed] [Google Scholar]
- 59.Yaglom J, Linskens M H K, Sadis S, Rubin D M, Futcher B, Finley D. p34Cdc28-mediated control of Cln3 cyclin degradation. Mol Cell Biol. 1995;15:731–741. doi: 10.1128/mcb.15.2.731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhou Z, Gartner A, Cade R, Ammerer G, Errede B. Pheromone-induced signal transduction in Saccharomyces cerevisiae requires the sequential function of three protein kinases. Mol Cell Biol. 1993;13:2069–2080. doi: 10.1128/mcb.13.4.2069. [DOI] [PMC free article] [PubMed] [Google Scholar]