

Abstract
We reported that Na/H exchanger isoform 1 (NHE1) activity in astrocytes is stimulated and leads to intracellular Na+ loading following oxygen and glucose deprivation (OGD) (13). However, the underlying mechanisms for this stimulation of NHE1 activity and its impact on astrocyte function are unknown. In the present study, we investigated the role for extracellular signal-regulatory protein kinase (ERK1/2) pathway in NHE1 activation. NHE1 activity was elevated by ~ 75% in NHE1+/+ astrocytes following 2 h OGD and 1 h reoxygenation (OGD/REOX). The OGD/REOX-mediated stimulation of NHE1 was partially blocked by 30 μM PD98059. Increased expression of phosphorylated ERK 1/2 was detected in NHE1+/+ astrocytes following OGD/REOX. Moreover, stimulation of NHE1 activity not only disrupted Na+ but also Ca2+ homeostasis via reverse-mode operation of Na+/Ca2+ exchange. OGD/REOX led to 103% increase in [Ca2+]i in NHE1+/+ astrocytes in the presence of thaspsigargin. Inhibition of NHE1 activity with NHE1 inhibitor HOE 642 decreased OGD/REOX-induced elevation of [Ca2+]i by 73%. To further investigate changes of Ca2+ signaling, bradykinin-mediated Ca2+ release was evaluated. Bradykinin-mediated Cai 2+ transient in NHE1+/+ astrocytes was increased by ~84% following OGD/REOX. However, in NHE1−/− astrocytes or NHE1+/+ astrocytes treated with HOE 642, the bradykinin-induced Ca2+ release was only increased by ~ 34%. Inhibition of the reverse mode of Na+/Ca2+ exchange abolished OGD/REOX-mediated Ca2+ rise. Taken together, our data suggest that ERK1/2 is involved in activation of NHE1 in astrocytes following in vitro ischemia. NHE1-mediated Na+ accumulation subsequently alters Ca2+ homeostasis via Na+/Ca2+ exchange.
Keywords: intracellular pH, cortical astrocytes, Na+, Ca2+ exchange, ischemia, intracellular Na+
INTRODUCTION
Na+/H+ exchanger isoform 1 (NHE1) mediates H+ efflux and Na+ influx through the cell membrane and plays a significant role in intracellular pH (pHi) regulation and recovery from acidosis (28). Inhibition of NHE1 reduces the resting pHi of cortical astrocytes of rats (23; 30) and mice (13). We recently reported that NHE1 activity in cortical astrocytes is stimulated by ~75% during reoxygenation following 2 h oxygen and glucose deprivation [OGD, (13)]. Stimulation of NHE1 activity leads to an ~5.6-fold increase in intracellular Na+ concentration ([Na+]i). However, the mechanisms underlying this stimulation of NHE1 activity and its impact on ion homeostasis of astrocytes are unknown.
NHE1 can be directly stimulated by acidosis via a H+-mediated allosteric effect (36). In addition, NHE1 is regulated by phosphorylation, which alters the affinity of the transmembrane internal H+ transport site of NHE1 (9; 24; 28). Activation of the extracellular signal-regulatory protein kinase (ERK) pathway and one of its downstream effectors, p90 ribosomal S6 kinase (p90RSK), has been reported to stimulate NHE1 activity during myocardial ischemia and reperfusion (24). There is growing evidence implicating ERK1/2 activation in promotion of cell death during oxidative neuronal injury (7). However, whether the ERK signaling cascade plays a role in stimulation of NHE1 activity in astrocytes following in vitro ischemia remains unknown.
Overstimulation of NHE1 activity may cause ischemic cell injury. Inhibition of NHE1 attenuates myocardial damage following ischemia and reperfusion (2). A specific NHE inhibitor SM-20220 (N-aminoiminomethyl-1-methyl-1-indole-2-carboxamide methanesulfonate) significantly reduces Na+ accumulation and edema in brain following transient cerebral ischemia in rats (14). Elevation of [Na+]i can trigger reverse-mode operation of Na+/Ca2+ exchanger and causes intracellular Ca2+ overload, which in turn may lead to Ca2+-dependent irreversible cell damage during anoxia and ischemia (6; 18). In the current study, we investigated whether NHE1-mediated accumulation of intracellular Na+ affects intracellular Ca2+ signaling in astrocytes following in vitro ischemia. We report here that stimulation of NHE1 activity in astrocytes following in vitro ischemia depends in part on activation of ERK1/2 signaling pathway. NHE1 activation not only causes Na+ accumulation but also alters Ca2+ signaling and loading of intracellular Ca2+ stores.
MATERIALS AND METHODS
Materials
Eagle’s modified essential medium (EMEM) and Hanks balanced salt solution (HBSS) were from Mediatech Cellgro (Herndon, VA). Fetal bovine serum (FBS) was obtained from Hyclone Laboratories (Logan, UT). Collagen-type I was from Collaborative Biomedical Products (Bedford, MA). The acetoxymethyl esters of 2′, 7′-bis(carboxyethyl)-5,6-carboxyfluorescein (BCECF-AM), fura-2(Fura-2 AM), and sodium-binding benzofuran isophthalate (SBFI-AM) were obtained from Molecular Probes (Eugene, OR). Pluronic acid was purchased from BASF (Ludwigshafen, Germany). Nigericin, dibutyryl cyclic AMP (dBcAMP), monensin, carbonyl cyanide p-trifluoromethoxyphenylhydrazone (FCCP), oligomycin, bradykinin, thapsigargin and gramicidin were purchased from Sigma (St. Louis, MO). HOE 642 was a kind gift from Aventis Pharma (Frankfurt, Germany). SEA0400 (2-[4-[2,5-diflurophenyl)methoxy]phenoxy]-5-ethoxyaniline) was from EMD Biosciences (San Diego, CA). PD98059 (2-(2-Amino-3-methoxyphenyl)-4H-1-benzopyran-4-one) was from Toris (Ellisville, MO). Antibody for phosphorylated ERK 1/2 (Tyr(P)-204/Thr(P)-202) was from Cell Signaling Technologies (Beverly, MA), and antibody for total ERK (K-23) from Santa Cruz Biotechnology (Santa Cruz, CA).
Animals and Genotype analysis
The NHE1 null mutant (NHE1−/−) mice used in the present study were generated as previously described (3). We obtained NHE1 homozygous mutant and wild-type (NHE1+/+) mice by breeding gene-targeted NHE1 heterozygous mutant mice. Tail biopsies were obtained from 1-day-old mice. Genotypes were determined by polymerase chain reaction (PCR) analysis of DNA from tail biopsies, as described in our previous study (13).
Primary culture of mouse cortical astrocytes
Dissociated cortical astrocyte cultures were established as described previously (31). In brief, cerebral cortices were removed from 1-day-old NHE1+/+ or NHE1−/−- mice. The cortices were incubated in a trypsin solution (0.25 mg/ml in HBSS) for 25 min at 37°C and the tissue was mechanically triturated. The dissociated cells were rinsed and resuspended in EMEM containing 10% FBS. Viable cells (1 × 104 cells/well) were plated in 6-well plates or on glass coverslips coated with collagen type-1. Cultures were maintained in a 5% CO2 atmosphere at 37oC and refed every three days throughout the study. To obtain morphologically differentiated astrocytes, confluent cultures (10 days in culture, DIV 10) were treated with EMEM containing 0.25 mM dBcAMP to induce differentiation. dBcAMP has been widely used to mimic neuronal influences on astrocyte differentiation (10; 33). Experiments were routinely performed in DIV 15–28 astrocytes. This culture age was chosen because immature astrocyte cultures are resistant to damage under ischemic conditions (12).
OGD treatment
NHE1+/+ or NHE1−/− cultures grown on coverslips were rinsed twice with an isotonic OGD solution containing (in mM, pH 7.4): 0 glucose, 20 NaHCO3, 120 NaCl, 5.36 KCl, 0.33 Na2HPO4, 0.44 KH2PO4, 1.27 CaCl2, 0.81 MgSO4. Cells were incubated in 1.0 ml of OGD solution in a hypoxic incubator at 37oC (Forma model 3130, Thermo Forma, Marietta, OH) equilibrated with 94% N2, 1% O2 and 5% CO2. The oxygen level in the medium of cultured cells was monitored with an oxygen probe (Model M1-730, Microelectrodes Inc., Bedford, NH) and decreased to ~2–3% after 60 min in the hypoxic incubator. Normoxic control cells were incubated in 5% CO2 and atmospheric air in isotonic control buffer containing 5.5 mM glucose with the rest of the components in the buffer identical to the isotonic OGD solution.
Measurement of pHi
pHi Measurement: The HEPES-buffered solution (pH 7.4) and pH calibration solutions were described in our previous study (13). Cells grown on coverslips were incubated with 2.5 μM BCECF-AM in HEPES-buffered solution for 10 min at ambient temperature, as described before (13). The coverslips were washed and placed in an open-bath imaging chamber (Model RC24, Warner Instruments, Hamden, CT) containing HEPES-buffered solution. The chamber was mounted on the stage of the TE 300 inverted epifluorescence microscope and the astrocytes visualized using a 40X Super Fluor oil immersion objective lens (13). The cells were excited every 10–30 s at 440 nm and 490 nm and the ratio of the emission fluorescence at 535 nm was recorded. Images were collected using a Princeton Instruments MicroMax CCD camera, and analyzed with MetaFluor image-processing software (Universal Imaging Corporation, Downington, PA). The ratio of the fluorescence emissions (F490/F440) was calibrated using the high K+/ nigericin technique (5). The background-corrected data were fit with a variant of the Michaelis-Menten equation (5).
Pre-pulse treatment:Cells were subjected to an acid load by the transient application (1–2 min) of a NH4+/NH3 solution as described before (13). NH4+/NH3 solutions was prepared by replacing 10 mM of NaCl in either the HEPES-buffered solution with an equimolar concentration of NH4Cl. pHi rose as NH4+ accumulated during the pre-pulse. Cells were subsequently returned to a HEPES-buffered solution. Acidification of the cytoplasm occurred when NH3 quickly diffused out of the cell.
Determination of buffer capacity (βi): βi was determined over a range of pHi by subjecting the cells to progressively decreasing concentrations of NH4+, based on βi = ΔNH4+/ΔpHi (22). These experiments were done in HCO3− and Na+-free buffers to block acid extrusion mechanisms. The change in [NH4+]i was determined from the [NH3]i, pK and pH [pHi = 9.3−log([NH4+]i/[NH3]i], with an assumption of [NH3]i = [NH3]o and Δ[NH4+]i = Δ[H+]i as reported previously (22). H+ flux rates (JH+: mM H+/min) were determined by multiplying βi by ΔpHi/Δt for each time interval during pHi recovery (1–2 min) following pre-pulse treatment.
Intracellular Ca2+ measurement
Astrocytes grown on coverslips were incubated with 5 μM fura-2 AM for 45 min (16). The cells were washed and the coverslips placed in the open-bath imaging chamber containing HEPES-MEM at ambient temperature. Using a Nikon TE 300 inverted epifluorescence microscope and a 40X Super Fluor oil immersion objective lens, astrocytes were excited every 10 seconds at 345 and 385 nm and the emission fluorescence at 510 nm recorded. Images were collected and analyzed with the MetaFluor image-processing software. At the end of each experiment, the cells were exposed to 1 mM MnCl2 in Ca2+-free HEPES-MEM. The Ca2+ insensitive fluorescence was subtracted from each wavelength before calculations (16). The MnCl2-corrected 345/385 emission ratios were converted to concentration using the Grynkiewicz equation (8), as described previously (16).
Bradykinin-induced Ca2+ release from intracellular stores was assessed by exposing astrocytes to 1 μM bradykinin in Ca2+-free HEPES-MEM (1 mM EGTA) to prevent entry of extracellular Ca2+. Peak Ca2+ release was measured following application of bradykinin. After washout of bradykinin, cells were exposed to 1 μM thapsigargin to assess residual Ca2+ release from bradykinin-insensitive Ca2+ stores. Thapsigargin-induced peak Ca2+ release from intracellular stores was measured in the presence of 1 μM thapsigargin in HEPES-MEM (1.2 mM Ca2+).
Reverse mode of Na+/Ca2+ exchange (NCX) was induced in astrocytes as described by Hoyt et al. (11). Astrocytes were loaded with Na+ by applying Ca2+-free HEPES buffer (2 μg/ml gramicidin) for one min. Reverse mode of NCX was then initiated by exposing cells to a Na+-free buffer (1.2 mM Ca2+) for 30–40 sec, which triggered a rise in [Ca2+]i. Cells quickly regulated Cai2+ to baseline values when control HEPES-MEM was reintroduced.
Gel Electrophoresis and Western Blotting
Cultured cells were washed with ice-cold phosphate-buffered saline (PBS, pH 7.4), which contained 2 mM EDTA and protease inhibitors (32). Cells were collected by scraping from the 6-well plates in an anti-phophatase (AP) buffer containing protease inhibitors (37). The protein content was measured with the bicinchoninic acid method (Pierce, Rockford, IL). Samples and pre-stained molecular mass markers (Bio-Rad, Hercules, CA) were denatured in SDS reducing buffer (1:2 by volume, Bio-Rad) and heated at 37 °C for 15 min before gel electrophoresis. The protein samples (10 μg lysate protein) were loaded and separated by SDS-PAGE (12%). After transferring to a PVDF membrane, the blots were incubated in 7.5% nonfat dry milk in Tris-buffered saline (TBS) and then incubated with a primary antibody, anti-phospho-ERK 1/2 (p-ERK1/2) polyclonal antibody. For a protein loading control, the blot was restripped and reprobed with the anti non phospho-ERK 1/2 polyclonal antibody, which recognized non-phosphosylated ERK 42/44 kDa proteins. Both antibodies were used at a concentration of 1:1000 in a solution of 7.5% milk/TBS buffer. After rinsing, the blots were incubated with horseradish peroxidase-conjugated secondary IgG. Bound antibody was visualized using the enhanced chemiluminescence assay (ECL, Amersham Corp.).
To elucidate a quantitative increase in p-ERK1/2, densitometry measurements of each protein band were performed. Protein bands on the blot were selected and average pixel was recorded with Un-Scan-It Gel (Silk Scientific, Orem, UT). A ratio of p-ERK 1/2 and non-phospho ERK1/2 were calculated in normoxic control or OGD/or REOX samples. The percent increase in p-ERK 1/2 expression was then evaluated by comparing values in normoxic control vs. OGD/or REOX samples.
Immunofluorescence staining
Cultured cells grown on coverslips were rinsed with PBS (pH 7.4) and fixed with 4% paraformaldehyde in PBS for 20 min at room temperature. After rinsing, cells were incubated with blocking solution (10% normal goat serum, 0.4% Triton X-100, and 1% bovine serum albumin in PBS) for 1 h. Cells were then incubated with anti-GFAP (glial fibrillary acidic protein) monoclonal antibody (1:100) or anti-p-ERK1/2 antibodies (1:100) in blocking solution for 1 h. Cells were rinsed with PBS and incubated with goat anti-mouse Alexa 488 or anti-rabbit Alexa 595-conjugated antibodies (1:200) for 1 h. Cell images were captured by a Nikon TE 300 inverted epifluorescence microscope (20X) using a Princeton Instruments MicroMax CCD camera and MetaMorph image-processing software (Universal Imaging Corp., Dowington, PA). Identical digital imaging acquisition parameters were used in both negative control and experimental images.
Measurement of cell swelling
Cell volume change in single cells was measured by using calcein as a marker of intracellular water volume as described previously (16). Briefly, astrocytes grown on coverslips were placed in a closed-bath imaging chamber and incubated with 2.5 μM calcein-AM. Calcein fluorescence in cells was monitored using a 40X Super Fluor oil immersion objective and a FITC filter set (excitation 480 nm, emission 535 nm, Chroma Technology, Rockingham, VT ) until the fluorescence plateaued ( ~ 30 min). Images were collected every 60 sec for 10 min to determine baseline data. The chamber was then switched to OGD buffer for 60 min. The fluorescence signals were corrected for baseline drift and calibration with the calibration standard buffers was performed at the end of each experiment as described before (16).
Statistics
Sigmastat (Systat Software, Point Richmond, CA) software was used for statistical analysis. The data are reported as means ± S.D. N values refer to the number of cells tested in each experimental condition. Significance between groups was tested with either a t-test or a Mann-Whitney test. F test was used for comparison of linear and non-linear regression fits.
RESULTS
βi in NHE+/+ astrocytes does not change following OGD/REOX
In order to investigate changes in proton flux to reflect NHE1 activity, we first examined whether the intrinsic buffer capacity βi of NHE1+/+ astrocytes was altered by either OGD/REOX or genetic ablation of NHE1. β i was determined in NHE1+/+ and NHE1−/− astrocytes under control conditions and in NHE1+/+ astrocytes under OGD/REOX conditions. A representative buffer capacity experiment from a single cell is shown in Figure 1A. Changes in pHi were induced by applying progressively decreasing concentrations of NH3/NH4+ (Figure 1A). In NHE1+/+ astrocytes βiincreased linearly from a value of about 7.8 mM/pH at a pHi of 7.0 to about 18 mM/pH at a pHi of 6.2 (Figure 1B). The fitted regression was βi = 98 + (−12.9* pHi), which is consistent with the reported values in rat astrocytes (4; 22; 30). βi was also determined in NHE1+/+ astrocytes following 2 h OGD and 1 h REOX (Figure 1B). OGD/REOX did not cause a significant change in the fitted line of βi vs. pHi [βi = 75 + (−9.6 * pHi), p > 0.05]. Likewise, the line fitted to the βi vs pHi in NHE1−/− astrocytes was not different from NHE1+/+ astrocytes under control conditions [βi = 103 + (−13.6 * pHi), p > 0.05].
Figure 1. Intrinsic buffer capacities (βi) in NHE1−/− and NHE1+/+ astrocytes are similar and unchanged following OGD/REOX.
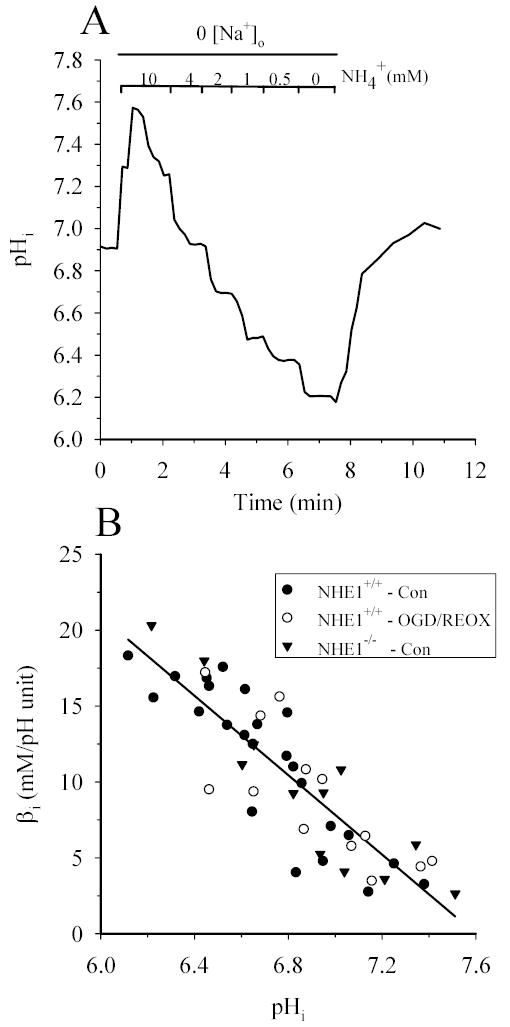
Intrinsic buffer capacity was determined in NHE1−/− and NHE1+/+ astrocytes and in NHE1+/+ astrocytes following 2 h OGD and 1 h REOX. Cells were sequentially exposed to decreasing concentration of NH4+ in Na+-free HEPES-MEM and the change in pHi determined. A. A representative trace of pHi in a single NHE1+/+ cell in response to changes in NH4+. B. βi was plotted as a function of pHi in normoxic NHE+/+ astrocytes, NHE1−/− astrocytes, and NHE1+/+ astrocytes following OGD/REOX (three cultures). There was no significant difference in the regression fits among these groups (p > 0.05).
Stimulation of NHE1 activity following OGD/REOX partially depends on activation of MEK-mediated pathways
Figure 2A shows representative pHi tracing from NHE1+/+ astrocyte subjected to an acid load. Following 2 h OGD and 15 min REOX there is an increase in pHi recovery rate in NHE1+/+ astrocytes (Figure 2A). This increase in pHi recovery reflects an increase in NHE1 activity (13). pHi recovery in the presence of MEK inhibitor PD98059 was not different from normoxic control (data not shown). In contrast, inhibition of MEK activity with 30 μM PD98059 significantly attenuated the pHi recovery rate following OGD and REOX. To further investigate the kinetics of NHE1 activity under these conditions, we calculated JH+ during the prepulse-induced pHi recovery and plotted it against the corresponding [H+]i (Figure 2B-D). In control NHE1+/+ astrocytes, the velocity of H+ efflux from cells increased as [H+]i increased and was best described by a sigmoid fit (Km of 0.264 ± 0.005 μM, Vmax of 7.21 ±0.23 mM/min; Figure 2B) with a Hill coefficient of 2.14 ± 0.11. The latter is consistent with other reports and suggests that there might be more than one binding site for H+ on the intracellular face of the protein (1). The values for Km and Vmax are comparable to those obtained for NHE1 expressed in fibroblasts as well as in C6 rat glioma cells (17, 29). When NHE1+/+ astrocytes were exposed to 2 h OGD plus 15 min REOX, the kinetics of NHE1 activity were dramatically changed (Figure 2C). Vmax increased about 3 times (28.6 ± 2.2 mM/min) while the Km shifted to a higher concentration of protons (0.71 ± 0.02 μM). The sigmoidal curve fit of the OGD/REOX data was significantly different from the curve fit to the normoxic control data in NHE1+/+ astrocytes (p < 0.05). The Hill coefficient for the OGD/REOX data was 2.03 ± 0.23. Inhibition of MEK1/2 activity with 30 μM PD98059 affected OGD/REOX-mediated changes on NHE1 kinetics (Figure 2D). Vmax was reduced by ~ 41% in the presence of 30 μM PD98059 (16.7 ± 0.4 mM/min), but still remained significantly higher than the normoxic control values. No further inhibition was found on Vmax with 60 μM PD98059 (data not shown). In addition, in the presence of PD98059, Km shifted back to a lower concentration of protons and was not significantly different from the normoxic control (0.377 ± 0.013 μM; p > 0.05). This suggests that the MEK1/2 and ERK1/2 pathways are partially responsible for activation of NHE1 activity in astrocytes following OGD/REOX.
Figure 2. OGD-mediated stimulation of NHE1+/+ activity is attenuated by PD98059.
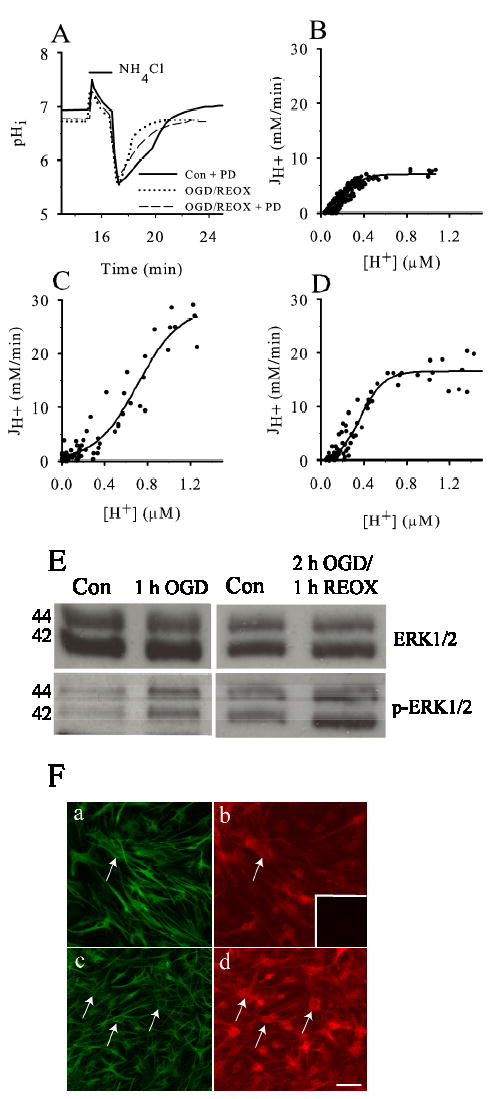
Following either normoxia or OGD/REOX in NHE1+/+ astrocytes, a NH3/NH4+ prepulse was applied and JH+ determined. In the PD98059 studies, 30 μM PD98059 was present during OGD and REOX. A. Representative pHi response in single cells to a prepulse following either normoxia (2.25 h), OGD (2 h)/REOX (15 min), or OGD/REOX in the presence of PD98059. B-D. During recovery from a prepulse, JH+ was determined and plotted. Data were fitted with a sigmoid curve to determine Km and Vmax. E. Following OGD or OGD/REOX, NHE1+/+ astrocytes were lysed and lysate was separated by 12% SDS-PAGE, and transferred to nitrocellulose membrane. The blot was probed with either anti-ERK antibody (ERK1/2) or anti-phospho-ERK1/2 antibody (p-ERK1/2). Representative images of three experiments is presented. F. Immunostaining for GFAP (a, c) and p-ERK1/2 (arrows, b, d) in NHE1+/+ astrocytes under normoxic control (a, b) or 1 h OGD (c, d). Scale bar: 120 μm. Representative image of three experiments is presented. Inset in F(b): image taken when anti-p-ERK1/2 antibody was omitted.
We further investigated the OGD/REOX-mediated increase in ERK 1/2 activity by measuring the phosphorylation status of ERK 1/2 in astrocytes. As shown in the upper panels of Figure 2E, when lysates of NHE1+/+ astrocytes were analyzed by immunoblotting, there were no significant differences in expression of total ERK1/2 proteins (~42–44 kDa) in response to either OGD or OGD/REOX conditions. In contrast, either 1 h OGD or 2 h OGD and 1 h REOX led to an increase in expression of phosphorylated ERK1/2 proteins (p-ERK1/2, ~42–44 kDa) in NHE1+/+ astrocytes (Figure 2E, lower panels). p-ERK1/2 expression was increased by 24 % following 1 h OGD and by 16 ± 1% following 2 h OGD and 1 h REOX. The increase in p-ERK1/2 protein expression was further demonstrated in NHE1+/+ astrocytes following 1 h OGD by immunofluorescence staining. As shown in Figure 2Fa, c, no significant morphological changes were found in NHE1+/+ astrocytes following 1 h OGD. However, immunoreactive signals of p-ERK1/2 protein was significantly increased in NHE1+/+ astrocytes at the end of 1 h OGD (Figure 2Fb, d). This signal was absent when the anti-p-ERK1/2 primary antibody was omitted from the procedures (inset in Figure 2Fb), which indicates that the immunofluorescent signal is specific to p-ERK1/2 protein.
To confirm that phosphorylation of ERKs correlates with NHE1 activation, we also determined NHE1 activity in astrocytes immediately following 1 h OGD. NHE1-mediated H+ extrusion rate in NHE1+/+ astrocytes was significantly increased to 0.48 ± 0.022 pH unit/min at 6.6 pHi following 1 h OGD, compared to a control of 0.33 ± 0.08 pH unit/min at pHi 6.4 (p < 0.05, n=3). Thus, the rise in ERK phosphorylation correlates with NHE1 activation following either 1 h OGD or 2 h OGD/REOX.
Prolonged acidosis is not responsible to NHE1 activation following OGD/REOX
It has been reported that prolonged acidosis of pHi = 6.55 ± 0.05 for 3 min in cultured rat ventricular cardiomyocytes led to ERK1/2-dependent stimulation of NHE1 activity (9). We found that 2 h OGD did reduce pHi to 6.80 ± 0.02 from a resting level of 7.09 ± 0.07 in NHE1+/+ astrocytes (13). Therefore, we investigated whether prolonged acidosis plays a role in activation of NHE1 activity following OGD. NHE1+/+ astrocytes were exposed to 30 mM NH3/NH4 for 1 min and were returned to the control HEPES-buffer containing Na+. JH+ was determined during pHi recovery (Figure 3A). The prolonged acidosis was induced by exposing cells to the second NH3/NH4+ treatment then returning them to a Na+-free HEPES buffer to maintain cellular acidosis (Figure 3A). pHi was reduced from 6.99 ± 0.03 to 6.25 ± 0.07. After 3 min, Na+o was reintroduced and the change of JH+ as pHi recovered was determined (Figure 3B). The control kinetics in these experiments were: Vmax = 7.93 ± 0.79 mM/min and Km = 0.30 ± 0.01 μM. Following prolonged acidosis at pHi of 6.25, Vmax was 7.77 ± 0.51 mM/min and Km was 0.28 ± 0.01 μM. The line fit to the control data was not significantly different from the line fit to the prolonged acidosis data (p > 0.05). This suggests that the response of NHE1 activity to prolonged acidosis in cortical astrocytes is different from that of cardiomyocytes (9). Because NHE1 activity is not altered by the prolonged acidosis in NHE1+/+ astrocytes, it is unlikely that the ERK1/2-dependent stimulation of NHE1 activity following OGD/REOX is initially triggered by an increase in [H+]i.
Figure 3. Prolonged acidosis does not lead to activation of NHE1.
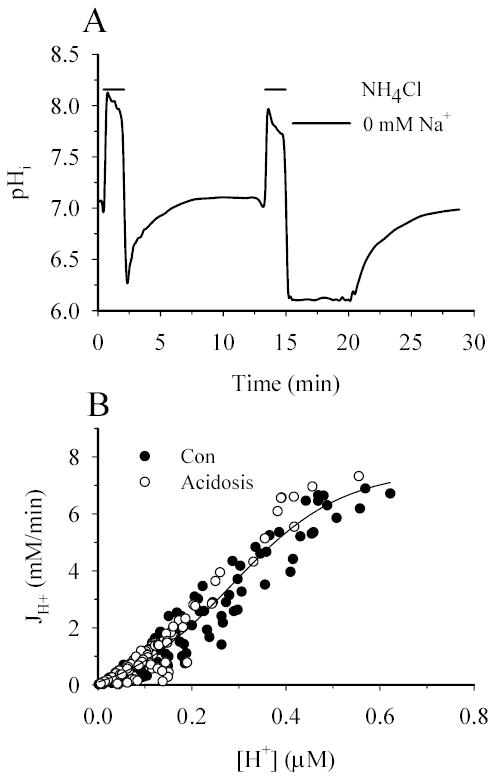
A. A representative pHi response in a single cell following a NH3/NH4+ prepulse in NHE1+/+ astrocytes. Following recovery, a second NH3/NH4+ prepulse in Na+-free buffer was applied to acidify the cell and prevent H+ efflux. Acidification was maintained for three minutes. Na+ was then reintroduced and NHE1 activity was determined. B. JH+ was calculated between each time point following control and post-acidification NH3/NH4+ prepulse. A sigmoid curve was fit to all data. Curves fit to the control- and post-acidification data were not different from each other (p > 0.05, three cultures).
Inhibition of ERK1/2 pathways attenuates NHE1-mediated increase in [Na+]i following OGD/REOX
We have previously observed that stimulation of NHE1 activity causes a substantial increase in [Na+]i in astrocytes at the end of 60 min REOX following 2 h OGD (13). In the present study, we further monitored NHE1-mediated Na+i accumulation every 5 min during 1 h REOX (Figure 4). As shown in Figure 4A, [Na+]i in NHE1+/+ astrocytes was 11.7 ± 3.4 mM at the end of 2 h OGD. It was not significantly different from normoxic control values (7.9 ± 1.5 mM). However, upon reoxygenation, [Na+]i rose sharply at a rate of 3.2 mM/min after 5 min REOX and it reached a plateau (71.4 ± 19.8 mM) at ~ 25 min REOX. In the presence of NHE1 inhibitor HOE 642 (Figure 4A), NHE1+/+ astrocytes exhibited a delayed rise in [Na+]i. A significant elevation of [Na+]i only occurred at 20 min REOX and its rate was much slower (0.8 mM/min). Moreover, the peak value of [Na+]i in the presence of HOE 642 was 37.7 ± 11 mM. Similar results were found in NHE1−/− astrocytes ([Na+]i =10.3 ± 3.4 mM at 0 min REOX and 39.3 ± 11.3 mM at 60 min REOX). These findings further confirm that NHE1 activation leads to accumulation of Na+ in astrocytes following OGD.
Figure 4. NHE1-mediated increase in [Na+]i is attenuated by PD98059.
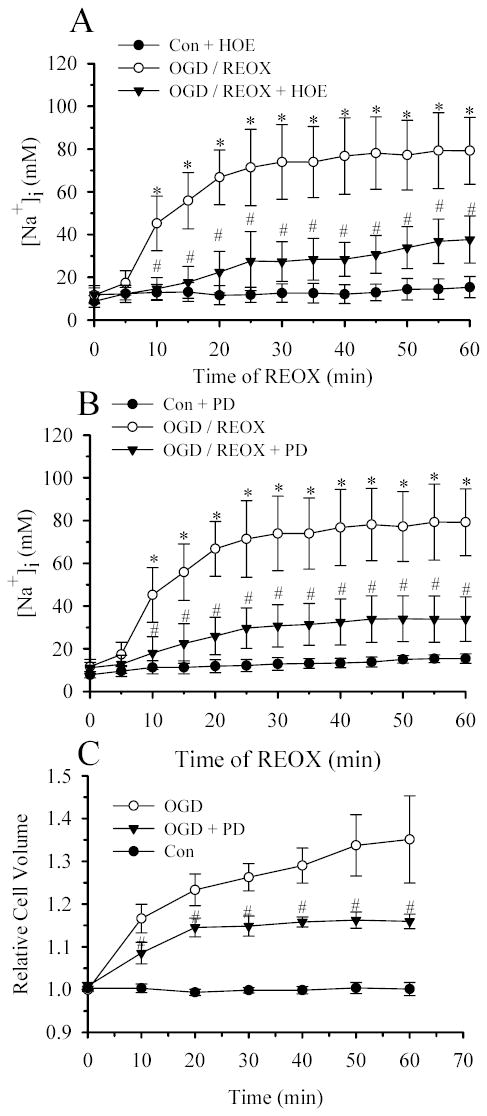
Following either 2 h normoxia or 2 h OGD, [Na+]i was determined every 5 min during 60 min REOX in NHE1+/+ astrocytes. A. 1.0 μM HOE 642 was present during either normoxia or OGD/REOX. B. 30 μM PD98059 was present during either normoxia or OGD/REOX. Data are expressed as mean ± SD (n = 60–100 cells, 3–5 cultures). C. Average changes in the relative cell volume in NHE1+/+ astrocytes were determined with the calcein method during 10 min control perfusion, followed by 60 min OGD. In the PD98059 experiments, cells were exposed to 30 μM PD98059 30 min prior to OGD and during all subsequent perfusions. Data are means ± SD (n = 3 cultures). Data of the control and OGD groups in panel C were compared here from (16). * p < 0.05 vs. normoxic control, # p < 0.001 vs. untreated NHE1+/+ OGD/or REOX.
Since data in Figure 2A suggest that activation of MEK1/2 and ERK1/2 pathways is involved in OGD/REOX-mediated stimulation of NHE1 activity, we next examined whether inhibition of MEK1/2 would block NHE1-mediated Na+ accumulation following OGD/REOX. As shown in Figure 4B, 30 μM PD98059 did not affect the basal levels of [Na+]i. In the presence of PD98059, [Na+]i following 2 h OGD did not change (11.3 ± 1.3 mM vs. 11.8 ± 3.3 mM). In contrast to a sharp rise in [Na+]i at 10 min of REOX in the absence of PD98059, [Na+]i increased at a markedly reduced rate (0.8 mM/min) in the treated astrocytes (p < 0.01). In the presence of PD98059, although [Na+]i continued to increase during REOX over the next 50 min, it reached a value 59% less than in untreated astrocytes (33.4 ±9.0 vs. 79.2± 17.2 mM, Figure 4B). Interestingly, the effects of PD98059 on the rate of Na+ increase and its peak value are similar to those observed in response to the NHE1 inhibitor HOE 642. The data imply that activation of MEK1/2 pathways may stimulate NHE1 activity and lead to a rise in Na+.
The OGD/REOX-mediated intracellular Na+ overload suggests that Na+ extrusion mediated by Na+/K+ATPase is either reduced or remains unchanged and thus fails to maintain Na+ homeostasis. OGD (8 h) reduced the Na+/K+ATPase activity by ~44% [measured by ouabain-sensitive Rb influx, (16)]. To further address this issue, we measured the Na+ accumulation rate in astrocytes during REOX in the absence or presence of Na+/K+ATPase inhibitor ouabain. The basal level of Na+ accumulation rate was 0.08 ± 0.07 mM/min under normoxic control conditions. Inhibition of Na+/K+ATPase activity with 1 mM ouabain increased the Na+ accumulation rate to 2.9 ± 0.5 mM/min in normoxic astrocytes. However, OGD/REOX alone significantly elevated the Na+ accumulation rate (3.95 ± 0.40 mM/min, n=3, p < 0.05) in the absence of ouabain. Blocking of Na+/K+ATPase activity with ouabain did not significantly change the Na+ accumulation rate (3.16 ± 0.86 mM/min). This implies that Na+/K+ATPase function is reduced following OGD/REOX and fails to maintain the physiological level of intracellular Na+. Thus, the loss of Na+ homeostasis following OGD/REOX results from accelerated Na+ influx and decreased Na+ extrusion by Na+/K+ATPase.
We have previously demonstrated that NHE1 activation led to intracellular Na+ overload and swelling in astrocytes (13). We then examined whether blocking of NHE1 activation by PD98059 affected cell swelling. As shown in Figure 4C, inhibition of ERK pathways with PD98059 significantly attenuated the OGD/REOX-induced swelling and this effect is similar to one mediated by NHE1 inhibition either by HOE 642 or genetic ablation (13). This further supports our view that OGD/REOX-triggered stimulation of NHE1 activation in part depends on ERK activation.
Stimulation of NHE1 activity contributes to an increase in [Ca2+]i in NHE1+/+ astrocytes following OGD/REOX
Figure 4 shows that activation of NHE1 activity following OGD/REOX led to ~ 5-fold increase in [Na+]i in NHE1+/+ astrocytes. We hypothesized that the NHE1-mediated Na+ overload could cause an influx of Ca2+ via a decrease and/or reversed operation of NCX. In this experiment, we measured changes in bulk [Ca2+]i following OGD/REOX. Basal [Ca2+]i under normoxic control conditions was 60 ± 5 nM.[Ca2+]i remained at 59 ± 12 nM in NHE1+/+ astrocytes following 2 h OGD and 1 h REOX. We speculated that a lack of increase in bulk [Ca2+]i following OGD/REOX might be due to sequesteration of Ca2+ into the endoplasmic reticulum (ER) (16). To investigate this possibility, we monitored changes of [Ca2+]i when the ER Ca2+-ATPase is inhibited. As shown in Figure 5A, application of 1 μM thapsigargin to NHE1+/+ astrocytes under normoxic conditions caused a transient increase in [Ca2+]i. The Ca2+ transient peaked at ~1 min (382 ± 233 nM, Figure 5A inset) and declined to baseline values over the next 6 min. Following OGD/REOX in NHE1+/+ astrocytes, the thapsigargin-induced Ca2+ transient increased to 777 ± 567 nM (Figure 5A, inset), which suggests an increase in ER Ca2+ loading during OGD/REOX. In contrast, when NHE1+/+ astrocytes were treated with 1 μM HOE 642 during OGD/REOX, the thapsigargin-induced Ca2+ transient was only 431 ± 194 nM (Figure 5A, inset), which was significantly lower than values in OGD/REOX (p < 0.001). We then examined whether a reduction in Ca2+ transient can be detected in NHE1−/− astrocytes (Figure 5B). The thapsigargin-induced peak Ca2+ transient in NHE1−/−astrocytes was 316 ± 204 nM under normoxia. Following OGD/REOX, it reached to 421 ± 226 nM, which was not significantly different from normoxic controls in NHE1+/+ astrocytes (p > 0.05). These results indicate that stimulation of NHE1 activity affects ER Ca2+ loading following OGD/REOX.
Figure 5. NHE1 contributes to an increase in [Ca2+]i in astrocytes following OGD/REOX.
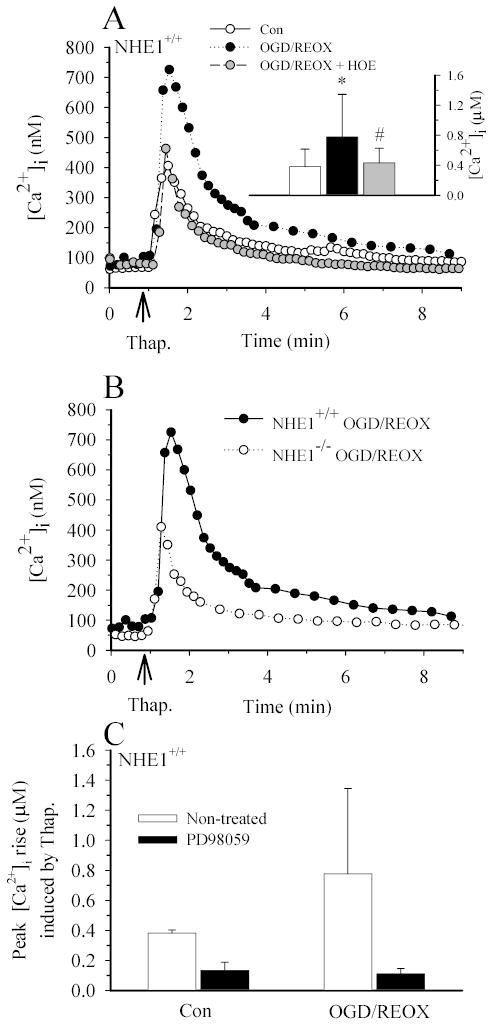
A. Thapsigargin-induced Ca2+ release was determined under control (1.2 mM) [Ca2+]o conditions in NHE1+/+ astrocytes following 3 h normoxia (CON) or 2 h OGD plus 1 h REOX (OGD/REOX). In some OGD/REOX studies, cells were exposed to 1 μM HOE 642 throughout the OGD/REOX and Ca2+ release experiment. Data points are the average of 20 cells from a representative experiment. Inset: Summary of peak thapsigargin-induced Ca2+ release data for CON or OGD/REOX with and without 1 μM HOE 642. Data are means ± SD (n = 80–120 cells, 4 cultures). * p < 0.001 vs. baseline control; # p < 0.001 vs. OGD/REOX. B. Thapsigargin-induced Ca2+ release was determined following OGD/REOX in NHE1+/+ and NHE1−/− astrocytes. C. Summary of PD98059 effects on thapsigargin-induced Ca2+ rise. Data points are means ± SD (20–60 cells from 1–3 cultures).
We performed some experiments to examine whether inhibition of ERK pathways via PD98059 would reduce Ca2+ overload following OGD/REOX. 30 μM of PD98059 significantly reduced the thapsigargin-mediated rise in Ca2+ in NHE1+/+ astrocytes under either control or OGD/REOX conditions (133 ± 16 nM, and 111 ± 36 nM; respectively, Figure 5C). Thus, ERK pathways are involved in Ca2+ loading under both normoxic and OGD/REOX conditions.
To further investigate a role for NHE1 in ER Ca2+ signaling following OGD/REOX, we measured releasable Ca2+ from the ER. As shown in Figure 6A & B, 1.0 μM bradykinin triggered Ca2+ release from intracellular Ca2+ stores and raised [Ca2+]i from a baseline of 69 ± 19 nM to 356 ± 151 nM in NHE1+/+ astrocytes under normoxic conditions. However, OGD/REOX caused an increase of bradykinin-induced Ca2+ release (654 ± 215 nM, p < 0.001) in NHE1+/+ astrocytes (Figure 6A & B). In NHE1−/− astrocytes, bradykinin evoked a 336 ± 79 nM Ca2+ release under normoxic control conditions, which was similar to that observed in NHE1+/+ cells (Figure 6C & D). In contrast, bradykinin triggered significantly less Ca2+ release in NHE1−/− astrocytes following OGD/REOX (450 ± 94 nM, p < 0.001, Figure 6C & D). This implies that OGD/REOX caused an increase in Ca2+ uptake by the intracellular Ca2+ stores only in NHE1+/+ astrocytes but not in NHE1−/− astrocytes.
Figure 6. An increase in bradykinin-evoked Ca2+ release in NHE1+/+ astrocytes but not in NHE1−/− astrocytes following OGD/REOX.
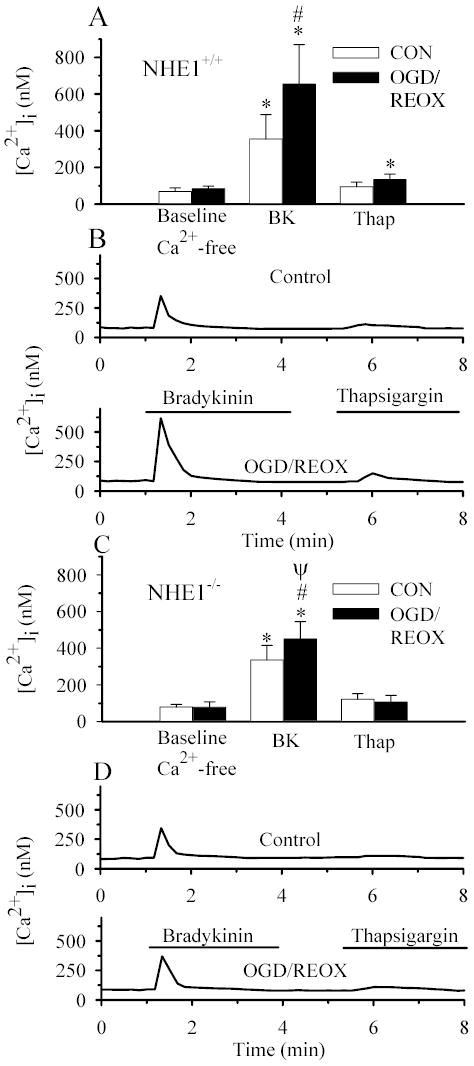
NHE1+/+ astrocytes (A, B) and NHE1−/− astrocytes (C, D) were treated for either 3 h normoxia (CON), or 2 h OGD and 1 h reoxygenation (OGD/REOX). Cells were exposed to [Ca2+]o-free buffer containing 1.0 μM bradykinin for 4.5 min, returned to [Ca2+]o-free buffer for 1 min, and then exposed to 1 μM thapsigargin for 3 min. A & C. Summary of peak bradykinin- and thapsigargin-induced Ca2+ release in NHE1+/+ and NHE1−/− astrocytes, respectively. B & D. Representative Ca2+ responses in single NHE1+/+ (B) and NHE1−/− (D) astrocytes to bradykinin and thapsigargin. Data are means ± SD (n = 60–80 cells, 4–6 cultures). * p < 0.001 vs. baseline control; # p < 0.001 vs. normoxic bradykinin; ψ p < 0.001 vs. NHE1+/+ OGD/REOX bradykinin.
We then confirmed this finding by analyzing the effect of the potent NHE1 inhibitor HOE 642 on Ca2+ signaling. Treatment of NHE1+/+ astrocytes with 1 μM HOE 642 did not change either baseline [Ca2+]i (84 ± 14 nM) or bradykinin-induced Ca2+ release (342 ± 96 nM) under normoxic conditions (Figure 7A & B). In contrast, treatment of NHE1+/+ cells with 1 μM HOE 642 during OGD/REOX significantly decreased the bradykinin-induced Ca2+ release (423 ± 120 vs. 654 ± 215 nM, Figure 7, p < 0.05). This is consistent with the observations in NHE1−/− astrocytes.
Figure 7. Inhibition of NHE1 activity attenuates the increase in bradykinin-evoked Ca2+ release following OGD/REOX.
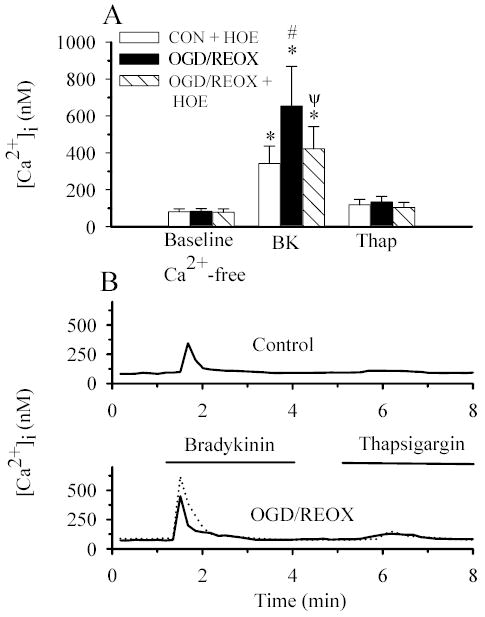
NHE1+/+ astrocytes were subjected to either 3 h normoxia (CON), or 2 h OGD and 1 h reoxygenation (OGD/REOX). In some experiments, OGD/REOX was performed in the presence of 1 μM HOE 642. Cells were exposed to 1.0 μM bradykinin (BK) and 1 μM thapsigargin (Thap) as described in Figure 6 legend. A. Summary of peak bradykinin- and thapsigargin-induced Ca2+ release in NHE1+/+ astrocytes following CON, OGD/REOX, and OGD/REOX with HOE 642. B. Representative Ca2+ responses in single NHE1+/+ astrocytes to bradykinin and thapsigargin under CON, OGD/REOX (the dotted tracing), or OGD/REOX treated with HOE 642 (the solid tracing). Data are means ± SD (n= 60–120 cells, 4–6 cultures). * p < 0.001 vs. baseline control; # p < 0.001 vs. bradykinin; ψ p < 0.001 vs. OGD/REOX bradykinin.
As shown in Figures 6 & 7, a small fraction of Ca2+ release was insensitive to bradykinin but triggered by thapsigargin. These results imply that astrocytes have both bradykinin-sensitive and bradykinin-insensitive Ca2+ stores. OGD/REOX led to an increase in thapsigargin-triggered Ca2+ release in NHE1+/+ but not in NHE1−/− astrocytes.
NHE1 activity contributes to mitochondrial Ca2+ accumulation in NHE1+/+ astrocytes
Our data indicate that bradykinin-sensitive Ca2+ stores contain higher levels of Ca2+ following OGD/REOX. However, mitochondria are also known to buffer increases in cytosolic Ca2+ following pathological stimuli. We therefore investigated whether OGD/REOX affected Ca2+ loading in mitochondria of NHE1+/+ astrocytes. NHE1+/+ astrocytes were exposed to 10 μM FCCP and 2.5 μg/ml oligomycin under Ca2+-free conditions, either following 3 h normoxia (Figure 8A) or 2 h OGD and 1 h REOX (Figure 8B). After a 10–20 sec delay, [Ca2+]i increased in response to FCCP/oligomycin in both normoxic astrocytes (166 ± 32 nM) and OGD/REOX-treated astrocytes (261 ± 43 nM, Figure 8A, B, C). Cytosolic Ca2+ was subsequently cleared (Figure 8A, B). When NHE1+/+ astrocytes were treated with 1 μM HOE 642 during OGD/REOX, the peak increase in [Ca2+]i was only 222 ± 47 nM and significantly smaller than in the absence of HOE 642 (Figure 8 B & C, p < 0.001). These results suggest that mitochondria do buffer increases in cytosolic Ca2+ following OGD/REOX, but to a smaller degree than ER Ca2+ stores. In addition, stimulation of NHE1 activity also increases Ca2+ loading in mitochondria.
Figure 8. NHE1 activity contributes to mitochondrial Ca2+ accumulation in NHE1+/+ astrocytes.
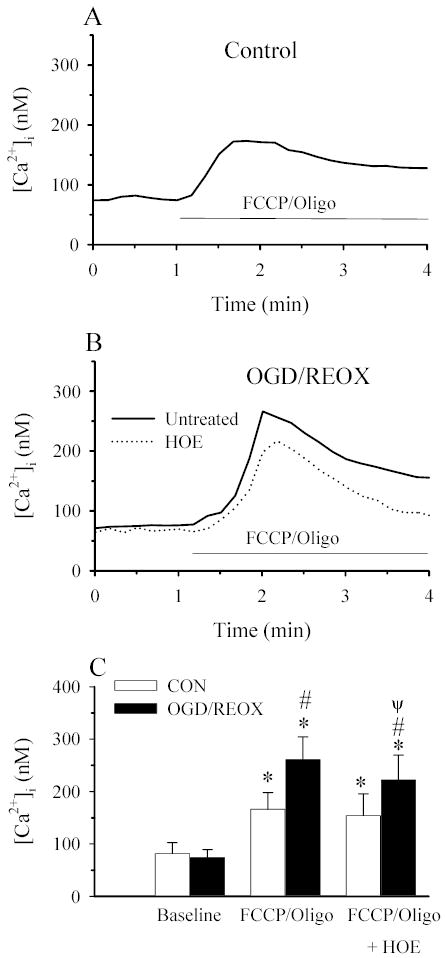
FCCP/Oligomycin-induced Ca2+ release was determined under [Ca2+]o-free conditions. NHE1+/+ astrocytes were subjected to either 3 h normoxia (CON), or 2 h OGD and 1 h reoxygenation (OGD/REOX). In some experiments, OGD/REOX was performed in the presence of 1 μM HOE 642. Cells were exposed to [Ca2+]o-free buffer containing 10 μM FCCP + 2.5 μg/ml oligomycin for 4 min to stimulate release of Ca2+ from mitochondria. A. Trace showing the representative Ca2+ response in cells following exposure to FCCP/Oligomycin. B. Representative traces of FCCP/Oligomycin-induced Ca2+ release in astrocytes subjected to OGD/REOX in the presence or absence of HOE 642. C. Summary graph of FCCP/Oligomycin-induced Ca2+ release in NHE1+/+ astrocytes under both CON and OGD/REOX conditions and in the absence or presence of HOE 642. Data are means ± SD (n= 60–80 cells, 3 cultures). * p < 0.001 vs. baseline; # p < 0.001 vs. CON; ψ p < 0.001 vs. FCCP/Oligo OGD/REOX.
An increase in [Ca2+]i is in part a result of reverse-mode operation of Na+/Ca2+ exchange
In our recent study, we demonstrated that overloading of intracellular Na+ via Na-K-Cl cotransporter-mediated Na+ influx resulted in reverse-mode operation of NCX and a rise in cytosolic Ca2+ (16). This increase in NCX activity is blocked by NCX blocker KB-R7943 (16). In the present study, we investigated whether reverse operation of NCX contributes to the increase in [Ca2+]i following stimulation of NHE1 activity shown in Figures 7 and 8. First, we established the effects of a potent and selective inhibitor SEA0400 on the reverse mode of NCX in NHE1+/+ astrocytes (Figure 9A). The reverse operation of NCX was triggered by removing extracellular Na+, which switches the NCX from the forward (Ca2+ exit) to the reverse (Ca2+ entry) mode. The reverse mode of NCX in NHE1+/+ astrocytes led to a large and repeatable increase in Ca2+i (Figure 9A). However, in the presence of 100 nM SEA0400, peak Ca2+ entry through NCX was reduced by ~ 90% (Figure 9A). This confirmed that SEA0400 is a potent inhibitor of the reverse operation of NCX in astrocytes, consistent with the report in other cell types (20).
Figure 9. NCX contributes to an increase in [Ca2+]i in astrocytes following OGD/REOX.
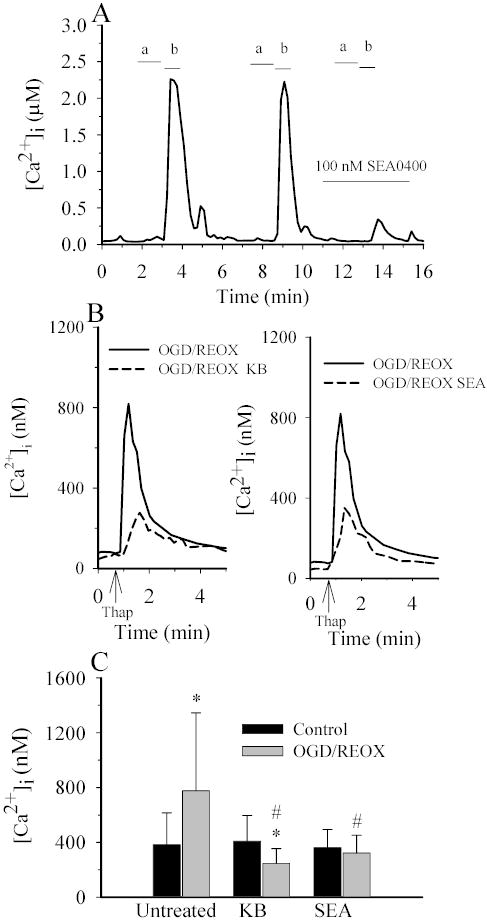
A. Reverse-mode of NCX was induced by exposing cells with Ca2+-free HEPES buffer containing gramicidin (2 mg/ml) for one min (a), and subsequently to a Na+-free buffer (1.2 mM Ca2+) for 30–40 sec (b). Cells were then returned to normal HEPES-MEM. The increases in [Ca2+]i via the reverse-mode of NCX were reproducible. However, it was decreased by ~90% in the presence of 0.1 μM SEA0400. B. Thapsigargin-induced Ca2+ release was determined under control (1.2 mM) [Ca2+]o conditions in NHE1+/+ astrocytes following 3 h normoxia (CON) or 2 h OGD plus 1 h REOX (OGD/REOX). In some OGD/REOX studies, cells were exposed to 3 μM KB-R7943 or 0.1 μM SEA0400 throughout the OGD/REOX and Ca2+ release experiment. Data points are the average of 20 cells from a representative experiment. C. Summary of peak thapsigargin-induced Ca2+ release data for CON or OGD/REOX with and without either 3 μM KB-R7943 or 0.1 μM SEA0400. Data are means ± SD (n = 80–120 cells, 3 cultures). * p < 0.001 vs. baseline control; # p < 0.001 vs. OGD/REOX.
We investigated whether the NHE1-mediated effect on the loading of Ca2+ stores during OGD/REOX depends on the reverse-mode operation of NCX. As shown in Figure 9B, both KB-R7943 (3μM) and SEA0400 (100 nM) significantly reduced the OGD/REOX-induced increase in the thapsigargin-induced Ca2+ transient. The untreated cells exhibited a rise in Ca2+ of 777 ± 567 nM following OGD/REOX (Figure 9B & C). In the presence of KB-R7943 or SEA0400, Ca2+ transient values were reduced to 247 ± 107 nM and 322 ±129 nM, respectively (Figure 9C). This result further suggests that ER Ca2+ loading following OGD/REOX is attributable to Ca2+ influx via the reverse mode of NCX.
To further support this finding, we performed the theoretical thermodynamic analysis of NCX in astrocytes. The dependence of NCX reversal potential on [Na+]i was modeled using LabHEART [Ver· 4.9.5 simulation software (27)]. The key variables, [Na+]o, [Ca2+]o, [Na+]i and [Ca2+]i, were modified using our experimental data under normoxic control or OGD/REOX conditions. An NCX Current-Voltage plot was generated as [Na+]i was varied from 10–80 mM. The membrane voltage at which each simulation generated a current of zero was then plotted vs. [Na+]i (Figure 10). Using this simulation, a reversal potential of − 43 mV was generated for control ionic conditions at [Ca2+]i of 100 nM. As [Na+]i increased, the reversal potential for NCX decreased. Resting plasma membrane potential in astrocytes has been reported to be about −60 mV (21), thus under these conditions NCX is in the forward mode and may help clear Ca2+ from the cell. The actual membrane potential during OGD/REOX is not measured in NHE1+/+ astrocytes and may remain either constant or depolarized. However, in either situation, the simulation predicts that an increase in [Na+]i during OGD/REOX will strongly favor an inwardly directed Ca2+ current (Figure 10). Under ischemic conditions, [Ca2+]i will rise and may reduce the reverse-mode operation of NCX. To take this factor into consideration, a simulation was performed when [Ca2+]i was elevated to 777 nM, a level that was reached following OGD/REOX in the presence of thaspsigargin. If plasma membrane potential remains constant during ischemia, then NCX is predicted to function in the reverse mode when [Na+]i increases above 25 mM with [Ca2+]i at 777 nM. In fact, [Na+]i was elevated to ~ 45 mM at 10 min REOX and reached to 80 mM at 60 min REOX. However, when NHE1 activity is inhibited by HOE 642, NHE1+/+ astrocytes exhibited ~ 50% less Na+i accumulation, which would tend to drive NCX in the forward mode. Taken together, this thermodynamic analysis further supports the view that NHE1-mediated Na+-overload triggers the reverse operation of NCX following OGD/REOX.
Figure 10. The reverse mode operation of NCX is modeled following OGD/REOX.
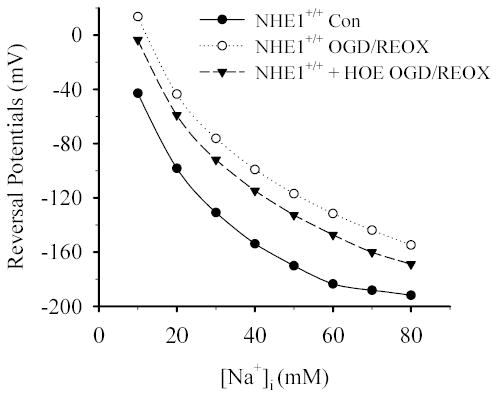
LabHeart V.4.9.5 was used to model NCX activity following OGD/REOX. It is assumed that NCX is an electrogenic mechanism that transports Na+ and Ca2+ with a stoichiometry of 3:1. The LabHeart simulation generated values of the NCX current for different voltages using known and assumed values for intracellular and extracellular values of Na+, Ca2+. Values for Na+, Ca2+ affinities, voltage saturation and the position of the energy barrier were set at the default values for the simulator. These current-voltage values were then used to create I-V curves for NCX under varying ionic conditions. The reversal potential for each ionic condition was the voltage when NCX current equaled zero. [Ca2+]i was set at 100, 777 or 431 nM for the normoxic control (Con) NHE1+/+, NHE1+/+ OGD/REOX or NHE1+/+ OGD/REOX + HOE 642, respectively.
DISCUSSION
The role for NHE1 in pHi regulation in cortical astrocytes
We reported recently that NHE1 plays a role in maintenance of resting pHi in mouse cortical astrocytes (13). In the absence of HCO3, pHi recovery rate following NH4+/NH3− mediated acid loading was increased by ~75% in NHE1+/+ astrocytes following 2 h OGD. Either inhibition of NHE1 activity with HOE 642 or genetic ablation of NHE1 abolished this elevated pHi regulation (13). We concluded that NHE1 activity is stimulated in astrocytes upon reoxygenation in response to in vitro transient ischemia. In this study, we further analyzed the kinetics of NHE1 in astrocytes following in vitro ischemia. First, we determined that the intrinsic buffering power was unaltered in NHE1+/+ astrocytes following OGD/REOX or in NHE1−/− astrocytes. At pHi 7.0, βi was 7.8 mM/pH in NHE1+/+ astrocytes under normoxic conditions. This βi value was unchanged in NHE1+/+ astrocytes following OGD/REOX. In addition, a similar βi value was found in NHE1−/− astrocytes under normoxic conditions. There were no significant changes in the slopes of the lines fitted to the βi vs pHi under these conditions. This finding suggests that not only is the cytosolic intrinsic buffering capacity unchanged in astrocytes following 2 h OGD but also that ablation of NHE1 does not trigger a compensatory effect on intrinsic buffering capacity. Therefore, the elevation of pHi recovery rate following OGD, which is sensitive to both Na+o and the NHE1 inhibitor HOE 642, is largely the result of an increase in NHE1 activity in NHE1+/+ astrocytes.
Our kinetic analysis revealed that when NHE1+/+ astrocytes were reoxygenated following 2 h OGD, the Vmax of Na+/H+ exchange was increased 3-fold and the Km for H+ was doubled. This implies that Na+/H+ exchange is more efficient in astrocytes following OGD. The causes of the significant elevation in Vmax are unknown. This could be due in part to ERK1/2-mediated phosphorylation of NHE1 or to increased expression of NHE1 in the surface of astrocytes via protein trafficking following OGD. A significant increase in NHE1 synthesis is unlikely considering the short term of OGD treatment.
Cell shrinkage results in stimulation of NHE1 activity in C6 glioma cells as a result of an alkaline shift in the pHi dependence of the exchanger without changing the Vmax (29). Serum-mediated growth signals also shift NHE1 towards the high-affinity form (15). In contrast, in the current study the NHE1 was less sensitive to acidification following OGD. This may imply that altering Vmax is a more efficient way for astrocytes to accelerate H+ extrusion than an alkaline shift of Km· 2 h OGD induced an intracellular acidosis in astrocytes [pHi of 6.8 vs. 7.10 of normoxic control (13)]. Therefore, an increase in Vmax and a decrease in H+ affinity would ensure that the NHE1 system would not be saturated under acidic conditions following OGD.
Activation of ERK1/2 signaling pathways is in part responsible for OGD/REOX-mediated NHE1 stimulation in astrocytes.
In addition to an NH2-terminal transmembrane ion translocation domain, NHE1 contains a COOH-terminal cytoplasmic regulatory domain that modulates transport activity most likely by altering the affinity of a H+ transport site in the transmembrane domain (28). A phosphorylation domain at the distal C-terminus of NHE1 contains a number of serine residues that are phosphorylated by serine/threonine kinases acting downstream of distinct signaling pathways.
It has been reported that a serine/threonine kinase p90RSK, a downstream substrate of ERK1/2, directly phosphorylates NHE1 and activates exchanger activity in response to growth factors (24; 34). MEK-dependent pathways, including p90RSK and ERK1/2, stimulate phosphorylation and activation of NHE1 in ischemic myocardium (24). The ERK signaling cascade is emerging as an important regulator of neuronal and glial responses to oxidative stress-related injury (7; 38). However, it is unknown whether ERK activation triggers NHE1 stimulation in astrocytes following OGD. In the present study, we found that 1 h OGD led to activation of ERK1/2 phosphorylation and that this stimulation was sustained in astrocytes following 2 h OGD and 1 h REOX. In addition, inhibition of MEK activity with PD98059 not only significantly reduced NHE1-mediated H+ efflux but also attenuated NHE1-mediated Na+ accumulation by ~60%. Therefore, we concluded that OGD/REOX-induced stimulation of NHE1 activity is in part dependent on MEK-ERK-phosphorylation in astrocytes. It remains to be established whether ERK1/2 activation leads to phosphorylation of p90RSK and whether this in turn directly regulates NHE1 activity in astrocytes following OGD/REOX.
A sustained intracellular acidosis (3 min, pHi 6.5) leads to stimulation of NHE1 activity in rat ventricular myocytes, which requires activation of ERK1/2 and p90RSK (9). We found that 2 h OGD caused a reduction in pHi from 7.09 to 6.80 (13). In the current study, we examined whether OGD/REOX-induced stimulation of NHE1 activity could be the result of OGD-mediated moderate and sustained acidosis. In cortical NHE1+/+ astrocytes, neither the Vmax nor the Km of NHE1 were altered following 3 min of acidosis at a pHi of 6.25. Thus, sustained acidosis failed to trigger NHE1 activation in mouse cortical NHE1+/+ astrocytes, suggesting that it might be a cell type-specific phenomenon. Activation of MEK-ERK-phosphorylation in astrocytes is unlikely to be the result of a sustained intracellular acidosis following OGD/REOX.
It is known that many signals can trigger activation of ERK pathways in response to oxidative stress (7). A 4-h period of glucose-deprivation in mouse cortical astrocytes caused ~ two-fold increase in ERK1/2 phosphorylation (38). In the present study, 2 h OGD in astrocytes led to ~ 25% reduction in cellular ATP level (16). Thus, an increase in intracellular Ca2+, reduction of ATP, glucose deprivation, and the generation of reactive oxygen species may subsequently stimulate MEK-ERK-phosphorylation in astrocytes following OGD/REOX.
On the other hand, it was recently observed in vascular smooth muscle cells that NHE1 activity regulates ERK phosphorylation (25). We have performed additional experiments in NHE1−/− astrocytes to investigate whether ERK phosphorylation was altered in the absence of NHE1 function. One hour OGD moderately stimulated ERK phosphorylation in NHE1−/− astrocytes (~ 10%).
Stimulation of NHE1 activity in astrocytes overloads Ca2+ stores in ER and mitochondria following OGD/REOX.
In the present study, there was a two-fold increase in releasable Ca2+ from ER Ca2+ stores when NHE1+/+ astrocytes underwent 2 h OGD and 1 h REOX. In addition, either inhibition of NHE1 activity or genetic ablation of NHE1 abolished the OGD/REOX-mediated Ca2+ loading of the ER. This finding was confirmed by two different measurements: thapsigargin-induced ER Ca2+ release in the presence of extracellular Ca2+ and bradykinin-mediated ER Ca2+ release in the absence of extracellular Ca2+. Moreover, we also evaluated the mitochondria’s response to the OGD/REOX-mediated Ca2+ loading. Mitochondria exhibited a moderate level of increase in Ca2+ loading following OGD/REOX. The ER plays a central role in the control of cellular Ca2+ homeostasis. ER dysfunction (depletion of ER Ca2+ stores, inhibition of ER protein folding and processing) is linked to ischemic cell damage (26). The ER has close physical and functional contacts to mitochondria and the coordination between Ca2+ signaling of the ER and mitochondria is facilitated by the strategic location of mitochondria at sites of ER Ca2+ release (6) (19). It has been reported that Ca2+ released during the ER stress response may promote mitochondrial Ca2+ overload, fragmentation and apoptosis (6). Therefore, during severe ischemic episodes in astrocytes, the NHE1-mediated ER stress response may have a significant impact on mitochondrial function. This view is further supported by a recent finding that inhibition of NHE1 activity with HOE 642 significantly suppressed mitochondrial Ca2+ overload and loss of mitochondrial membrane potential induced by H2O2-mediated oxidative stress (35).
Stimulation of NHE1 activity triggers a reverse-mode operation of NCX.
The present study indicates that an increase in Ca2+ entry during OGD/REOX, which is buffered by ER and mitochondrial Ca2+stores, is linked to NHE1 activity. We reported recently that intracellular Na+ loading mediated by Na+-dependent Cl− transport triggers Ca2+ influx via a reverse-mode operation of NCX following OGD (16). In this study, 3 μM KB-R7943, which blocked ~90 % of the NCX activity (16), abolished the OGD/REOX-induced enhancement in ER Ca2+ stores. A more selective inhibitor of NCX1, SEA0400 [IC50 of 5 nM in astrocytes, (20)], also blocked the OGD/REOX-mediated increase in Ca2+ transient. The data support our view that OGD/REOX initiates the reverse-mode operation of NCX in astrocytes and causes Ca2+ transient influx. We believe that this process is triggered by NHE1-mediated Na+ overload. Our thermodynamic analysis revealed that the reverse-mode operation of NCX would occur when [Na+]i increases above 25 mM. At the end of 1 h REOX following 2 h OGD, [Na+]i in NHE1+/+ astrocytes was increased to ~80 mM as a result of accelerated Na+ influx via NHE1 and decreased Na+ extrusion via Na+/K+ATPase. In contrast, NHE1−/− astrocytes exhibited a 50% reduction in Na+ overload. These data leads us to conclude that stimulation of NHE1 activity overloads astrocytes with intracellular Na+ and subsequently triggers Ca2+ influx via the reverse-mode of Na+/Ca2+ exchange.
In summary, we found that NHE1 activity was significantly stimulated in astrocytes in response to in vitro ischemia. This stimulation was in part dependent on activation of ERK1/2 signaling pathways. NHE1 activity led to intracellular Na+ overload and affected Ca2+ stores in ER and mitochondria. Therefore, overstimulation of NHE1 in astrocytes may contribute to ER stress and mitochondrial dysfunction under ischemic conditions.
Acknowledgments
This work was supported in part by NIH grants R01NS38118 and R01NS048216 (D. Sun), and R01HL61974 (G. E. Shull), and an AHA Established-Investigator award grant N0540154 (D. Sun). The authors would like to thank Dr. Tom Cook for consultation on the statistical analysis in this study.
References
- 1.Aronson PS, Nee J, Suhm MA. Modifier role of internal H+ in activating the Na+-H+ exchanger in renal microvillus membrane vesicles. Nature. 1982;299:161–163. doi: 10.1038/299161a0. [DOI] [PubMed] [Google Scholar]
- 2.Avkiran M. Protection of the ischaemic myocardium by Na+/H+ exchange inhibitors: potential mechanisms of action. Basic Res Cardiol. 2001;96:306–311. doi: 10.1007/s003950170037. [DOI] [PubMed] [Google Scholar]
- 3.Bell SM, Schreiner CM, Schultheis PJ, Miller ML, Evans RL, Vorhees CV, Shull GE, Scott WJ. Targeted disruption of the murine Nhe1 locus induces ataxia, growth retardation, and seizures. Am J Physiol. 1999;276:C788–C795. doi: 10.1152/ajpcell.1999.276.4.C788. [DOI] [PubMed] [Google Scholar]
- 4.Bevensee MO, Weed RA, Boron WF. Intracellular pH regulation in cultured astrocytes from rat hippocampus. I. Role Of HCO3−. J Gen Physiol. 1997;110:453–465. doi: 10.1085/jgp.110.4.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boyarsky G, Ransom B, Schlue WR, Davis MB, Boron WF. Intracellular pH regulation in single cultured astrocytes from rat forebrain. Glia. 1993;8:241–248. doi: 10.1002/glia.440080404. [DOI] [PubMed] [Google Scholar]
- 6.Brookes PS, Yoon Y, Robotham JL, Anders MW, Sheu SS. Calcium, ATP, and ROS: a mitochondrial love-hate triangle. Am J Physiol Cell Physiol. 2004;287:C817–C833. doi: 10.1152/ajpcell.00139.2004. [DOI] [PubMed] [Google Scholar]
- 7.Chu CT, Levinthal DJ, Kulich SM, Chalovich EM, DeFranco DB. Oxidative neuronal injury. The dark side of ERK1/2. Eur J Biochem. 2004;271:2060–2066. doi: 10.1111/j.1432-1033.2004.04132.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- 9.Haworth RS, McCann C, Snabaitis AK, Roberts NA, Avkiran M. Stimulation of the plasma membrane Na+/H+ exchanger NHE1 by sustained intracellular acidosis. Evidence for a novel mechanism mediated by the ERK pathway. J Biol Chem. 2003;278:31676–31684. doi: 10.1074/jbc.M304400200. [DOI] [PubMed] [Google Scholar]
- 10.Hertz L. Dibutyryl cyclic AMP treatment of astrocytes in primary cultures as a substitute for normal morphogenic and ‘functiogenic’ transmitter signals. Adv Exp Med Biol. 1990;265:227–243. doi: 10.1007/978-1-4757-5876-4_22. [DOI] [PubMed] [Google Scholar]
- 11.Hoyt KR, Arden SR, Aizenman E, Reynolds IJ. Reverse Na+/Ca2+ exchange contributes to glutamate-induced intracellular Ca2+ concentration increases in cultured rat forebrain neurons. Mol Pharmacol. 1998;53:742–749. [PubMed] [Google Scholar]
- 12.Juurlink BH, Hertz L, Yager JY. Astrocyte maturation and susceptibility to ischaemia or substrate deprivation. Neuroreport. 1992;3:1135–1137. doi: 10.1097/00001756-199212000-00026. [DOI] [PubMed] [Google Scholar]
- 13.Kintner DB, Su G, Lenart B, Ballard AJ, Meyer JW, Ng LL, Shull GE, Sun D. Increased tolerance to oxygen and glucose deprivation in astrocytes from Na+/H+ exchanger isoform 1 null mice. Am J Physiol Cell Physiol. 2004;287:C12–C21. doi: 10.1152/ajpcell.00560.2003. [DOI] [PubMed] [Google Scholar]
- 14.Kuribayashi Y, Itoh N, Kitano M, Ohashi N. Cerebroprotective properties of SM-20220, a potent Na+/H+ exchange inhibitor, in transient cerebral ischemia in rats. Eur J Pharmacol. 1999;383:163–168. doi: 10.1016/s0014-2999(99)00645-7. [DOI] [PubMed] [Google Scholar]
- 15.Lacroix J, Poet M, Maehrel C, Counillon L. A mechanism for the activation of the Na/H exchanger NHE-1 by cytoplasmic acidification and mitogens. EMBO Rep. 2004;5:91–96. doi: 10.1038/sj.embor.7400035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lenart B, Kintner DB, Shull GE, Sun D. Na-K-Cl cotransporter-mediated intracellular Na+ accumulation affects Ca2+ signaling in astrocytes in an in vitro ischemic model. J Neurosci. 2004;24:9585–9597. doi: 10.1523/JNEUROSCI.2569-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Levine SA, Montrose MH, Tse CM, Donowitz M. Kinetics and regulation of three cloned mammalian Na+/H+ exchangers stably expressed in a fibroblast cell line. J Biol Chem. 1993;268:25527–25535. [PubMed] [Google Scholar]
- 18.Li S, Jiang Q, Stys PK. Important role of reverse Na+-Ca2+ exchange in spinal cord white matter injury at physiological temperature. J Neurophysiol. 2000;84:1116–1119. doi: 10.1152/jn.2000.84.2.1116. [DOI] [PubMed] [Google Scholar]
- 19.Mannella CA, Buttle K, Rath BK, Marko M. Electron microscopic tomography of rat-liver mitochondria and their interaction with the endoplasmic reticulum. Biofactors. 1998;8:225–228. doi: 10.1002/biof.5520080309. [DOI] [PubMed] [Google Scholar]
- 20.Matsuda T, Arakawa N, Takuma K, Kishida Y, Kawasaki Y, Sakaue M, Takahashi K, Takahashi T, Suzuki T, Ota T, Hamano-Takahashi A, Onishi M, Tanaka Y, Kameo K, Baba A. SEA0400, a novel and selective inhibitor of the Na+-Ca2+ exchanger, attenuates reperfusion injury in the in vitro and in vivo cerebral ischemic models. J Pharmacol Exp Ther. 2001;298:249–256. [PubMed] [Google Scholar]
- 21.McKhann GM, D'Ambrosio R, Janigro D. Heterogeneity of astrocyte resting membrane potentials and intercellular coupling revealed by whole-cell and gramicidin-perforated patch recordings from cultured neocortical and hippocampal slice astrocytes. J Neurosci. 1997;17:6850–6863. doi: 10.1523/JNEUROSCI.17-18-06850.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McLean LA, Roscoe J, Jorgensen NK, Gorin FA, Cala PM. Malignant gliomas display altered pH regulation by NHE1 compared with nontransformed astrocytes. Am J Physiol Cell Physiol. 2000;278:C676–C688. doi: 10.1152/ajpcell.2000.278.4.C676. [DOI] [PubMed] [Google Scholar]
- 23.Mellergard P, Ouyang YB, Siesjö BK. Intracellular pH regulation in cultured rat astrocytes in CO2/HCO3− containing media. Exp Brain Res. 1993;95:371–380. doi: 10.1007/BF00227129. [DOI] [PubMed] [Google Scholar]
- 24.Moor AN, Gan XT, Karmazyn M, Fliegel L. Activation of Na+/H+ exchanger-directed protein kinases in the ischemic and ischemic-reperfused rat myocardium. J Biol Chem. 2001;276:16113–16122. doi: 10.1074/jbc.M100519200. [DOI] [PubMed] [Google Scholar]
- 25.Mukhin YV, Garnovskaya MN, Ullian ME, Raymond JR. ERK is regulated by sodium-proton exchanger in rat aortic vascular smooth muscle cells. J Biol Chem. 2004;279:1845–1852. doi: 10.1074/jbc.M304907200. [DOI] [PubMed] [Google Scholar]
- 26.Paschen W, Frandsen A. Endoplasmic reticulum dysfunction--a common denominator for cell injury in acute and degenerative diseases of the brain? J Neurochem. 2001;79:719–725. doi: 10.1046/j.1471-4159.2001.00623.x. [DOI] [PubMed] [Google Scholar]
- 27.Puglisi JL, Bers DM. LabHEART: an interactive computer model of rabbit ventricular myocyte ion channels and Ca transport. Am J Physiol Cell Physiol. 2001;281:C2049–C2060. doi: 10.1152/ajpcell.2001.281.6.C2049. [DOI] [PubMed] [Google Scholar]
- 28.Putney LK, Denker SP, Barber DL. The changing face of the Na+/H+ exchanger, NHE1: structure, regulation, and cellular actions. Annu Rev Pharmacol Toxicol. 2002;42:527–552. doi: 10.1146/annurev.pharmtox.42.092001.143801. [DOI] [PubMed] [Google Scholar]
- 29.Shrode LD, Klein JD, Douglas PB, O'Neill WC, Putnam RW. Shrinkage-induced activation of Na+/H+ exchange: role of cell density and myosin light chain phosphorylation. Am J Physiol. 1997;272:C1968–C1979. doi: 10.1152/ajpcell.1997.272.6.C1968. [DOI] [PubMed] [Google Scholar]
- 30.Shrode LD, Putnam RW. Intracellular pH regulation in primary rat astrocytes and C6 glioma cells. Glia. 1994;12:196–210. doi: 10.1002/glia.440120305. [DOI] [PubMed] [Google Scholar]
- 31.Su G, Kintner DB, Flagella M, Shull GE, Sun D. Astrocytes from Na+-K+-Cl− cotransporter-null mice exhibit absence of swelling and decrease in EAA release. Am J Physiol Cell Physiol. 2002;282:C1147–C1160. doi: 10.1152/ajpcell.00538.2001. [DOI] [PubMed] [Google Scholar]
- 32.Sun D, Murali SG. Na+-K+-2Cl− cotransporter in immature cortical neurons: A role in intracellular Cl- regulation. J Neurophysiol. 1999;81:1939–1948. doi: 10.1152/jn.1999.81.4.1939. [DOI] [PubMed] [Google Scholar]
- 33.Swanson RA, Liu J, Miller JW, Rothstein JD, Farrell K, Stein BA, Longuemare MC. Neuronal regulation of glutamate transporter subtype expression in astrocytes. J Neurosci. 1997;17:932–940. doi: 10.1523/JNEUROSCI.17-03-00932.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Takahashi E, Abe J, Gallis B, Aebersold R, Spring DJ, Krebs EG, Berk BC. p90(RSK) is a serum-stimulated Na+/H+ exchanger isoform-1 kinase. Regulatory phosphorylation of serine 703 of Na+/H+ exchanger isoform-1. J Biol Chem. 1999;274:20206–20214. doi: 10.1074/jbc.274.29.20206. [DOI] [PubMed] [Google Scholar]
- 35.Teshima Y, Akao M, Jones SP, Marban E. Cariporide (HOE642), a selective Na+-H+ exchange inhibitor, inhibits the mitochondrial death pathway. Circulation. 2003;108:2275–2281. doi: 10.1161/01.CIR.0000093277.20968.C7. [DOI] [PubMed] [Google Scholar]
- 36.Wakabayashi S, Ikeda T, Iwamoto T, Pouyssegur J, Shigekawa M. Calmodulin-binding autoinhibitory domain controls “pH-sensing” in the Na+/H+ exchanger NHE1 through sequence-specific interaction. Biochemistry. 1997;36:12854–12861. doi: 10.1021/bi9715472. [DOI] [PubMed] [Google Scholar]
- 37.Yan Y, Dempsey RJ, Flemmer A, Forbush B, Sun D. Inhibition of Na(+)-K(+)-Cl(−) cotransporter during focal cerebral ischemia decreases edema and neuronal damage. Brain Res. 2003;961:22–31. doi: 10.1016/s0006-8993(02)03832-5. [DOI] [PubMed] [Google Scholar]
- 38.Yung HW, Tolkovsky AM. Erasure of kinase phosphorylation in astrocytes during oxygen-glucose deprivation is controlled by ATP levels and activation of phosphatases. J Neurochem. 2003;86:1281–1288. doi: 10.1046/j.1471-4159.2003.01946.x. [DOI] [PubMed] [Google Scholar]