

Abstract
Subtle changes in protein sequence can equate to large changes in function, such as enabling pathogens to evade the immune system, hindering antibody recognition of antigens, or conferring antibiotic resistance. Even single amino acid substitutions may alter ligand binding affinity, enzymatic activity, and protein stability. Yet, due to limitations in time and resources, proteins closely related in sequence to those already characterized often remain unexamined. AlphaFold has emerged as a promising tool for protein structure prediction, though its utility in modeling single amino acid substitutions remains uncertain. In this study, we assessed AlphaFold 3’s accuracy in modeling natural variants of the Helicobacter pylori catalase KatA by comparing its predictions to a novel high-resolution crystal structure of KatA from strain SS1. This variant contains key substitutions at residues 234, 237, 255, and 421 relative to the well-characterized strain 26695. AlphaFold 3 models accurately reproduced the global structure and local conformations of most variant residues, with high fidelity in conservative substitutions but variable accuracy in more flexible or interface-exposed sites. We further explored how user inputs, such as incorrect oligomeric states or sequence modifications, influence prediction quality. While AlphaFold 3 consistently produced high-quality models, deviations at variant sites occurred when incorrect oligomeric states were specified. Our findings highlight both the strengths and limitations of AlphaFold 3 in modeling natural protein variants and underscore the importance of accurate user input for reliable structural predictions.
Keywords: Alphafold, natural variants, Helicobacter, catalase
Introduction
Proteins in nature exist as a spectrum of amino acid variants 1–4. These sequence differences may result from genetic drift, which over evolutionary timescales can lead to gain- or loss-of-function events 5. In some cases, however, single amino acid changes can immediately influence protein function by altering ligand-binding specificity, enzymatic efficiency, conformational dynamics, thermostability, immunogenicity, or other properties 6–9. Amino acid variation underlies positive selection in host-pathogen evolutionary relationships and antimicrobial resistance 10–12. The structural and functional consequences of such amino acid changes can be studied and understood using experimental structure-determination methods like protein crystallography, which can reveal unexpected alterations not readily predicted by sequence analysis alone 13,14. However, structural studies of proteins similar in sequence, such as natural protein variants, are not commonly undertaken due to constraints in time and resources, limiting our understanding of how natural variances affect structure and function.
AlphaFold has emerged as a promising tool to address some of these challenges 4,15,16. Widely recognized as a significant advancement in protein structure prediction, AlphaFold has been used successfully to model new protein structures ahead of experimental validation, and is continually being updated and developed with improved features 15–17. There is disagreement in the literature about the application of AlphaFold modeling of single amino acid substitutions. Studies suggest that AlphaFold’s predictive accuracy is system-dependent and heavily influenced by the quality of experimental data used during its training 18,19. The developers explicitly caution against using AlphaFold to predict the structural impact of single amino acid substitutions, such as destabilizing mutations (https://alphafold.ebi.ac.uk/faq) 20. A recent study showed that AlphaFold’s predictions for the energetic consequences of single mutations poorly correlate with experimentally determined protein stabilities 9,20. However, new ways to circumvent some of these limitations have been reported 4. As a result, the utility of AlphaFold in accurately modeling local structural variations remains uncertain.
Despite these limitations, AlphaFold has significantly broadened access to protein structure prediction, enabling researchers without structural biology expertise to model proteins and explore mechanistic hypotheses 21. The online AlphaFold 3 server, for example, only requires users to input a primary amino acid sequence and oligomerization state and returns a model within minutes 22. However, this ease of access also introduces risks. Users unfamiliar with structural biology may provide suboptimal input—such as incorrect oligomeric states—or misinterpret the resulting models, potentially compromising prediction accuracy. Thus, user input represents another variable affecting AlphaFold’s performance.
In this study, we evaluated the ability of AlphaFold 3 to model previously uncharacterized natural variants of the catalase KatA from the gastric pathogen Helicobacter pylori. This system is well-suited as a case study because H. pylori has a notoriously high mutation rate, leading to numerous naturally occurring protein variants that may contribute to its pathogenicity and resistance to antibacterial drugs 23,24. High-resolution crystal structures exist for KatA, though they are limited to a single strain (26695), providing AlphaFold with accurate experimental data as a basis for modeling 25,26. We performed a straightforward test by first solving a novel KatA crystal structure from strain SS1, which contains several sequence variations from the published crystal structures, and then assessed the accuracy of predictions made by the Alphafold 3 server 22. We used the AlphaFold 3 server under conditions that mimic how a non-structural biologist might approach modeling, and tested how slightly incorrect inputs impact the resulting models. This study serves as a test case for AlphaFold’s capability to predict the structures of naturally occurring variants and offers insights into how user-provided input may influence modeling accuracy.
Results & Discussion
Characterization of natural KatA variants
KatA is the only catalase produced by H. pylori, is monofunctional, and, like other catalases, converts hydrogen peroxide (H2O2) to water and oxygen through a heme b prosthetic group, thereby protecting the bacterium against oxidative stress 25–27. The enzyme is 505 amino acids in length and forms a tetramer, with one active site per monomer and solvent channels that enable the buried heme groups to access and dismutate H2O2 substrate (Fig. 1A) 25. To our knowledge, no prior work has evaluated the natural variants of H. pylori KatA, and so we conducted searches of the published H. pylori genomes for KatA orthologues, retrieving 1,931 sequences, with 1,922 being unique (Data S1).
Fig. 1.

Natural variants of H. pylori KatA. A. The biologically-relevant KatA tetramer, colored by chain (white, light gray, dark gray, yellow). B. Sequence identity at each amino acid position over 1,931 H. pylori KatA sequences. Natural variants of interest in this study highlighted in pink, with the residue for SS1 strain in bold and corresponding residue for strain 26695 in parentheses. C. Relatedness tree for H. pylori KatA sequences with commonly-used model strains noted. A small number of divergent sequences, which could potentially be misannotations from other species, are not shown (*). D. The position within the H. pylori KatA monomer for the variants of this study are indicated in pink, along with SeqLogo plots showing conservation patterns at each site across all identified sequences. Single Asterix (*) indicates the residue present in strain 26695 and double Asterix (**) indicates the residue present in strain SS1.
To compare variation at each amino acid site, we performed multi-sequence alignment and assessed sequence conservation at each position. While KatA is overall highly conserved, approximately 20 sites show less than 90% sequence identity (Fig. 1B). Among these, we discovered that residues 234, 237, 255, and 421 are common sites of variation, which for the previously-characterized KatA from strain 26695 are Ile, His, Tyr, and Asp, respectively (Fig. 1B) 25. Position 234 and 237 constitute part of the solvent channel opening, 255 is solvent exposed and makes no other notable interactions, and 421 is involved in the stabilizing tetrameric interactions with the N-terminus of another partner chain (PDB: 1qwl, 1qwm,2a9e, 2iqf) 25,26.
Strain 26695 is a clinical isolate used as a research model, as are the strains G27, J99, and SS1, the latter being a mouse-adapted strain commonly employed in animal infection studies 28–33. To visualize sequence variation of KatA across H. pylori strains, we generated a relatedness tree for KatA variants, and noted that these aforementioned strains apparently represent different clades (Fig. 1C). Examining the variation across all H. pylori strains at the four sites mentioned above, shows each to be dominated by two different amino acids; 234 (Ile/Val), 237 (His/Tyr), 255 (Phe/Tyr), and 421 (Asp/Glu) (Fig. 1D). Interestingly, we noted that while the earlier KatA 26695 crystal structures represent the subpopulation of Ile, His, Tyr, and Asp at these sites, the SS1 strain possesses the complementary Val, Tyr, Phe, and Glu (Fig. 1D). Hence, we decided to pursue structural studies of KatA SS1 to obtain the first experimental characterization of the structure of these variants; we reasoned this also would provide us with a small but straightforward set of standards for Alphafold 3 modeling spanning a few different residue types and locations across the protein (Fig. 1D).
Structure of H. pylori strain SS1 KatA positions of variant residues
The crystal structure of KatA from strain SS1, which we henceforth refer to as KatASS1, was solved at 1.87 Å resolution in space group P21 2 21 with two chains in the asymmetric unit, i.e. one half of the biologically-relevant tetramer. This was serendipitous since this novel structure represents a new crystal form with different crystal packing interactions than previously-published structures 25,26, and the two non-crystallographic symmetry-related chains offer two separate views for the amino acids of interest. The electron density was clear and easy to interpret for the majority of the structure, and only portions of the N- and C-terminus were not modeled (see Methods). An overlay of the two chains of KatASS1 with the four structures from strain 26695 (KatA26695) showed high similarity, as expected, with no major differences in global architecture (Fig. 2A).
Fig. 2.

Comparisons of crystal structure and Alphafold 3 models of KatASS1. A. Overlay of crystal structure KatASS1 (2 chains, yellow), and crystal structures of KatA26659 (light gray, 4 chains). Variant sites are noted in pink. B-E. Poses of variant residues within KatASS1, as indicated. KatASS1 crystal structure chains are shown in yellow and KatA26995 crystal structure chains in light gray. Green mesh is Fo-Fc omit map electron density at 3.5 σ averaged over the two non-crystallographic symmetry (NCS) chains, and dark blue mesh is 2Fo-Fc NCS-averaged electron density at 1.0 σ of the final model. F. Overlay of the KatASS1 crystal structure (yellow) with three Alphafold 3 models (light blue, blue, dark blue) using native sequence and tetramer oligomerization as input parameters. G-J. Overlay of each variant residue comparing crystal structure (yellow) positions to Alphafold 3 predictions (blue). K. Global root-mean-square-deviation (RMSD) values for each structure and model in relation to crystal structure KatASS1 chain A.
To assess the positions of the variant sites, we deleted positions 234, 237, 255, and 421 to generate an omit map, which reduces modeling bias in the electron density, and also calculated non-crystallographic symmetry maps averaged across the two chains in the asymmetric unit (Fig. 2B-E). For residue 234 (Ile234 in KatA26695), the density can be clearly interpreted as a Val, positioned similar to Ile234 without the Cδ (Fig. 2B). For Tyr237, the density is much weaker, and small difference density peaks near Cβ suggest it may adopt multiple conformations. The dominant residue position is well-defined through Cβ, and the general plane of the phenol ring is apparent, but the side chain O is not resolved (Fig. 2C). The Tyr237 residues in the two KatASS1 chains also adopt slightly different positions (Fig. 2C). The electron density is unambiguous for both residue 255, which can be readily modeled as a Phe, and for residue 421, a Glu, with all atoms of these residues clearly visible in the electron density (Fig. 2D, Fig. 2E). Overall, the positions of these variants in the KatASS1 structure are well-supported by the electron density and adopt conformations that could be reasonably predicted by an experienced structural biologist.
Alphafold 3 accurately predicts KatASS1 variant poses
Having experimentally-determined the positions of the four KatASS1 variant sites, for which no previous structure has been reported, and therefore are not part of the Alphafold training model, we next utilized the Alphafold 3 server to generate models of KatASS1 in its native tetrameric form, supplying no other information or adjustments to the modeling protocol besides the amino acid sequence. Because we had only four variant sites to use for predictions, and basic chemical knowledge heavily restricts the amount of variability in structural position that would be reasonable at these sites, we limited the number of models we generated to three, which would at least allow us to sample a few different starting seeds and understand the degree to which models might vary. Given the high-quality structural information provided by the four earlier crystal structures, we were unsurprised to find that the global architecture of these Alphafold 3 models mapped well to our new crystal structure (Fig. 2F). However, overlays of these models with our structure also revealed a high degree of accuracy at all four of the variant positions (Fig. 2G-J). Indeed, the models all report high confidence for all regions except for the termini.
After confirming that the AlphaFold 3 server accurately modeled both the global structure of KatASS1 and the variant positions of interest, we investigated whether small changes in user input could affect the results. We generated a new set of models with the following input modifications: (1) single amino acid substitutions at each variant site (with the remainder of the sequence identical to KatA26695), (2) insertion of a Trp at either the N-terminus or C-terminus, expected to have minimal impact on global structure or variant positions, and (3) intentional misassignment of the oligomerization state as monomer, dimer, or trimer. To assess similarity in a quantitative way we employed root-mean-square deviation (RMSD) calculations between structural overlays performed across all Cα in a single chain. The two chains of the KatASS1 crystal structure are nearly identical, with an RMSD of 0.14 Å across 488 Cα atoms (Fig. 2K). Comparisons between our KatASS1 crystal structure and AlphaFold 3 models showed RMSD similar to the four KatA26695 crystal structures, ranging from 0.27–0.3 Å across 487–488 Cα atoms (Fig. 2K). All models were highly accurate in terms of global RMSD, with the worst, though only by a small margin, being one of the ‘correct’ input models with the true KatASS1 sequence and tetrameric state (Fig. 2K). Hence, small changes to user input, in this case, did not manifest into any substantial changes to global structure.
Dubious user-input may lower prediction quality for variant sites
Next, we evaluated these models for their ability to accurately predict poses for Val234, Tyr237, Phe255, and Glu421. Overall, the input changes still resulted in poses similar to the KatASS1 crystal structure, but for a couple models the Tyr237 and Glu421 positions had deviations (Fig. 3A-L). For example, of the models with the single position changes, and modeled as the biologically-relevant tetramer, only the Glu421 side chain carboxyl was misplaced (Fig. 3A-D). In the crystal structure the Glu421 Oε2 atom forms a hydrogen bond with the guanidinium group of Arg28 from another chain of the tetramer, at a distance of 2.73 Å between the heavy atoms; the shift seen in the AlphaFold3 model is unjustified, moving it to a less favorable 3.14 Å (Fig. 3D). Neither the N- nor C-term Trp additions resulted in any notable decrease in predictive accuracy (Fig. 3E-H).
Fig. 3.
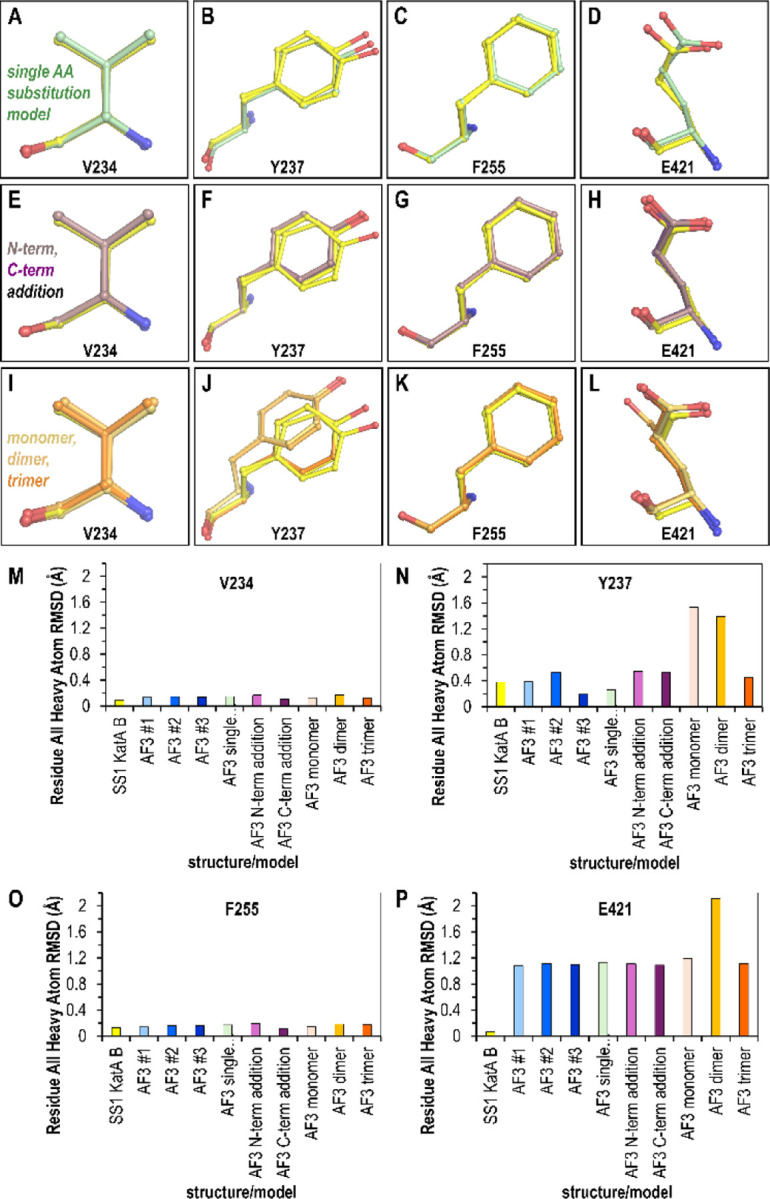
Impact of erroneous user input on Alphafold 3 modeling of variant sites. Overlays show variant sites between the KatASS1 crystal structure (yellow) and Alphafold 3 models generated with: (A–D) single-site mutations (green); (E–H) a single distant Trp insertion at the N-terminus (light purple) or C-terminus (dark purple); and (I–L) the correct sequence modeled with incorrect oligomeric states: monomer (light orange), dimer (orange), and trimer (dark orange). (M–P) Root-mean-square deviation (RMSD) calculations for all heavy atoms compare the Alphafold 3 models to the crystal structure.
However, the set of models most prone to deviation were those with non-native oligomeric states. The Tyr237 position, likely the most challenging site to model, was shifted in the placement of the phenol ring for both the monomer and dimer (Fig. 3J). Since even the experimental structure is poorly resolved at this site, and the residue is solvent-exposed and likely adopts multiple conformations, these deviations may simply reflect that there are multiple energetically-feasible poses. Interestingly, the dimer model contains an entirely different rotamer for the Glu421 position (Fig. 3L). Upon further examination, this could be due to the lack of the Arg28 from the partner chain mentioned above, which would otherwise provide a direct steric clash inhibiting this pose, and the dimer model has made the reasonable guess to shift the Glu421 side chain to be within hydrogen-bonding distance of the nearby His276. Yet, it is unclear why the monomer model, which also is blind to the residues of the tetrameric interface, would not be similarly adjusted, and we presume that this arises from variability in Alphafold 3 outputs.
Examining the RMSD for all heavy atoms between the KatASS1 crystal structure and the Alphafold3 models provides a quantitative comparison for the different strategies (Fig. 3M-P). Across all models, low deviation was seen for the Val234 and Phe255 sites (Fig. 3M, Fig. 3O). Although these sites represent conservative substitutions of similar amino acids, and their structures are arguably the easiest to predict, it is still notable that the deviation from the crystal structure by the Alphafold 3 models is on the same order as the deviation between the two chains within the crystal structure, i.e. these models can be thought of as being at experimental quality (Fig. 3M, Fig. 3O). The Tyr237 site showed the greatest variability in RMSD variation across different models. Striking here is that for some reason the monomer and dimer models, but not the trimer, were the highest deviators, about 2.9-fold greater than other models (Fig. 3N), though there is no clear structural rationale. Lastly, while both chains of the crystal structure have highly similar poses (RMSD of 0.07 Å), the Glu421 site was overall the most challenging for Alphafold 3 to generate highly accurate models, with most near 1.13 Å RMSD, with the dimer, and its different rotamer, the highest at 2.11 Å RMSD (Fig. 3P). Importantly, we note that the AlphaFold model of H. pylori KatASS1 currently reported on the UniProt website as part of the ModBase database is a monomer, and shares the incorrect positioning of Glu421 as seen in our AlphaFold 3 monomer model, despite that the per-residue confidence score (predicted local distance difference test, pLDDT) is >90% (AF-F4ZZ52-F1) 22,34.
Taken together, the Alphafold 3 models were generally of high quality both globally and at the level of single amino acid prediction for these KatASS1 variants. Variability in model quality does occur even with identical, and correct, user input (Fig. 3M-P). It also seems there may be some risk of lowering model quality if the incorrect oligomeric state is supplied.
Interpretations of this modeling case study and caveats
In this study, I evaluated how AlphaFold 3 performs in modeling naturally occurring variants of H. pylori catalase using only basic input parameters. This system benefits from existing high-quality structural data, likely included in AlphaFold 3’s training set, and the analyzed variants primarily involved conservative amino acid substitutions, presenting relatively straightforward modeling challenges. My key finding is that AlphaFold 3 can produce highly accurate structural predictions of variant residues when provided with the correct sequence and native oligomeric state of the target protein. This is encouraging for researchers without deep expertise in structural biology who rely on AlphaFold 3 for structural insights. However, users should be mindful of specifying the correct oligomeric state. Monomeric models should not be used as proxies for oligomeric structures, as missing tertiary interactions can lead to inaccuracies at the single-residue level.
It is important to note that I used a protein crystal structure as the benchmark for accuracy. Crystallography remains one of the most precise methods for protein structure determination and is well-supported by complementary structural and biophysical approaches 35–38. Crystal structures are generally considered reliable representations of proteins in solution and are useful for applications like structure-based drug design 39–41. In support of this idea, the various KatA crystal structures, including the novel KatASS1 structure reported here, were solved under different crystallization conditions and crystal forms, yet they maintain a remarkably consistent architecture (Fig. 2A). Crystal structures represent a time- and space-averaged conformation under specific crystallization and data collection conditions 37,38. Our structure, like most, was solved under cryocooled conditions, meaning that side chain dynamics at physiological temperatures are likely underrepresented 42. Nevertheless, most structures in the Protein Data Bank, used in AlphaFold 3’s training, share these biases 22,37,38,42. Without additional experimental evidence, we cannot definitively state whether the deviations observed in our AlphaFold 3 models are biologically relevant, though this possibility remains open (Fig. 3).
Overall, our findings further support the utility of AlphaFold 3 in modeling of protein variants. As the developers and the broader structural biology community continue to focus on improving variant modeling, as reflected in recent studies, this area is poised for further advancement 4,9,20. As others have suggested, the most rigorous application of AlphaFold modeling involves using it to test specific hypotheses and interpreting results within the constraints of experimental validation 18,43.
Materials & Methods
Bioinformatics
The sequence for H. pylori KatA SS1 (WP_077231901.1) was retrieved from Uniprot. Geneious was used to perform BLAST searches using standard parameters and retrieve H. pylori sequences from the non-redundant amino acid database. Partial sequences were eliminated from further analyses, resulting in 1,931 total sequences, with 1,922 unique (Data S1). MUSCLE 44 was used to perform multi-sequence alignment. The resulting aligned sequences were used for analyses of conservation at variant sites of interest and tree-building using Geneious.
Cloning and protein purification
H. pylori KatA from strain SS1 was recombinantly expressed and purified following established protocols with minor modifications 45. The katA gene was cloned into a pBH vector containing an N-terminal 6×His affinity tag and a TEV protease cleavage site. The construct was transformed into Arctic cells (Agilent) for protein expression. Cells were grown to mid-log phase, induced with 0.4 mM IPTG, and incubated overnight at 15 °C. The protein was purified using nickel affinity chromatography, followed by TEV protease digestion (0.1 mg) to remove the His tag. A reverse His affinity step was performed to isolate the cleaved, untagged KatA. The protein was further purified by size-exclusion chromatography using an S200 column. Enzymatic activity was verified by mixing 10 µl of eluted protein fractions with 100 µl of 30% H2O2 and 100 µl of Triton X-100, confirming catalase function. The final protein was concentrated in a buffer of 20 mM Tris pH 8, 25 mM NaCl to 10.3 mg/ml and flash-frozen for storage.
Protein crystallography
Protein crystals were grown using the vapor diffusion hanging drop method. Optimal crystallization conditions for KatA were found to be a 1:1 ratio of protein solution and 0.1 M sodium acetate, pH 4.5, 25% PEG3350. After two weeks of growth at room temperature, large angular crystals appeared suitable for x-ray diffraction analysis. Crystals were incubated with 20% glycerol as a cryoprotectant and then flash frozen in liquid N2 for data collection. Diffraction data were collected at the Berkeley Advanced Light Source (ALS) Beamline 5.0.2 with the source at 12398.4 eV. The best-diffracting crystal was chosen for structure determination. Data were processed using DIALS 46, and the resolution cutoff set to 1.87 Å. Using a single chain from the crystal structure of PDB: 2a9e the structure was solved through molecular replacement using Phaser 47 and found to be in the space group P 21 2 21 with a dimer in the asymmetric unit, which forms the biologically-relevant tetramer with symmetry mates. To alleviate potential bias in the starting model, the coordinates of the placed solution were randomized by 0.05 Å and the B-factors set to be isotropic and 30 Å2. So that the two chains would not be biased to be similar, we did not utilize non-crystallographic symmetry weights. Subsequent model building in Coot 48 and refinement with Phenix 49, with use of individual B-factors, TLS groups, and riding hydrogens, yielded a final model R/Rfree of 14.2/19.0%. The final structure has a Molprobity clash score of 2.24, putting it in the 99th percentile for structures of comparable resolution, and uploaded to the PDB as entry 9nh3. Crystallographic statistics are reported in Table S1.
Alphafold 3 modeling
The Alphafold 3 server at url: https://alphafoldserver.com was used for modeling, using only basic parameters and supplying the sequence of interest and desired oligomer as inputs.
Structural overlays and RMSD calculations
Overlays and RMSD calculations were performed using the MatchMaker function in ChimeraX. Comparisons of global structure and variant positions were made using overlays in which the entire length of Chain A from crystal structure KatASS1 was used as the reference (across 488 Cα atoms).
Supplementary Material
Description of Supplementary Materials: Includes Excel file with H. pylori KatA variant sequences (Data S1) and crystallographic statistics (Table S1).
Importance:
Experimental structure determination is rarely performed for natural protein variants possessing only minor amino acid differences from published structures, even though small substitutions can significantly impact structure and function. Here, we present a case study showing that AlphaFold 3 can accurately model the structures of natural protein variants. However, providing an incorrect oligomeric state can reduce model accuracy—an error that non-expert users may easily make.
Acknowledgments
Funding for this research was provided by NIAID through awards 1K99AI148587 and 4R00AI148587-03, and funding from the College of Veterinary Medicine at Washington State University to AB.
Footnotes
Declaration of Interests
A.B. owns Amethyst Antimicrobials, LLC.
References
- 1.Vihinen M. Functional effects of protein variants. Biochimie 180, 104–120 (2021). [DOI] [PubMed] [Google Scholar]
- 2.Laskowski R. A., Stephenson J. D., Sillitoe I., Orengo C. A. & Thornton J. M. VarSite: Disease variants and protein structure. Protein Science 29, 111–119 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stein A., Fowler D. M., Hartmann-Petersen R. & Lindorff-Larsen K. Biophysical and Mechanistic Models for Disease-Causing Protein Variants. Trends in Biochemical Sciences 44, 575–588 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Accurate proteome-wide missense variant effect prediction with AlphaMissense | Science. https://www.science.org/doi/10.1126/science.adg7492. [DOI] [PubMed] [Google Scholar]
- 5.Park Y., Metzger B. P. H. & Thornton J. W. Epistatic drift causes gradual decay of predictability in protein evolution. Science 376, 823–830 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Takahashi Y., Nishiyama S., Kawagishi I. & Imada K. Structural basis of the binding affinity of chemoreceptors Mlp24p and Mlp37p for various amino acids. Biochemical and Biophysical Research Communications 523, 233–238 (2020). [DOI] [PubMed] [Google Scholar]
- 7.Gavira J. A., Matilla M. A., Fernández M. & Krell T. The structural basis for signal promiscuity in a bacterial chemoreceptor. The FEBS Journal 288, 2294–2310 (2021). [DOI] [PubMed] [Google Scholar]
- 8.Wheeler L. C., Perkins A., Wong C. E. & Harms M. J. Learning peptide recognition rules for a low-specificity protein. Protein Sci 29, 2259–2273 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pak M. A. et al. Using AlphaFold to predict the impact of single mutations on protein stability and function. PLoS One 18, e0282689 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baker E. P. et al. Evolution of host-microbe cell adherence by receptor domain shuffling. eLife 11, e73330 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hu Y. et al. Naturally Occurring Mutations of SARS-CoV-2 Main Protease Confer Drug Resistance to Nirmatrelvir. ACS Cent. Sci. 9, 1658–1669 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cho H. & Misra R. Mutational Activation of Antibiotic-Resistant Mechanisms in the Absence of Major Drug Efflux Systems of Escherichia coli. J Bacteriol 203, e00109–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Perkins A. et al. The Sensitive Balance between the Fully Folded and Locally Unfolded Conformations of a Model Peroxiredoxin. Biochemistry 52, 8708–8721 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nelson K. J. et al. Experimentally Dissecting the Origins of Peroxiredoxin Catalysis. Antioxidants & Redox Signaling 28, 521–536 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jumper J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Varadi M. et al. AlphaFold Protein Structure Database in 2024: providing structure coverage for over 214 million protein sequences. Nucleic Acids Research 52, D368–D375 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bertoline L. M., Lima A. N., Krieger J. E. & Teixeira S. K. Before and after AlphaFold2: An overview of protein structure prediction. Frontiers in bioinformatics 3, 1120370 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Agarwal V. & McShan A. C. The power and pitfalls of AlphaFold2 for structure prediction beyond rigid globular proteins. Nat Chem Biol 20, 950–959 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang Z., Zeng X., Zhao Y. & Chen R. AlphaFold2 and its applications in the fields of biology and medicine. Sig Transduct Target Ther 8, 1–14 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Buel G. R. & Walters K. J. Can AlphaFold2 predict the impact of missense mutations on structure? Nat Struct Mol Biol 29, 1–2 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Szczepski K. & Jaremko Ł. AlphaFold and what is next: bridging functional, systems and structural biology. Expert Review of Proteomics 22, 45–58 (2025). [DOI] [PubMed] [Google Scholar]
- 22.Abramson J. et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 630, 493–500 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Suerbaum S. & Ailloud F. Genome and population dynamics during chronic infection with Helicobacter pylori. Current Opinion in Immunology 82, 102304 (2023). [DOI] [PubMed] [Google Scholar]
- 24.Correa P. & Piazuelo M. B. Evolutionary History of the Helicobacter pylori Genome: Implications for Gastric Carcinogenesis. Gut Liver 6, 21–28 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Loewen P. C. et al. Structure of Helicobacter pylori Catalase, with and without Formic Acid Bound, at 1.6 Å Resolution. Biochemistry 43, 3089–3103 (2004). [DOI] [PubMed] [Google Scholar]
- 26.Alfonso-Prieto M. et al. The Structures and Electronic Configuration of Compound I Intermediates of Helicobacter pylori and Penicillium vitale Catalases Determined by X-ray Crystallography and QM/MM Density Functional Theory Calculations. J. Am. Chem. Soc. 129, 4193–4205 (2007). [DOI] [PubMed] [Google Scholar]
- 27.Benoit S. L. & Maier R. J. Helicobacter Catalase Devoid of Catalytic Activity Protects the Bacterium against Oxidative Stress♦. J Biol Chem 291, 23366–23373 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kronsteiner B. et al. Helicobacter pylori Infection in a Pig Model Is Dominated by Th1 and Cytotoxic CD8+ T Cell Responses. Infect Immun 81, 3803–3813 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thompson L. J. et al. Chronic Helicobacter pylori Infection with Sydney Strain 1 and a Newly Identified Mouse-Adapted Strain (Sydney Strain 2000) in C57BL/6 and BALB/c Mice. Infect Immun 72, 4668–4679 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ferrero R. L., Thiberge J.-M., Huerre M. & Labigne A. Immune Responses of Specific-Pathogen-Free Mice to Chronic Helicobacter pylori (Strain SS1) Infection. Infection and Immunity 66, 1349–1355 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Burt A. et al. Complete structure of the chemosensory array core signalling unit in an E. coli minicell strain. Nat Commun 11, 743 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Baltrus D. A. et al. The Complete Genome Sequence of Helicobacter pylori Strain G27. J Bacteriol 191, 447–448 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kao C.-Y., Chen J.-W., Wang S., Sheu B.-S. & Wu J.-J. The Helicobacter pylori J99 jhp0106 Gene, under the Control of the CsrA/RpoN Regulatory System, Modulates Flagella Formation and Motility. Front. Microbiol. 8, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pieper U. et al. ModBase, a database of annotated comparative protein structure models and associated resources. Nucleic Acids Research 42, D336–D346 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fowler N. J. & Williamson M. P. The accuracy of protein structures in solution determined by AlphaFold and NMR. Structure 30, 925–933.e2 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brereton A. E. & Karplus P. A. Ensemblator v3: Robust atom level comparative analyses and classification of protein structure ensembles. Protein Sci 27, 41–50 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Deller M. C. & Rupp B. Models of protein–ligand crystal structures: trust, but verify. J Comput Aided Mol Des 29, 817–836 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Acharya K. R. & Lloyd M. D. The advantages and limitations of protein crystal structures. Trends in Pharmacological Sciences 26, 10–14 (2005). [DOI] [PubMed] [Google Scholar]
- 39.Lionta E., Spyrou G., Vassilatis D. K. & Cournia Z. Structure-Based Virtual Screening for Drug Discovery: Principles, Applications and Recent Advances. Curr Top Med Chem 14, 1923–1938 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Batool M., Ahmad B. & Choi S. A Structure-Based Drug Discovery Paradigm. Int J Mol Sci 20, 2783 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Aplin C., Milano S. K., Zielinski K. A., Pollack L. & Cerione R. A. Evolving Experimental Techniques for Structure-Based Drug Design. J. Phys. Chem. B 126, 6599–6607 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fraser J. S. et al. Accessing protein conformational ensembles using room-temperature X-ray crystallography. Proceedings of the National Academy of Sciences 108, 16247–16252 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Terwilliger T. C. et al. AlphaFold predictions are valuable hypotheses and accelerate but do not replace experimental structure determination. Nat Methods 21, 110–116 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Edgar R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research 32, 1792–1797 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Perkins A., Tudorica D. A., Amieva M. R., Remington S. J. & Guillemin K. Helicobacter pylori senses bleach (HOCl) as a chemoattractant using a cytosolic chemoreceptor. PLoS Biol 17, e3000395 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Winter G. et al. DIALS: implementation and evaluation of a new integration package. Acta Cryst D 74, 85–97 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McCoy A. J. et al. Phaser crystallographic software. J Appl Cryst 40, 658–674 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Emsley P. & Cowtan K. Coot: model-building tools for molecular graphics. Acta Cryst D 60, 2126–2132 (2004). [DOI] [PubMed] [Google Scholar]
- 49.Adams P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Cryst D 66, 213–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.