

Abstract
The cytoplasmic regions of the receptors for epidermal growth factor (EGF) and platelet-derived growth factor (PDGF) bind and activate phospholipase C-γ1 (PLC-γ1) and other signaling proteins in response to ligand binding outside the cell. Receptor binding by PLC-γ1 is a function of its SH2 domains and is required for growth factor-induced cell cycle progression into the S phase. Microinjection into MDCK epithelial cells and NIH 3T3 fibroblasts of a polypeptide corresponding to the noncatalytic SH2-SH2-SH3 domains of PLC-γ1 (PLC-γ1 SH2-SH2-SH3) blocked growth factor-induced S-phase entry. Treatment of cells with diacylglycerol (DAG) or DAG and microinjected inositol-1,4,5-triphosphate (IP3), the products of activated PLC-γ1, did not stimulate cellular DNA synthesis by themselves but did suppress the inhibitory effects of the PLC-γ1 SH2-SH2-SH3 polypeptide but not the cell cycle block imposed by inhibition of the adapter protein Grb2 or p21 Ras. Two c-fos serum response element (SRE)-chloramphenicol acetyltransferase (CAT) reporter plasmids, a wild-type version, wtSRE-CAT, and a mutant, pm18, were used to investigate the function of PLC-γ1 in EGF- and PDGF-induced mitogenesis. wtSRE-CAT responds to both protein kinase C (PKC)-dependent and -independent signals, while the mutant, pm18, responds only to PKC-independent signals. Microinjection of the dominant-negative PLC-γ1 SH2-SH2-SH3 polypeptide greatly reduced the responses of wtSRE-CAT to EGF stimulation in MDCK cells and to PDGF stimulation in NIH 3T3 cells but had no effect on the responses of mutant pm18. These results indicate that in addition to Grb2-mediated activation of Ras, PLC-γ1-mediated DAG production is required for EGF- and PDGF-induced S-phase entry and gene expression, possibly through activation of PKC.
Cell proliferation plays a fundamental role in the development and maintenance of organisms. A variety of biological factors including epidermal growth factor (EGF) and platelet-derived growth factor (PDGF) are known to influence cell proliferation. The growth-stimulatory signals of growth factors are mediated by their cognate growth factor receptors. Ligand binding induces receptor dimerization and consequent trans phosphorylation of the EGF receptors (EGFRs) and PDGF receptors (PDGFRs) at several sites (28). These phosphorylation sites serve as binding sites for various SH2-containing proteins (18, 21). The SH2-containing proteins which bind to both EGFRs and PDGFRs include intracellular enzymes such as phospholipase C-γ1 (PLC-γ1) and Ras GTPase activating protein (1, 11, 12, 16, 36) and nonenzymatic adapter proteins such as the p85α subunit of phosphatidylinositol 3-kinase (7, 14), SHC (23), Grb2 (10), and Nck (9, 15, 20). All of these SH2-containing proteins have been implicated in transducing the mitogenic signals of EGF and PDGF.
PLC is a family of cellular proteins believed to play a significant role in the intracellular signaling mechanisms utilized by diverse hormones. Certain growth factors appear to stimulate cellular PLC activity by selective, receptor-mediated tyrosine phosphorylation of the PLC-γ1 isozyme (12, 35). PLC-γ1, a 145-kDa protein, contains two SH2 domains and one SH3 domain and hydrolyzes phosphatidylinositol-4,5-bis-phosphate to form inositol-1,4,5-triphosphate (IP3) and diacylglycerol (DAG). These second messengers are known to stimulate the release of Ca2+ from internal stores and activate protein kinase C (PKC), respectively (19, 35). PLC-γ1 forms a complex in vivo with both PDGFR and EGFR. Complex formation leads to the phosphorylation of PLC-γ1 on tyrosine residues 771, 783, and 1254 (8) and to an increase in its enzymatic activity (8, 26, 27). PLC-γ1 binds most strongly with Y992 on the EGFR and interacts less strongly with Y1173 and Y1086 (27). PLC-γ1 has a major binding site at Y1021 and a minor binding site at Y1009 in PDGFR (26). Injection of antibodies to PLC-γ1 blocks serum- and Ras-stimulated DNA synthesis (31). A PDGFR mutant lacking the PLC-γ1 binding site (Y1021) is still mitogenically active. However, when multiple phosphorylation sites are removed, readdition of Y1021 correlates with a gain of mitogenic signaling capacity (34). Recently, it was reported that PLC-γ1 was required for PDGF-induced DNA synthesis and c-Fos expression, a Ras-dependent event important for signaling (25).
The role and mechanism of action of PLC-γ1 in growth factor-induced mitogenesis are far from clear. It is not known whether PLC-γ1 is required for EGF-induced DNA synthesis and c-fos activation or whether the enzymatic activity of PLC-γ1 is required for PDGF-induced DNA synthesis and c-fos activation. It is not known how PLC-γ1 mediates growth factor-induced mitogenesis. As an intracellular enzyme containing two SH2 domains and one SH3 domain, PLC-γ1 may play various roles in growth factor signaling. In response to growth factor stimulation, PLC-γ1 may act as an enzyme to generate DAG and IP3, which can then activate downstream signaling molecules such as PKC. PLC-γ1 also has the potential to act as an adapter protein to bind both an activated growth factor receptor via its SH2 domains and a downstream molecule via its SH3 domain. For example, PLC-γ1 binds dynamin with its SH3 domain both in vitro and in vivo in response to EGF stimulation (5, 29). In this communication, we demonstrate that enzymatic activity of PLC-γ1 is required for both EGF- and PDGF-induced DNA synthesis and activation of a serum response element (SRE)-containing reporter plasmid and that the role of PLC-γ1 in these responses is to produce DAG and likely to activate PKC.
MATERIALS AND METHODS
Cells.
MDCK cells and NIH 3T3 cells were grown at 37°C in Dulbecco’s modified Eagle’s medium containing 10% fetal bovine serum, penicillin, and streptomycin and maintained in a 5% CO2 atmosphere.
Antibodies and chemicals.
Mouse monoclonal antibromodeoxyuridine (anti-BrdU) antibody was purchased from Amersham. Rabbit polyclonal anti-EGFR antibody was purchased from Santa Cruz. Rabbit polyclonal anti-Sos antibody and mouse monoclonal antidynamin antibody were purchased from Transduction Lab. Rabbit anti-chloramphenicol acetyltransferase (anti-CAT) antibody was purchased from 5 Prime→3 Prime. All of the tetramethyl rhodamine isocyanate (TRITC)- and fluorescein isothiocyanate (FITC)-conjugated secondary antibodies were purchased from Jackson Immunology Lab. Recombinant human EGF and PDGF were purchased from Upstate Biotechnology Inc. All of the other chemicals, including IP3, 1,2-dioleoyl-sn-glycerol (DAG1), and 1,2-dioctanoyl-sn-glycerol (DAG2), were from Sigma. DAG1 and DAG2 were dissolved in chloroform and stored at −20°C. Just before use, the chloroform was evaporated. The dried residues of DAG1 and DAG2 were resuspended in phosphate-buffered saline (PBS) and sonicated for 3 min at 0°C.
Purification of GST fusion proteins.
Generation of pGEX vectors that encode glutathione S-transferase (GST) fusion proteins and the purification of the expressed proteins have been described previously (2, 27, 33). Briefly, with the cDNA of PLC-γ1 used as the template, DNA fragments corresponding to the various domains were synthesized by using the PCR and oligonucleotides that contained appropriate restriction sites flanking the domains of interest. The amplified DNA was cloned into the pGEX-3X bacterial expression plasmid, which was then used to transform Escherichia coli. The expression of GST proteins in E. coli was induced with isopropyl-β-d-thiogalactopyranoside (IPTG) for 3 h. The cells were then centrifuged and lysed in PBS by sonication. Triton X-100 was added, and the lysate was clarified by centrifugation at 12,000 × g. GST fusion proteins were bound to glutathione-Sepharose beads (Sigma) and washed three times in PBS–1% Triton X-100 to remove nonspecific bound proteins. The GST fusion proteins were then biotinylated as described previously (37). Biotinylated GST fusion proteins were then eluted with an excess of reduced glutathione. Fractions were dialyzed against PBS to remove glutathione and concentrated to 4 mg/ml.
Preparation of c-fos SRE-CAT plasmids.
The two c-fos SRE-CAT reporter plasmids, a wild-type version, wtSRE-CAT, and a mutant, pm18, were provided by M. Gilman. The methods for constructing these plasmids were described previously (3, 4, 6).
Binding assay.
Cell lysates were made as described previously (38). Briefly, three 150-mm-diameter plates of MDCK cells were treated with EGF at 37°C for 15 min, washed with ice-cold PBS, lysed with 0.5 ml of RIPA buffer [PBS, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate, 1 mM sodium orthovanadate, 0.1 mM 4-(2-aminoethyl)-benzenesulfonyl fluoride, 10 μg of aprotinin per ml, 1 μM pepstatin A (pH 7.4)], and centrifuged at 12,000 × g for 10 min. Supernatants were used in the binding assay. For binding assays, GST fusion proteins, bound to glutathione-Sepharose beads, were incubated with the MDCK cell lysates at 4°C overnight. After extensive washing with RIPA buffer, the bound proteins were separated on a 7.5% polyacrylamide gel and transferred to a nitrocellulose membrane. The filters were incubated with rabbit polyclonal anti-EGFR antibody, rabbit polyclonal anti-Sos antibody, or mouse monoclonal antidynamin antibody (dilution, 1:1,000). Detection was performed by incubation of the filters with horseradish peroxidase-conjugated goat anti-rabbit antibody (for EGFR and Sos) or goat anti-mouse antibody (for dynamin) followed by enhanced chemiluminescence development (Pierce).
Microinjection and DNA synthesis assay.
The microinjection experiments were carried out by the method described by Wang and Moran (37). MDCK and NIH 3T3 cells were grown on glass coverslips to subconfluence and then serum starved for 24 h. The cells were then microinjected with biotinylated GST fusion proteins or biotinylated GST (control). The cells were injected with approximately 1 fl of 0.5× PBS-containing GST fusion proteins (2 mg/ml) and/or IP3 (20 μM). Following microinjection, the cells were incubated at 37°C for 1 h, and then BrdU and EGF (100 ng/ml) or BrdU and PDGF (20 ng/ml) were added to the medium. In some experiments, DAG1 or DAG2 was added to the incubation medium to a final concentration of 20 μM. The cells were fixed 16 h after injection in acidic alcohol (ethanol-water-acetic acid, 90:5:5) at room temperature for 30 min. After the cells were blocked with 3% bovine serum albumin for 30 min, the coverslips were incubated with a mouse anti-BrdU antibody (cell proliferation kit; Amersham) and, finally, with a mixture of FITC-conjugated avidin (to stain cells microinjected with biotinylated fusion proteins) and TRITC-conjugated donkey anti-mouse immunoglobulin G (to stain BrdU). DNA synthesis was calculated by the following formula: percent BrdU-positive cells = [(number of BrdU-positive injected cells)/(total number of injected cells)] × 100. For each experiment, 200 to 300 cells were microinjected.
Microinjection and CAT expression assay.
MDCK and NIH 3T3 cells were grown on glass coverslips to subconfluence and then serum starved for 24 h. A mixture containing a c-fos SRE-CAT expression plasmid (50 μg/ml) and a specified GST fusion protein (2 mg/ml) in microinjection buffer (50 mM HEPES [pH 7.2], 100 mM KCl, 5 mM Na2HPO4) was injected into the nuclei of the cells. Following microinjection, the cells were incubated at 37°C for 1 h, and then EGF (100 ng/ml) or PDGF (20 ng/ml) was added to the medium. In some experiments, DAG1 or DAG2 was added to the incubation medium to a final concentration of 20 μM. After 15 h of incubation, the cells were fixed with methanol at −20°C for 5 min and stained with a rabbit anti-CAT antibody followed by TRITC-conjugated donkey anti-rabbit antibody. The cells were also treated with FITC-conjugated avidin to stain cells microinjected with biotinylated fusion protein. CAT expression was calculated by the following formula: percent CAT expression-positive cells = [(number of CAT-positive injected cells)/(total number of injected cells)] × 100. For each experiment, 200 to 300 cells were microinjected.
RESULTS
Requirement for PLC-γ1 in EGF-induced S-phase entry.
To examine the function of PLC-γ1 in EGF-induced S-phase entry, a dominant-negative approach was established as described previously (37). In this approach, an excess of a dominant-negative form of the cytoplasmic signaling protein was microinjected into cells in an attempt to competitively inhibit the protein-protein interactions of the endogenous signaling molecule. The dominant-negative peptides used in this research included various SH2 and/or SH3 domain-containing forms of PLC-γ1 fused to GST (Fig. 1).
FIG. 1.
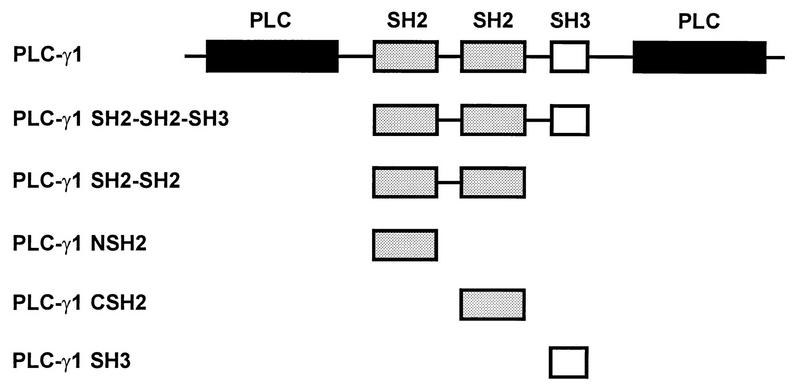
GST fusion PLC-γ1 proteins used in this study. SH2 domains (shaded bars), SH3 domains (empty bars), and catalytic phospholipase domains (solid bars) are indicated.
The GST fusion proteins were purified by adsorbtion to glutathione-Sepharose beads, and their abilities to coprecipitate activated EGFR, dynamin, and Sos were tested. MDCK cells treated with EGF for 5 min at 37°C were lysed, and the cell lysates were incubated with immobilized GST fusion proteins. After extensive washing, bound EGFR, dynamin, and Sos were detected by immunoblotting with their respective antibodies (Fig. 2). PLC-γ1 NSH2, CSH2, SH2-SH2, and SH2-SH2-SH3 were able to associate with activated EGFR (Fig. 2A), whereas PLC-γ1 SH3 and SH2-SH2-SH3 were able to associate with both dynamin and Sos (Fig. 2B and C). These results indicate that the individual SH3 and SH2 domains of PLC-γ1 are functional binding domains which still retain their binding abilities, that either one of the SH2 domains can bind the activated EGFR, and that the SH3 domain is sufficient to bind Sos and dynamin.
FIG. 2.

Association of GST fusion PLC-γ1 proteins with the activated EGFRs, Sos, and dynamin in vitro. GST fusion PLC-γ1 proteins were bound to glutathione-Sepharose beads and incubated with lysates from EGF-stimulated MDCK cells. Bound proteins were detected by Western immunoblotting with anti-EGFR, anti-Sos, or antidynamin antibodies. Lanes: 1, PLC-γ1 SH2-SH2-SH3; 2, PLC-γ1 SH2-SH2; 3, PLC-γ1 NSH2; 4, PLC-γ1 CSH2; 5, PLC-γ1 SH3; 6, GST.
The effect of microinjection into cells of purified PLC-γ1 SH2 domains on EGF-induced S-phase entry was analyzed. Quiescent MDCK cells were seeded onto coverslips, and GST fusion proteins (2 mg/ml) were injected into the cytoplasm. The cells were then stimulated with EGF (100 ng/ml), and BrdU was added to the medium. After 16 h of incubation at 37°C, the cells were fixed and immunostained for BrdU that had been metabolically incorporated into DNA during the S phase. Most of the cells microinjected with PLC-γ1 NSH2, CSH2, and SH2-SH2 did not enter the S phase, whereas most of the cells microinjected with GST alone and the nonmicroinjected cells did. Quantification of the results showed that 35% of the MDCK cells microinjected with PLC-γ1 CSH2, 19% of the MDCK cells microinjected with PLC-γ1 NSH2, and only 7% of the MDCK cells microinjected with PLC-γ1 SH2-SH2 entered the S phase, while approximately 70% of the MDCK cells microinjected with PLC-γ1 SH3 or GST entered the S phase (Fig. 3). These results showed that the SH2 domains of PLC-γ1 were effective inhibitors of S-phase entry, as measured by BrdU incorporation into microinjected cells.
FIG. 3.
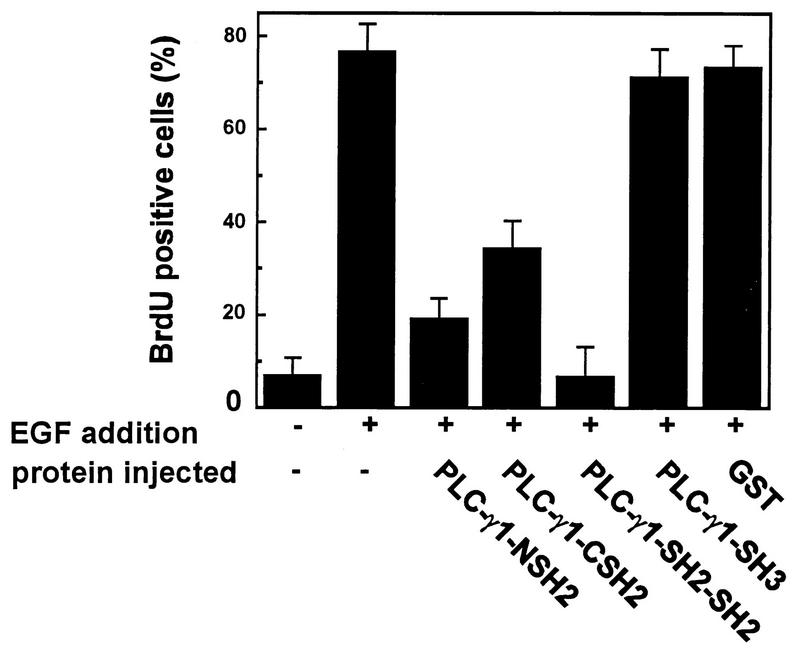
Inhibition of EGF-induced DNA synthesis by GST fusion PLC-γ1 SH3 and SH2 domains. Quiescent MDCK cells were microinjected with the indicated GST fusion proteins, and EGF (100 ng/ml) and BrdU were added to the incubation medium. The cells were fixed and stained after 16 h of treatment. The percentage of BrdU-positive cells was calculated as described in Materials and Methods. Data are means + standard errors of three independent experiments.
Requirement for PLC-γ1 enzymatic activity in EGF-induced S-phase entry.
If PLC-γ1 functions as an adapter to mediate EGF-induced mitogenesis independent of its enzymatic functions, a truncated PLC-γ1 containing the two SH2 domains and one SH3 domain (PLC-γ1 SH2-SH2-SH3) might be functional instead of functioning as a dominant-negative protein similar to PLC-γ1 SH2-SH2. However, microinjection of PLC-γ1 SH2-SH2-SH3 blocked EGF-induced DNA synthesis to the same extent as microinjection of PLC-γ1 SH2-SH2 (Fig. 4 and 5). These results suggest that merely retaining the adapter ability to form a large complex via its SH2 and SH3 domains is not sufficient for a truncated PLC-γ1 to mediate EGF-induced S-phase entry and that the catalytic domain of PLC-γ1 is required. If the injected PLC-γ1 domains are inhibitory because they block the function of endogenous PLC-γ1, then provision of IP3 and/or DAG, the products of PLC-γ1, might relieve these inhibitory constraints. This prompted us to test whether IP3 and DAG could relieve the inhibition of EGF-induced DNA synthesis elicited by PLC-γ1 SH2-SH2-SH3.
FIG. 4.
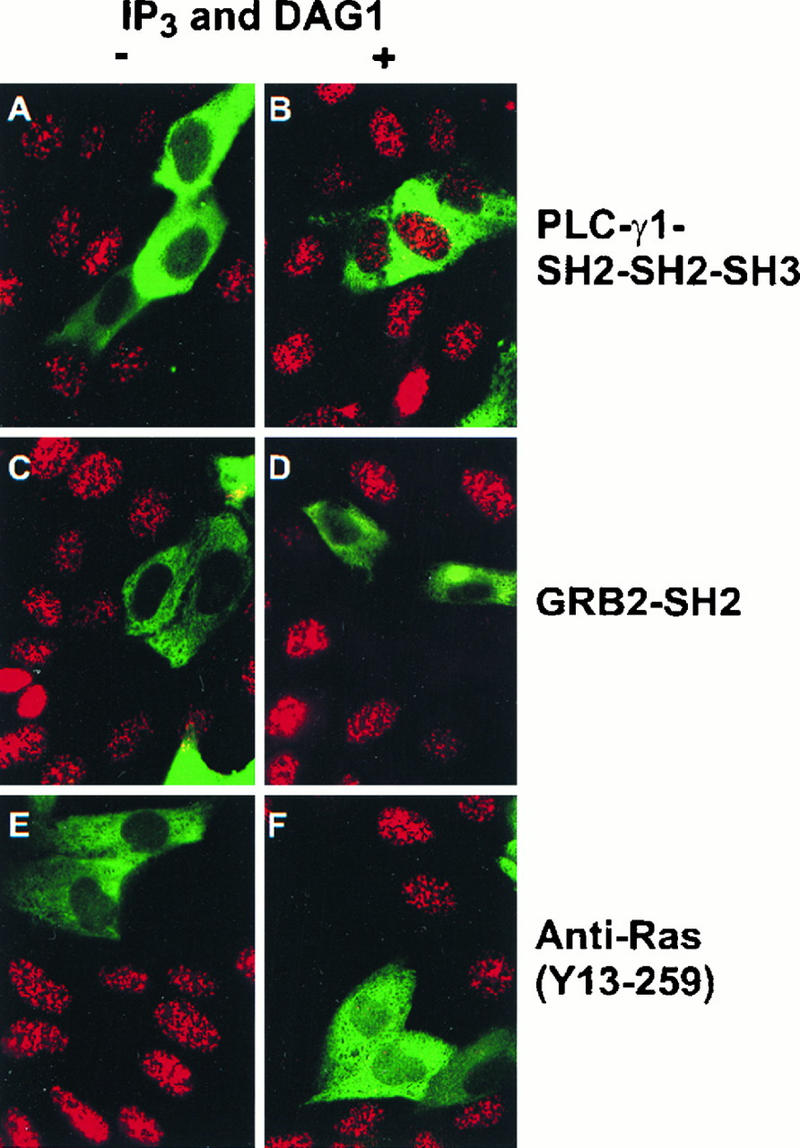
The inhibition of S-phase entry by microinjection of PLC-γ1 SH2-SH2-SH3 but not Grb2 SH2 or anti-Ras antibody is relieved by IP3 and DAG. Quiescent MDCK cells were injected with the proteins indicated below, incubated at 37°C for 1 h, and then incubated with EGF (100 ng/ml) and BrdU with or without DAG for 15 h. Following fixation, the microinjected cells were identified by use of either FITC-conjugated avidin (A to D) or FITC-conjugated antirat antibody (E and F), and BrdU incorporation was detected by use of mouse anti-BrdU antibody followed by TRITC-conjugated antimouse antibody. (A) Cells injected with PLC-γ1 SH2-SH2-SH3; (B) cells injected with PLC-γ1 SH2-SH2-SH3 and IP3 and treated with DAG1; (C) cells injected with Grb2 SH2; (D) cells injected with Grb2 SH2 and IP3 and treated with DAG1; (E) cells injected with anti-Ras Y13-259; (F) cells injected with anti-Ras Y13-259 and IP3 and treated with DAG1. Magnification, ×120.
FIG. 5.
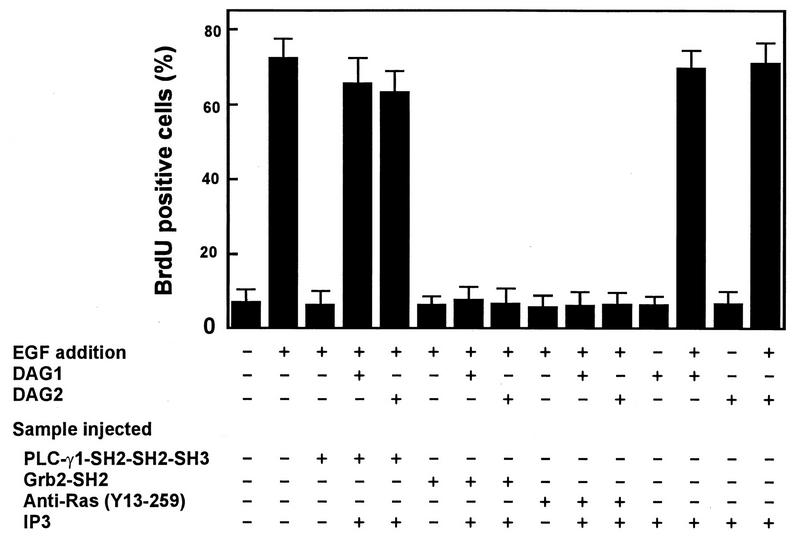
EGF-induced S-phase entry is restored by IP3 and DAG in cells injected with PLC-γ1 SH2-SH2-SH3 but not in cells injected with Grb2 SH2 or anti-Ras. Quiescent MDCK cells were injected with the proteins indicated at the bottom of the figure, incubated at 37°C for 1 h, incubated with EGF (100 ng/ml) and BrdU with or without DAG for 15 h, and then processed for immunofluorescence. The percent BrdU-positive cells was calculated as described in Materials and Methods. Data are means + standard errors of three independent experiments.
Two membrane-permeative DAGs, DAG1 and DAG2, were tested in these experiments. Comicroinjection of IP3 with PLC-γ1 SH2-SH3-SH3 followed by treatment of the cells with DAG1 or DAG2 was sufficient to overcome the inhibition caused by PLC-γ1 SH2-SH2-SH3 on EGF-induced S-phase entry (Fig. 4 and 5); however, microinjection of IP3 alone followed by treatment with DAG1 or DAG2 did not stimulate S-phase entry (Fig. 4 and 5). These results indicate that comicroinjection of IP3 and treatment of the cells with DAG are able to relieve the inhibition of PLC-γ1 SH2-SH2-SH3 but IP3 and DAG are not sufficient to stimulate S-phase entry. Microinjection of the Grb2 SH2 domain or a neutralizing anti-Ras antibody, Y13-259, also inhibited EGF-induced S-phase entry. However, the inhibition caused by these two agents was not relieved by comicroinjection with IP3 and treatment of the cells with DAG1 or DAG2 (Fig. 4 and 5). Similar results were obtained with Rat2 cells (data not shown). These results suggest that both Grb2-mediated Ras activation and PLC-γ1 enzymatic activity are required for EGF-induced S-phase entry.
Requirement for PLC-γ1 enzymatic activity in PDGF-induced DNA synthesis.
It has been reported that PLC-γ1 is required for PDGF-induced S-phase entry in NIH 3T3 cells (25). In agreement with these findings, microinjection of PLC-γ1 SH2-SH2-SH3 inhibited PDGF-induced S-phase entry in NIH 3T3 cells and the inhibition was overcome by comicroinjection with IP3 followed by treatment of the cells with DAG1 or DAG2 (data not shown). Similar to the results described above, comicroinjection of IP3 followed by treatment of the cells with DAGs did not relieve the inhibition of PDGF-induced S-phase entry elicited by the Grb2 SH2 domain or anti-Ras antibody Y13-259 (data not shown). Similar results were obtained with MDCK cells (data not shown).
The function of PLC-γ1 in EGF- and PDGF-induced S-phase entry is through a PKC-dependent pathway.
Experiments were carried out to elucidate the mechanism by which PLC-γ1 mediates EGF- and PDGF-induced S-phase entry. To determine which second messenger, IP3 or DAG, is responsible for EGF- and PDGF-induced S-phase entry, two experiments were conducted. In one experiment, we comicroinjected IP3 with PLC-γ1 SH2-SH2-SH3 in the absence of DAG. In the other experiment, we microinjected the cells with PLC-γ1 SH2-SH2-SH3 alone and then treated the cells with DAG. The results showed that DAG1 and DAG2 were sufficient to relieve the inhibition of EGF-induced S-phase entry (Fig. 6) and PDGF-induced S-phase entry (data not shown) while IP3 was not (data not shown). These results suggest that increased production of DAG and subsequent activation of PKC may be the link between EGF- and PDGF-induced activation of PLC-γ1 enzymatic activity and S-phase entry. To further test the role of PLC-γ1-induced activation of PKC in EGF- and PDGF-induced S-phase entry, we took advantage of the two previously described c-fos SRE-CAT reporter plasmids, a wild-type version, wtSRE-CAT, and a mutant, pm18 (3, 4, 6). wtSRE-CAT responds to both PKC-dependent and -independent signals, while pm18 responds only to PKC-independent signals (6).
FIG. 6.

Relief of the PLC-γ1 SH2-SH2-SH3 inhibition of EGF-induced S-phase entry by DAG. Quiescent MDCK cells were injected with PLC-γ1 SH2-SH2-SH3, incubated at 37°C for 1 h, and then incubated with EGF (100 ng/ml) and BrdU with or without DAG for 15 h. The cells were fixed and processed for immunofluorescence, and the percentage of BrdU-positive cells was calculated as described in Materials and Methods. Data are means + standard errors of three independent experiments.
MDCK cells microinjected with the wild-type reporter, wtSRE-CAT, showed a strong response to EGF stimulation, as visualized by immunofluorescence staining of CAT and coinjected GST (Fig. 7A and B and 8A), while cells containing the mutated plasmid and GST showed a weaker response to EGF stimulation, as reflected in both a weaker CAT signal and a lower ratio of CAT-positive cells to microinjected cells (Fig. 7G and H and 8A). Comicroinjection of PLC-γ1 SH2-SH2-SH3 and wtSRE-CAT greatly reduced EGF-induced CAT expression, and the inhibition was overcome by treatment with DAG1 or DAG2 (Fig. 7C to F and 8A). Comicroinjection of PLC-γ1 SH2-SH2-SH3 and the mutant plasmid, pm18, had no effect on EGF-induced expression of CAT (Fig. 7I to L and 8A). Similar results were obtained for NIH 3T3 cells stimulated with PDGF (Fig. 8B). These results indicate that inhibition of PLC-γ1 enzymatic activity reduces PKC-dependent, but not PKC-independent, activation of the c-fos promoter in response to EGF and PDGF stimulation.
FIG. 7.

Microinjection of PLC-γ1 SH2-SH2-SH3 inhibits EGF-induced PKC-dependent, but not PKC-independent, activation of c-fos promoter plasmids. Quiescent MDCK cells were injected with wtSRE-CAT (A to F) or mutant pm18 (G to L) together with PLC-γ1 SH2-SH2-SH3 (C to F and I to L) or GST (A, B, G, and H) and incubated at 37°C for 1 h, incubated with EGF (100 ng/ml) with (E, F, K, and L) or without (A to D and G to J) DAG for 15 h, and then fixed and stained by immunofluorescence. Microinjected cells were identified with an FITC-avidin stain (left panels), and CAT was assayed with polyclonal anti-CAT antibody followed by a TRITC-conjugated antirabbit antibody (right panels). Magnification, ×120.
FIG. 8.
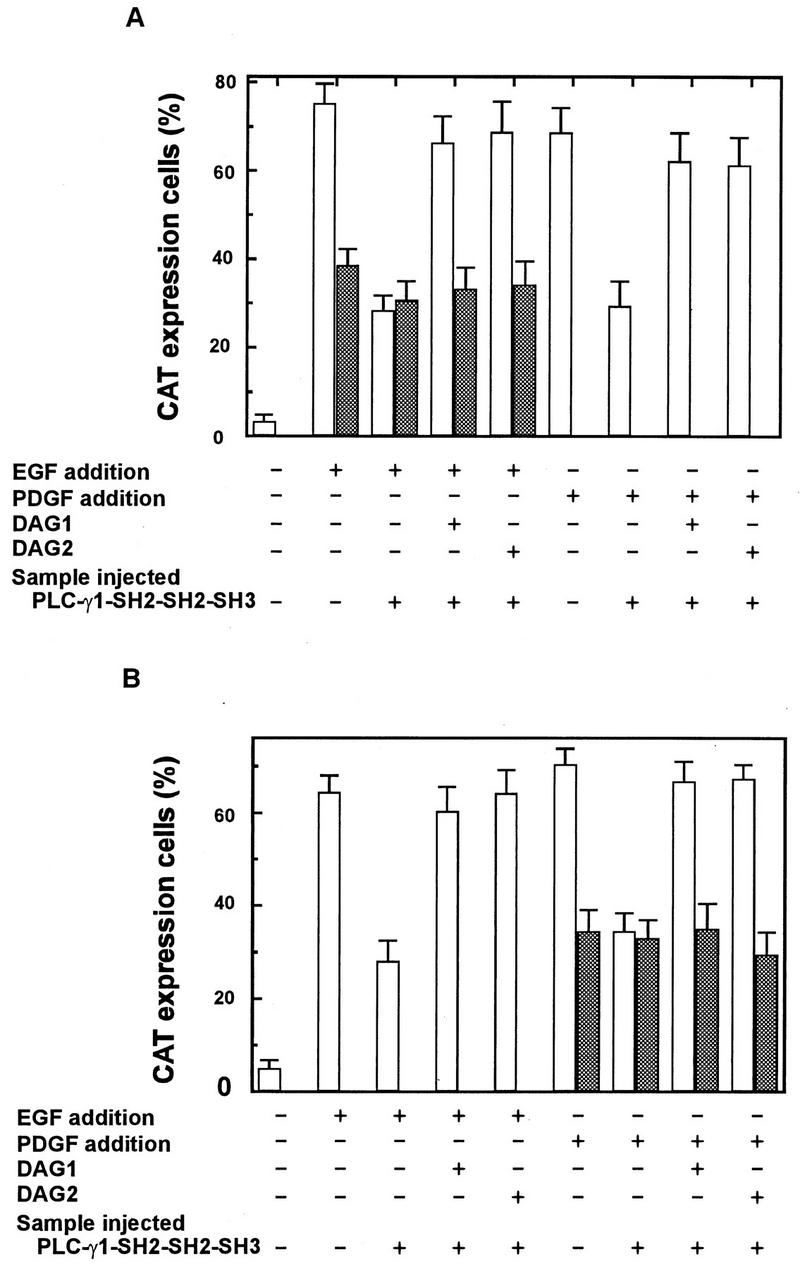
Microinjection of PLC-γ1 SH2-SH2-SH3 inhibits EGF- and PDGF-induced PKC-dependent, but not PKC-independent, activation of c-fos promoter plasmids. Quiescent MDCK cells (A) or NIH 3T3 cells (B) were injected with wtSRE-CAT (unshaded) or mutant pm18 (shaded) plasmids together with GST or PLC-γ1 SH2-SH2-SH3 and incubated at 37°C for 1 h, incubated with EGF (100 ng/ml) or PDGF (20 ng/ml) with or without DAG for 15 h, and then fixed and stained by immunofluorescence. Microinjected cells were identified with an FITC-avidin stain, and CAT was assayed with a polyclonal anti-CAT antibody followed by a TRITC-conjugated antirabbit antibody. The percentage of cells positive for CAT expression was calculated as described in Materials and Methods. Data are means + standard errors of three independent experiments.
Activation of the pm18 reporter was too weak to be assayed in MDCK cells treated with PDGF and in NIH 3T3 cells treated with EGF. The signal from wtSRE-CAT was weak in response to PDGF in MDCK cells and to EGF in NIH 3T3 cells, and these responses were inhibited by microinjection of PLC-γ1 SH2-SH2-SH3 (Fig. 8).
DISCUSSION
In this study, we used a well-established dominant-negative approach (37) to examine the function of PLC-γ1 in growth factor-induced mitogenesis. A dominant-negative PLC-γ1 protein which contains two SH2 domains and one SH3 domain but lacks the phospholipase domains (PLC-γ1 SH2-SH2-SH3) was generated, and its ability to inhibit EGF- and PDGF-induced mitogenesis was investigated. It is impossible to directly measure the enzymatic activity of PLC-γ1 in microinjected cells. However, in vitro, PLC-γ1 SH2-SH2-SH3 was able to associate with activated EGFRs through its SH2 domains and to associate with both dynamin and Sos through its SH3 domain (Fig. 2). While SH2 domains display sequence-specific binding to phosphotyrosine-containing sequences (21), their affinities are such that at concentrations high enough, binding to nonphysiological sites might occur (22). This fact, plus the inherent redundancy of SH2 binding sites in the activated EGFRs (32), means that one cannot be sure that the effects of microinjected SH2 domains are due solely to competition for binding with the corresponding endogenous protein.
Our results showed that injection of PLC-γ1 SH2-SH2-SH3 into MDCK cells completely blocked EGF-induced S-phase entry and that this inhibitory effect was relieved by injection of IP3 and DAG treatment or just DAG treatment alone. Since these products of PLC-γ1 activity were able to overcome the cell cycle block caused by injected PLC-γ1 SH2-SH2-SH3, we conclude that the injected PLC-γ1 SH2-SH2-SH3 construct is functioning as an inhibitor of endogenous PLC-γ1. PLC-γ1 by itself is not sufficient for cell cycle progression to the S phase, however, since IP3 and DAG are not mitogenic. This latter conclusion is based on the assumption that microinjection of IP3 is a valid method to introduce this compound into cells. Since IP3 is not required to suppress the inhibitory effects of the microinjected PLC-γ1 SH2-SH2-SH3 protein, we have no formal evidence that microinjected IP3 is functional in injected cells.
As we showed previously (37), microinjected Grb2 SH2 domains and the neutralizing antibody to Ras Y13-259 were also able to block S-phase entry but their inhibitory effects were not relieved by DAG and IP3. This result indicates that the relief of the inhibitory effect of PLC-γ1 SH2-SH2-SH3 by DAG is specific. Similar results were obtained with different cell types and when PDGF was used instead of EGF.
Our results suggest that in addition to Grb2-mediated Ras activation, PLC-γ1-mediated production of DAG is an essential event following EGFR and PDGFR activation, which promotes cell cycle progression to the S phase. The mechanism of action of PLC-γ1 in this pathway is therefore likely to be the stimulation of DAG-dependent PKC activity or some other DAG-dependent signaling pathway. The apparent lack of a requirement for IP3 downstream of PLC-γ1 in this signal suggests that IP3-induced Ca2+ mobilization may not be essential for S-phase entry following activation of receptor tyrosine kinases. This result is in agreement with that of Margolis et al. (13), i.e., that PDGF-induced generation of IP3 in cells overexpressing PLC-γ1 did not influence PDGF-induced DNA synthesis.
wtSRE-CAT contains a c-fos SRE placed adjacent to a basic promoter element and linked to CAT. SRE is a primary nuclear target for intracellular signal transduction pathways triggered by growth factors. It is the target for both PKC-dependent and -independent signals. Function of the SRE requires binding of a cellular protein, termed serum response factor (SRF). A second protein, p62TCF, recognizes the SRE-SRF complex and forms a ternary complex. The mutant construct, pm18, which contains a single base substitution in the SRE, can still bind SRF but fails to form the ternary complex. wtSRE-CAT responds to both PKC-dependent and -independent signals, while pm18 responds only to PKC-independent signals (6).
Both wtSRE-CAT and mutant pm18 responded to EGF stimulation in MDCK cells and to PDGF stimulation in NIH 3T3 cells. The response of wtSRE-CAT was much stronger than that of mutant pm18 (Fig. 7 and 8), suggesting that EGF and PDGF initiate both PKC-dependent and -independent mitogenic signals. This result is consistent with the previous report that both wtSRE-CAT and pm18 reporters were activated by purified recombinant c-Sis (a form of PDGF) and that the response of mutant pm18 to c-Sis was weaker than that of wtSRE-CAT (6).
Microinjection of the dominant-negative PLC-γ1 SH2-SH2-SH3 protein greatly reduced the responses of wtSRE-CAT to EGF stimulation in MDCK cells and to PDGF stimulation in NIH 3T3 cells, whereas it had no effect on the responses of mutant pm18. Furthermore, the inhibition of the wtSRE-CAT expression was relieved by treatment with DAG (Fig. 7 and 8). These results further indicate that the role of PLC-γ1 in EGF- and PDGF-induced mitogenesis is to produce DAG and subsequently activate PKC. Our results also indicate that growth factor-induced mitogenic signals are different in different cell types. The failure of mutant pm18 to respond to EGF in NIH 3T3 cells and to PDGF in MDCK cells may suggest that EGF-induced mitogenic signals in NIH 3T3 and PDGF-induced mitogenic signals in MDCK cells are more PKC dependent. Another explanation is that the EGFR concentration in NIH 3T3 cells is much lower than that of PDGFR and the EGFR concentration in MDCK cells is much higher than that of PDGFR (36a). Similar cell type differences in response to serum stimulation were observed previously (6, 30).
Our findings with the EGF and PDGF receptors distinguish them from the fibroblast growth factor receptor which also binds and activates PLC-γ1. PLC-γ1 activation is dispensable for fibroblast growth factor-induced DNA synthesis (17, 24). Clearly, there are some differences in the signaling pathways used by various receptor tyrosine kinases to stimulate completion of the G1 phase and entry into the S phase. It is likely that these differences reflect the signaling proteins recruited to the cytoplasmic domains of different, activated, growth factor receptor. The ability to selectively inhibit SH2 domain interaction as described in this study should help to better our understanding of the signaling events controlled by these protein-protein interactions.
ACKNOWLEDGMENTS
We thank M. Gilman for the c-fos SRE-CAT constructs, J. Schlessinger for the Grb2 reagents, J. Knopf for the PLC-γ1 cDNA, and R. Lafrenie and E. Gauthier for critical reading of the manuscript.
This work was supported by funds from the Ontario Cancer Treatment and Research Foundation and from the Northern Cancer Research Foundation (to Z.W.), the Northern Ontario Heritage Fund Corporation (to S.G.), and the National Cancer Institute of Canada (to M.F.M.). M.F.M. is a Medical Research Council of Canada Scientist.
REFERENCES
- 1.Anderson D, Koch C A, Grey L, Ellis C, Moran M F, Pawson T. Binding of SH2 domains of phospholipase Cγ1, GAP, and Src to activated growth factor receptors. Science. 1990;250:979–982. doi: 10.1126/science.2173144. [DOI] [PubMed] [Google Scholar]
- 2.Bar-Sagi D, Rotin D, Batzer A, Mandiyan V, Schlessinger J. SH3 domains direct cellular localization of signaling molecules. Cell. 1993;74:83–91. doi: 10.1016/0092-8674(93)90296-3. [DOI] [PubMed] [Google Scholar]
- 3.Gilman M Z. The c-fos serum response element responds to protein kinase C-dependent and -independent signals but not to cyclic AMP. Genes Dev. 1988;2:394–402. doi: 10.1101/gad.2.4.394. [DOI] [PubMed] [Google Scholar]
- 4.Gilman M Z, Wilson R N, Weinberg R A. Multiple protein-binding sites in the 5′-flanking region regulate c-fos expression. Mol Cell Biol. 1986;6:4305–4316. doi: 10.1128/mcb.6.12.4305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gout I, Dhand R, Hiles I D, Fry M J, Panayotou G, Das P, Truong O, Totty N F, Hsuan J, Booker G W. The GTPase dynamin binds to and is activated by a subset of SH3 domains. Cell. 1993;75:25–36. [PubMed] [Google Scholar]
- 6.Graham R, Gilman M. Distinct protein targets for signals acting at the c-fos serum response element. Science. 1991;251:189–192. doi: 10.1126/science.1898992. [DOI] [PubMed] [Google Scholar]
- 7.Hu P, Margolis B, Skolnik E Y, Lammers R, Ullrich A, Schlessinger J. Interaction of phosphatidylinositol 3-kinase-associated p85 with epidermal growth factor and platelet-derived growth factor receptors. Mol Cell Biol. 1992;12:981–990. doi: 10.1128/mcb.12.3.981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim H K, Kim J W, Zilberstein A, Margolis B, Kim J G, Schlessinger J, Rhee S G. PDGF stimulation of inositol phospholipid hydrolysis requires PLC-γ1 phosphorylation on tyrosine residues 783 and 1254. Cell. 1991;65:435–441. doi: 10.1016/0092-8674(91)90461-7. [DOI] [PubMed] [Google Scholar]
- 9.Li W, Hu P, Skolnik E Y, Ullrich A, Schlessinger J. The SH2 and SH3 domain-containing Nck protein is oncogenic and a common target for phosphorylation by different surface receptors. Mol Cell Biol. 1992;12:5824–5833. doi: 10.1128/mcb.12.12.5824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lowenstein E J, Daly R J, Batzer A G, Li W, Margolis B, Lammers R, Ullrich A, Skolnik E Y, Bar-Sagi D, Schlessinger J. The SH2 and SH3 domain-containing protein GRB2 links receptor tyrosine kinases to ras signaling. Cell. 1992;70:431–442. doi: 10.1016/0092-8674(92)90167-b. [DOI] [PubMed] [Google Scholar]
- 11.Margolis B, Li N, Koch A, Mohammadi M, Hurwitz D R, Zilberstein A, Ullrich A, Pawson T, Schlessinger J. The tyrosine phosphorylated carboxyterminus of the EGF receptor is a binding site for GAP and PLC-γ. EMBO J. 1990;9:4375–4380. doi: 10.1002/j.1460-2075.1990.tb07887.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Margolis B, Rhee S G, Felder S, Mervic M, Lyall R, Levitzki A, Ullrich A, Zilberstein A, Schlessinger J. EGF induces tyrosine phosphorylation of phospholipase C-II: a potential mechanism for EGF receptor signaling. Cell. 1989;57:1101–1107. doi: 10.1016/0092-8674(89)90047-0. [DOI] [PubMed] [Google Scholar]
- 13.Margolis B, Zilberstein A, Franks C, Felder S, Kremer S, Ullrich A, Rhee S G, Skorecki K, Schlessinger J. Effect of phospholipase C-γ overexpression on PDGF-induced second messengers and mitogenesis. Science. 1990;248:607–610. doi: 10.1126/science.2333512. [DOI] [PubMed] [Google Scholar]
- 14.McGlade C J, Ellis C, Reedijk M, Anderson D, Mbamalu G, Reith A D, Panayotou G, End P, Bernstein A, Kazlauskas A. SH2 domains of the p85 alpha subunit of phosphatidylinositol 3-kinase regulate binding to growth factor receptors. Mol Cell Biol. 1992;12:991–997. doi: 10.1128/mcb.12.3.991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meisenhelder J, Hunter T. The SH2/SH3 domain-containing protein Nck is recognized by certain anti-phospholipase C-γ1 monoclonal antibodies, and its phosphorylation on tyrosine is stimulated by platelet-derived growth factor and epidermal growth factor treatment. Mol Cell Biol. 1992;12:5843–5856. doi: 10.1128/mcb.12.12.5843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Meisenhelder J, Suh P G, Rhee S G, Hunter T. Phospholipase C-γ is a substrate for the PDGF and EGF receptor protein-tyrosine kinases in vivo and in vitro. Cell. 1989;57:1109–1122. doi: 10.1016/0092-8674(89)90048-2. [DOI] [PubMed] [Google Scholar]
- 17.Mohammadi M, Dionne C A, Li W, Li N, Spivak T, Honegger A M, Jaye M, Schlessinger J. Point mutation in FGF receptor eliminates phosphatidylinositol hydrolysis without affecting mitogenesis. Nature. 1992;358:681–684. doi: 10.1038/358681a0. [DOI] [PubMed] [Google Scholar]
- 18.Moran M, Koch C, Anderson D, Ellis C, England L, Martin G, Pawson T. Src homology region 2 domains direct protein-protein interactions in signal transduction. Proc Natl Acad Sci USA. 1990;87:8622–8626. doi: 10.1073/pnas.87.21.8622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nishizuka Y. Intracellular signaling by hydrolysis of phospholipids and activation of protein kinase C. Science. 1992;258:607–614. doi: 10.1126/science.1411571. [DOI] [PubMed] [Google Scholar]
- 20.Park D, Rhee S G. Phosphorylation of Nck in response to a variety of receptors, phorbol myristate acetate, and cyclic AMP. Mol Cell Biol. 1992;12:5816–5823. doi: 10.1128/mcb.12.12.5816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pawson T. Protein modules and signalling networks. Nature. 1995;373:573–580. doi: 10.1038/373573a0. [DOI] [PubMed] [Google Scholar]
- 22.Payne G, Stolz L A, Pei D, Band H, Shoelson S E, Walsh C T. The phosphopeptide binding specificity of Src family SH2 domains. Chem Biol. 1994;1:99–105. doi: 10.1016/1074-5521(94)90047-7. [DOI] [PubMed] [Google Scholar]
- 23.Pelicci G, Lanfrancone L, Grignani F, McGlade J, Cavallo F, Forni G, Nicoletti I, Pawson T, Pelicci P G. A novel transforming protein (SHC) with an SH2 domain is implicated in mitogenic signal transduction. Cell. 1992;70:93–104. doi: 10.1016/0092-8674(92)90536-l. [DOI] [PubMed] [Google Scholar]
- 24.Peters K G, Marie J, Wilson E, Ives H E, Escobedo J, Del-Rosairo M, Mirda D, Williams L T. Point mutation of an FGF receptor abolishes phosphatidylinositol turnover and Ca2+ flux but not mitogenesis. Nature. 1992;358:678–681. doi: 10.1038/358678a0. [DOI] [PubMed] [Google Scholar]
- 25.Roche S, McGlade J, Jones M, Gish G D, Pawson T, Courtneidge S A. Requirement of phospholipase Cγ, the tyrosine phosphatase Syp and the adaptor proteins Shc and Nck for PDGF-induced DNA synthesis: evidence for the existence of Ras-dependent and Ras-independent pathways. EMBO J. 1996;15:4940–4948. [PMC free article] [PubMed] [Google Scholar]
- 26.Ronnstrand L, Mori S, Arridsson A K, Eriksson A, Wernstedt C, Hellman U, Claesson-Welsh L, Heldin C H. Identification of two C-terminal autophosphorylation sites in the PDGF beta-receptor: involvement in the interaction with phospholipase C-γ. EMBO J. 1992;11:3911–3919. doi: 10.1002/j.1460-2075.1992.tb05484.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rotin D, Margolis B, Mohammadi M, Daly R J, Daum G, Li N, Fischer E H, Burgess W H, Ullrich A, Schlessinger J. SH2 domains prevent tyrosine dephosphorylation of the EGF receptor: identification of Tyr992 as the high-affinity binding site for SH2 domains of phospholipase Cγ. EMBO J. 1992;11:559–567. doi: 10.1002/j.1460-2075.1992.tb05087.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schlessinger J, Ullrich A. Growth factor signaling by receptor tyrosine kinases. Neuron. 1992;9:383–391. doi: 10.1016/0896-6273(92)90177-f. [DOI] [PubMed] [Google Scholar]
- 29.Seedorf K, Kostka G, Lammers R, Bashkin P, Daly R, Burgess W H, van der Bliek A M, Schlessinger J, Ullrich A. Dynamin binds to SH3 domains of phospholipase Cγ and GRB-2. J Biol Chem. 1994;269:16009–16014. [PubMed] [Google Scholar]
- 30.Shaw P E, Schroter H, Nordheim A. The ability of a ternary complex to form over the serum response element correlates with serum inducibility of the human c-fos promoter. Cell. 1989;56:563–572. doi: 10.1016/0092-8674(89)90579-5. [DOI] [PubMed] [Google Scholar]
- 31.Smith M R, Liu Y L, Kim H, Rhee S G, Kung H F. Inhibition of serum- and ras-stimulated DNA synthesis by antibodies to phospholipase C. Science. 1990;247:1074–1077. doi: 10.1126/science.2408147. [DOI] [PubMed] [Google Scholar]
- 32.Soler C, Beguinot L, Carpenter G. Individual epidermal growth factor receptor autophosphorylation sites do not stringently define association motifs for several SH2-containing proteins. J Biol Chem. 1994;269:12320–12324. [PubMed] [Google Scholar]
- 33.Songyang Z, Shoelson S E, Chaudhuri M, Gish G, Pawson T, Haser W G, King F, Roberts T, Ratnofsky S, Lechleider R J. SH2 domains recognize specific phosphopeptide sequences. Cell. 1993;72:767–778. doi: 10.1016/0092-8674(93)90404-e. [DOI] [PubMed] [Google Scholar]
- 34.Valius M, Kazlauskas A. Phospholipase C-γ1 and phosphatidylinositol 3 kinase are the downstream mediators of the PDGF receptor’s mitogenic signal. Cell. 1993;73:321–334. doi: 10.1016/0092-8674(93)90232-f. [DOI] [PubMed] [Google Scholar]
- 35.Wahl M, Carpenter G. Selective phospholipase C activation. Bioessays. 1991;13:107–113. doi: 10.1002/bies.950130303. [DOI] [PubMed] [Google Scholar]
- 36.Wahl M I, Daniel T O, Carpenter G. Antiphosphotyrosine recovery of phospholipase C activity after EGF treatment of A-431 cells. Science. 1988;241:968–970. doi: 10.1126/science.2457254. [DOI] [PubMed] [Google Scholar]
- 36a.Wang, Z. Unpublished observations.
- 37.Wang Z, Moran M F. Requirement for the adapter protein GRB2 in EGF receptor endocytosis. Science. 1996;272:1935–1939. doi: 10.1126/science.272.5270.1935. [DOI] [PubMed] [Google Scholar]
- 38.Wang Z, Tung P S, Moran M F. Association of p120 ras GAP with endocytic components and colocalization with epidermal growth factor (EGF) receptor in response to EGF stimulation. Cell Growth Differ. 1996;7:123–133. [PubMed] [Google Scholar]