

Summary
In insects, the circulating lipoproteins vitellogenin and lipophorin function as yolk protein precursors during oogenesis. Here, we present a protocol for tracing vitellogenin and lipophorin pathways during egg development in three species of Chagas disease vectors (Triatominae, Hemiptera: Reduviidae). We describe steps for purifying and fluorescent labeling of lipoproteins, their injection into vitellogenic females, and analysis using confocal microscopy. This approach enables the fate of lipoprotein particles and their lipid cargo in ovarian tissues to be tracked at the subcellular level.
For complete details on the use and execution of this protocol, refer to Fruttero et al.1 and Leyria et al.2
Subject areas: Cell Biology, Microscopy, Protein Biochemistry, Protocols in Entomology
Graphical abstract
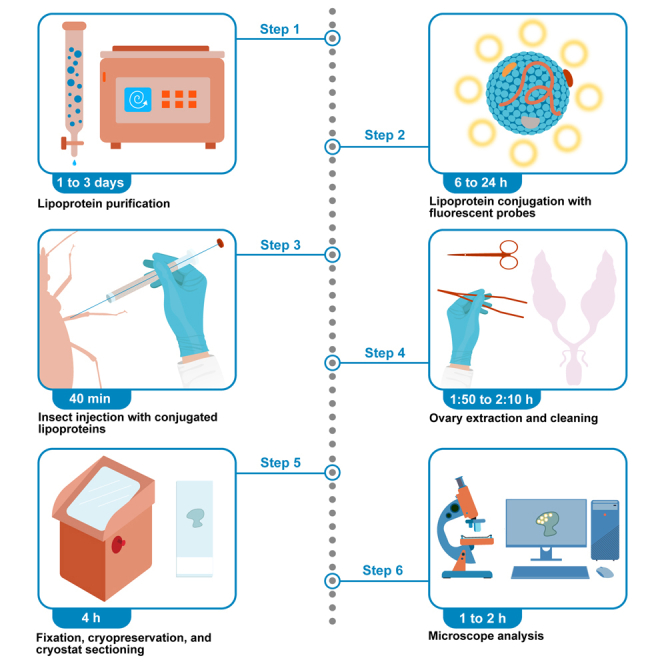
Highlights
-
•
Protocol for studying the lipoprotein pathway in triatomines using fluorescent probes
-
•
Steps for lipoprotein isolation and preparation for fluorescent labeling
-
•
Guidance on conjugation, lipoprotein injection, and tissue processing
-
•
Optimized for triatomines but adaptable to other insect species
Publisher’s note: Undertaking any experimental protocol requires adherence to local institutional guidelines for laboratory safety and ethics.
In insects, the circulating lipoproteins vitellogenin and lipophorin function as yolk protein precursors during oogenesis. Here, we present a protocol for tracing vitellogenin and lipophorin pathways during egg development in three species of Chagas disease vectors (Triatominae, Hemiptera: Reduviidae). We describe steps for purifying and fluorescent labeling of lipoproteins, their injection into vitellogenic females, and analysis using confocal microscopy. This approach enables the fate of lipoprotein particles and their lipid cargo in ovarian tissues to be tracked at the subcellular level.
Before you begin
Insect lipoproteins are macromolecular complexes primarily responsible for transporting and delivering lipids to target tissues.3 The main circulating lipoprotein in insects is lipophorin (Lp), while vitellogenin (Vg) serves as the primary precursor of yolk proteins.4 Both lipoproteins are synthesized in the fat body and secreted into the hemolymph. Vitellogenin is internalized by oocytes through endocytosis, whereas Lp can follow either endocytic or non-endocytic pathways in ovarian tissue, depending on the species.5 Lipoproteins can be traced when conjugated with radiolabeled lipids. However, compared to radioactive lipid labeling, fluorescently labeled lipoproteins offer several advantages: radioactive labeling is generally time-consuming, requires specific biosafety measures and regulations that may not always be available, and, more importantly, is less suitable for assessing cellular distribution in in vivo studies.6,7,8,9 Moreover, fluorescence labeling enables real-time tracking and high-resolution imaging, which is challenging with radioactive tracers. On the other hand, by combining exchangeable and non-exchangeable fluorescent markers for the protein and lipid moieties of lipoproteins, their pathways to ovarian tissue can be tracked using in vivo injection assays.1,2 This protocol provides detailed steps for Lp and Vg purification and labeling, enabling their use in in vivo assays to study their interactions with ovarian tissue and their intracellular processing. Although optimized for the triatomines Panstrongylus megistus, Dipetalogaster maxima, and Rhodnius prolixus, this protocol can be adapted for other insect species.
Institutional permissions
Prior to conducting experiments, the necessary institutional approvals for animal housing and maintenance must be obtained. Ethics statements regarding the blood source for rearing triatomines should address key aspects, including the use of commercial blood if applicable, the housing and handling of animals used as a blood source, protocols to minimize stress, and the frequency of bug-feeding sessions, among others.
In our case, insects were obtained from colonies fed on hen blood, following the recommendations of the National Institute of Parasitology (Health Ministry, Argentina).10 As described by Leyria et al.,11 housing and handling of hens adhered to protocols approved by the animal facility of the Centro de Investigaciones en Bioquímica Clínica e Inmunología (CIBICI-CONICET-Universidad Nacional de Córdoba, Res. Dec.-2021-737), in compliance with the Canadian Council on Animal Care guidelines (Assurance Number: A5802-01, Office of Laboratory Animal Welfare, NIH). The CIBICI animal facility operates under the Argentine National Ministry of Science (Sistema Nacional de Bioterios, MINCyT; www.bioterios.mincyt.gob.ar).
Lipophorin purification by density gradient ultracentrifugation
Timing: 24 h
-
1.Hemolymph collection.Note: Hemolymph is collected from vitellogenic females of D. maxima, P. megistus, and R. prolixus. The insects are maintained under standardized conditions (28°C, 70% relative humidity, 12:12 h light:dark cycle) and fed at species-appropriate intervals using an adequate blood source, such as hen's blood.11
-
a.Place the insects on ice for 10–15 min to immobilize them.
-
b.Cut one of the hind legs and, while applying gentle pressure, collect the hemolymph using a Hamilton syringe. The hemolymph is pooled into pre-chilled, pre-labeled microcentrifuge tubes with a fixed volume of 500 μL, which already contain Na2EDTA (final concentration: 10 mM), dithiothreitol (final concentration: 5 mM), and a protease inhibitor cocktail (final dilution: 1:250). Although the volume of hemolymph collected may vary, the final concentrations must be maintained.Note: The final concentration of the protease inhibitor cocktail should be 8 μM 4-(2-aminoethyl) benzenesulfonyl (AEBSF), 1.3 nM aprotinin, 0.46 μM bestatin, 56 nM E-64, 4 μM Na2EDTA, 1 nM leupeptin.
 CRITICAL: Avoid contamination with gut content by applying minimal pressure to the insect.
CRITICAL: Avoid contamination with gut content by applying minimal pressure to the insect. -
c.Centrifuge the collected hemolymph at 10,000 × g for 10 min at 4°C to remove hemocytes and debris. Transfer the supernatant to a new 1.5 mL microcentrifuge tube.
 Pause point: The supernatant can be stored at −80°C for up to three months before proceeding to the next step.
Pause point: The supernatant can be stored at −80°C for up to three months before proceeding to the next step.
-
a.
-
2.Lipophorin purification.
-
a.Preparation of hemocyte-free hemolymph for ultracentrifugation.
-
i.Adjust the hemocyte-free hemolymph to a density of 1.31 g/mL by adding anhydrous KBr (0.89 g of KBr to 2 mL of hemolymph).
-
ii.Measure the sample density and adjust, if necessary, using 0.15 M NaCl or additional KBr.Note: Density is best measured by refractometry for precise and rapid control (using for instance an Abbe refractometer 2WA-J), or alternatively by gravimetry if a refractometer is unavailable.
-
iii.Carefully layer the 1.31 g/mL hemolymph at the bottom of an appropriate ultracentrifuge tube, for instance a standard polypropylene Quick-Seal tube, overlay it with 0.15 M NaCl and seal using the hand-held sealing tool.
-
i.
-
b.Ultracentrifugation.
-
i.Centrifuge the sample at 212,000 × g (4°C) for 4 h using an ultracentrifuge equipped with a vertical rotor Beckman VTi 50 or similar.
-
ii.Carefully collect the Lp fraction, which appears as a distinct yellow band at the top of the gradient.
-
iii.Measure the density of the Lp fraction, which should be approximately 1.14 g/mL.Alternatives: The same method has been successfully tested using the Optima MAX-XP micro-ultracentrifuge, yielding comparable results:
-
iv.Prepare a KBr solution with a density of 1.12 g/mL.
-
v.Adjust hemocyte-free hemolymph to a density of 1.33 g/mL.
-
vi.Carefully layer 250 μL of the 1.33 g/mL hemolymph at the bottom of a 0.5 mL Open-Top Thickwall Polycarbonate tube (Beckman-Coulter, Cat#343776) and overlay it with 250 μL of the 1.12 g/mL KBr solution.
-
vii.Centrifuge at 435,400 × g (4°C) for 4 h using an Optima MAX-XP Ultracentrifuge with a TLA-120.1 rotor.
-
viii.After centrifugation, carefully collect the Lp fraction from the top of the tube.Pause point: Lipophorin with KBr can be stored at 4°C for up to one week.
-
i.
-
c.Desalt Lp.
-
i.Load the Lp fraction onto a PD-10 Desalting column (or an equivalent device) equilibrated with PBS to remove excess KBr.
-
ii.Since the PD-10 Desalting column is designed for sample volumes of 2.5 mL, dilute Lp with PBS to reach that volume.
-
iii.Load 2.5 mL of the sample, allowing it to enter the column completely before adding the elution buffer.
-
iv.Add the elution buffer (PBS) and collect 3.5 mL.Alternatives: To prevent sample dilution, a Thermo Scientific Zeba Spin Desalting Column can be used instead of a PD-10 Desalting Column.Alternatives: Another option to remove KBr is dialysis using membranes; however, it is less efficient in terms of time and yield compared to column-based methods. While dialysis can be useful for certain applications, column-based approaches provide a faster and more effective solution for buffer exchange and sample purification, making them preferable in most cases. For dialysis, use a dialysis membrane with a molecular weight cutoff (MWCO) <100 kDa and dialyze purified Lp against PBS. Perform at least three buffer exchanges, each lasting 3 h, under continuous stirring, using a 200:1 buffer-to-sample volume ratio. This dialysis protocol can be adapted for volumes below 600 μl.12
-
i.
-
d.Use an Amicon Ultra Centrifugal Filter 100 kDa MWCO (or an equivalent device) according to the manufacturer’s protocol to concentrate the eluate containing Lp to the desired concentration. For fluorophore conjugation, the concentration should be between 1 and 5 μg/μL.
-
i.Use the Bradford13 assay to determine the final protein concentration of the purified Lp.
-
ii.Check purity and integrity by running samples on PAGE under both denaturing and non-denaturing conditions and visualize the bands using Coomassie Blue staining.Optional: Although Lp isolation by ultracentrifugation gradient is suitable for most applications, some studies may require an additional step to obtain highly purified Lp for labeling:
-
iii.Pack a Sepharose 6B column (Pharmacia Fine Chemicals, 90 cm length, 2 cm diameter) and equilibrate it with PBS.
-
iv.Apply the concentrated Lp sample to the column and elute with PBS.
-
v.Collect fractions and monitor absorbance at 280 nm for proteins.
-
vi.Pool the fractions containing Lp based on 280 nm recorded profile and purity.Note: After running a 7.5% acrylamide SDS-PAGE gel, purified Lp should display two main bands corresponding to apolipophorin I (∼240 kDa) and apolipophorin II (∼80 kDa) as shown in Figure 1. Depending on the insect species and metabolic status, a third band corresponding to apolipophorin III (∼15 kDa) may be present. Its detection by SDS-PAGE will be determined by the acrylamide gel percentage used.Note: Fraction collection can be aided by a peristaltic pump.Note: For long-term storage, aliquot the Lp and keep it at −80°C. Avoid repeated freeze-thaw cycles to maintain stability.
-
i.
-
a.
Figure 1.

Electrophoretic profile of purified lipophorin from Panstrongylus megistus
(A) 5 μg of unconjugated lipophorin subjected to 7.5% SDS-PAGE and stained with Coomassie blue.
(B) 5, 2.5, and 1 μg of FITC-labeled lipophorin subjected to 7.5% SDS-PAGE and visualized under a UV transilluminator (negative image). The original fluorescent lane loaded with 5 μg of FITC-labeled lipophorin is shown in (B′).
Vitellogenin purification by ion-exchange chromatography
-
3.Preparation of the ion exchange column.
-
a.Pack the column with DEAE Trisacryl 3M.
-
b.Activate the matrix with 1 M NaCl. The column volume is 20 mL, and the flow rate should be approximately one drop every 6–7 s.
-
a.
-
4.Column equilibration.
-
a.Before purification, prepare the column as follows:
-
i.Wash with 2 column volumes of 100 mM Tris-HCl buffer (pH 8.3).
-
ii.Wash with 2-3 column volumes of 50 mM Tris-HCl buffer (pH 8.3).
-
iii.Finally, wash with at least 4 column volumes of 20 mM Tris-HCl buffer (pH 8.3).
-
i.
-
a.
Note: During this step, monitor the pH of the eluate at multiple points to ensure it remains between 8.2 and 8.3.
-
5.Ion-exchange chromatography.
-
a.Load the column with ∼15 mg of hemolymph, obtained as described in the “lipophorin purification by density gradient ultracentrifugation” section.
-
b.For the low-salt buffer elution step, use 20 mM Tris-HCl buffer (pH 8.3) and collect ∼2 mL fractions (40 drops) using a tube collector.
-
c.Measure absorbance at 280 nm in each tube, continuing collection until several consecutive tubes show very low readings (Figure 2).Note: This step allows for the selective removal of non-vitellogenin proteins that do not bind to the matrix.
-
d.For elution, use a 300 mM NaCl gradient in 20 mM Tris-HCl buffer (pH 8.3):
-
i.Load 150 mL of 300 mM NaCl and 150 mL of 20 mM Tris-HCl buffer into a gradient maker.
-
ii.Collect ∼2 mL fractions in test tubes and measure absorbance at 280 nm.Note: Two elution peaks (PI and PII) are commonly obtained (see Figure 2 for reference).
-
i.
-
e.Pool the fractions containing Vg based on 280 nm recorded profile and purity.
-
f.Use an Amicon Ultra Centrifugal Filter 100 kDa MWCO or an equivalent device to concentrate the eluate containing Vg to the desired concentration (1–5 μg/μL for fluorophore conjugation).
-
g.Determine the final protein concentration using the Bradford13 assay.
-
h.Assess Vg purity and integrity by running samples on SDS-PAGE under both denaturing and non-denaturing conditions, and visualize bands using Coomassie Blue staining.Note: After running a 7.5% acrylamide SDS-PAGE gel, purified Vg should appear as two major subunits (∼174 and ∼170 kDa, arranged in doublets) and two minor components (∼50 and ∼44 kDa).1Note: For long-term storage, aliquot the Vg and keep it at −80°C. Avoid repeated freeze-thaw cycles to maintain stability.Note: This protocol can also be used for purification of vitellin, the deposited form of Vg. In this case, the starting biological material is an egg homogenate.14
-
a.
Figure 2.

Vitellogenin purification by ion-exchange chromatography
The figure shows the absorbance (ABS) measured at 280 nm for test tubes (∼2 mL each) collected during elution. The two eluted peaks (PI: fractions 19–25; PII: fractions 30–36) are highlighted in blue.
Lipoprotein conjugation with different fluorescent labels
-
6.
Lipoprotein conjugation with fluorescein isothiocyanate (FITC).
FITC selectively labels the protein moiety of Lp and Vg, enabling the tracking of the entire particle’s fate.Note: This protocol is optimized for conjugating ∼1 mg of either Lp or Vg, regardless of the volume, as long as the concentration remains within the range of 1 to 5 μg/μL, to achieve an optimal fluorophore-to-lipoprotein ratio. For different ligand quantities, scale accordingly.-
a.Using a dialysis membrane with a MWCO <100 kDa, dialyze purified Lp or Vg against 50 mM borate buffer (pH 8.5) at a concentration of 1–5 μg/μL. Perform at least three buffer exchanges, each lasting 3 h, under continuous stirring, using a 200:1 buffer-to-sample volume ratio. This dialysis protocol can be adapted for volumes below 600 μL.12Pause point: The final buffer exchange can be left several hours.Note: To expedite the process, a desalting column can be used.
-
b.Prepare a 1 mg/mL FITC solution in DMSO.
-
c.Retrieve the lipoprotein from the dialysis membrane and mix it with 15 μL of the FITC solution.
-
d.Incubate in a flat-bottomed container under continuous stirring for 2 h at 20°C–25°C.CRITICAL: Protect all fluorophore-containing solutions from light.
-
e.While conjugation is in progress, equilibrate a desalting column with PBS or the desired buffer as described in step 2.c.
-
f.Pass the conjugation mixture through the column to remove unconjugated fluorophore.
-
g.The eluate can be concentrated to the desired final concentration (e.g., 0.5–1 μg/μL) using an Amicon Ultra Centrifugal Filter 100 kDa MWCO or equivalent. Protein concentration should be determined using the Bradford13 assay or an equivalent method.Note: Lp-FITC or Vg-FITC conjugates can be aliquoted and stored in microcentrifuge tubes at −20°C for up to one year.Note: To monitor the relative fluorescence intensity and the association of the fluorophore with the particle, it is advisable to subject the conjugate to 7.5% acrylamide SDS-PAGE and analyze the resulting gel under a UV transilluminator (Figure 1B).Note: The final concentration of conjugates can also be adjusted by diluting with PBS if needed.
-
a.
-
7.
Lipoprotein conjugation with 1,1′-Dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI).
DiI is a lipophilic, non-exchangeable dye that intercalates into the phospholipid fraction of lipoproteins,15 allowing the entire particle to be tracked.Note: This protocol is optimized for conjugating ∼1 mg of either Lp or Vg, regardless of the volume, as long as the concentration remains within the range of 1 to 5 μg/μL, to achieve an optimal fluorophore-to-lipoprotein ratio. For different ligand quantities, scale accordingly.-
a.Prepare a 3 mg/mL DiI solution in DMSO. Set up an appropriate agitation system at 37°C.
-
b.In a glass tube, while vortexing, add 75 μL of the DiI solution to 1 mL of lipoprotein in PBS. Incubate for 16 h at 37°C under constant agitation.CRITICAL: Prolonged agitation at this temperature using a magnetic stirrer may denature the lipoprotein. It is recommended to use a metabolic Dubnoff incubator or equivalent equipment.
-
c.Follow step 2.c. to remove unconjugated fluorophore.
-
a.
-
8.
Lipoprotein conjugation with 4,4-Difluoro-5,7-Dimethyl-4-Bora-3a,4a-Diaza-s-Indacene-3-Hexadecanoic Acid (Bodipy FL C16 or Bodipy-FA).
Bodipy-FA is a fluorescent palmitic acid analog that labels the exchangeable lipid fraction of lipoproteins,16 enabling the tracking of transported lipids.Note: This protocol is optimized for conjugating ∼1 mg of either Lp or Vg, regardless of the volume, as long as the concentration remains within the range of 1 to 5 μg/μL, to achieve an optimal fluorophore-to-lipoprotein ratio. For different ligand quantities, scale accordingly.-
a.Prepare a chloroform/methanol (2:1, v/v) mixture and dissolve 0.33 mg of Bodipy-FA in 1 mL of this solvent system.
-
b.Weigh 30 mg of glass beads, transfer them to a vial, and add the solubilized Bodipy-FA. Agitate gently on an orbital shaker (∼100 rpm) for 15 min at 20°C–25°C.
-
c.Evaporate the chloroform/methanol mixture using a gentle nitrogen stream, ensuring uniform distribution of the fluorophore at the bottom of the vial.
-
d.Once dried, add ∼1 mL of lipoprotein in PBS and stir gently for 2 h at 20°C–25°C.
-
e.Centrifuge the mixture at 10,000 × g for 5 min at 20°C–25°C to pellet the beads.
-
f.Collect the supernatant and follow step 2.c. to remove unconjugated fluorophore from the lipoprotein.
-
a.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| BODIPY FL C16 | Invitrogen | Cat#D3821; CAS: 158757-82-5 |
| Bradford Reagent | Sigma-Aldrich | Cat#B6916 |
| Chloroform | Sigma-Aldrich | Cat#650498; CAS: 67-66-3 |
| Coomassie blue R 250 | Fluka | Cat#27816; CAS: 6104-59-2 |
| Cryoplast | Biopack | Cat#1204.05 |
| DEAE-Trisacryl M | Sepracor | Cat#250771; CAS: 97930-01-3 |
| DiI | Sigma-Aldrich | Cat#468495; CAS: 41085-99-8 |
| Dithiothreitol | Millipore | Cat#10197777001; CAS: 3483-12-3 |
| DMSO | Millipore | Cat#D2650; CAS: 67-68-5 |
| Na2EDTA | Mallinckrodt | Cat#4931; CAS: 60-00-4 |
| FluorSave Reagent | Millipore | Cat#345789 |
| Formaldehyde | Sigma-Aldrich | Cat#F8775; CAS: 50-00-0 |
| FITC | Sigma-Aldrich | Cat#F7250; CAS: 27072-45-3 |
| Hydrochloric acid | Mallinckrodt | Cat#7647-01-0; CAS: 7647-01-0 |
| Methanol | Sigma-Aldrich | Cat#34860; CAS: 67-56-1 |
| NaCl | Millipore | Cat#116224; CAS: 7647-14-5 |
| Potassium bromide | Mallinckrodt | Cat#0505; CAS: 7758-02-3 |
| Monobasic potassium phosphate | Sigma-Aldrich | Cat#P5655; CAS: 7778-77-0 |
| Protease inhibitor cocktail | Sigma | Cat#P2714-1BTL |
| Sepharose 6B | Sigma | Cat#6B-100 |
| Sodium phosphate dibasic | Sigma-Aldrich | Cat#S9763; CAS 7558-79-4 |
| Sucrose | Mallinckrodt | Cat#8360; CAS: 57-50-1 |
| Tris | Bio Basic | Cat#TB0194; CAS: 126-72-7 |
| Experimental models: Organisms/strains | ||
| Dipetalogaster maxima (vitellogenic females) | Supplied by CIBICI-CONICET-UNC (Argentina) | RRID:NCBITaxon_72496 |
| Rhodnius prolixus (vitellogenic females) | Supplied by CIBICI-CONICET-UNC (Argentina) | RRID:NCBITaxon_13249 |
| Panstrongylus megistus (vitellogenic females) | Supplied by CIBICI-CONICET-UNC (Argentina) | RRID:NCBITaxon_65343 |
| Software and algorithms | ||
| Adobe Illustrator | Adobe Systems Incorporated | Released 2020. RRID:SCR_010279 |
| Adobe PhotoShop | Adobe Systems Incorporated | Released 2021. RRID:SCR_014199 |
| ImageJ (Fiji version) | Schindelin et al.13 | https://imagej.nih.gov/ij/ RRID:SCR_002285 |
| Other | ||
| Abbe refractometer | WYA | 2WA-J |
| Amber glass vials | N/A | N/A |
| Amicon ultra centrifugal filter, 100 kDa MWCO | Millipore | Cat#UFC9100 |
| Carbon steel edge blade | Treet | N/A |
| Chromatography column | Pharmacia Fine Chemicals | N/A |
| Clear nail polish | N/A | N/A |
| Corning PYREX Reusable Petri dishes | Fisher Scientific | Cat#08-747A |
| Cover glass 24 × 30 mm; thickness: 0.12 mm to 0.16 mm | Corning | Cat#2975-243 |
| Cryostat | Thermo Scientific | Model: Shadow Cryotome E |
| Dressing tissue forceps | Merck | Cat#F4642 |
| Metabolic shaking incubator | Vicking | Model: DUBNOFF |
| Extra fine Bonn scissors | FST | Cat#14084-08 |
| Fiber optic low-noise illuminator | Cole-Parmer 9741-50 | Model: 41510-00 |
| Fraction collector | LKB BROMMA | Model: 2112 Redirac |
| Glass beads, acid-washed | Sigma | Cat#G1145 |
| Hamilton syringe (Microliter #701) | Sigma-Aldrich | Cat#HAM80075 |
| Gradient maker | Hoefer | Cat#10049309 |
| Jewelers forceps, Dumont No. 5 | Merck | Cat#F6521 |
| Laser scanning confocal microscopy | Olympus | Model: FV1200 |
| Magnetic Plate Stirrer | Dauerhaft | Model: Mag D-320 digital |
| Micro-ultracentrifuge tube | Beckman Coulter | Cat#343776 |
| Micro-ultracentrifuge | Beckman Coulter | Model: Optima MAX-XP |
| Microscope slides | HDA | Cat#HDAS001 |
| Positive charged microscope slides | HDA | Cat#HDAS001 |
| Microtome blades | Feather | Cat#A35 |
| Mini-PROTEAN 3 cell for polyacrylamide gel electrophoresis (PAGE) setup | Bio-Rad | Cat#165-3301 |
| Nitrogen gas stream setup | N/A | N/A |
| Orbital shaker | Vicking | Model: M-23 |
| Paper masking tape | N/A | N/A |
| PD-10 Desalting columns | Cytiva | Cat#GE17-0851-01 |
| pH meter | Corning | Model: 430 |
| PowerPac Basic Power Supply | Bio-Rad | Cat#164-5050 |
| Spectrophotometer | Agilent | Model: BioTek Synergy HTX |
| Standard cellulose dialysis tubing | Fisher Scientific | Cat#0867139 |
| Stereoscopic microscope | Schonfeld Optik microscope | Model: LBX |
| Tabletop centrifuge | Eppendorf | Model: 5810/5810R (EP022628168) |
| Ultracentrifuge tubes (standard polypropylene Quick-Seal tube) | Beckman Coulter | Cat#342414 |
| Zeba Spin desalting columns and plates, 40K MWCO, 75 μL–10 mL | Thermo Scientific | Cat#A57756 |
Materials and equipment
PBS
-
•
Mix 15.2 mL of solution A, 84.8 mL of solution B, and 100 mL of 1.5 M NaCl. Add H2O to a final volume of 1000 mL. Adjust the pH to 7.4 if necessary.
Note: Prepare solution A, solution B and 1.5 M NaCl as indicated:
-
•
Solution A: Dissolve 4.49 g of KH2PO4 in H2O to a final volume of 500 mL (66 mM).
-
•
Solution B: Dissolve 4.68 g of anhydrous Na2HPO4 in H2O to a final volume of 500 mL (66 mM).
-
•
1.5 M NaCl: Dissolve 87.7 g of NaCl in H2O to a final volume of 1000 mL.
30%, 20%, and 10% sucrose in PBS
-
•
30% sucrose - Final volume 6 mL: Dissolve 1.8 g of sucrose in PBS to a final volume of 6 mL.
-
•
20% sucrose - Final volume 3 mL: Mix 2 mL of 30% sucrose with 1 mL of PBS.
-
•
10% sucrose - Final volume 3 mL: Mix 1 mL of 30% sucrose with 2 mL of PBS.
3.7% formaldehyde solution in PBS
Protocol standardized for starting from a 37% formaldehyde stock solution.
-
•
Dilute 1 mL of formaldehyde stock with PBS (pH 7.4) to a final volume of 10 mL. Leave this solution at 20°C–25°C.
Note: Always prepare and use a fresh 3.7% formaldehyde solution.
Step-by-step method details
In vivo injection of triatomines
This step describes the procedure for injecting triatomines with a Hamilton syringe. Proper immobilization and careful handling are essential to prevent injuries and ensure reproducibility.
Note: The estimated time required is calculated for a single insect. Additional time will be required when working with multiple insects.
Note: For all injections, mated females from the following triatomine species are used: D. maxima (∼35–40 mm in length), P. megistus (∼25–30 mm in length), and R. prolixus (∼17–25 mm in length). All are reared in a standardized colony under controlled laboratory conditions (28°C, 70% humidity, 12:12 h light:dark photoperiod).
Note: It is advisable to analyze lipoprotein interaction with ovarian tissue in early vitellogenesis. While the exact timelines vary by species, they typically range as follows, measured in days post-blood feeding: 4 to 6 days for D. maxima, 3 to 5 days for P. megistus, and 2 to 4 days for R. prolixus.
Note: Before starting the procedure, ensure a clean and organized workspace. Prepare all necessary materials, including a sterilized Hamilton syringe (or an alternative insulin syringe), and keep them readily accessible. Work on a stable surface with adequate lighting to facilitate precise syringe handling and accurate injection into the insect.
Note: PBS is sterilized by autoclaving at 121°C for 30 min.
-
1.
Disinfect the work surface with 70% ethanol and allow it to dry.
-
2.
Disinfect the Hamilton syringe by rinsing it with 70% ethanol, followed by sterile PBS to remove any contaminants. Repeat the PBS rinse 3 times.
-
3.
Fill the syringe with the injection solution, ensuring no air bubbles remain.
Note: As an alternative, insulin syringes (1 mL capacity, 31G gauge, 8 mm needle length) can be used. To draw up the conjugate, pipette the desired volume onto a flat, sterile surface (e.g., a microcentrifuge tube cap) and carefully aspirate, ensuring no air is drawn in. Perform this step under a stereoscopic microscope for precision.
-
4.Immobilization and injection procedure.
-
a.Carefully immobilize the non-anesthetized insect by gently placing it on a piece of paper masking tape on a microscope glass slide, ensuring minimal movement (Figure 3A).Note: Handle the insect with care to avoid injury; dressing forceps can be used for this step.
-
b.Place the microscope glass slide under a stereoscopic microscope (e.g., Schonfeld Optik LBX), using 7.5× to 10× magnification for precise visualization.
-
c.Identify the injection site: the membrane between the posterior coxa and trochanter.
-
d.Using fine forceps, carefully position the legs to expose this membrane, where the syringe is then inserted (Figure 3B).
- e.
-
f.Slowly inject the desired volume (e.g., 0.5–10 μL) over 5 s.
-
g.Carefully withdraw the needle after counting 5 s, applying minimal force to prevent hemolymph leakage.CRITICAL: If solution reflux occurs, reduce injection volume or slow down the injection speed.
-
a.
-
5.Post-injection recovery and handling.
-
a.Carefully remove the injected insect from the paper masking tape, ensuring it is not injured. Transfer it to a recovery container at 20°C–25°C.
-
b.Monitor the insect for 10–15 min until it regains full movement.
-
c.Once recovered, return the insect to its housing conditions, maintaining appropriate temperature and humidity settings.
-
a.
Note: The injection should not cause visible physical damage or excessive leakage. Under proper conditions, insects should recover quickly and resume normal movement.
Figure 3.

Immobilization and injection procedure
(A) Immobilization of a non-anesthetized insect using a piece of paper masking tape. The dashed square indicates the injection area.
(B) Injection sites marked with white arrows.
(C) Injection into the membrane between the posterior coxa and trochanter.
(D) Injection into the membrane between the third thoracic sternite and the first abdominal sternite. Rhodnius prolixus was used as the model for this figure. Bars: 1 mm.
Tissue dissection (ovaries)
This section describes the procedure for dissecting the ovaries of triatomines. Proper handling and precise dissection techniques are essential to preserve tissue integrity and ensure reliable downstream analyses.
Note: The recommended dissection time window for observing the label in ovarian tissue is 2 to 24 h post-injection.
-
6.Initial procedure for insect dissection (preparation).
-
a.Hold the insect with one hand and, using fine scissors in the other hand, remove the head, legs, and wings. Then, cut along the connexivum (Figure 4A).
-
b.Place the insect dorsal side up in a clean Petri dish.
-
c.Position the Petri dish containing the insect under the stereoscopic microscope.
-
d.Using a piece of razor blade, make an incision in the dorsal abdominal cuticle near the thorax.
-
e.Carefully lift and separate the cuticle to expose the underlying tissues (Figure 4A).
-
f.Add fresh, sterile, and cold PBS to the exposed abdominal cavity.
-
a.
-
7.Digestive system removal.
-
a.Carefully remove the superficial fat body to expose the digestive system.
-
i.Using forceps, grasp the anterior part of the digestive tract (foregut) and gently pull upward (Figure 4B), assisting with a second pair of forceps as needed.
-
ii.Remove the entire digestive system in one piece, from the esophagus to the rectum.Note: After this step, the internal organs, including the reproductive tissues, will be exposed (Figure 4C).Note: The digestive system of triatomines comprises three main regions: foregut (which includes the esophagus), the midgut (the primary site for digestion and nutrient absorption), and hindgut (involved in water absorption and waste processing, terminating at the rectum where excretion occurs).17CRITICAL: To prevent potential contamination of the abdominal cavity with ingested blood, ensure the digestive system is gently extracted in one piece.
-
i.
-
a.
-
8.Ovary extraction.
-
a.Using fine scissors, make an incision near the terminal filaments of the ovaries (Figure 4C).
-
b.To minimize damage to the oocytes, grasp the ovarian tissue by the common oviducts before detaching it (Figure 4C).
-
c.Carefully cut near the end of the common oviduct.
-
d.Gently lift the reproductive system from the common oviduct and transfer it to a clean Petri dish.
-
e.Add 100 μL of fresh, sterile PBS.
-
f.Place the Petri dish containing the reproductive tissue under the stereoscopic microscope.
-
a.
-
9.Tissue cleaning.
-
a.Using two forceps, one in each hand, carefully clean the tissue by removing the surrounding muscle sheath and tracheoles (Figure 4D).
-
a.
Note: The reproductive system of triatomines consists of two ovaries, each with 7 ovarioles, two lateral oviducts, and one common oviduct (Figure 4D). In R. prolixus, the ovaries appear pink due to Rhodnius heme-binding protein (RHBP), a yolk protein precursor that binds heme groups released from digested red blood cells, aiding in ovary recognition.18
-
10.Fixation.
-
a.Place the cleaned ovaries in a 2 mL amber glass vial.
-
b.Add 1 mL of freshly prepared 3.7% formaldehyde solution (in PBS, pH 7.4) and let the samples fix for 1 h at 20°C–25°C.
-
a.
Alternatives: Fixation can be performed for several hours at 4°C.
-
11.Cryopreservation, mounting and slide preparation.Note: Cryopreservation prevents ice crystal formation, which can damage cellular structures.
-
a.Immerse the fixed ovaries sequentially at 4°C for 30 min each in a 2 mL amber glass vial containing 1 mL of sucrose in PBS at 10%, followed by 20%, and finally 30%.Pause point: Alternatively, the fixed and sucrose-treated tissue can be stored at 20°C–25°C for up to 10 days with the addition of 0.01% NaN3 or at −80°C for at least a year.CRITICAL: Before proceeding, ensure the cryostat is set to the desired temperature (−20°C to −30°C for most tissues).
-
b.Place the fixed and sucrose-treated ovaries onto a cryocassette using Cryoplast (or equivalent) as the embedding medium.
-
c.Orient them appropriately and add more Cryoplast as needed.
-
d.Leave the samples inside the closed cryostat (e.g., Thermo Scientific, Shadow Cryotome E) for 20–30 min.CRITICAL: Always wear cold-resistant gloves to prevent cold burns.
-
e.Adjust the blade (e.g., Microtome Blade, Feather A35) angle and set the desired section thickness (preferably 10–12 μm).
-
f.Using a standard razor blade, trim excess tissue until smooth sections are obtained.
-
g.Collect sections with a brush or fine forceps and place them on positive charged microscope slides (e.g., Cat#HDAS001, 1 mm thick, from HDA).
-
h.Allow the sections to dry at 20°C–25°C.
-
i.Store the slides at −80°C or proceed with the microscopic analysis.
-
a.
Figure 4.

Procedure for insect dissection
(A) Removal of the head, legs, wings (indicated with asterisks), and connexivum (dashed line). The incision in the dorsal abdominal cuticle is indicated by a white line with a scissor-shaped arrowhead.
(B) Visualization of the digestive system. The grasp sites at the upper end of the digestive system and the rectum are marked with white arrows.
(C) Internal organs exposed after removal of the digestive system. The incision site near the terminal filaments of the ovaries is marked with a white arrow, while the grasp site of the ovarian tissue at the common oviduct is indicated with asterisks.
(D) Ovarian tissue inside the dashed circle. The left ovary shows the attached tracheoles and sheaths, whereas the right ovary shows seven cleaned ovarioles. Note: one terminal oocyte appears pink due to the Rhodnius heme-binding protein (RHBP). Rhodnius prolixus was used as the model for this figure. Bars: 1 mm.
Microscopic analysis
-
12.
Cryostat sections are mounted using an aqueous mounting medium compatible with fluorescent tracers (e.g., FluorSave Reagent, Cat#345789, Millipore from Merck).
-
13.
Carefully place a coverslip over the section and seal it on the slide by applying clear nail polish along all edges and corners.
-
14.
The samples are then examined and analyzed using laser scanning confocal microscopy (e.g., Olympus FV1200).
-
15.
Images should be acquired and processed using image analysis software (e.g., FluoView FV1200 software or FIJI, a pre-packaged version of ImageJ).19
-
16.
Once processed, images should be exported as .tiff files, and final panels assembled using Adobe Photoshop or Illustrator.
Note: Acquire at least 5 regions of interest per each slide from independent experiments.
Alternatives: To obtain semi-quantitative data on Bodipy-FA incorporation, dissected tissues can be homogenized, total lipids extracted, and fluorescence measured using a fluorometer, as already described.20,21,22
Expected outcomes
In insects, pioneering studies using fluorescently labeled lipoproteins to assess their tissue biodistribution were conducted in Locusta migratoria6,7 and in the mosquitoes Aedes aegypti and Anopheles gambiae.8,9 In all species studied to date, Vg is endocytosed and stored in yolk granules until utilized for embryo development. However, in vitellogenic females, Lp can follow both endocytic and non-endocytic pathways in oocytes, depending on the species.5
This protocol outlines a step-by-step approach for in vivo assays to track the histological and intracellular fate of lipoproteins, whether as entire particles or their lipid cargo towards developing triatomine oocytes. While Lp endocytosis does not occur in R. prolixus,23 these techniques have demonstrated that, in P. megistus, Lp is endocytosed in the oocyte and co-localizes with Vg in yolk granules (Figure 5, upper panel), highlighting the capacity of these techniques to distinguish processes and enable deeper analysis. Additionally, the use of fluorescently labeled Lp also demonstrated that, as observed in D. maxima, Lp transfers its lipid cargo to lipid droplets (Figure 5, bottom panel).
Figure 5.

Lipoprotein pathways in developing triatomine oocytes
Upper panel: Pathway of vitellogenin (Vg) and lipophorin (Lp) in the terminal follicles of Panstrongylus megistus, visualized using fluorescently labeled lipoproteins. Lower panel: Tracking the fate of Lp-lipid cargo in developing oocytes of Dipetalogaster maxima using fluorescent Lp-Bodipy-FA and Lp-DiI probes. Arrows indicate lipid droplets, while arrowheads mark yolk granules. Merged images show probe co-localization in yolk granules (asterisks). H, hemolymph; FE, follicular epithelium; Oo, oocyte; DIC, differential interference contrast microscopy. Scale bars: 20 μm.
Limitations
While this protocol offers several advantages, particularly as an in vivo approach that provides otherwise unattainable information, it also has limitations. Dissecting triatomine ovaries and injecting vitellogenic females are delicate procedures that require skilled operators. Improper handling can damage ovaries, rendering the tissue unusable for further analysis.
Precision is also crucial during injection to avoid puncturing the anterior midgut, which, in vitellogenic fed females, occupies a large portion of the body cavity. Accidental puncture can be fatal. Additionally, the injected volume should be minimized, as excessive amounts increase mortality and may alter the biological response under study.
Finally, this protocol requires specialized equipment, including ultracentrifuges, cryostats and confocal microscopes, which may not be readily available in all laboratories.
Troubleshooting
Problem 1
Insect mortality.
Potential solutions
-
•
Use lower injection volumes to minimize impact: no more than 2 μL for R. prolixus, 5 μL for P. megistus, and 10 μL for D. maxima.
-
•
Use a finer needle to reduce tissue damage.
-
•
Consider species and developmental stage sensitivity, as some insects are more vulnerable to injection. Use sterile solutions by autoclaving or filter solutions through a 0.22 μm pore.
-
•
Delay injection after feeding to allow the anterior midgut to shrink, reducing the risk of puncture.
-
•
After injection, place insects for ∼5 min at 4°C to minimize fluid expulsion.
Problem 2
No fluorescence or weak signal observed when examining slides under the microscope. This may result from issues related to the lipoprotein conjugate, the slide, or the insect.
Potential solutions
-
•
Variability in lipoprotein conjugation: This can result from factors such as lipoprotein and fluorophore concentrations, as well as incubation conditions. An excess of fluorophore may compromise lipoprotein functionality, particularly receptor binding. Optimize conjugation conditions by assessing fluorophore and lipoprotein concentrations. Avoid fluorophore excess and ensure consistent incubation parameters. Test the functional integrity of the conjugate through receptor-binding assays or other functional tests.
-
•
Altered lipoprotein conjugate: The lipoprotein may be denatured or the fluorophore bleached. Assess its integrity by running non-denaturing gel electrophoresis. Use a different aliquot, ensuring it is protected from light to prevent bleaching.
-
•
Bleached slide: Keep slides protected from light and use a mounting medium with an anti-bleaching agent.
-
•
Undeveloped follicle: If the follicle is not undergoing vitellogenesis, it will not actively take up lipids and proteins. Verify the mating status of females before injection and exclude virgins. Ensure that the analyzed section corresponds to a developing follicle. For a more representative assessment, examine multiple ovarian sections.
Problem 3
Autofluorescence.
Potential solutions
-
•
To differentiate autofluorescence from true sample fluorescence, include a negative control, such as an ovary from a PBS-injected female.
-
•
Increase the concentration of the lipoprotein conjugate.
-
•
Use fluorophores with higher brightness and quantum yield to enhance signal detection.
- •
Resource availability
Lead contact
Further information or requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Lilian E. Canavoso (lilian.canavoso@unc.edu.ar).
Technical contact
For protocol details or additional inquiries, email the technical contacts: Leonardo L. Fruttero (lfruttero@unc.edu.ar) and Jimena Leyria (jimena.leyria@unc.edu.ar).
Materials availability
This study did not generate any new unique reagents.
Data and code availability
This study did not generate or analyze datasets or code.
Acknowledgments
Work in the laboratory was supported by grants from CONICET, FONCyT, and SECyT-UNC. L.L.F., J.L., and L.E.C. are members of the National Scientific and Technical Research Council (CONICET-Argentina). The authors thank Dr. David Rojas Marquez (@darwid_illustration) for illustrating the graphical abstract. Microscopical processing and analysis were conducted at the Center for Microscopy and Nanoscopy of Córdoba (CEMINCO, CONICET-UNC, https://ceminco.conicet.unc.edu.ar/).
Author contributions
All authors participated in the optimization of the protocols and wrote the manuscript.
Declaration of interests
The authors declare no competing interests.
Contributor Information
Leonardo L. Fruttero, Email: lfruttero@unc.edu.ar.
Jimena Leyria, Email: jimena.leyria@unc.edu.ar.
Lilián E. Canavoso, Email: lilian.canavoso@unc.edu.ar.
References
- 1.Fruttero L.L., Frede S., Rubiolo E.R., Canavoso L.E. The storage of nutritional resources during vitellogenesis of Panstrongylus megistus (Hemiptera: Reduviidae): The pathways of lipophorin in lipid delivery to developing oocytes. J. Insect Physiol. 2011;57:475–486. doi: 10.1016/j.jinsphys.2011.01.009. [DOI] [PubMed] [Google Scholar]
- 2.Leyria J., Fruttero L.L., Aguirre S.A., Canavoso L.E. Ovarian nutritional resources during the reproductive cycle of the hematophagous Dipetalogaster maxima (Hemiptera: Reduviidae): Focus on lipid metabolism. Arch. Insect Biochem. Physiol. 2014;87:148–163. doi: 10.1002/arch.21186. [DOI] [PubMed] [Google Scholar]
- 3.Soulages J.L., Wells M.A. Lipophorin: The structure of an insect lipoprotein and its role in lipid transport in insects. Adv. Protein Chem. 1994;45:371–415. doi: 10.1016/s0065-3233(08)60644-0. [DOI] [PubMed] [Google Scholar]
- 4.Canavoso L.E., Jouni Z.E., Karnas K.J., Pennington J.E., Wells M.A. Fat metabolism in insects. Annu. Rev. Nutr. 2001;21:23–46. doi: 10.1146/annurev.nutr.21.1.23. [DOI] [PubMed] [Google Scholar]
- 5.Leyria J., Fruttero L.L., Canavoso L.E. Lipids in Insect Reproduction: Where, How, and Why. Adv. Exp. Med. Biol. 2024 doi: 10.1007/5584_2024_809. Published online June 15, 2024. [DOI] [PubMed] [Google Scholar]
- 6.Dantuma N.P., Pijnenburg M.A., Diederen J.H., Van der Horst D.J. Developmental down-regulation of receptor-mediated endocytosis of an insect lipoprotein. J. Lipid Res. 1997;38:254–265. [PubMed] [Google Scholar]
- 7.Dantuma N.P., Potters M., De Winther M.P., Tensen C.P., Kooiman F.P., Bogerd J., Van der Horst D.J. An insect homolog of the vertebrate very low density lipoprotein receptor mediates endocytosis of lipophorins. J. Lipid Res. 1999;40:973–978. doi: 10.1016/s0022-2275(20)32134-9. [DOI] [PubMed] [Google Scholar]
- 8.Atella G.C., Shahabuddin M. Differential partitioning of maternal fatty acid and phospholipid in neonate mosquito larvae. J. Exp. Biol. 2002;205:3623–3630. doi: 10.1242/jeb.205.23.3623. [DOI] [PubMed] [Google Scholar]
- 9.Atella G.C., Silva-Neto M.A.C., Golodne D.M., Arefin S., Shahabuddin M. Anopheles gambiae lipophorin: characterization and role in lipid transport to developing oocyte. Insect Biochem. Mol. Biol. 2006;36:375–386. doi: 10.1016/j.ibmb.2006.01.019. [DOI] [PubMed] [Google Scholar]
- 10.Nuñez J.A., Segura E.L. In: Chagas’ Disease Vectors. Volume II. Anatomic and Physiological Aspects. Brenner R.R., Stoka A.M., editors. CRC Press; 1987. Rearing of Triatominae; pp. 31–40. [Google Scholar]
- 11.Leyria J., Fruttero L.L., Nazar M., Canavoso L.E. The role of DmCatD, a cathepsin D-like peptidase, and acid phosphatase in the process of follicular atresia in Dipetalogaster maxima (Hemiptera: Reduviidae), a vector of Chagas’ disease. PLoS One. 2015;10 doi: 10.1371/journal.pone.0130144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Overall C.M. A microtechnique for dialysis of small volume solutions with quantitative recoveries. Anal. Biochem. 1987;165:208–214. doi: 10.1016/0003-2697(87)90221-1. [DOI] [PubMed] [Google Scholar]
- 13.Bradford M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 14.Aguirre S.A., Frede S., Rubiolo E.R., Canavoso L.E. Vitellogenesis in the hematophagous Dipetalogaster maxima (Hemiptera: Reduviidae), a vector of Chagas’ disease. J. Insect Physiol. 2008;54:393–402. doi: 10.1016/j.jinsphys.2007.10.012. [DOI] [PubMed] [Google Scholar]
- 15.Via D.P., Smith L.C. Fluorescent labeling of lipoproteins. Methods Enzymol. 1986;129:848–857. doi: 10.1016/0076-6879(86)29108-9. [DOI] [PubMed] [Google Scholar]
- 16.Martin-Nizard F., Richard B., Torpier G., Nouvelot A., Fruchart J.C., Duthilleul P., Delbart C. Analysis of phospholipid transfer during HDL binding to platelets using a fluorescent analog of phosphatidylcholine. Thromb. Res. 1987;46:811–825. doi: 10.1016/0049-3848(87)90073-9. [DOI] [PubMed] [Google Scholar]
- 17.Nation J.L. In: Encyclopedia of Entomology. Capinera J.L., editor. Springer Netherlands; 2008. Alimentary Canal and Digestion; pp. 111–118. [DOI] [Google Scholar]
- 18.Braz G.R.C., Moreira M.F., Masuda H., Oliveira P.L. Rhodnius heme-binding protein (RHBP) is a heme source for embryonic development in the blood-sucking bug Rhodnius prolixus (Hemiptera, Reduviidae) Insect Biochem. Mol. Biol. 2002;32:361–367. doi: 10.1016/s0965-1748(01)00163-1. [DOI] [PubMed] [Google Scholar]
- 19.Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fruttero L.L., Demartini D.R., Rubiolo E.R., Carlini C.R., Canavoso L.E. β-chain of ATP synthase as a lipophorin binding protein and its role in lipid transfer in the midgut of Panstrongylus megistus (Hemiptera: Reduviidae) Insect Biochem. Mol. Biol. 2014;52:1–12. doi: 10.1016/j.ibmb.2014.06.002. [DOI] [PubMed] [Google Scholar]
- 21.Fruttero L.L., Leyria J., Ramos F.O., Stariolo R., Settembrini B.P., Canavoso L.E. The process of lipid storage in insect oocytes: The involvement of β-chain of ATP synthase in lipophorin-mediated lipid transfer in the chagas’ disease vector Panstrongylus megistus (Hemiptera: Reduviidae) J. Insect Physiol. 2017;96:82–92. doi: 10.1016/j.jinsphys.2016.10.014. [DOI] [PubMed] [Google Scholar]
- 22.Fruttero L.L., Leyria J., Moyetta N.R., Ramos F.O., Settembrini B.P., Canavoso L.E. The fat body of the hematophagous insect, Panstrongylus megistus (Hemiptera: Reduviidae): Histological features and participation of the β-chain of ATP synthase in the lipophorin-mediated lipid transfer. J. Insect Sci. 2019;19:16. doi: 10.1093/jisesa/iez078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gondim K.C., Oliveira P.L., Masuda H. Lipophorin and oögenesis in Rhodnius prolixus: Transfer of phospholipids. J. Insect Physiol. 1989;35:19–27. doi: 10.1016/0022-1910(89)90032-2. [DOI] [Google Scholar]
- 24.Wan C.P., Park C.S., Lau B.H. A rapid and simple microfluorometric phagocytosis assay. J. Immunol. Methods. 1993;162:1–7. doi: 10.1016/0022-1759(93)90400-2. [DOI] [PubMed] [Google Scholar]
- 25.Mosiman V.L., Patterson B.K., Canterero L., Goolsby C.L. Reducing cellular autofluorescence in flow cytometry: an in situ method. Cytometry. 1997;30:151–156. [PubMed] [Google Scholar]
- 26.Neumann M., Gabel D. Simple method for reduction of autofluorescence in fluorescence microscopy. J. Histochem. Cytochem. 2002;50:437–439. doi: 10.1177/002215540205000315. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This study did not generate or analyze datasets or code.