

Abstract
DNA gyrase is an essential type II topoisomerase that is conserved across bacteria species and has an essential function of resolving overwound (positive supercoiled) DNA and introducing negative supercoils into relaxed DNA. The overall catalytic activity of gyrase can be determined using in vitro assays utilizing purified enzyme subunits and a DNA substrate. As gyrase is the only topoisomerase that can introduce negative supercoils into relaxed DNA, the inhibition of this catalytic activity is a good indicator of the efficacy and potency of potential antibacterial compounds. This chapter outlines a protocol for a purified enzyme assay with relaxed DNA that utilizes gel electrophoresis to monitor the ability of gyrase to introduce negative supercoils into DNA. The protocol focuses on the DNA supercoiling activity of Escherichia coli wild-type gyrase. However, it can be easily modified for use with gyrase from other bacterial species.
1. Introduction:
The helical nature of DNA, which is further compressed by coiling to fit into the cell, requires enzymes that will allow access to the genetic function for essential functions such as replication and transcription [1,2]. Topoisomerases are enzymes that alter the topological state of DNA and are conserved across all domains of life. Type II topoisomerases can alter DNA under- and over-wounding by passing a double helix through a transient double-stranded break that they generate in a separate segment of DNA. To maintain genomic integrity during the cleavage event, the double-stranded breaks are anchored to the enzyme via a covalent bond [3,4].
Most bacteria encode two type II topoisomerases, DNA gyrase and topoisomerase IV. Some organisms, such as those in the Mycobacterium genus express only gyrase, underscoring the importance of this enzyme[3,5,6]. It is important to note, however, that in organisms that encode gyrase and topoisomerase IV, both enzymes are essential [3,6]. Gyrase and topoisomerase IV each have specific roles in the cell. The primary function of gyrase in bacteria is to remove positive supercoils that accumulate ahead of transcription complexes and replication bubbles and to introduce negative supercoils into the chromosome [7–9]. This latter activity is critical, as most bacterial species maintain a genome that is ~6% underwound [2,3,7]. Gyrase is the only topoisomerase that can independently introduce negative supercoils into DNA [3,10]. Conversely, the primary function of topoisomerase IV is to decatenate daughter chromosomes after replication and to resolve knots introduced by recombination events [3,10,11].
Most type II topoisomerase-targeting drugs work via two mechanisms: either by stabilizing the covalent enzyme cleavage-DNA intermediate, which leads to formation of persistent double-stranded breaks or by inhibiting the catalytic activity of the enzymes [3,12,13]. Fluoroquinolones, one of the most efficacious and broad-spectrum antibacterial classes currently in clinical use, act through both mechanisms [3,10,13].
The activity of compounds that target type II topoisomerases can be determined using whole cell and infection models. However, in vitro assays using purified proteins and different DNA substrates allow for greater ease and higher level of control across multiple factors. By varying the enzyme and DNA concentration as well as reaction times, a host of information can be gathered via a simple gel-based technique. As gyrase is the only topoisomerase that can introduce negative supercoils into relaxed DNA, the in vitro DNA supercoiling assay can be used to monitor the activity and efficiency of the enzyme. The assay below utilizes wild-type Escherichia coli gyrase. However, it can be easily modified for gyrase from any bacterial species. Additionally, the assay can be used to assess the inhibition of gyrase-catalyzed DNA supercoiling by antibacterial agents.
2. Materials:
The following materials for the Escherichia coli supercoiling assay should be made using ultra-pure deionized water and analytical reagent grade chemicals. All reagents are stored at 4°C unless otherwise noted.
2.1. Supercoiling Assay:
-
Preparation of Relaxed DNA:
Reaction Buffer: 5x topoisomerase I relaxation buffer: 250 mM Tris-Cl pH 7.5, 250 mM KCl, 50 mM MgCl2, 2.5 mM DTT, 0.5 mM Na2EDTA, and 150µg/mL BSA [14]. To make the buffer, add 2.5 mL of 1 M Tris-Cl pH 7.5, 1.25 mL of 2 M KCl, 500 µL of MgCl2, 50 µL of 0.5 M DTT, 20 µLNa2EDTA, and 150 µL of 10 mg/mL BSA in a 10 mL graduate cylinder. Bring the volume up to 10 mL, cover with parafilm, and mix by inversion.
pBR322 plasmid: Dilute to 1 mg/mL concentration in 5 mM Tris-HCl (pH 7.5), 0.5 mM Na2EDTA. (see Note 1)
Calf Thymus Topoisomerase I – reagent grade (see Note 2)- To make the reaction mix, add 50 µL of 1 mg/mL pBR322 DNA plasmid (50 µg), 30 µL of the 5x topoisomerase I relaxation buffer, and 5 µL of calf thymus topoisomerase I. Add ultrapure water (65 µL) to bring the final volume to 150 µL.
- Incubate the reaction for 30 min at 37˚C.
- Increase the temperature to 75˚C, and allow the mixture to incubate for an additional 10 min. The increased temperature will inactivate topoisomerase I.
- Cool the reaction mixture to room temperature. This usually takes about 10 min.
- Use a spectrophotometer to determine the final DNA concentration (see Note 3).
- To determine if the DNA is fully relaxed, subject the sample to electrophoresis on a 1% agarose TBE (Tris-Borate-EDTA) gel at 150 V for 2.5 h (refer to 2.2: Agarose gel electrophoresis reagents).
- To stain to the gel, add 10 µL of 10 mg/mL ethidium bromide to 200 mL of ultra-pure water. Add the gel to the mixture and allow the gel to stain under gentle shaking for 30 min.
- Destain the gel by adding the gel to 200 mL of ultrapure water, and gently shake for 15 min.
-
Image the gel under a medium range UV (see Figure 1).Relaxed DNA can be stored at −80˚C. After being thawed, the DNA should be stored at 4˚C to prevent excessive freeze thaw cycles. The aliquot of relaxed DNA is stable at 4˚C for up to 1 month after one freeze thaw cycle.
E. coli gyrase subunits: Each of the gyrase subunits, Gyr A and Gyr B, are isolated from BL21 cells that were transformed with Kanamycin-resistant plasmid with each subunit as outlined in Aldred et. al. [14] or Chan et. al. [15]. The gyrase subunits can also be purchased commercially. Following purification, Gyr A and Gyr B are aliquoted and stored in 200 mM NaCl, 50 mM Tris-HCl (pH 7.5), 20% glycerol at −80˚C or liquid nitrogen. Further freeze thaw cycles should be avoided to prevent loss of enzyme activity.
Enzyme diluent: E. coli gyrase dilution buffer: 40 mM HEPES-KOH, pH 7.6, 1 mM Na2EDTA, 150 mM potassium glutamate, 40% glycerol [16]. To make the buffer, add 400 µL of 1M HEPES-KOH pH 7.6, 40 µL of 250 mM Na2EDTA, 600 µL of 2.5M potassium glutamate, 8 mL of 50% glycerol, and then bring up the volume to 10 mL. Mix well before use.
Reaction Buffer: 5x E. coli gyrase supercoiling buffer: 50 mM Tris-HCl pH 7.5, 5 mM MgCl2, 175 mM potassium glutamate, and 50 μg/mL bovine serum albumin (BSA) [16]. Store at 4˚C. To make the buffer, add 500 µL of 1 M Tris-HCl pH 7.5, 50 µL of 1 M MgCl2, 700 μL of 2.5 M potassium glutamate, and 500 µL of 1 mg/mL BSA a 10 mL graduate cylinder. Bring up the volume to 10 mL. Mix well before use. The reaction buffer will differ for enzymes from different species (see Note 4).
20 mM adenosine triphosphate (ATP) in water. Should be aliquoted and stored at −20˚C.
Supercoiling stop solution: 77.5 mM Na2EDTA, 0.77% SDS. To make the solution, add 155 µL of Na2EDTA, 77 µL of 5% SDS (made in water), and 268 µL of ultrapure water. Mix well by vortexing before use. Store at room temperature. (see Note 5)
Proteinase K: 0.8 mg/mL in 50 mM Tris-HCl, pH 7.9, 1 mM CaCl2. Make a stock of the buffer by adding 500 µL of 1 M Tris-HCL pH 7.9, 10 µL of CaCl2 and bring it up to 10 mL. Mix well before use. To a 1 mL of aliquot of the buffer, add 0.8 mg of proteinase K.
Figure 1:

Representative gel showing negatively supercoiled [(−) SC] and relaxed DNA.
2.2. Agarose Gel Electrophoresis Reagents
Agarose gel loading buffer: 600 mg/mL sucrose, 10 mM Tris-HCl, pH 7.9, 5 mg/mL bromophenol blue, 5 mg/mL xylene cyanole. To make the buffer, add 6 g of sucrose, 100 μL of 1 M Tris-Cl, pH 7.9, 50 mg of bromophenol blue, 50 mg of xylene cyanole to a 15 mL conical tube. Slowly add ultrapure water to a total volume of 10 mL. To allow the sucrose to dissolve faster, the buffer can be heated at 37 °C for 20 min. The buffer is stored for long term use at −20°C and for short term use (up to 1 month) at room temperature.
Agarose Type I (molecular biology/electrophoresis grade).
Commercial gel electrophoresis apparatus and power source.
10x Tris-Borate-EDTA (TBE) buffer: 1 M Tris base, 1 M boric acid, 20 mM ethylenediaminetetraacetic acid (EDTA). To make the buffer, dissolve 121.1 g of Tris base, 61.8g of boric acid and 5.8g of EDTA in ~800 mL of ultrapure water. Once the reagents are dissolved, transfer the contents to a graduated cylinder. Add ultrapure water to a final volume of 1 L. Filter sterilize and store at room temperature.
1% Agarose TBE gel: Make 800 mL of 1x TBE (80 mL of 10x TBE and 720 mL of ultrapure water) and mix well. Add 1 g of agarose to 100 mL of 1x TBE in a microwave-safe flask. This volume is sufficient to pour a 12 × 14 cm agarose gel in a commercial gel box. Heat to boiling in a microwave oven (see Note 6) until no agarose particles are visible upon swirling. Allow to cool to approximately 60°C before casting with a 16 or 20 well comb. The comb should be 1 cm from the anode edge of the gel. Allow the gel to completely cool, before covering with the remaining 700 mL of 1x TBE buffer. The buffer should completely immerse the gel and have the same width of liquid above it as the gel itself. Carefully, remove the comb before loading the gel.
Ethidium Bromide – 10 mg/mL solution of ethidium bromide in water. Store in a dark container, as it is light sensitive (see Note 7).
UV light source (medium range).
Gel imaging system.
3. Methods:
3.1. Time Course of DNA Supercoiling:
Determine the number of time points to be monitored and label the reaction tubes. Additionally, label an extra tube as the DNA control. To each tube, except the DNA, control, add 3 μL of the supercoiling stop solution.
Determine the desired number of timepoints and calculate the volume of a master assay mixture that includes per reaction: 4 μL of 5x E. coli gyrase supercoiling buffer, X μL of relaxed DNA (X μL represents a volume that is equivalent to 0.3 μg of relaxed DNA), and (11 – X) μL ultrapure water, for a final volume of 15 μL per reaction. The master assay mixture should be calculated to include one reaction for each timepoint plus a DNA control that contains no enzyme (n reactions). The assay mixture should also include one extra reaction to ensure there is sufficient volume for accurate pipetting (n + 1 reactions). For example, a time course with 10 timepoints would require a master assay mixture for 12 total reactions (n = 11, n + 1 = 12). This would result in a final assay mixture of 180 μL total volume.
To a pre-chilled tube (chilled on ice for 3–5 min on a metal block) add all the above reagents and vortex lightly to mix thoroughly, and store on ice.
To set up the DNA control tube, add 16 μL of the master assay mixture to 4 μL of ultrapure water for a final volume of 20 μL.
Each enzyme subunit should be diluted to 20-fold of the desired concentration using the enzyme diluent, so that 2 μL of each subunit will be added per tube. The E. coli gyrase subunits are added to the master assay mixture at a ratio of Gyr A2: Gyr B2 to 1:1. The final concertation of gyrase in the DNA supercoiling reaction will be 5 nM. Consequently, each subunit should be diluted to 100 nM.
To the master assay mixture, add 2 × n μL of each enzyme subunit and mix lightly and keep on ice to prevent the catalytic activity from starting. This is because the master assay mixture was originally (n+1) and after the removal of 1 reaction for the DNA control, there are ‘n’ reactions left in the tube. Keep the gyrase subunit at −80°C until ready to add to the master assay mixture. Mix the assay mixture by pipetting and gently vortexing.
Incubate the master assay mixture tube at 37°C. At the desired timepoints, remove 20 μL samples (swirl and mix before removing) and add to the prepared tube with 3 μL of the supercoiling stop solution. Mix thoroughly by pipetting vigorously (at least 3 times). Keep samples at room temperature until the end of the timepoints.
At the end of the time course, stop the DNA control with 3 μL of the stop solution.
After the time course has been completed, add 2 μL of agarose gel loading buffer to each tube.
Stopping point: If you want to store the samples to be analyzed later, add 2 μL of proteinase K to each tube. Incubate at 45°C for 30 min before adding the loading buffer. These tubes can be stored at −20°C for several days before gel electrophoresis. To load the samples, thaw the tubes at 45°C for 20 min. Cool to room temperature before loading.
Carefully load 20 μL of each sample into wells of the 1% agarose TBE gel and run the gel at 150V for 2.5 h (see Notes 8 and 9).
Stain the gel in a mixture of 200 mL of water and 20 μL of 10 mg/mL ethidium bromide for 30 min with agitation.
Destain with plain water and agitation for 15 min.
The DNA bands in the gel are visualized using medium-range UV light and an imaging system (see Figure 2).
Figure 2:
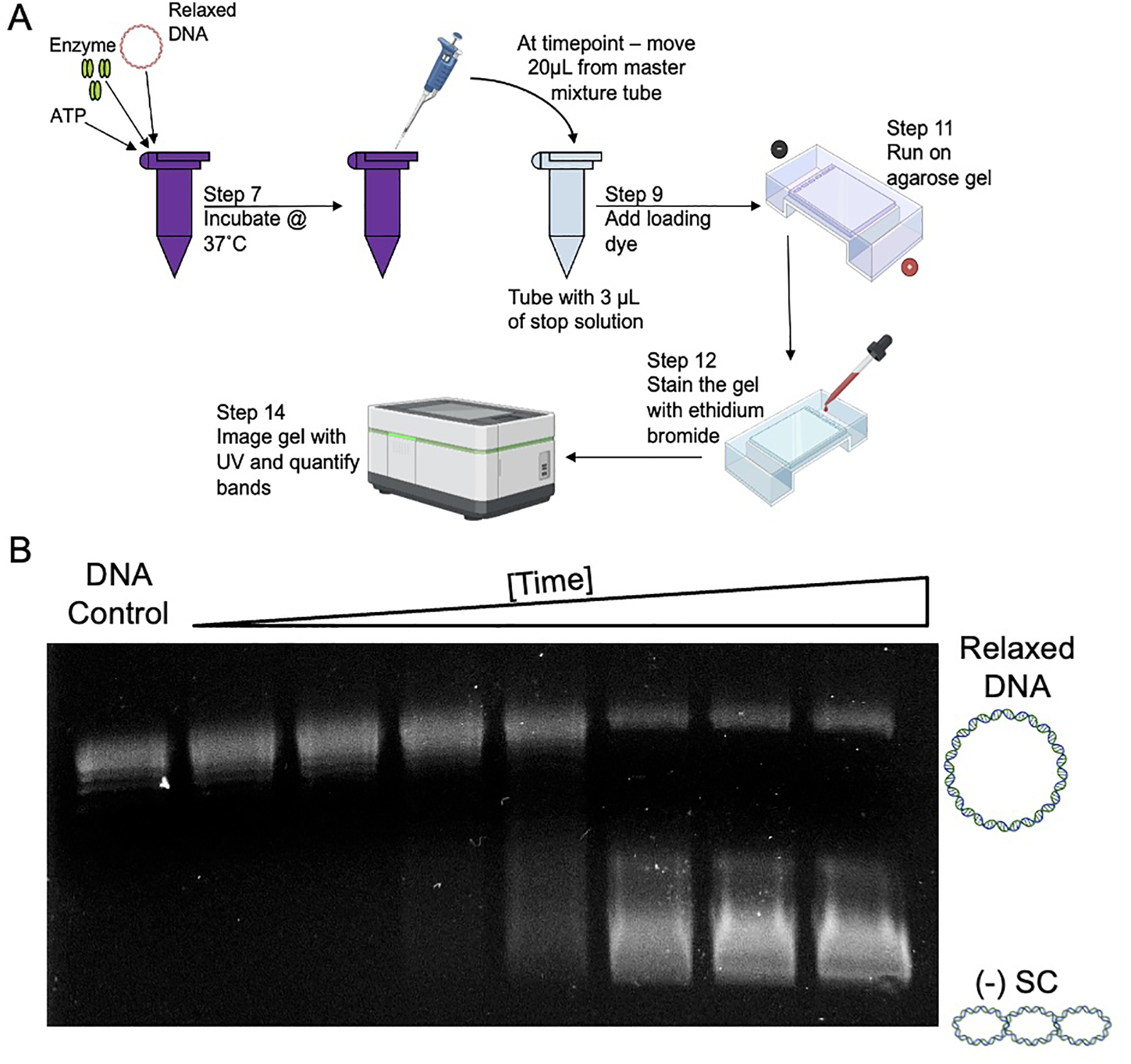
A) Overview of the time course experimental set up. B) Representative gel of a time course showing the DNA supercoiling activity of gyrase. The first lane contains a DNA control with just relaxed DNA. The second lane is a 0 min time point that is taken immediately after incubating the master mixture assay tube at 37°C. Each successive lane has increased levels of negative supercoiled [(−) SC] DNA as the reaction time is increased.
3.2: Inhibition of DNA Supercoiling by Antibacterial Drugs
The above protocol can be adapted to examine the effects of antibacterial compounds on gyrase-catalyzed DNA supercoiling. To evaluate the inhibitory effect, you can monitor the effects of a single concentration of a compound over a time course (point 3.2.2) or monitor the effects of a titration of the compound at a single time point (point 3.2.3)
Single concentration of compound over a time course: Pick a concentration of the compound desired to be tested. Follow the DNA supercoiling protocol as described in 3.1 with some minor changes. Replace 2 μL of the ultrapure water per reaction with 10x of the desired compound in the assay mix. It is recommended to add the drug right before step 7 as it reduces error associated compound-enzyme interaction, as it is a factor of time. Follow the rest of the steps as outlined in protocol 3.1.
- Compound titration: This assay will help determine the effects of a drug/compound on the supercoiling activity of DNA gyrase. The results can be used to calculate the inhibitory effects of the drug on the enzyme.
- Optimize for enzyme concentration and reaction time such that 80–90% of the relaxed DNA is negatively supercoiled by gyrase in the absence of compound. These can be optimized by a series of enzyme titrations and time courses (see Notes 10 and 11). The conditions optimal for the following assay with E. coli wild-type gyrase are 5 nM enzyme and a 60 min incubation.
- Decide the titration range for a given compound. Label tubes for the titration, a no compound control, and a DNA control.
- Serially dilute the compound to concentrations 10x the desired final concentration. When diluting the compound, an appropriate diluent must be chosen. A common compound diluent is DMSO (dimethyl sulfoxide). However, its concentration should not exceed 5% in the final reaction mixture as it is a cryo-preservative and has cytotoxic effects (see Note 12).
- Aliquot 2 μL of the 10x compound into pre-chilled labelled reaction tubes. Add 2 μL of the compound diluent to the no drug control and DNA control. Also, add 4 μL of enzyme diluent to the DNA control.
- The number of tubes, which includes the compound titration, DNA control, and no drug control, will be called ‘n’. Each reaction will be composed of 4 μL of the 5X E. coli DNA supercoiling reaction buffer, 1 μL of ATP, X μL of relaxed DNA (final concentration of 0.3μg), and (9 – X) mL ultrapure water to bring the final volume to up 14 μL. Like the protocol in 3.1, make the master assay mixture with enough volume for (n+1) reactions.
- Combine the master assay mixture reagent in pre-chilled tube and mix vigorously. Add 14 μL from the master assay mixture to the DNA control tube, to bring up the final volume to 20 μL.
- Dilute the gyrase subunits to 20x of the desired concentration, such that 2 μL of each subunit is required per tube. Add 2 μL × ‘n’ (as DNA control has been removed) of each subunit to the master assay mixture. The master assay mixture should vortexed lightly while keeping on ice to prevent the enzymatic activity from starting.
- Add 18 μL of the master assay mixture to the no drug control, and the titration tubes (with 2 μL of the 10x drug) and mix well by pipetting and incubate at 37°C. Stagger the start of each reaction tube by 10–30 s to allow time to sequentially stop each individual tube.
- After samples are removed at the desired times (as decided in step1), reactions are stopped by adding 3 μL of the supercoiling stop solution and mixed by pipetting and/or vortexing.
- Follow the protocol 3.1 from step 9 for the gel electrophoresis or step 10 to store the reaction at −20°C (see Figure 3).
Figure 3:
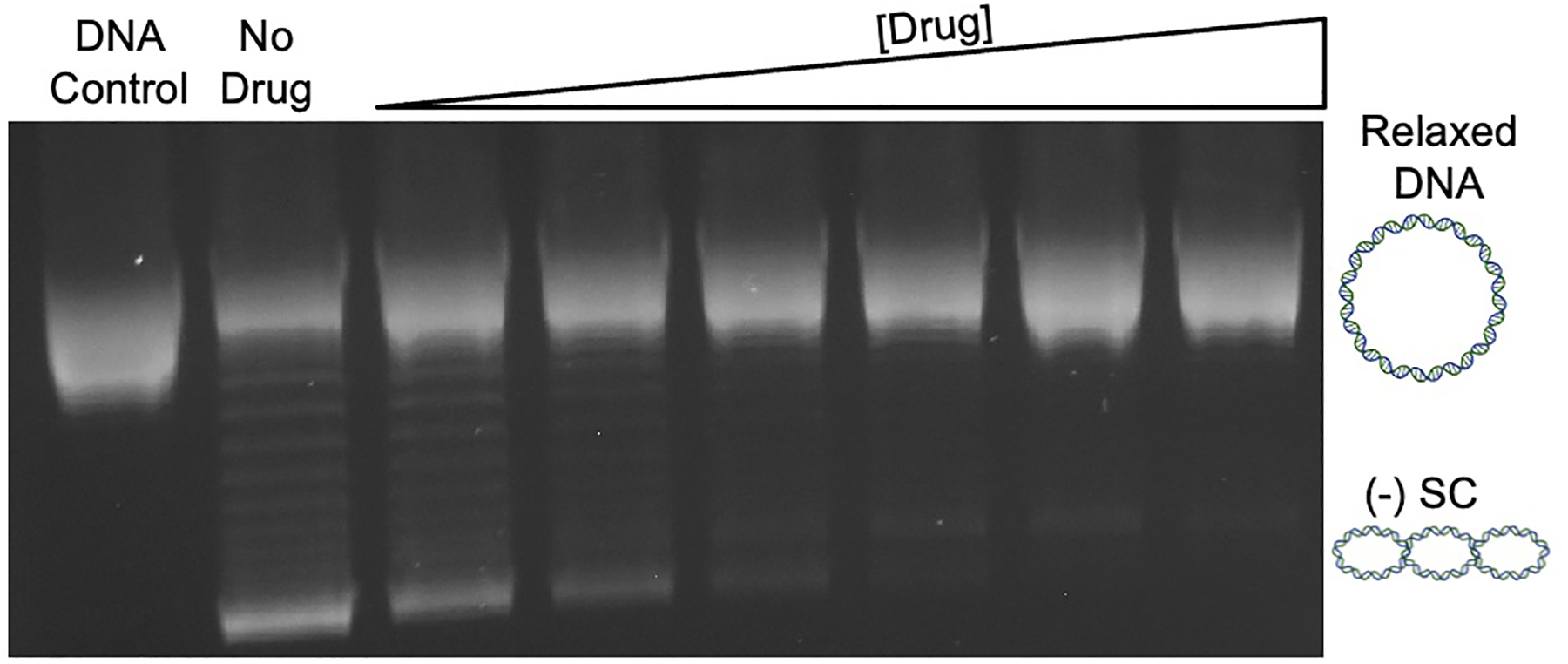
Representative gel showing a drug titration for the inhibition of the DNA supercoiling activity of gyrase. The first lane shows a DNA control that contains only relaxed DNA, and the second lane contains relaxed DNA and the enzyme without any drug. The next lanes contain an increasing concentration of drug, which inhibits the ability of gyrase to introduce negative supercoils [(−) SC] into the relaxed DNA.
4. Notes:
pBR322 can be purified [14,16,17] or commercially purchased. The plasmid can be purified using commercially available kits.
The activity of calf thymus topoisomerase I varies based on batches when ordered from different suppliers. It is recommended to test the batch for activity and modify the amount of calf thymus topoisomerase to make sure that all the DNA has been completely relaxed.
-
The concentration of DNA using the A260/A280 can be calculated by the following formula.
Concentration (μg/mL) = A260 × dilution factor × 50 μg/mL (A260 of 1.0 = 50 μg/ml pure double-stranded DNA)
The A260/A280 ratio is an indicator of DNA purity. Good quality DNA has a ratio between 1.7 – 2.0. However, a lower ratio does not render the DNA unusable; it merely indicates the presence of additional contaminants. Additional information can be found in Green and Sambrook [17].
- The reaction buffer specific to the organism and needs to be modified accordingly. Common modifications to the buffers include:
- Addition of glycerol. If using glycerol, it is recommended that it has been sterile filtered.
- Fresh dithiothreitol (DTT) can be included in the DNA supercoiling assay buffer. DTT can be used to improve the efficiency of the enzyme. However, if used, it is recommended that the DTT not be added to the 5x supercoiling buffer (which can be stored up to a year), but added when making up the master assay mixture. It is also recommended to avoid using DTT solutions that are older than 3 months.
It is recommended to use 5% SDS to make up the stop solution, as this reduces the amount of precipitation.
When making the 1% agarose gel, bringing the agarose in 100 mL of TBE buffer takes 1–2 min in a conventional microwave. Flasks can be loosely covered while boiling but should never be sealed, as this can cause pressure to build up inside the flask and explode. Watch the flask when heating to prevent excessive boiling and stop when all particles of agarose have been dissolved upon swirling.
Ethidium bromide is a carcinogen. Caution should be used when handling and disposing of it.
The 60% sucrose in the agarose loading buffer is to prevent the samples from floating out of the wells. The two dyes added, xylene cyanole (light blue) and bromophenol blue (dark blue), visibly migrate through the gel during electrophoresis and can be used to approximate the progress of DNA migration.
Sometimes DNA bands can be wavy or streaked due to precipitation. Running the agarose gels without any sample for ~10 min before loading warms up the gel. This prevents SDS precipitation from interfering with the banding patterns of the DNA.
Incubation times can vary between species and between enzyme preparations. The optimal time is determined by a series of time course experiments (can use the protocol above in the absence of drug) to define the minimum time required to supercoil 80−90% of the DNA.
Gyrase subunits from other species can require different concentrations. When looking to determine the optimal concentration, it is important to keep in mind that the holoenzyme concentration (A2B2) and the optimal ratio of the GyrA: GyrB subunits can vary.
The physiochemical properties of compounds can vary and affect their solubility in various solutions. A solubility assessment under a variety of conditions is crucial. Some common diluents include water, DMSO, or a basic solution.
Acknowledgements:
Work in the laboratory of the senior author (N.O.) was supported by National Institutes of Health grants R01 GM126363 and R01 AI170546.
Citations:
- 1.Watson JD, Crick FH (1953) Genetical implications of the structure of deoxyribonucleic acid. Nature 171 (4361):964–967. doi: 10.1038/171964b0 [DOI] [PubMed] [Google Scholar]
- 2.Ashley RE, Osheroff N Regulation of DNA Topology by Topoisomerases: Mathematics at the Molecular Level. In, Cham, 2019. Knots, Low-Dimensional Topology and Applications. Springer International Publishing, pp 411–433 [Google Scholar]
- 3.Collins JA, Osheroff N (2024) Gyrase and Topoisomerase IV: Recycling Old Targets for New Antibacterials to Combat Fluoroquinolone Resistance. ACS Infect Dis 10 (4):1097–1115. doi: 10.1021/acsinfecdis.4c00128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vann KR, Oviatt AA, Osheroff N (2021) Topoisomerase II Poisons: Converting Essential Enzymes into Molecular Scissors. Biochemistry 60 (21):1630–1641. doi: 10.1021/acs.biochem.1c00240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gibson EG, Blower TR, Cacho M, Bax B, Berger JM, Osheroff N (2018) Mechanism of Action of Mycobacterium tuberculosis Gyrase Inhibitors: A Novel Class of Gyrase Poisons. ACS Infect Dis 4 (8):1211–1222. doi: 10.1021/acsinfecdis.8b00035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hirsch J, Klostermeier D (2021) What makes a type IIA topoisomerase a gyrase or a Topo IV? Nucleic Acids Res 49 (11):6027–6042. doi: 10.1093/nar/gkab270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tadesse S, Graumann PL (2006) Differential and dynamic localization of topoisomerases in Bacillus subtilis. J Bacteriol 188 (8):3002–3011. doi: 10.1128/JB.188.8.3002-3011.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ahmed W, Sala C, Hegde SR, Jha RK, Cole ST, Nagaraja V (2017) Transcription facilitated genome-wide recruitment of topoisomerase I and DNA gyrase. PLoS Genet 13 (5):e1006754. doi: 10.1371/journal.pgen.1006754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stracy M, Wollman AJM, Kaja E, Gapinski J, Lee JE, Leek VA, McKie SJ, Mitchenall LA, Maxwell A, Sherratt DJ, Leake MC, Zawadzki P (2019) Single-molecule imaging of DNA gyrase activity in living Escherichia coli. Nucleic Acids Res 47 (1):210–220. doi: 10.1093/nar/gky1143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gibson EG, Ashley RE, Kerns RJ, Osheroff N (2018) Bacterial Type II Topoisomerases and Target-Mediated Drug Resistance. In: Fong IW, Shlaes D, Drlica K (eds) Antimicrobial Resistance in the 21st Century. Springer International Publishing, Cham, pp 507–529. doi: 10.1007/978-3-319-78538-7_16 [DOI] [Google Scholar]
- 11.Wang X, Reyes-Lamothe R, Sherratt DJ (2008) Modulation of Escherichia coli sister chromosome cohesion by topoisomerase IV. Genes Dev 22 (17):2426–2433. doi: 10.1101/gad.487508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bax BD, Murshudov G, Maxwell A, Germe T (2019) DNA Topoisomerase Inhibitors: Trapping a DNA-Cleaving Machine in Motion. J Mol Biol 431 (18):3427–3449. doi: 10.1016/j.jmb.2019.07.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aldred KJ, Kerns RJ, Osheroff N (2014) Mechanism of quinolone action and resistance. Biochemistry 53 (10):1565–1574. doi: 10.1021/bi5000564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aldred KJ, Blower TR, Kerns RJ, Berger JM, Osheroff N (2016) Fluoroquinolone interactions with Mycobacterium tuberculosis gyrase: Enhancing drug activity against wild-type and resistant gyrase. Proc Natl Acad Sci U S A 113 (7):E839–846. doi: 10.1073/pnas.1525055113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chan PF, Srikannathasan V, Huang J, Cui H, Fosberry AP, Gu M, Hann MM, Hibbs M, Homes P, Ingraham K, Pizzollo J, Shen C, Shillings AJ, Spitzfaden CE, Tanner R, Theobald AJ, Stavenger RA, Bax BD, Gwynn MN (2015) Structural basis of DNA gyrase inhibition by antibacterial QPT-1, anticancer drug etoposide and moxifloxacin. Nat Commun 6:10048. doi: 10.1038/ncomms10048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Oviatt AA, Gibson EG, Huang J, Mattern K, Neuman KC, Chan PF, Osheroff N (2024) Interactions between Gepotidacin and Escherichia coli Gyrase and Topoisomerase IV: Genetic and Biochemical Evidence for Well-Balanced Dual-Targeting. ACS Infect Dis 10 (4):1137–1151. doi: 10.1021/acsinfecdis.3c00346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Green MR, Sambrook J (2018) Isolation and Quantification of DNA. Cold Spring Harb Protoc 2018 (6). doi: 10.1101/pdb.top093336 [DOI] [PubMed] [Google Scholar]