

Abstract
Progressive aggregation of α-synuclein (α-Syn), a small cytosolic protein involved in cell vesicle trafficking, in the midbrain, hypothalamus, and thalamus is linked to Parkinson’s disease (PD). Amyloid oligomers and fibrils formed as a result of such aggregation are highly toxic to neurons. However, it remains unclear whether amyloid-induced toxicity of neurons is the primary mechanism of the progressive neurodegeneration observed upon PD. In the current study, we investigated cytotoxicity exerted by α-Syn fibrils formed in the lipid-free environment, as well as in the presence of two phospholipids, on macrophages, dendritic cells, and microglia. We found that α-Syn fibrils are far more toxic to dendritic cells and microglia compared to neurons. We also observe low toxicity levels of such amyloids to macrophages. Real-time polymerase chain reaction (RT-PCR) results suggest that toxicity of amyloids aggregates is linked to the levels of autophagy in cells. These results suggest that a strong impairment of the immune system in the brain may be the first stop of neurodegenerative processes that are taking place upon the onset of PD.
Keywords: α-Synuclein, Macrophages, Dendritic cells, Neurons, Microglia, Phospholipids
1. Introduction
α-Synuclein (α-Syn) is a small cytosolic protein involved in vesicle trafficking in synaptic clefts [1–4]. Its progressive aggregation is associated with several diseases, including Parkinson’s disease (PD). α-Syn aggregates observed in substantia nigra pars compacta (SCpc) upon PD have high structural and morphological heterogeneity [5–11]. A growing body of evidence indicate that α-Syn oligomers can propagate from cells to cells, causing the spread of PD across the midbrain, hypothalamus and thalamus. Some of the formed α-Syn oligomers initiate template association of misfolded α-Syn, which results in the formation of mature fibrils that have β-sheet secondary structure [12,13].
Microscopic analysis of α-Syn deposits in the brain of PD patients revealed the presence of fragments of lipid membranes [14]. These findings, as well as numerous in vitro experiments, suggest that lipid membranes can alter the rate of α-Syn aggregation, as well as modify the secondary structure of both oligomers and fibrils [6,15–19]. Recently reported results from our and other research groups indicate that anionic lipids strongly accelerate α-Syn aggregation, whereas zwitterionic phosphatidylcholine (PC), on the opposite, strongly inhibited protein aggregation [15,16]. Furthermore, the change in the rate of α-Syn aggregation directly depends on protein-to-lipid ratio [18,20,21]. With an increase in the concentration of lipid vesicles relative to the concentration of α-Syn, an increase in the protein aggregation rate is observed [18,20,21]. Galvagnion and co-workers hypothesized that surface of lipid vesicles facilitated protein-protein interactions that were critical for the oligomer formation. However, with the following increase in the concentration of lipids, a decrease in rate of α-Syn aggregation was observed [18,20,21]. This effect was explained by a drastic increase in the surface of lipid membranes relative to the concentration of protein [18,22,23]. NMR and fluorescence methods demonstrated that lipid membranes not only act as aggregation sites for α-Syn, but also provide lipids that interact with the protein. In this case, charged headgroups of lipids interact with charged amino acid residues of α-Syn, such as lysine and glutamic acid, that are highly abundant in the N-terminus (aa 1–60) of α-Syn [24]. In parallel, protein-lipid complexes are also stabilized by hydrophobic interactions that are taking place between the central part (aa 61–95) of α-Syn, also known as the non-amyloid component (NAC) domain, and fatty acids of lipids [25,26]. Utilization of nano-Infrared spectroscopy showed that such protein-lipid complexes form structurally different fibrils compared to the lipid-free conditions [27,28]. Furthermore, oligomers and fibrils formed in the presence of lipids exert drastically different cytotoxicity to rat dopaminergic neurons [15]. Similar findings were reported by Matveyenka and co-workers for other amyloidogenic proteins, such as insulin and lysozyme [29–32]. It should be noted that such studies are focused on elucidation of the toxic effect of protein aggregates on neurons [6,17–19,29–36]. However, several research groups showed that other cells in the brain, including astrocytes, can be affected by α-Syn aggregates [37–40]. In such cases, neurons become extremely vulnerable, which leads to their progressive degeneration.
Expanding upon this, we investigated cytotoxicity exerted by α-Syn fibrils formed in the lipid-free environment, as well as in the presence of zwitterionic phosphatidylethanolamine (PE) and anionic phosphatidylglycerol (PG) on macrophages, dendritic cells (DC) and microglia. PE was chosen to determine the effect of zwitterionic lipids on the aggregation of α-Syn, while PG represents the effect of anionic lipids on the aggregation of the protein. Macrophages are key players in our immune system. They endocytose and rapidly digest viruses and microorganisms, as well as remove dead cells. Unlike macrophages, DC cells preserve the captured antigens for their later presentation on the major histocompatibility complex (MHC) [41]. Antigen presentation by DCs initiate T cell immune response and other cascades are required for the activation of immune response [42]. Brain macrophages, also known as non-parenchymal macrophages, and microglia are essential for the development, homeostasis, and diseases of the central nervous system mononuclear phagocytes [43]. These cells constitute 5–10 % of total brain cells [44]. Microglia cells express transmembrane protein 119 (TMEM119), P2Y purinoceptor 12 (P2RY12), and Sal-like protein 1 (SALL1), which non-parenchymal macrophages express CD45 and MHC class II molecules [43]. Brain macrophages and microglia interact with almost all cell types in the brain. Brain macrophages and microglia phagocytose apoptotic and pre-apoptotic cells in the developing and adult brain [45]. Thus, these cells are also at the frontline of the brain defense against viruses and microorganisms. Several reported to date pieces of experimental evidence indicate that microglial directly and indirectly contributes to the onset and progression of neurodegenerative processes in the brain [46–48]. However, it remains unclear to what extent microglia, macrophages, and DC cells themselves are affected by amyloid aggregates.
2. Results and discussion
Kinetic analysis of protein aggregation showed that neither PG nor PE changed the lag-phase of α-Syn aggregation. We only observed a small, however, statistically insignificant increase in the rate of protein aggregation in the presence of PE, Fig. 1, A and B. After 120 h of α-Syn aggregation in the lipid-free environment at 37 °C, we observed the presence of long 6–21 nm in height fibrillar species, Fig. 1, C and D. We also found that PG facilitated lateral assembly of amyloid fibrils without substantial changes in their height (6–15 nm). At the same time, the presence of PE at the stage of α-Syn aggregation resulted in the formation of thin filaments (9–12 nm in height) and large aggregates (18–21 nm in height), Fig. 1, C and D. Based on these results, we can conclude that PG and PE alter morphology of α-Syn aggregates formed in their presence. We also used FTIR spectroscopy to examine the extent to which PE and PG alter the secondary structure of α-Syn aggregates, Fig. 1, E. FITR spectra acquired from these samples exhibited strong amide I and II bands. Amide I band in all acquired spectra has a peak at 1630 cm−1, which indicates the predominance of parallel β-sheet in α-Syn, α-Syn:PE, and α-Syn:PG fibrils. We also observed a shoulder at ~ 1660 cm−1, which points on the presence of the small amount of unordered protein in all analyzed samples. Finally, in the IR spectrum acquired from α-Syn: PG fibrils, we observed a vibrational band centered at 1735 cm-1, which originated from C=O vibration of PG.
Fig. 1.

Phospholipids alter aggregation properties of α-Syn. ThT kinetics (A) of α-Syn aggregation in the presence of PG (red) and PE (blue), as well as in the lipid-free environment (green) with corresponding lag-phase (tlag) and half-time (t1/2) that represent 10 % and 50 % increase in the ThT intensity, respectively; n = 3 (B). According to one-way ANOVA, *p < 0.05, NS is non-significant difference. AFM images (C) with corresponding height histograms (D) of protein aggregates formed at 120 h of α-Syn aggregation at 37 °C under 510 rpm shaking, pH 7.4. For each sample, at least 30 individual aggregates were measured. Scale bars are 250 nm. IR spectra (E) of samples incubated at 37 °C under 510 rpm shaking, pH 7.4 for 120 h. Final protein final concentration of 40 μM, protein: lipid ratio was 1:1.
Next, we performed cell proliferation assay to reveal the extent to which α-Syn fibrils formed in the lipid-free environment, as well as in the presence of PE and PG (α-Syn:PE and α-Syn:PG), altered the proliferation of macrophages, DC, microglia, and neurons, Fig. 2. We found that after 24 h of exposition of all cell types to the amyloid aggregates, substantial decrease in the cell proliferation was observed for high concentrations (20 μM) of fibrils formed in the lipid-free environment (α-Syn), Fig. 2, A. We also found that α-Syn:PE exerted a much greater magnitude of proliferation impairment on macrophages, DC cells and microglia, whereas nearly no changes in the proliferation of neurons were observed in the presence of α-Syn:PE. At the same time, α-Syn:PG aggregates at all examined concentrations caused no noticeable changes on the proliferation of macrophages and microglia, whereas substantial decrease in the proliferation of DC and neuronal cells were observed in the presence of 10–20 μM of α-Syn:PG. It should be noted that PE and PG themselves caused no substantial changes on proliferation of any of the tested cell types.
Fig. 2.
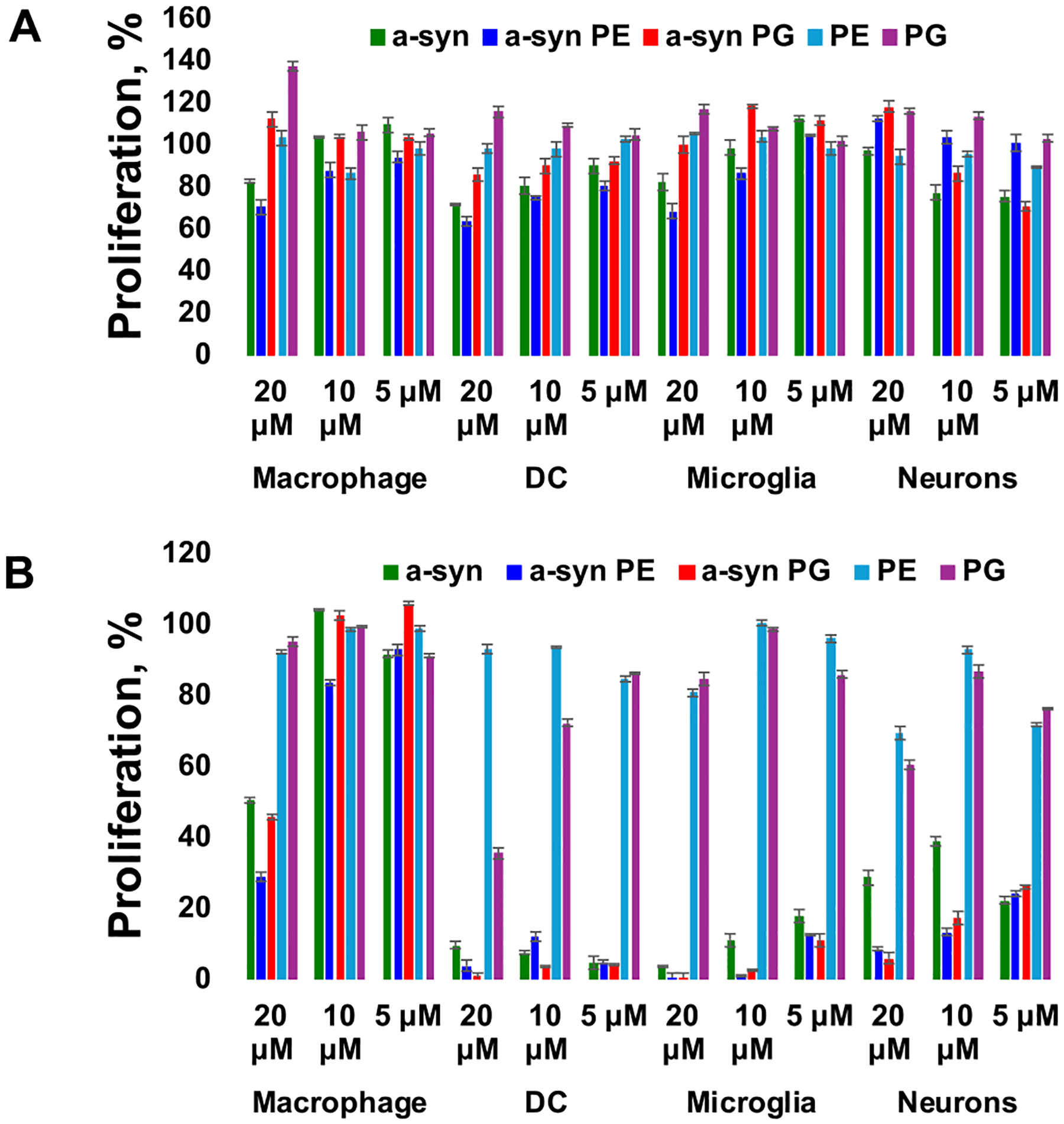
Proliferation of macrophages, DC, microglia and neurons exposed to 5 μM, 10 μM, and 20 μM of α-Syn, α-Syn:PE and α-Syn:PG aggregates as well as lipids themselves (PE and PG) for 24 h (A) and 48 h (B). ±SD with n = 3. Final protein final concentration of 40 μM, protein: lipid ratio was 1:1.
A cross-comparison of magnitude of exerted suppression in the cell proliferation during 24 h caused by α-Syn, α-Syn:PE, and α-Syn:PG fibrils revealed that DC were the most vulnerable while macrophages were the least vulnerable to the toxic effect exerted by these amyloids. This conclusion is further supported by the analysis of cell proliferation after 48 h of macrophages, DC, microglia, and neurons to amyloid aggregates, Fig. 2, B. We found that DC and microglia cells’ proliferation was strongly impaired by α-Syn fibrils formed in the lipid-free environment, as well as in the presence of PE and PG. Nearly all microglia and DC cells exposed to amyloid aggregates died by 48 h. We also found that only high (20 μM) concentration of amyloid aggregates caused substantial changes in the proliferation of neurons and macrophages. Macrophages were able to fully recover after exposition to medium (10 μM) and low (5 μM) concentrations of α-Syn fibrils formed in the lipid-free environment, as well as in the presence of PE and PG. However, such an efficient recovery was not evident for neurons. Although we did not find as detrimental effects of amyloid aggregates on neurons as were observed for microglia and DC, only near 30 % of the proliferation was evident for neurons exposed to α-Syn fibrils and ~ 20 % for α-Syn:PE and α-Syn:PG present at medium and low concentrations. These results indicate that amyloid aggregates formed in the presence of lipids caused stronger impairment of neuronal proliferation compared to amyloid fibrils formed in the lipid-free environment. These results are consistent with experimental findings previously reported by Dou and co-workers [16]. We can also conclude that DC, neurons, and microglia cells appeared to be highly vulnerable to amyloid aggregates, whereas macrophages were able to recover if exposed to medium and low concentrations of amyloid aggregates formed in the presence and absence of lipids. These results are consistent with experimental findings previously reported by Matveyenka and co-workers [49].
We also used lactate dehydrogenase (LDH) assay to validate the discussed above results of cell proliferation. Our results show (Fig. S1) that α-Syn fibrils formed in the presence and absence of lipids exert weaker cytotoxic effects in macrophages, DC cells, and microglia compared to neurons after 24 h of cell exposition to amyloid aggregates. We also observed a substantial decrease in the viability in all cells except macrophages after 48 h of cell exposition to amyloid aggregates. These results confirmed the discussed above conclusions about high vulnerability of DC, neurons, and microglia cells to amyloid aggregates.
We hypothesized that amyloid aggregates could alter cytokine profiles of macrophages, microglia, DC, and neurons. TNFα, INF, interleukin-1 beta (IL-1β), and interleukin 6 (IL-6) are key inflammatory cytokines in macrophages [50–52]. The release of these cytokines initiates a potent defensive inflammatory response [53–56]. In addition to these interleukins, macrophage colony-stimulating factor (G-CSF) activates macrophage differentiation into inflammatory cells (M1) [57]. We found that α-Syn fibrils formed in the lipid-free environment, as well as in the presence of PE and PG (α-Syn:PE and α-Syn:PG), activated both IL-6 and TNFα in macrophages, whereas no significant changes in the expression of INF were observed. We also observed a strong expression of G-CSF and IL-1β in macrophages exposed to α-Syn fibrils formed in the lipid-free environment, whereas expression of these interleukins was not observed by these cells as a response to α-Syn:PE and α-Syn:PG aggregates, Fig. 3, A. It should be noted that lipids themselves caused no changes in the expression of G-CSF, IL-1β, IL-6, TNFα, and INF. These results indicate that only amyloid aggregates trigger strong changes in the cytokine profile of macrophages. Our results also indicate that macrophages strongly differentiate α-Syn fibrils formed in the lipid-free environment from α-Syn fibrils formed in the presence of lipids.
Fig. 3.

Changes in the cytokine profiles of macrophages (A), microglia (B), DC (C), and neurons (D), as a result of the cell exposure to α-Syn fibrils formed in the lipid-free environment, α-Syn:PE and α-Syn:PG, as well as lipids themselves (PE and PG). Final protein final concentration of 40 μM, protein: lipid ratio was 1:1. Fluorescence measurements were recorded using a flow cytometer. For each reading, 10,000 cells were analyzed.
Such amyloid-specific response was not evident, however, in microglia, Fig. 3, B. We found that microglia cells expressed G-CSF, IL-6, and TNFα interleukins as a response to α-Syn fibrils formed in the lipid-free environment, as well as in the presence of PE and PG. We did not observe the expression of INF or IL-1β in these cells. At the same time, amyloid-specific response was observed in DC cells that expressed G-CSF, IL-6, and TNFα interleukins as a response to α-Syn fibrils formed in the lipid-free environment, whereas DC cells exposed to α-Syn:PE expressed only IL-1β and TNFα, as well as cells exposed to α-Syn:PG expressed only IL-6 and TNFα, Fig. 3, C. Finally, our results indicate that neurons do not express G-CSF, IL-1β, IL-6, TNFα, and INF interleukins upon exposition to amyloid aggregates and lipid vesicles, Fig. 3, D.
Autophagy is an intracellular degradation process that is essential for the survival of eukaryotic cells [58,59]. To analyze the extent to which the amyloid aggregates were internalized by macrophages, microglia, DC, and neurons, we quantified changes in the expression of LC3b and p62 proteins, Table 1. LC3 protein is required for autophagosome formation and, therefore, has been widely used to monitor the number of autophagosomes, as well as autophagic activity [60]. Moreover, emerging evidence has shown that during selective autophagy, LC3 functions as an adaptor protein to recruit selective cargo to the autophagosome via interaction with cargo receptors [61,62]. P62, also called sequestosome 1 (SQSTM1), is a ubiquitin-binding scaffold protein that colocalizes with ubiquitinated protein aggregates in many neurodegenerative diseases and proteinopathies of the liver [63,64].
Table 1.
Heat-map of LC3b and p62 RNA expression in macrophages (A), microglia (B), DC (C) and neurons (D) at 24 after cells were exposed to α-Syn, α-Syn:PE, and α-Syn:PG, as well as lipids themselves (PE and PG). After 24 h, cells were washed away from the aggregates and incubated another 24 h (48 h). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used to normalize the data.
| A | ||||||
|---|---|---|---|---|---|---|
| α-Syn | α-Syn:PE | α-Syn:PG | PE | PG | ||
| LC3b | 24 h | 1.2 | 2.3 | 1.9 | 2.5 | 1.1 |
| 48 h | 0.9 | 0.8 | 0.8 | 1.2 | 0.9 | |
| p62 | 24 h | 0.6 | 1.3 | 1.4 | 1.2 | 1.4 |
| 48 h | 1.2 | 1.6 | 1.2 | 1.0 | 0.3 | |
| B | ||||||
| α-Syn | α-Syn:PE | α-Syn:PG | PE | PG | ||
| LC3b | 24 h | 3.8 | 0.7 | 0.8 | 0.8 | 1.0 |
| 48 h | 0.8 | 0.4 | 0.2 | 0.2 | 0.2 | |
| p62 | 24 h | 7.6 | 3.8 | 3.9 | 4.0 | 3.6 |
| 48 h | 3.9 | 2.2 | 1.4 | 1.1 | 1.2 | |
| C | ||||||
| α-Syn | α-Syn:PE | α-Syn:PG | PE | PG | ||
| LC3b | 24 h | 1.9 | 1.8 | 1.0 | 2.2 | 1.3 |
| 48 h | 0.6 | 0.8 | 0.7 | 0.8 | 0.4 | |
| p62 | 24 h | 3.1 | 4.4 | 5.4 | 8.7 | 4.6 |
| 48 h | 4.5 | 3.4 | 4.4 | 8.3 | 5.7 | |
| D | ||||||
| α-Syn | α-Syn:PE | α-Syn:PG | PE | PG | ||
| LC3b | 24 h | 1.3 | 1.2 | 2.3 | 2.5 | 1.4 |
| 48 h | 0.7 | 0.8 | 1.1 | 1.3 | 1.0 | |
| p62 | 24 h | 2.6 | 3.1 | 2.7 | 3.4 | 2.1 |
| 48 h | 4.2 | 2.8 | 2.4 | 3.1 | 1.4 |
Our results indicate that co-incubation of neurons with α-Syn fibrils formed in the lipid-free environment, as well as in the presence of PE and PG, results in ~3-fold activation of the expression of p62, Table 1. We found that removal of amyloid aggregates does not substantially decrease the expression of p62 after 48 h. It should be noted that lipids themselves trigger the increase in the expression of p62 that also remains relatively high (3.1 and 1.4 folds for PE and PG, respectively) after 48 h in cells washed from the lipid vesicles. α-Syn fibrils formed in the lipid-free environment, as well as in the presence of PE and PG, caused only small increases in LC3b expression that nearly completely leveled back to the normal level in the cells after 48 h. Similar to p62, expression levels of LC3b were found elevated in the neuronal cells exposed to both PE and PG.
A strong activation of the expression of p62 was observed for DC cells as a result of the cell exposition to α-Syn fibrils formed in the lipid-free environment, as well as in the presence of PE and PG, as well as lipids themselves. However, substantially weaker activation of LC3b were evident for DC cells exposed to α-Syn:PE and α-Syn:PG aggregates. An increase in the expression of this autophagosome marker was observed only in the DC cells exposed to α-Syn fibrils formed in the lipid-free environment that was ~4-fold higher even after aggregates were removed and cells were incubated in the amyloid-free environment for 24 h. Similar conclusions could be made about microglia cells. We observed significant increases in the expression of p62 in the microglia cells exposed to α-Syn fibrils formed in the lipid-free environment, as well as in the presence of PE and PG. It should be noted that α-Syn and α-Syn:PE fibrils caused a strong activation of LC3b that was not evident for α-Syn:PG fibrils. However, expression levels of LC3b were brought to the normal levels after cells were incubated in the amyloid-free environment for 24 h. It should be noted that both PE and PG caused a very strong upregulation of LC3b and p62 in microglia cells.
Finally, we observed a strong upregulation in the expression of LC3b and p62 in macrophages exposed to α-Syn:PE and α-Syn:PG aggregates. However, no changes in the expression of these autophagosome markers were observed for the cells exposed to α-Syn fibrils formed in the lipid-free environment. Similar to microglia, PE vesicles caused a strong upregulation of LC3b in macrophages, whereas PG, on the other hand, triggered an activation of the expression of p62. The expression levels of both of these markers were normalized after cells were incubated in the lipid-free environment for 24 h. These results indicate that both amyloid aggregates and lipids themselves caused a strong increase in the autophagosome activity in macrophages, microglia, DC and neurons. Our results show that magnitude of the autophagosome activity is determined by the lipid. These results suggest that lipid control toxicity of amyloid aggregates by changing the levels of cell autophagosome activity, and consequently, their ability to clear out amyloid aggregates.
Finally, we used ELISA to prove that both cells and exosomes possess amyloid aggregates, Fig. 4. Our results showed that macrophages, microglia, DC, and neurons possessed high concentrations of α-Syn aggregates after 24 h of their exposition to amyloids, Fig. 4, A. Our results also show that concentrations of α-Syn aggregates in all cells have only statistically insignificant variability. These results are in good agreement with the discussed above qPCR-based evidence of amyloid internalization by cells. We also found that after 30 h and 48 h, only neurons possessed statistically significant amount of α-Syn, whereas no evidence of α-Syn was observed in macrophages, DC, or microglia cells. As was previously reported by our group, macrophages have innate mechanisms that degrade amyloids. This conclusion is further supported by their strong proliferation under these experimental conditions. We infer that no amyloids in DC and microglia cells were evident due to high mortality of these cells.
Fig. 4.

ELISA of α-Syn in (A) macrophages, microglia, DC and neurons, as well as in the cell exosomes (B) at 24 h, 30 h and 48 h. According to one-way ANOVA, **p < 0.01, NS is non-significant difference; n = 3. Final protein final concentration of 4 μM, protein: lipid ratio was 1:1.
Our results also show that exosomes extracted from macrophages, microglia, DC, and neurons after 24 h of cell incubation with α-Syn fibrils possess a high concentration of protein, Fig. 4, B. These results indicated that macrophages, microglia, DC, and neurons actively exocytosed α-Syn, which is in a good agreement with the previously discussed results on cell autophagy. We also found no α-Syn in exosomes extracted from all types of cells past 24 h. Active autophagy explains a decrease in the protein in neurons at 24 and 30 h, as well as absence of significant changes in the concentration of α-Syn possessed by neurons between 30 h and 48 h.
3. Conclusions
Our results indicate that α-Syn fibrils formed in the lipid-free environment, as well as in the presence of PE and PG exerted substantially greater cytotoxicity to microglia and DC cell compared to neurons. It should be noted that PE and PG neither altered the rate of α-Syn aggregation, nor changed the morphology and structure of α-Syn fibrils. We also found that macrophages could quickly recover from the toxic effects of amyloid aggregates. Our data also indicates that PE and PG uniquely alter amyloid uptake by microglia, DC, macrophages, and neurons, as well as determine the change in cytokine profile of microglia, DC, and macrophages, while causing no expression of G-CSF, IL-1β, IL-6, TNFα, and INF interleukins in neurons. After their internalization, cells exocytose amyloid aggregate via autophagy. This mechanism likely aims to reduce cytotoxic effects of amyloid aggregates.
4. Experimental Procedures:
4.1. Materials
1,2-Dimyristoyl-sn-glycero-3-phospho-(1′-rac-glycerol) (14:0 PG) and 1,2-dimyristoyl-sn-glycero-3-phosphoethanolamine (14:0 PE) were purchased from Avanti (Alabaster, AL, USA).
4.2. Protein expression and purification α-syn
To express protein, pET21a-α-synuclein plasmid was transformed into BL21 Escherichia coli (DE3), strain Rosetta. Transfomred cells were exposed to 1 mM of IPTG to induce α-Syn expression. Once appropriate optical density of the cell culture was reached, bacterial culture was centrifuged for 10 min at 8, 000 rpm. The formed pellet was resuspended in Tris buffer (10 mM EDT, 150 mM NaCl, 50 mM Tris, pH 7.5) and boiled for 30 min to lyse bacterial cells. Sample fraction enriched with α-Syn were centrifuged for 40 min at 16,000 g and the supernatant was collected. Next, we added streptomycin sulfate (10 % solution, 136 μL/mL) in glacial acetic acid (228 μL/mL) to the supernatant to precipitate bacterial proteins and contaminants. Next, samples were centrifuged at 4 °C for 10 min at 16,000 g. α-Syn in the supernatant was precipitated by saturated ammonium sulfate ((NH4)2SO4). The pellet was re-suspended in 100 mM NH4(CH3COO) under constant stirring for 5 min and absolute ethanol was added to the resuspended harvested α-Syn and dissolved in PBS.
4.3. Size Exclusion Chromatography (SEC)
Protein samples were exposed to 14,000 g for 30 min prior to SEC. For each SEC run, 500 μL of the concentrated α-syn was injected in AKTA pure (GE Healthcare) FPLC system equipped with a gel-filtration column (Superdex 200 10/300). The system and the column were kept at 4 °C. Samples were eluted isocratically using PBS, pH 7.4 at a flow rate of 0.5 mL/min. Gel image of purified α-syn is shown in Fig. S2, while SEC profile of protein purification is shown in Fig. S3.
4.4. Liposome preparation
PG and PE were dissolved in chloroform. Once all solvent was dried, lipids were dissolved in PBS, pH 7.4. Next, lipid solution was heated in a water bath to ~50 °C for 30 min and cooled in liquid nitrogen for 3–5 min. This procedure was repeated 10 times. After that, lipid solution was processed using an extruder equipped with a 100 nm membrane (Avanti, Alabaster, AL, USA). Dynamic light scattering was used to ensure that the size of PG and PE LUVs was within 90 ± 10 nm, Fig. S4.
4.5. Protein aggregation
To perform protein aggregation in the lipid-free environment, α-Syn was dissolved in PBS to reach the final protein final concentration of 40 μM. For α-Syn:PG and α-Syn:PE, 40 μM of α-Syn and LUVs were mixed at 1:1 M ratio in PBS. Next, samples were placed into 96 well-plate that was kept in the plate reader (Tecan, Männedorf, Switzerland) at 37 °C for 150 h under 510 rpm agitation.
4.6. Kinetic measurements
Rates of α-Syn, α-Syn:PG and α-Syn:PE aggregation were measured using thioflavin T (ThT) fluorescence assay. For this, samples were mixed with 2 mM of ThT solution to reach 300 μM the final concentration of ThT and placed in into 96 well-plate that was kept in the plate reader (Tecan, Männedorf, Switzer-land) at 37 °C for 24 h under 510 rpm agitation. Fluorescence measurements were taken every 10 min (excitation 450 nm; emission 488 nm).
4.7. AFM imaging
AIST-NT-HORIBA system (Edison, NJ) was used to acquire all AFM images. Silicon AFM probes (force constant 2.7 N/m; resonance frequency 50–80 kHz) purchased from Appnano (Mountain View, CA, USA) were utilized. Scar removal and noise filtering of the collected AFM images were made using AIST-NT software (Edison, NJ, USA).
4.8. Attenuated total reflectance Fourier-transform Infrared (ATR-FTIR) spectroscopy
FTIR spectra were acquired on Spectrum 100 FTIR spectrometer (Perkin-Elmer, Waltham, MA, USA) equipped with ATR module at room temperature. Spectra were analyzed using Thermo Grams Suite software (Thermo-Fisher Scientific, Waltham, MA, USA).
4.9. Cell culturing
Rat midbrain N27 neuronal cells were purchased from ATCC and grown in RPMI 1640 Medium (Thermo Fisher Scientific, Waltham, MA, USA) with 10 % fetal bovine serum (FBS) (Invitrogen, Waltham, MA, USA) in 96 well-plate (50,000 cells per well) at 37 °C and 5 % CO2. Macrophages H36.12j [Pixie 12j] were purchased from ATCC and grown in RPMI-1640 (Thermo Fisher Scientific, Waltham, MA, USA) with 10 % fetal bovine serum (FBS) (Invitrogen, Waltham, MA, USA) in 96 well-plate (50,000 cells per well) at 37 °C and 5 % CO2. Dendritic cells (DC2.4) were purchased from ATCC and RPMI-1640 with 10 % fetal bovine serum (FBS) (Invitrogen, Waltham, MA, USA) in 96 well-plate (50,000 cells per well) at 37 °C and 5 % CO2. Microglia cells (SIM-A9) were purchased from ATCC and grown in mix of medium DMEM and F12 in ratio 1:1 and 10 % fetal bovine serum (FBS) (Invitrogen, Waltham, MA, USA), 5 % horse serum (HS) (Invitrogen, Waltham, MA, USA) at 37 °C and 5 % CO2. Antibiotic Normocin (InvivoGen, CA, USA) was added to all cell cultures to prevent bacterial contaminations.
4.10. Proliferation assay
After 24 h of incubation with the sample of protein aggregates, cells were stained using alamarBlue HS Cell Viability Reagent (Thermo Fisher Scientific, Waltham, MA, USA). Cells were incubated with 10 μL the dye for 3 h at 37 °C under 5 % CO2. Fluorescence measurements were made in plate reader (Tecan, Männedorf, Switzerland) at 560 nm excitation. Emission signal was collected at 590 nm. Every well was measured 25 times in different locations.
4.11. Lactate dehydrogenase assay
After 24 h of cell incubation with the sample of protein aggregates, CytoTox 96 non-radioactive cytotoxicity assay (G1781, Promega, Madison, WI, USA) was used to quantify LDH levels in the cell media. An increase in the concentration of LDH indicates a decrease in cell viability. Absorption measurements were made in plate reader (Tecan, Männedorf, Switzerland) at 490 nm. Every well was measured 25 times in different locations.
4.12. Expression of cytokines
After incubation cells with samples, RayPlex Inflammation Bead Kit (RayBiotech Life, GA, USA) was used. Prepare dilutions for standard and dilutions for samples. Obtain 6 clean 1.5 ml microcentrifuge tubes. Pipette 30 μl Std1 into tube Std2 and mix gently. Perform 5 more serial dilutions by adding 30 μl of Std2 to tube Std3, mix, and so on. Add 60 μl 1× Assay Diluent or RayBio Serum Diluent to another tube labeled as negative. Add 60 μl 1× Assay Diluent or RayBio Serum Diluent as applicable to each tube. Add 25 μl of RayPlex Multiplex Bead Cocktail to 25 μl each well that will contain the negative, standard, or sample. Shake at 1,000 rpm at room temperature for 2 h or overnight at 4 °C for which may increase the signal-to-noise ratio, particularly for proteins with low concentrations. Wash the beads by adding 200 μl 1× Wash Buffer. Add 25 μl of 1× Biotinylated Detection Antibody Cocktail to each well. Resuspend the beads by gently pipetting and incubate on an orbital shaker at 1,000 rpm at room temperature for 1 h and wash twice. Add 50 μl of 1× Streptavidin-PE to each well, incubate on an orbital shaker at 1,000 rpm at room temperature for 30 min. Wash plate once. Resuspend in 150 μl of 1× Wash Buffer. Analyze samples on BD LSR II Flow Cytometer (Becton Dickinson, USA).
4.13. Enzyme-linked immunoassay (ELISA)
ELISA assay was performed by using a α-Syn ELISA Kit (Invitrogen, cat.N. KHB0061). Cells were concentrated by centrifugation and lysed using Cell Extraction Buffer (10 mM Tris (pH 7.4), 100 mM NaCl, 1 mM EGTA, 1 mM NaF, 20 mM Na2P2O7, 2 mM Na3VO4, 1 % Triton X-100, 10 % glycerol, 0.1 % SDS, and 0.5 % deoxycholate) for 30 min, on ice, with mixing on vortex at 10-mim intervals. Then lysate was centrifuged at 13,000 rpm for 10 min at 4°С, supernatant was saved.
After standards preparation, 50 μL of standards, controls, and samples were added to wells, except for chromogen blanks. Then 50 μL of Hu-α-Synuclein Detection Antibody solution was added to each well except for the chromogen blanks. After incubation for 3 h at room temperature solution was aspirated from wells, and the wells were washed 4 times with Wash Buffer. Next 100 μL Anti-Rabbit IgC HRP was added into each well except for the chromogen blanks. The plate was incubated at room temperature for 30 min. After that solution was aspirated, and the wells were washed 4 times with Wash Buffer. Then, 100 μL of Stabilized Chromogen was added to each well. After incubation for 30 min in the dark 100 μL Stop Solution was added to each well. Absorbance was read at 450 nm.
4.14. Gene Expression
RNA was first extracted from the cells exposed to amyloid aggregates, as well as the cells that were treated using the same volume of PBS using a GeneJET RNA Purification Kit (K0732, Thermo Scientific). NanoDrop One (Thermo Scientific) was used to determine the concentration of extracted RNA. Next, synthesis of cDNA was performed with Super-Script™ III Reverse Transcriptase (18080093, Invitrogen) and random primers (48190011, Invitrogen). Specific primers were designed for p62 and LC-3 genes. qPCR were performed on C1000 Touch Thermal Cycler (BioRad). Each reaction mixture contained cDNAs and gene-specific primers, as well as SYBR Green PCR master mix (4309155, Applied Biosystems). PCRs were performed to 40 cycles. Beta-actin was used as a reference housekeeping gene to normalize the expression levels of p62 and LC-3 genes. The quantification of the relative expression of p62 and LC-3 genes was determined using the comparative Ct method (2-ΔΔCt). In this approach, ΔCt represents the difference in threshold cycles between the target gene and the housekeeping gene (GAPDH), while ΔΔCt represents the difference in ΔCt values between the cells exposed to amyloid samples and the control.
Supplementary Material
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.bbapap.2024.141061.
Acknowledgment
We are grateful to the National Institute of Health for the provided financial support (R35GM142869).
Declaration of competing interest
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests. Dmitry Kurouski reports financial support was provided by Texas A&M University. If there are other authors, they declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Abbreviations:
- α-Syn
α-Synuclein
- DC
dendritic cells
- PE
phosphatidylethanolamine
- PG
phosphatidylglycerol
- LUVs
large unilamellar vesicles
- ThT
thioflavin T
Footnotes
CRediT authorship contribution statement
Mikhail Matveyenka: Writing – review & editing, Writing – original draft, Visualization, Methodology, Investigation, Conceptualization. Abid Ali: Writing – review & editing, Methodology, Investigation. Charles L. Mitchell: Writing – review & editing, Methodology, Investigation. Mikhail Sholukh: Writing – review & editing, Project administration. Dmitry Kurouski: Writing – review & editing, Writing – original draft, Supervision, Resources, Project administration, Funding acquisition, Conceptualization.
Data availability
Data will be made available on request.
References
- [1].Auluck PK, Caraveo G, Lindquist S, α-Synuclein: membrane interactions and toxicity in Parkinson’s disease, Annu. Rev. Cell Dev. Biol 26 (1) (2010) 211–233. [DOI] [PubMed] [Google Scholar]
- [2].Burré J, Sharma M, Südhof TC, α-Synuclein assembles into higher-order multimers upon membrane binding to promote SNARE complex formation, Proc. Natl. Acad. Sci. USA 111 (40) (2014) E4274–E4283, 10.1073/pnas.1416598111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Burŕe J, Sharma M, Tsetsenis T, Buchman V, Etherton MR, Südhof TC, Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro, Science 329 (5999) (2010) 1663–1667, 10.1126/science.1195227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Diao J, Burré J, Vivona S, Cipriano DJ, Sharma M, Kyoung M, Südhof TC, Brunger AT, Native α-synuclein induces clustering of synaptic-vesicle mimics via binding to phospholipids and synaptobrevin-2/VAMP2, eLife 2 (2013) e00592, 10.7554/eLife.00592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Pieri L, Madiona K, Melki R, Structural and functional properties of prefibrillar α-synuclein oligomers, Sci. Rep 6 (2016) 24526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Chen SW, Drakulic S, Deas E, Ouberai M, Aprile FA, Arranz R, Ness S, Roodveldt C, Guilliams T, De-Genst EJ, et al. , Structural characterization of toxic oligomers that are kinetically trapped during alpha-synuclein fibril formation, Proc. Natl. Acad. Sci. USA 112 (16) (2015) E1994–E2003, 10.1073/pnas.1421204112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Cremades N, Cohen SI, Deas E, Abramov AY, Chen AY, Orte A, Sandal M, Clarke RW, Dunne P, Aprile FA, et al. , Direct observation of the interconversion of normal and toxic forms of alpha-synuclein, Cell 149 (5) (2012) 1048–1059, 10.1016/j.cell.2012.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Apetri MM, Maiti NC, Zagorski MG, Carey PR, Anderson VE, Secondary structure of alpha-synuclein oligomers: characterization by raman and atomic force microscopy, J. Mol. Biol 355 (1) (2006) 63–71. [DOI] [PubMed] [Google Scholar]
- [9].O’Leary EI, Lee JC, Interplay between alpha-synuclein amyloid formation and membrane structure, Biochim. Biophys. Acta, Proteins Proteomics 1867 (5) (2019) 483–491, 10.1016/j.bbapap.2018.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Kurouski D, Van Duyne RP, Lednev IK, Exploring the structure and formation mechanism of amyloid fibrils by Raman spectroscopy: a review, Analyst 140 (15) (2015) 4967–4980, 10.1039/c5an00342c. [DOI] [PubMed] [Google Scholar]
- [11].Hong DP, Han S, Fink AL, Uversky VN, Characterization of the non-fibrillar alpha-synuclein oligomers, Protein Pept. Lett 18 (3) (2011) 230–240. [DOI] [PubMed] [Google Scholar]
- [12].Li B, Ge P, Murray KA, Sheth P, Zhang M, Nair G, Sawaya MR, Shin WS, Boyer DR, Ye S, et al. , Cryo-EM of full-length alpha-synuclein reveals fibril polymorphs with a common structural kernel, Nat. Commun 9 (1) (2018) 3609, 10.1038/s41467-018-05971-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Guerrero-Ferreira R, Taylor NM, Mona D, Ringler P, Lauer ME, Riek R, Britschgi M, Stahlberg H, Cryo-EM structure of alpha-synuclein fibrils, Elife (2018) 7, 10.7554/eLife.36402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Shahmoradian SH, Lewis AJ, Genoud C, Hench J, Moors TE, Navarro PP, Castano-Diez D, Schweighauser G, Graff-Meyer A, Goldie KN, et al. , Lewy pathology in Parkinson’s disease consists of crowded organelles and lipid membranes, Nat. Neurosci 22 (7) (2019) 1099–1109, 10.1038/s41593-019-0423-2. [DOI] [PubMed] [Google Scholar]
- [15].Dou T, Kurouski D, Phosphatidylcholine and phosphatidylserine uniquely modify the secondary structure of alpha-Synuclein oligomers formed in their presence at the early stages of protein aggregation, ACS Chem. Neurosci 13 (16) (2022) 2380–2385, 10.1021/acschemneuro.2c00355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Dou T, Matveyenka M, Kurouski D, Elucidation of secondary structure and toxicity of alpha-Synuclein oligomers and fibrils grown in the presence of phosphatidylcholine and phosphatidylserine, ACS Chem. Neurosci 14 (17) (2023) 3183–3191, 10.1021/acschemneuro.3c00314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Chiti F, Dobson CM, Protein Misfolding, amyloid formation, and human disease: a summary of Progress over the last decade, Annu. Rev. Biochem 86 (1) (2017) 27–68, 10.1146/annurev-biochem-061516-045115. [DOI] [PubMed] [Google Scholar]
- [18].Galvagnion C, Brown JW, Ouberai MM, Flagmeier P, Vendruscolo M, Buell AK, Sparr E, Dobson CM, Chemical properties of lipids strongly affect the kinetics of the membrane-induced aggregation of alpha-synuclein, Proc. Natl. Acad. Sci. USA 113 (26) (2016) 7065–7070, 10.1073/pnas.1601899113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Hardenberg MC, Sinnige T, Casford S, Dada S, Poudel C, Robinson EA, Fuxreiter M, Kaminksi C, Kaminski-Schierle GS, Nollen EAA, et al. , Observation of an alpha-synuclein liquid droplet state and its maturation into Lewy body-like assemblies, J. Mol. Cell Biol (2021), 10.1093/jmcb/mjaa075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Galvagnion C, The role of lipids interacting with -Synuclein in the pathogenesis of Parkinson’s disease, J. Parkinsons Dis 7 (2017) 433–450. [DOI] [PubMed] [Google Scholar]
- [21].Galvagnion C, Buell AK, Meisl G, Michaels TC, Vendruscolo M, Knowles TP, Dobson CM, Lipid vesicles trigger alpha-synuclein aggregation by stimulating primary nucleation, Nat. Chem. Biol 11 (3) (2015) 229–234, 10.1038/nchembio.1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Alza NP, Iglesias Gonzalez PA, Conde MA, Uranga RM, Salvador GA, Lipids at the crossroad of alpha-Synuclein function and dysfunction: biological and pathological implications, Front. Cell. Neurosci 13 (2019) 175, 10.3389/fncel.2019.00175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Galvagnion C, The role of lipids interacting with -Synuclein in the pathogenesis of Parkinson’s disease, J. Parkinsons Dis 7 (2017) 433–450. [DOI] [PubMed] [Google Scholar]
- [24].Viennet T, Wordehoff MM, Uluca B, Poojari C, Shaykhalishahi H, Willbold D, Strodel B, Heise H, Buell AK, Hoyer W, et al. , Structural insights from lipid-bilayer nanodiscs link alpha-Synuclein membrane-binding modes to amyloid fibril formation, Commun Biol 1 (2018) 44, 10.1038/s42003-018-0049-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Giasson BI, Murray IV, Trojanowski JQ, Lee VM, A hydrophobic stretch of 12 amino acid residues in the middle of alpha-synuclein is essential for filament assembly, J. Biol. Chem 276 (4) (2001) 2380–2386, 10.1074/jbc.M008919200. [DOI] [PubMed] [Google Scholar]
- [26].Ueda K, Fukushima H, Masliah E, Xia Y, Iwai A, Yoshimoto M, Otero DA, Kondo J, Ihara Y, Saitoh T, Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease, Proc. Natl. Acad. Sci. USA 90 (23) (1993) 11282–11286, 10.1073/pnas.90.23.11282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Jakubec M, Barias E, Furse S, Govasli ML, George V, Turcu D, Iashchishyn IA, Morozova-Roche LA, Halskau O, Cholesterol-containing lipid nanodiscs promote an alpha-synuclein binding mode that accelerates oligomerization, FEBS J. 288 (6) (2021) 1887–1905, 10.1111/febs.15551. [DOI] [PubMed] [Google Scholar]
- [28].Dou T, Zhou L, Kurouski D, Unravelling the structural Organization of Individual alpha-Synuclein oligomers grown in the presence of phospholipids, J. Phys. Chem. Lett 12 (18) (2021) 4407–4414, 10.1021/acs.jpclett.1c00820. [DOI] [PubMed] [Google Scholar]
- [29].Matveyenka M, Rizevsky S, Kurouski D, Unsaturation in the fatty acids of phospholipids drastically alters the structure and toxicity of insulin aggregates grown in their presence, J. Phys. Chem. Lett (2022) 4563–4569, 10.1021/acs.jpclett.2c00559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Matveyenka M, Rizevsky S, Kurouski D, The degree of unsaturation of fatty acids in phosphatidylserine alters the rate of insulin aggregation and the structure and toxicity of amyloid aggregates, FEBS Lett. 596 (11) (2022) 1424–1433, 10.1002/1873-3468.14369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Matveyenka M, Rizevsky S, Kurouski D, Length and unsaturation of fatty acids of phosphatidic acid determines the aggregation rate of insulin and modifies the structure and toxicity of insulin aggregates, ACS Chem. Neurosci 13 (16) (2022) 2483–2489, 10.1021/acschemneuro.2c00330. [DOI] [PubMed] [Google Scholar]
- [32].Matveyenka M, Rizevsky S, Kurouski D, Amyloid aggregates exert cell toxicity causing irreversible damages in the endoplasmic reticulum, Biochim. Biophys. Acta Mol. basis Dis 1868 (11) (2022) 166485, 10.1016/j.bbadis.2022.166485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Matveyenka M, Rizevsky S, Kurouski D, Elucidation of the effect of phospholipid charge on the rate of insulin aggregation and structure and toxicity of amyloid fibrils, ACS Omega 8 (13) (2023) 12379–12386, 10.1021/acsomega.3c00159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Matveyenka M, Rizevsky S, Pellois JP, Kurouski D, Lipids uniquely alter rates of insulin aggregation and lower toxicity of amyloid aggregates, Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1868 (1) (2023) 159247, 10.1016/j.bbalip.2022.159247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Matveyenka M, Zhaliazka K, Kurouski D, Concentration of phosphatidylserine influence rates of insulin aggregation and toxicity of amyloid aggregates in vitro, ACS Chem. Neurosci 14 (12) (2023) 2396–2404, 10.1021/acschemneuro.3c00277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Matveyenka M, Zhaliazka K, Kurouski D, Unsaturated fatty acids uniquely Alter aggregation rate of α-Synuclein and insulin and modify secondary structure and toxicity of amyloid aggregates formed in their presence, FASEB J. (2023), 10.1096/fj.202300003R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Ozoran H, Srinivasan R, Astrocytes and alpha-Synuclein: friend or foe? J. Parkinsons Dis 13 (8) (2023) 1289–1301, 10.3233/JPD-230284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Tsunemi T, Ishiguro Y, Yoroisaka A, Valdez C, Miyamoto K, Ishikawa K, Saiki S, Akamatsu W, Hattori N, Krainc D, Astrocytes protect human dopaminergic neurons from alpha-Synuclein accumulation and propagation, J. Neurosci 40 (45) (2020) 8618–8628, 10.1523/JNEUROSCI.0954-20.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Chou TW, Chang NP, Krishnagiri M, Patel AP, Lindman M, Angel JP, Kung PL, Atkins C, Daniels BP, Fibrillar alpha-synuclein induces neurotoxic astrocyte activation via RIP kinase signaling and NF-kappaB, Cell Death Dis. 12 (8) (2021) 756, 10.1038/s41419-021-04049-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Yang Y, Song JJ, Choi YR, Kim SH, Seok MJ, Wulansari N, Darsono WHW, Kwon OC, Chang MY, Park SM, et al. , Therapeutic functions of astrocytes to treat alpha-synuclein pathology in Parkinson’s disease, Proc. Natl. Acad. Sci. USA 119 (29) (2022) e2110746119, 10.1073/pnas.2110746119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Liu K, Dendritic Cells, Encyclopedia of Cell Biol. (2016) 741–749. [Google Scholar]
- [42].Delamarre L, Pack M, Chang H, Mellman I, Trombetta ES, Differential lysosomal proteolysis in antigen-presenting cells determines antigen fate, Science 307 (5715) (2005) 1630–1634, 10.1126/science.1108003. [DOI] [PubMed] [Google Scholar]
- [43].Li Q, Barres BA, Microglia and macrophages in brain homeostasis and disease, Nat. Rev. Immunol 18 (4) (2018) 225–242, 10.1038/nri.2017.125. [DOI] [PubMed] [Google Scholar]
- [44].Aguzzi A, Barres BA, Bennett ML, Microglia: scapegoat, saboteur, or something else? Science 339 (6116) (2013) 156–161, 10.1126/science.1227901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Marin-Teva JL, Dusart I, Colin C, Gervais A, van Rooijen N, Mallat M, Microglia promote the death of developing Purkinje cells, Neuron 41 (4) (2004) 535–547, 10.1016/s0896-6273(04)00069-8. [DOI] [PubMed] [Google Scholar]
- [46].Asai H, Ikezu S, Tsunoda S, Medalla M, Luebke J, Haydar T, Wolozin B, Butovsky O, Kugler S, Ikezu T, Depletion of microglia and inhibition of exosome synthesis halt tau propagation, Nat. Neurosci 18 (11) (2015) 1584–1593, 10.1038/nn.4132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TC, Maeda J, Suhara T, Trojanowski JQ, Lee VM, Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model, Neuron 53 (3) (2007) 337–351, 10.1016/j.neuron.2007.01.010. [DOI] [PubMed] [Google Scholar]
- [48].Maphis N, Xu G, Kokiko-Cochran ON, Jiang S, Cardona A, Ransohoff RM, Lamb BT, Bhaskar K, Reactive microglia drive tau pathology and contribute to the spreading of pathological tau in the brain, Brain 138 (Pt 6) (2015) 1738–1755, 10.1093/brain/awv081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Matveyenka M, Zhaliazka K, Kurouski D, Macrophages and natural killers degrade alpha-Synuclein aggregates, Mol. Pharm 21 (5) (2024) 2565–2576, 10.1021/acs.molpharmaceut.4c00160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Murray RZ, Stow JL, Cytokine secretion in macrophages: SNAREs, Rabs, and membrane trafficking, Front. Immunol 5 (2014) 538, 10.3389/fimmu.2014.00538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Dinarello CA, Immunological and inflammatory functions of the Interleukin-1 family, Annu. Rev. Immunol 27 (2009) 519–550. [DOI] [PubMed] [Google Scholar]
- [52].Sawaya BE, Deshmane SL, Mukerjee R, Fan S, Khalili K, TNF alpha production in morphine-treated human neural cells is NF-kappaB-dependent, J. NeuroImmune Pharmacol 4 (1) (2009) 140–149, 10.1007/s11481-008-9137-z. [DOI] [PubMed] [Google Scholar]
- [53].Novick D, Elbirt D, Dinarello CA, Rubinstein M, Sthoeger ZM, Interleukin-18 binding protein in the sera of patients with Wegener’s granulomatosis, J. Clin. Immunol 29 (1) (2009) 38–45, 10.1007/s10875-008-9217-0. [DOI] [PubMed] [Google Scholar]
- [54].Wang M, Tan J, Wang Y, Meldrum KK, Dinarello CA, Meldrum DR, IL-18 binding protein-expressing mesenchymal stem cells improve myocardial protection after ischemia or infarction, Proc. Natl. Acad. Sci. USA 106 (41) (2009) 17499–17504, 10.1073/pnas.0908924106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Ipseiz N, Pickering RJ, Rosas M, Tyrrell VJ, Davies LC, Orr SJ, Czubala MA, Fathalla D, Robertson AA, Bryant CE, et al. , Tissue-resident macrophages actively suppress IL-1beta release via a reactive prostanoid/IL-10 pathway, EMBO J. 39 (14) (2020) e103454, 10.15252/embj.2019103454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Deshmane SL, Kremlev S, Amini S, Sawaya BE, Monocyte chemoattractant protein-1 (MCP-1): an overview, J. Interf. Cytokine Res 29 (6) (2009) 313–326, 10.1089/jir.2008.0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Lotfi N, Thome R, Rezaei N, Zhang GX, Rezaei A, Rostami A, Esmaeil N, Roles of GM-CSF in the pathogenesis of autoimmune diseases: an update, Front. Immunol 10 (2019) 1265, 10.3389/fimmu.2019.01265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Barth S, Glick D, Macleod KF, Autophagy: assays and artifacts, J. Pathol 221 (2) (2010) 117–124, 10.1002/path.2694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Glick D, Barth S, Macleod KF, Autophagy: cellular and molecular mechanisms, J. Pathol 221 (1) (2010) 3–12, 10.1002/path.2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Jung S, Jeong H, Yu SW, Autophagy as a decisive process for cell death, Exp. Mol. Med 52 (6) (2020) 921–930, 10.1038/s12276-020-0455-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Johansen T, Lamark T, Selective autophagy: ATG8 family proteins, LIR motifs and cargo receptors, J. Mol. Biol 432 (1) (2020) 80–103, 10.1016/j.jmb.2019.07.016. [DOI] [PubMed] [Google Scholar]
- [62].Lamark T, Johansen T, Mechanisms of selective autophagy, Annu. Rev. Cell Dev. Biol 37 (2021) 143–169, 10.1146/annurev-cellbio-120219-035530. [DOI] [PubMed] [Google Scholar]
- [63].Mensah TNA, Shroff A, Nazarko TY, Ubiquitin-binding autophagic receptors in yeast: Cue5 and beyond, Autophagy 19 (9) (2023) 2590–2594, 10.1080/15548627.2023.2196878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Shroff A, Nazarko TY, SQSTM1, lipid droplets and current state of their lipophagy affairs, Autophagy 19 (2) (2023) 720–723, 10.1080/15548627.2022.2094606. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data will be made available on request.