

Abstract
The relatively short history of linkage studies in bipolar disorders (BPs) has produced inconsistent findings. Implicated regions have been large, with reduced levels of significance and modest effect sizes. Both phenotypic and genetic heterogeneity may have contributed to the failure to define risk loci. BP is part of a spectrum of apparently familial affective disorders, which have been organized by severity. Heterogeneity may arise because of insufficient data to define the spectrum boundaries, and, in general, the less-severe disorders are more difficult to diagnose reliably. To address the inherent complexities in detecting BP susceptibility loci, we have used restricted diagnostic classifications and a genetically more homogeneous (Ashkenazi Jewish) family collection to perform a 9-cM autosomal genomewide linkage scan. Although they are genetically more homogeneous, there are no data to suggest that the rate of illness in the Ashkenazim differs from that in other populations. In a genome scan of 41 Ashkenazi pedigrees with a proband affected with bipolar I disorder (BPI) and at least one other member affected with BPI or bipolar II disorder (BPII), we identified four regions suggestive of linkage on chromosomes 1, 3, 11, and 18. Follow-up genotyping showed that the regions on chromosomes 1, 3, and 18 are also suggestive of linkage in a subset of pedigrees limited to relative pairs affected with BPI. Furthermore, our chromosome 18q22 signal (D18S541 and D18S477) overlaps with previous BP findings. This research is being conducted in parallel with our companion study of schizophrenia, in which, by use of an identical approach, we recently reported significant evidence for a schizophrenia susceptibility locus in the Ashkenazim on chromosome 10q22.
Introduction
Bipolar disorders (BPs [MIM 125480]) are relatively common and disabling diseases of variable expression, with apparent phenotypic as well as etiologic heterogeneity. The Diagnostic and Statistical Manual, fourth edition (DSM-IV), recognizes several forms of BP (i.e., bipolar I disorder [BPI], bipolar II disorder [BPII], cyclothymia, and BP not otherwise specified [BP NOS] [American Psychiatric Association 1994]). These vary in the extent of disability and the characterization of the periods with manic and/or depressive episodes. Estimates of lifetime prevalence for BPI have been reported to vary between 0.4% and 1.6%, and, for BPII, the prevalence has been reported to be ∼0.5% (American Psychiatric Association 1994), although some controversy exists, with other estimates as high as 5%, depending on the diagnostic criteria used (Akiskal 2002). Evidence from family, twin, and adoption studies strongly supports a genetic component of BP, with heritability estimates of 58%–74% (Tsuang and Faraone 1990) and risk to first-degree relatives of 5%–10% (Craddock and Jones 1999). The mean age at onset is 28 years; however, onset has also been observed in children, as well as in adults in their 6th and 7th decades (Stone 1989; Goodwin and Jamison 1990). Relatives of probands with earlier ages at onset appear to be at increased risk for BP (Weissman et al. 1984; Coryell et al. 2001; Grigoroiu-Serbanescu et al. 2001; Todd 2002). Although segregation analyses have been inconclusive, they suggest a complex and multigenic susceptibility to BP, involving both genes and environment. Intensive efforts to identify or localize susceptibility genes for BP have yielded neither significant associations for disease genes themselves nor strong consensus about linkage regions. Genetic-linkage findings in at least 11 independent regions have reached genomewide significance (P<.05) in single studies: 4p16 (Blackwood et al. 1996), 5q (Sklar et al. 2003), 6q16 and 17q25 (Dick et al. 2003), 6q22 (Middleton et al. 2004), 8q24 (McInnis et al. 2003b), 12q (Morissette et al. 1999), 13q (Detera-Wadleigh et al. 1999), 16p (Ekholm et al. 2003; McInnis et al. 2003a), 18q (Schulze et al. 2003), and 22q12 (Kelsoe et al. 2001). However, these findings do not consistently replicate across the 29 genome scans reported in the past 2 decades. The two meta-analyses conducted thus far also failed to support overlapping regions: Badner and Gershon (2002) identified regions on chromosomes 13q and 22q, whereas Segurado et al. (2003) identified regions on chromosomes 9p, 10q, and 14q (Segurado et al. 2003).
The reasons for such inconsistencies are likely the complex etiology of BP and the genetic heterogeneity present in most linkage samples. Several genes acting in concert may be responsible for BP in some families, whereas other genetic combinations may be the source of the susceptibility among others. If true, linkage studies to date have unwittingly combined families of different genetic etiologies into one analysis. One approach to reduce heterogeneity is to study families from a relatively genetically isolated population that has emerged from a small number of founders (McKusick 1973; Ekelund et al. 2000; McGinnis et al. 2002). This increases the likelihood that the families in the sample will have a similar underlying genetic predisposition, improving the ability to detect the effects of a particular gene. Previous genetic linkage studies of BP have employed this approach, analyzing a limited number of multiplex pedigrees from, for example, the Old Order Amish of Pennsylvania, Portugal, Costa Rica, Finland, Scotland, and Canada (Saguenay–Lac-St.-Jean) and such population isolates are recommended for both linkage disequilibrium (LD)–based screening and fine mapping of BP genes, if reasonably large samples can be collected (Escamilla 2001).
An additional strategy to reduce heterogeneity is to improve the precision of phenotype classification and thus reduce misclassification. For example, studies of BP have often included BPI, BPII, recurrent unipolar depression, and schizoaffective disorder diagnoses as “affected,” under an assumption that these diagnoses reflect a common genetic predisposition. If they do, this broad-phenotype approach will provide the greatest power. However, this may be detrimental for at least two reasons: (1) the etiologies underlying each classification may be distinct, and “lumping” washes out effects of any particular signal; and (2) the misclassification rates for each diagnosis category may be quite different. Restricting the affected phenotype to the diagnosis that provides the greatest confidence can reduce noise.
With these considerations of heterogeneity in mind, we have conducted a genome scan in a set of Ashkenazi Jewish families who are affected with BP, first considering as affected all individuals with BPI and their relatives with either BPI or BPII and then restricting our analyses to only pedigrees with relative pairs affected with BPI (DSM-IV). The current Ashkenazi Jewish population lives mostly in central and eastern Europe and the United States and is descended from a small founder population established ∼500 years ago (Ostrer 2001). The close genetic relationship of Ashkenazi descendants has been documented through traces of several Mendelian genetic disorders, as well as through Y-chromosome and mitochondrial similarities (Tikochinski et al. 1991; Santachiara Benerecetti et al. 1993; Hammer et al. 2000; Nebel et al. 2000, 2001). Genetic studies among the Ashkenazim have been productive in the identification of susceptibility genes for several disorders, given the reduced genetic variation. Founder mutations have been shown to be important as causes of colorectal cancer (Foulkes et al. 2002; Niell et al. 2003), breast cancer (Struewing et al. 1997), and prostate cancer (Foulkes et al. 2002) in the Ashkenazim, and mutations in the identified genes (MSH2, BRCA1, and BRCA2) have been shown to be important for disease susceptibility in other non-Ashkenazi populations (Neuhausen et al. 1998; Lynch et al. 2004). Even though the rates of BPI and BPII among the Ashkenazim do not appear to differ from other populations, focusing on this isolate can reduce heterogeneity in linkage analyses and increase the utility of association analyses. Our analysis of all 41 families, as well as a BPI-restricted subset, allowed us to gain information from the entire set, under the assumption of shared genetic vulnerability, and to gain signal in the subset, given the reliability of the BPI diagnosis and the potential for increased genetic homogeneity.
Material and Methods
Ascertainment of Study Subjects
Ashkenazi Jewish families with members affected with BPI were recruited nationally over a 6-year period through advertisements in newspapers and Jewish newsletters, talks to community organizations, letters to leaders of the Jewish community, and a study Web site (Johns Hopkins Epidemiology/Genetics Program in Psychiatry Home Page). Families were eligible for inclusion in these analyses if the proband met DSM-IV criteria for a BPI diagnosis and a first- or second-degree relative met DSM-IV criteria for BPI or BPII. Our ascertainment strategy for the multiply affected pedigrees was to examine directly all potentially affected individuals (including those with BPI, BPII, any psychotic disorder, and those with any hospitalization for a psychiatric problem), the parents of those individuals, and any other family members connecting the affected pairs. Rules for extending a pedigree included initially obtaining family history information about all first- and second-degree relatives of the proband and all first-degree relatives of other affected individuals in the family. When any parent of an affected individual was unavailable, we sought at least one unaffected sibling for DNA samples.
Diagnostic Instruments and Procedures
Potentially affected individuals and their parents were examined in person by a doctoral-level clinical psychologist (clinical examiner) and were asked for a blood sample. All recruitment methods and protocols for the collection of blood samples and clinical data were approved by the Johns Hopkins University institutional review board, and appropriate informed consent was obtained from all human subjects. The data collection for this study was done in tandem with a similar study focusing on Ashkenazi Jewish families with schizophrenia (SZ [MIM 181500]) (Fallin et al. 2003). All diagnostic procedures for data collection were identical. Examiners were blind to the proband’s diagnosis. Therefore, they did not know if the family was being recruited for the study of BP or SZ. Most of the subjects were seen in their homes. The Diagnostic Interview for Genetic Studies, version 2.0 (DIGS; revised for DSM-IV) (see National Institutes of Mental Health [NIMH] Center for Genetic Studies Web site), a semistructured interview to elicit information about lifetime history of psychiatric symptoms and behaviors, was used. As part of the interview process, a psychiatric-treatment history was prepared, and each subject was asked to sign release forms allowing copies of his or her psychiatric-treatment records to be made available to our research team. Interviews were tape-recorded for quality-control purposes and for review by members of a Consensus Diagnostic Committee, who assigned the final diagnosis for each subject (see below). To decrease measurement error, in addition to interviewing the subject, the clinical examiner also interviewed an informant about the subject. The clinical examiner completed a written diagnostic formulation for each case, describing prominent features and the course of illness.
Final diagnoses were assigned through a consensus procedure. Information available about each subject (e.g., the tape-recorded interview, interview booklets, a summary of information obtained from informant, the clinical examiner’s diagnostic formulation, and psychiatric-treatment records) was reviewed independently by two members (psychiatrists or doctoral-level clinical psychologists) of a Consensus Diagnostic Committee, who each filled out a DSM-IV diagnostic checklist. The checklist contained each necessary criterion for 26 Axis I DSM-IV disorders. Disorders were rated as absent, possibly present, probably present, definitely present, or unknown. The age at onset for disorders rated as present (for any level of certainty) was also recorded. Ratings were assigned independently by the two members of the committee and were then compared. If disagreement existed with respect to (1) the ratings of any of the 26 diagnoses (including certainty levels assigned), (2) the age at onset (>4-year discrepancy) for any of the disorders that were rated as positive, or (3) course-of-illness ratings for individuals with psychotic symptoms, then the two members of the committee met to resolve the discrepancies.
Demographic and Clinical Characteristics
Ancestry questionnaires were completed for each proband to establish the country or region of origin of the proband's parents and four grandparents and to reduce the possibility of non-Ashkenazi grandparents or founders in our sample. Eastern Europe (Russia, Poland, Latvia, Lithuania, and Estonia) and central Europe (Austria, Germany, Hungary, the Czech Republic, Slovakia, Serbia, and Romania) account for >99% of the known countries of origin among grandparents in the 41 pedigrees. Families were excluded from these analyses if any grandparent of an affected subject was known to be of non-Ashkenazi descent.
The family structures and phenotypes for our data set are provided in figure 1. The families consisted mostly of affected sibling pairs (ASPs). Four families contained affected cousin pairs, six families contained affected avuncular pairs, and one family had a grandparent-grandchild affected pair. Among the total set of 41 families, affected individuals included those with BPI (76.3%) and BPII (23.7%). Twenty-six families contained at least two members with BPI. Of these 26, 22 families contained relative pairs with BPI that were informative for linkage. These 22 families were used in the BPI-restricted subanalysis, denoted “BPI subset,” and are shown in the top half of figure 1. Of the affected individuals in the 41 families, 46% were male. The average age at onset for all affected individuals was 25.5 years (range, 5–76 years), and the average age at onset among the 22 families in the BPI subset was 24.7 years (range, 14–69 years). In the larger data set of 41 pedigrees, 34 individuals were classified as “phenotype unknown” in all analyses, because they were given one of the following consensus diagnoses: major depression without comorbid anxiety disorders (n=17), major depression with comorbid anxiety disorders (n=6), cyclothymia (n=1), possible BPII (n=3), schizophrenia/schizoaffective disorder (n=6), or schizophrenia spectrum personality disorder (n=1). For the smaller BPI subset analyses, there was one family with a sibling who had a diagnosis of BPII, in addition to the two siblings with BPI. This BPII sibling was classified as “phenotype unknown.”
Figure 1.

Structure and phenotypes of Ashkenazi pedigrees with BP. “*dna*” indicates DNA available for the subject. Phenotypes are indicated as follows: BPI = bipolar I disorder; BPII = bipolar II disorder; SZ = schizophrenia; SZA = schizoaffective disorder; FH = diagnosis based on family history data; UP = unipolar depression; and UNK = unknown phenotype.
Genotyping
All genotyping for this scan was performed at the Australian Genome Research Facility (AGRF). The autosomal scan included 382 markers (ABI Prism Linkage Mapping Set, version 2 [MD-10]), with an average spacing of 8.85 cM, covering 3,381 cM, and an average heterozygosity of 0.785. The largest gaps were 24.1 cM on chromosome 8q12.1 and 23.8 cM on 6p21.31. Map order and distances were determined from the Généthon map. Markers were genotyped in 28 multiplexed panels. All PCR reactions were performed under standard conditions in a total volume of 6 μl by use of a PTC-225 DNA Engine Tetrad (MJ Research). Primers were labeled with the fluorescent dyes FAM, HEX, and NED (Applied Biosystems LMSV2). PCR products were then pooled into multiplex panels ranging from 10 to 20 markers and electrophoresed for 2.8 h (0.2 mm denaturing polyacrylamide gels, 4.5%) on an Applied Biosystems 377 DNA Sequencer. GeneScan software (Applied Biosystems) assigns tracking for each sample lane. Files are then imported into Genotyper (Applied Biosystems) software that interprets the electropherogram and assigns genotypes.
When possible, follow-up markers were chosen from the ABI linkage mapping set (LMS-HD5); otherwise, they were chosen from the Marshfield map (Marshfield Center for Medical Genetics Web site) or from published literature (Brzustowicz et al. 2000; Schulze et al. 2003). For each region, markers were chosen to achieve doubled density in the 1-LOD linkage interval (increasing information content to ∼90%, on average). Priority was given to markers with the highest heterozygosity. Map order and distance were based on the July 2003 human genome assembly available through the University of California–Santa Cruz Genome Browser (UCSC Genome Bioinformatics Web site). In total, 27 follow-up markers were successfully genotyped (8 each on chromosomes 1 and 11, 6 on chromosome 3, and 5 on chromosome 18).
Statistical Analyses
Mendelian inconsistencies and potential relationship errors were evaluated and corrected prior to data analysis by use of the Pedmanager (version 0.9) interface at AGRF (Ewen et al. 2000). The AGRF software was also used for binning of alleles, and Genehunter, version 2.0, was used to identify double (flanking) crossovers in haplotypes. Markers were removed from subsequent analyses for the entire family when Mendelian inconsistencies were apparent and were removed for individuals when apparent double crossovers were identified in Genehunter haplotypes. Parametric and model-free allele-sharing linkage analyses were performed by use of the software Genehunter (Kruglyak et al. 1996). Parametric LODs and heterogeneity LOD (HLOD) scores were calculated for both dominant and recessive models. Parameters for these models are noted in table 1. Model-free allele sharing was assessed via the nonparametric linkage (NPL) statistic on the basis of estimated allele sharing for all affected relative pairs (ARPs) in the data set. Marker-allele frequencies among the Ashkenazim were estimated on the basis of founders from 60 Ashkenazi pedigrees collected for our psychiatric-genetics studies (101 parents from families with BPI or SZ, according to DSM-IV criteria). This allowed a more precise estimate of frequencies than relying on founders in the 41 families with BPI. However, parents were available for most of our families (see fig. 1), so the impact of these allele-frequency estimates should not be large.
Table 1.
Maximum Linkage Signal for Each Chromosome at Initial Genome Scan for 41 Multiplex BPI-BPII Ashkenazi Pedigrees[Note]
|
Results of Parametric Analysis for |
|||||||||||
|
Results of Model-Free Analysis |
Dominant Modela |
Recessive Modelb |
|||||||||
| Chromosome | Location(cM) | PeakMarker | InformationContentat Peak | NPL | P | Location(cM) | MaximumHLOD | α | Location(cM) | MaximumHLOD | α |
| 1 | 167.3 | D1S484 | .74 | 2.464 | .0072 | 167.3 | 1.73 | .65 | 219.4 | 1.01 | .27 |
| 2 | 151.0 | D2S151 | .72 | 2.165 | .0157 | 153.32 | 1.54 | .61 | 138.88 | .72 | .28 |
| 3 | 57.1 | D3S1277 | .79 | 2.223 | .0135 | 59.7 | 1.44 | .61 | 119.6 | .80 | .28 |
| 4 | 31.3 | D4S419 | .74 | 1.890 | .0300 | 33.7 | .71 | .43 | 38.5 | 1.31 | .41 |
| 5 | 202.7 | D5S408 | .62 | 1.413 | .0796 | 202.7 | .33 | .27 | 202.7 | .49 | .24 |
| 6 | 117.16 | D6S276 | .74 | 2.066 | .0203 | 113.28 | 1.48 | .55 | 22.5 | .51 | .21 |
| 7 | 3.9 | D7S531 | .85 | 1.915 | .0284 | 137.28 | .25 | .27 | 139.32 | .37 | .24 |
| 8 | 67.5 | D8S285 | .73 | 1.166 | .1224 | 67.5 | .30 | .25 | 67.5 | .97 | .31 |
| 9 | 9.3 | D9S286 | .85 | 1.233 | .1094 | 5.58 | .69 | .37 | 156.8 | 1.23 | .39 |
| 10 | 117.8 | D10S185 | .81 | 1.970 | .0251 | 127.7 | .99 | .43 | 26.02 | .56 | .26 |
| 11 | 58.0 | D11S4191 | .86 | 2.347 | .0099 | 58.00 | .37 | .27 | 59.26 | 1.96 | .44 |
| 12 | 31.9 | D12S364 | .87 | 1.085 | .1394 | 35.14 | .38 | .30 | 31.9 | .48 | .22 |
| 13 | 15.10 | D13S171 | .78 | 1.358 | .0881 | 15.10 | .66 | .37 | 88.2 | .03 | .05 |
| 14 | 40.2 | D14S288 | .82 | 1.358 | .0880 | 40.20 | .44 | .30 | 40.2 | .63 | .23 |
| 15 | 69.2 | D15S131 | .85 | 1.086 | .1394 | 58.06 | .19 | .21 | 69.2 | .73 | .27 |
| 16 | 41.6 | D16S3068 | .79 | 1.201 | .1156 | 41.60 | .16 | .19 | 38.0 | .72 | .27 |
| 17 | 125.0 | D17S784 | .76 | .068 | .4706 | … | … | … | … | … | … |
| 18 | 109.8 | D18S61 | .77 | 2.224 | .0136 | 103.2 | .36 | .33 | 109.8 | 1.08 | .38 |
| 19 | 29.2 | D19S226 | .81 | 1.338 | .0912 | 31.72 | .60 | .39 | 21.9 | .10 | .09 |
| 20 | 51.6 | D20S195 | .82 | 1.326 | .0932 | 51.60 | .37 | .27 | … | … | … |
| 21 | 41.3 | D21S266 | .65 | .883 | .1887 | 33.74 | .38 | .33 | 0 | .50 | .29 |
| 22 | 49.7 | D22S274 | .72 | .383 | .3496 | … | … | … | 49.7 | .54 | .23 |
Note.— NPL scores >2.2 are shown in bold italics. α = estimated proportion of linked families at this location.
Dominant parametric HLOD scores, calculated in Genehunter, version 2.0, assumed a disease allele frequency of .005 and penetrances of .65, .65, and .0096 for homozygotes, heterozygotes, and noncarriers, respectively.
Recessive parametric HLOD scores, calculated in Genehunter, version 2.0, assumed a disease allele frequency of .11 and penetrances of .65, .0096, and .0096 for homozygotes, heterozygotes, and noncarriers, respectively.
Two data sets and phenotype classifications were used for the linkage scan. First, all 41 families were included. For these analyses, individuals with a definite or probable diagnosis of BPI or BPII were considered “affected”; individuals in these families with certain other psychiatric diagnoses (specified above) were classified as “phenotype unknown.” This resulted in 97 affected individuals and 54 ARPs (38 ASPs). The second data set was a subset that included only the families with at least one informative ARP in which both individuals were identified as having BPI. This subset contained 22 families, with 52 individuals affected with BPI and 33 ARPs (19 ASPs). For these analyses, only those with a definite or probable diagnosis of BPI were considered “affected”; individuals affected with BPII and the other above-specified psychiatric disorders were classified as “phenotype unknown.”
Parent-of-origin effects were incorporated into the parametric linkage analyses by use of the Genehunter imprinting (GHI) software, which allows different heterozygote penetrances depending on parental inheritance of the disease allele (Strauch et al. 2000). In this software, “maternal imprinting” refers to masking the maternal alleles, such that the paternal allele is expressed in the offspring (i.e., paternal inheritance), and “paternal imprinting” reflects the opposite. Model-free parent-of-origin effects for ASPs were estimated via the method of Holmans (2003) by use of parent-specific identical-by-descent (IBD)–sharing estimates derived from output from the GENIBD component of the SAGE software package, version 4.5 (SAGE 2003).
Empirical P values were calculated for the NPLall scores via simulation. The program MERLIN (Multipoint Engine for Rapid Likelihood Inference) (Abecasis et al. 2002) was used to generate 50,000 replicates of families identical to those in our sample. Markers with similar allele sizes and frequencies were also generated under the assumption of no linkage. Linkage analyses were then performed on these unlinked replicates (following the procedure of Li and Haghighi [1999]), and genomewide empirical P values were estimated by extrapolating results for chromosome 1 to the whole-genome level, assuming chromosome 1 represents 1/10 of the genome. For fine mapping, chromosomewide empirical P values were calculated as the proportion of 50,000 replicates showing an equal or more extreme NPL score at any point on the chromosome. Although recessive and dominant model parametric analyses were also performed for each chromosome, our main focus has been on model-free NPL-score results, and we therefore focused on this statistic for P-value estimation. Furthermore, these additional parametric tests are correlated (on average) with the NPL results and are not likely to increase the overall type 1 error greatly.
The location of the unobserved trait locus, along with the 95% CI, was estimated for regions of follow-up genotyping on chromosomes 1, 3, 11, and 18 via GeneFinder software (Liang et al. 2001), which uses IBD information from all available ASPs. The estimated mean proportion of alleles that were shared IBD (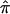 ) and the SEs for this estimation were obtained by use of the GENIBD and SIBPAL components of the SAGE software package (SAGE 2003).
) and the SEs for this estimation were obtained by use of the GENIBD and SIBPAL components of the SAGE software package (SAGE 2003).
Results
Initial Autosomal Scan Linkage Analyses
NPL and HLOD plots across the entire genome for the full set of 41 families and for the subset of 22 BPI-restricted families are shown in figure 2. No initial linkage signal reached “significant” evidence for linkage, according to the criteria of Lander and Kruglyak (1995), in the 41 families or in the subset of BPI-BPI pairs. The highest NPL and parametric LOD signals on each chromosome are shown in tables 1 and 2 for the overall sample and BPI-restricted set. The highest signal across the autosomes among all families was observed on chromosome 1q23.3 (D1S484), with an NPL score of 2.46 and a dominant maximum HLOD score of 1.73. This NPL score corresponds to a genomewide P value of .8 on the basis of simulations. Given the limited power of our study, an NPL score of ⩾3.29 would be needed to achieve empirical genomewide significance at the .05 level in these data, according to simulations. Considering this, we chose a liberal threshold of NPL >2.2 for genotyping additional markers in a region to increase the marker density in our data set. Three other peaks met this threshold in the overall set and were considered for follow-up of additional markers: chromosomes 3p23 (NPL=2.22 at D3S1277), 11q12.1 (NPL=2.35 at D11S4191), and 18q22.2 (NPL=2.22 at D18S61). In the BPI subset, chromosome 3p23 had an NPL score of 2.46 (also at D3S1277), corresponding to an empirical genomewide P value of .77. No other peaks met our threshold for follow-up in the subset analysis.
Figure 2.

Genome scan for all of the families with BPI or BPII and for the BPI-restricted subset. NPL score is shown in black, dominant HLOD in red, and recessive HLOD in blue.
Table 2.
Maximum Linkage Signal for Each Chromosome at Initial Genome Scan for 22 Multiplex BPI-Restricted Ashkenazi Pedigrees[Note]
|
Results of Parametric Analysis for |
|||||||||||
|
Results of Model-Free Analysis |
Dominant Modela |
Recessive Modelb |
|||||||||
| Chromosome | Location(cM) | PeakMarker | InformationContentat Peak | NPL | P | Location(cM) | MaximumHLOD | α | Location(cM) | MaximumHLOD | α |
| 1 | 167.3 | D1S484 | .72 | 1.867 | .0324 | 167.3 | 1.18 | .82 | 32.9 | .94 | .44 |
| 2 | 140.9 | D2S112 | .72 | 1.852 | .0336 | 142.92 | .89 | .64 | 136.86 | 1.07 | .50 |
| 3 | 57.1 | D3S1277 | .79 | 2.457 | .0077 | 59.7 | 1.36 | .82 | 53.10 | 1.52 | .70 |
| 4 | 43.3 | D4S391 | .81 | 1.419 | .0797 | 57.3 | .23 | .29 | 36.10 | .48 | .38 |
| 5 | 202.7 | D5S408 | .63 | 1.189 | .1186 | 40.3 | .26 | .35 | 202.70 | .66 | .36 |
| 6 | 164.3 | D6S1581 | .64 | 1.606 | .0558 | 162.3 | .84 | .53 | 154.30 | .70 | .36 |
| 7 | 143.4 | D7S530 | .82 | 1.509 | .0673 | 144.8 | .22 | .33 | 127.92 | .82 | .51 |
| 8 | 0 | D8S264 | .72 | 1.385 | .0846 | 0 | .46 | .43 | 0 | .67 | .34 |
| 9 | 9.3 | D9S286 | .83 | 1.546 | .0629 | 9.3 | .71 | .48 | 156.8 | .88 | .45 |
| 10 | 117.8 | D10S185 | .82 | 1.074 | .1424 | 153.24 | .35 | .33 | 63.86 | .39 | .31 |
| 11 | 10.6 | D11S1338 | .79 | 1.403 | .0820 | 10.6 | .23 | .31 | 146.7 | .40 | .31 |
| 12 | 31.9 | D12S364 | .85 | 1.739 | .0427 | 35.14 | .57 | .47 | 31.9 | 1.32 | .52 |
| 13 | 24.1 | D13S218 | .75 | 1.290 | .1000 | 24.10 | .71 | .50 | 48.9 | .09 | .14 |
| 14 | 40.2 | D14S288 | .82 | 1.538 | .0637 | 96.9 | .50 | .51 | 40.2 | .85 | .43 |
| 15 | 69.2 | D15S131 | .84 | 1.234 | .1101 | 63.68 | .09 | .19 | 67.82 | .41 | .33 |
| 16 | 60.6 | D16S415 | .84 | 1.845 | .0340 | 60.6 | .39 | .43 | 60.6 | .57 | .35 |
| 17 | 125.0 | D17S784 | .76 | −.071 | .5240 | 15.0 | .05 | .09 | 90.0 | .05 | .06 |
| 18c | 6.0 | D18S63 | .75 | 1.796 | .03802 | 6.0 | .71 | .50 | 47.76 | .97 | .52 |
| 19 | 29.2 | D19S226 | .83 | 2.060 | .0210 | 36.76 | 1.20 | .81 | 21.9 | .71 | .40 |
| 20 | 51.6 | D20S195 | .84 | 1.400 | .0823 | 52.78 | .83 | .52 | 49.08 | .07 | .15 |
| 21 | 41.3 | D21S266 | .67 | .654 | .2558 | 22.00 | .21 | .29 | 0 | .19 | .21 |
| 22 | 9.00 | D22S539 | .77 | −.495 | .6862 | … | 0 | 0 | 49.7 | .02 | .08 |
Note.— NPL scores >2.2 are shown in bold italics. α = estimated proportion of linked families at this location.
Dominant parametric HLOD scores, calculated in Genehunter, version 2.0, assumed a disease allele frequency of .005 and penetrances of .65, .65, and .0096 for homozygotes, heterozygotes, and noncarriers, respectively.
Recessive parametric HLOD scores, calculated in Genehunter, version 2.0, assumed a disease allele frequency of .11 and penetrances of .65, .0096, and .0096 for homozygotes, heterozygotes, and noncarriers, respectively.
Secondary peak at 109.8 cM (peak marker, D18S61; information content at peak, .72; NPL score, 1.792; P = .0382).
Additional Linkage Analyses, with Increased Marker Density in Four Regions
Additional markers in four regions were genotyped and analyzed in both the larger data set of 41 pedigrees and in the subset of 22 pedigrees. Results after addition of markers to double the density in each of these regions are shown in table 3. Although, as expected, the information content improved by ∼10% in each region, we found that there was little change in the results in the overall set of 41 families, except for an increase of 0.58 in the NPL score on 1q23 (top half of table 3) (estimated location 171.8 cM [95% CI 160.0–183.7 cM]). However, several changes are worth noting in the 22-family BPI subset (bottom half of table 3). The NPL score on 1q23.3 from the initial analyses in the 22 families was only 1.87. However, since the whole set of families was genotyped at additional markers in this region, we were able to run follow-up linkage in the subset as well. On follow-up, the increased marker density provided an NPL score of 3.05 (estimated location 169.4 cM [95% CI 154.6–184.2 cM]). A similar increase was observed for the parametric analyses (the HLOD score increased from 1.18 to 2.03) on chromosome 1q23.3. Also, the signal on chromosome 3 improved in the BPI subset (estimated location 57.4 cM [95% CI 49.0–65.8 cM]) but did not increase in the full data set. Linkage in chromosome 18 in the BPI subset also increased substantially: the NPL score increased from 1.79 to 2.44 (estimated location 106.2 cM [95% CI 96.5–115.8 cM]). In addition, CIs were computed for the secondary peak on chromosome 18 at 18p11 for BPI-BPII pedigrees; the peak CI was 9.84 cM (0.0–30.37 cM). These observed increases in evidence for linkage, especially for chromosomes 1 and 18, most likely demonstrate an increase in power as a result of increased marker density when relying on a small data set.
Table 3.
Maximum Linkage Signal for Each Chromosome, with Follow-Up (FU) Markers[Note]
|
Results of Parametric Analysis for |
||||||||||||
|
Results of Model-Free Analysis |
Dominant Modela |
Recessive Modelb |
||||||||||
| Data Setand Chromosome | Bandc | Location(cM) | PeakMarker | InformationContentat Peakd | NPL | P | Location(cM) | MaximumHLOD | α | Location(cM) | MaximumHLOD | α |
| Families with BPI-BPII (n = 41): | ||||||||||||
| 1 | 1q23.3 | 167.3 | D1S484 | .74 | 2.464 | .0072 | 167.3 | 1.73 | .65 | 219.4 | 1.01 | .27 |
| 1-FU | 1q23.3 | 169.2 | D1S2675 | .88 | 3.047 | .0012 | 169.2 | 2.22 | .64 | 271.6 | .79 | .28 |
| 3 | 3p23 | 57.1 | D3S1277 | .79 | 2.223 | .0135 | 59.7 | 1.44 | .61 | 119.6 | .80 | .28 |
| 3-FU | 3p22.1 | 60.94 | D3S3685 | .87 | 2.177 | .0153 | 59.98 | 1.55 | .52 | 113.91 | .80 | .28 |
| 11 | 11q12.1 | 58.0 | D11S4191 | .86 | 2.347 | .0099 | 58.0 | .37 | .27 | 59.26 | 1.96 | .44 |
| 11-FU | 11q12.1 | 72.74 | D11S4191e | .93 | 2.445 | .0076 | 58.75 | .39 | .27 | 72.74 | 1.74 | .40 |
| 18 | 18q22.2 | 109.8 | D18S61 | .77 | 2.224 | .0136 | 103.2 | .36 | .33 | 109.8 | 1.08 | .38 |
| 18-FU | 18q22.3 | 109.6 | D18S541 | .88 | 2.267 | .012 | 111.0 | .94 | .43 | 108.02 | .75 | .30 |
| Families with BPI (n = 22): | ||||||||||||
| 1 | 1q23.3 | 167.3 | D1S484 | .72 | 1.867 | .0324 | 167.3 | 1.18 | .82 | 32.9 | .94 | .44 |
| 1-FU | 1q23.3 | 169.2 | D1S2675 | .86 | 3.045 | .0014 | 169.2 | 2.03 | .93 | 32.9 | .94 | .44 |
| 3 | 3p23 | 57.1 | D3S1277 | .79 | 2.457 | .0077 | 59.7 | 1.36 | .82 | 53.1 | 1.52 | .7 |
| 3-FU | 3p22.1 | 60.94 | D3S3685 | .88 | 2.850 | .0025 | 59.98 | 2.23 | .88 | 49.06 | 1.01 | .47 |
| 11 | 11p15.4 | 10.6 | D11S1338 | .79 | 1.403 | .0820 | 10.6 | .23 | .31 | 146.7 | .40 | .31 |
| 11-FU | 11p15.4 | 10.6 | D11S1338 | .79 | 1.403 | .0820 | 10.6 | .23 | .30 | 102.5 | .39 | .28 |
| 18f | 18p11.31 | 6.0 | D18S63 | .75 | 1.796 | .0380 | 6.0 | .71 | .50 | 47.76 | .97 | .52 |
| 18-FU | 18q22.1 | 103.0 | D18S477 | .88 | 2.439 | .0089 | 103.0 | 1.34 | .78 | 47.76 | .97 | .53 |
Note.— NPL scores >2.2 are shown in bold italics. α = estimated proportion of linked families at this location.
Dominant parametric HLOD scores, calculated in Genehunter, version 2.0, assumed a disease allele frequency of .005 and penetrances of .65, .65, and .0096 for homozygotes, heterozygotes, and noncarriers, respectively.
Recessive parametric HLOD scores, calculated in Genehunter, version 2.0, assumed a disease allele frequency of .11 and penetrances of .65, .0096, and .0096 for homozygotes, heterozygotes, and noncarriers, respectively.
Band designation obtained from National Center for Biotechnology Information (NCBI) build 34 of the human genome, July 2003 (UCSC Genome Bioinformatics Web site).
Information content from Genehunter, version 2.0.
This is the same location as the original scan peak. Location estimates have changed because of additional information.
Secondary peak at band 18q22.2 (location, 109.8 cM; peak marker, D18S61; information content at peak, .72; NPL score, 1.792; P = .0382).
It is important to point out that, despite the smaller sample size, these improvements occurred among the restricted BPI-only analyses. For the chromosome 1, 3, and 18 signals, the mean IBD estimates ( ) were actually higher for this restricted set than for the overall set of families: chromosome 1 location,
) were actually higher for this restricted set than for the overall set of families: chromosome 1 location,  versus 0.63; chromosome 3 location,
versus 0.63; chromosome 3 location,  versus 0.61; chromosome 18p location,
versus 0.61; chromosome 18p location, 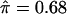 versus 0.59; and chromosome 18q location,
versus 0.59; and chromosome 18q location,  versus 0.60. This supports our strategy of increasing homogeneity in phenotype.
versus 0.60. This supports our strategy of increasing homogeneity in phenotype.
Parent-of-Origin (P-O-O) Analyses of Chromosome 18
Non-Mendelian P-O-O effects have been observed for rare disorders such as Prader-Willi and Angelman syndromes, and there is evidence that P-O-O effects contribute to common complex diseases such as diabetes (Huxtable et al. 2000) and Alzheimer disease (Bassett et al. 2002). P-O-O effects for BP have been proposed, both on the basis of family data that suggest an increased risk of illness for maternal relatives of a BP proband (McMahon et al. 1995) and on the basis of genetic data that suggests paternal imprinting (McMahon et al. 2001; McInnis et al. 2003a).
Considering the previous evidence for a BP locus on 18q22, as well as previous reports of P-O-O effects for this signal (McInnis et al. 2003c), we also performed P-O-O analyses in our data. Parametric analyses incorporating parental origin for penetrance of heterozygotes (see fig. 3) showed strong evidence for paternal inheritance (labeled as “maternal imprinting” in the figure) across the 18p11.3 region that has been previously linked to BP, but only weak evidence in the chromosome 18q22 region. Allele-sharing analyses incorporating P-O-O effects were conducted on 38 ASPs in 34 pedigrees and also showed evidence for paternal inheritance along most of the chromosome, including the 18p region. It is interesting that the best P-O-O signal was near the centromere (D18S1102, chromosome 18q12), at a region showing no linkage when P-O-O was not considered. The signal at this site results from strong oversharing of paternal alleles (estimated proportion of paternal/maternal alleles shared IBD, 0.74/0.26). Similar model-free P-O-O results were obtained by use of the “parent of origin” option for concordant ASPs in the LODPAL component of SAGE (data not shown).
Figure 3.

P-O-O analyses of chromosome 18 for all of the families with BPI or BPII. For the GHI analyses, dominant models identical to those in table 3 were used, with the exception that alleles from the imprinted parent were given a penetrance equal to that of noncarriers. “Maternal imprinting” for parametric analyses denotes increased penetrance of the paternal allele and therefore is analogous to paternal sharing.
Discussion
We have performed the first genome scan for BP-susceptibility loci among an Ashkenazi Jewish sample of multiplex pedigrees, in hopes of reducing the underlying heterogeneity among BP linkage samples. Although our strategy restricted recruitment to a relatively small number of families, we have potentially attained a more homogeneous group for detection of linkage. These families consisted of probands with BPI, the most severe form of the illness, and other affected members with either BPI or the clinically less-severe form of the illness, BPII. Given potential genetic heterogeneity among these diagnostic classifications, we also analyzed a BPI-only subset of families (n=22) with ARPs that were informative for linkage, which should have further reduced heterogeneity.
Although none of the analyses provided a linkage signal with a genomewide P value <.05, four chromosomes showed NPL signals >2.2 in the larger 41-family analysis: chromosome 1q23.3 (NPL=2.46 [D1S484]), chromosome 3p23 (NPL=2.22 [D3S1277]), chromosome 11q12 (NPL=2.35 [D11S4191]), and chromosome 18q22.2 (NPL=2.22 [D18S61]). For the BPI-only subset of 22 pedigrees, our strongest signal was achieved at chromosome 3p23 (D3S1277), with an NPL score of 2.46. Follow-up genotyping with additional markers had the greatest impact on signals observed in the 22-family BPI-only subset: in chromosome 1q23.3, the NPL score increased from 1.867 to 3.045; in chromosome 3p23, the NPL score increased from 2.457 to 2.850; and in chromosome 18q, the NPL score increased from 1.792 to 2.439.
We should emphasize the choices made for recruitment and phenotype restrictions. The field of BP genetic discovery has been fraught with weak and inconsistent findings. Realizing that a major source of this problem is likely to be heterogeneity across families, we pursued strategies to maximize the similarity of our families with respect to genetic etiology. Thus, the first feature of our study was to focus on the Ashkenazi population, since there may be a reduced number of founders compared with outbred populations and therefore potentially less allelic and locus heterogeneity for the same phenotype.
The second feature is more fundamental: that of phenotype identification and classification. Most previous studies have included several diagnostic categories at once, often considering BPI, BPII, and, sometimes, recurrent unipolar depression or schizoaffective disorders as “affected” for linkage analyses, with no tabulation of the distribution of these diagnoses within families. BPI is characterized by at least one full manic or mixed episode and usually at least one full major depressive episode. Psychotic symptoms occur in ∼60% of individuals with BP (Goodwin and Jamison 1990; Potash et al. 2001, 2003; A. E. Pulver, unpublished data) and can occur during the depressive, manic, or mixed episodes in BPI. BPII is characterized by at least one full depressive episode, and one or more hypomanic episodes. The DSM-IV defines a hypomanic episode as “a distinct period during which there is an abnormally and persistently elevated, expansive or irritable mood that lasts at least 4 days” (American Psychiatric Association 1994). A hypomanic episode differs from a manic episode in the following respects: (1) it does not cause marked impairment in social or work functioning; (2) it does not require hospitalization; and (3) it does not include psychotic features. Patients frequently have experienced a feeling of well-being and thus do not identify the episode as part of a disorder (Cassano et al. 1999; Benazzi 2001). Therefore, information from informants about the affected individual can be particularly important to diagnosing BPII, and this has been our approach. However, considering these differences between BPI and BPII criteria, diagnosis of BPII is often more difficult than for BPI, and the diagnoses have different reliabilities and sensitivities, making misclassification rates variable across those designated as “affected” in an analysis. In general, the less-severe disorders are more difficult to diagnose reliably (Andreasen et al. 1981). For this reason, we feel it is important to attempt subset analyses restricted to the BPI phenotype, which may have a distinct genetic etiology and which also has the least misclassification potential among these diagnoses.
The advantages and disadvantages to this approach depend on the underlying nature of the genetic etiology. If there are genes that predispose to general phenotypes common across the BPI and BPII diagnoses, then a “lumping” approach may be more powerful. However, families typically have not been selected for a particular subphenotype, which would be the most efficient approach if this scenario were true. The advantages of BPI-only subset analyses are that a more reliable phenotype should reduce “noise” introduced by misclassification and, furthermore, that particular genes may predispose to particular features that distinguish these diagnoses. In this scenario, the “lumping” approach would introduce genetic heterogeneity and limit resolution of any particular gene.
The linkage signals in these Ashkenazi families do not overlap well with the meta-analyses of previous BP genome scans, consisting mostly of outbred populations (Badner and Gershon 2002; Segurado et al. 2003), except for the 18q finding, which lies in a region previously linked to a BP locus in several other studies (see fig. 4). Also, although not as strong as our 18q peak, a linkage signal in the 18p region was observed that has also been implicated in other studies (fig. 4). Both putative chromosome 18 loci have been associated with P-O-O linkage effects, suggesting imprinting or some other parent-specific effect. The pattern and mechanism of parental inheritance are unclear, and such determination is complicated by the often-confused notions of parental origin based on phenotype, versus parental origin based on genotype. Many previous studies have stratified families according to unilateral inheritance from a particular parent on the basis of the diagnosis of the parent, as well as on the basis of shown linkage to a chromosome 18 locus in one or the other stratified group. This is quite different from observing which parental chromosomes are shared in affected individuals, regardless of the observable phenotype in a parent. We must be clear in our discussion of P-O-O effects to distinguish these methods. We believe that the parental chromosome–sharing approach is more specific to the hypothesis of P-O-O effects, and thus we evaluated the potential for parent-specific effects in our sample through both parametric and model-free analyses that focused on the parental origin of marker alleles. We found no compelling evidence for a P-O-O effect in the 18q linkage region, but we did observe paternal inheritance (maternal imprinting) in 18p. Our inability to distinguish a P-O-O effect at 18q22 is consistent with other studies, in that results have been ambivalent. For example, McMahon et al. found evidence of linkage to paternal chromosomes at 18q via one IBD analysis, but they also found evidence supporting maternal inheritance in stratified IBD analyses of the same data (McMahon et al. 1997)
Figure 4.

Summary of linkage findings for BP on chromosome 18
Our chromosome 1q, 3p, 11p, and 18q findings are each in the proximity of previous linkage to BP and/or SZ. Kelsoe et al. found linkage to a BP locus on 3p21 in a set of 20 North American families (Kelsoe et al. 2001). The peak signal in that study was ∼20 Mb from the peak in the present study, but the intervals of signal overlap well between these two studies. Several reports have shown linkage to 11p15 (Egeland et al. 1987; Smyth et al. 1996; Craddock and Jones 1999; Serretti et al. 2000; Sklar et al. 2002; Dick et al. 2003; McInnis et al. 2003a; Zandi et al. 2003), which is near the linkage signal observed in our BPI-restricted set. Most recently, Zandi et al. (2003) found a peak NPL score of 2.96 at D11S1923, ∼3 Mb from our signal, with largely overlapping linkage intervals. This region harbors the candidate genes tyrosine hydroxylase (TH), which has been inconsistently implicated in BP (Smyth et al. 1996), and brain-derived neurotrophic factor (BDNF), also inconsistently implicated through candidate-gene studies as a susceptibility locus for both SZ and BP (Sklar et al. 2002; Green and Craddock 2003; Hong et al. 2003; Fanous et al. 2004).
On the q arm of chromosome 11, several linkage findings for SZ and affective disorders have been reported, although many are more telomeric than our signal in the overall set of 41 families (St. Clair et al. 1990; Nanko et al. 1992; Maziade et al. 1995; Blackwood et al. 2001; Gurling et al. 2001). Our chromosome 1 signal overlaps with a putative SZ locus identified in 22 Celtic Canadian families at 1q21 (Brzustowicz et al. 2000, 2002). This region has also been implicated in two other SZ linkage studies of American and European families (Shaw et al. 1998; Gurling et al. 2001), as well as in a study of Taiwanese families (Hwu et al. 2003). At least two candidate genes in this region have shown evidence for association with SZ and/or psychosis, although not conclusively: the neuronal small conductance calcium-activated potassium channel (KCNN3) gene (Ritsner et al. 2003) and the G-protein signaling 4 (RGS4) gene (Chowdari et al. 2002). It is interesting to note that the RGS4 gene has been shown to be underexpressed in the cerebral cortex of schizophrenics (Mirnics et al. 2001). The 1q region has also been implicated in SZ and affective disorders on the basis of a 1:11 translocation in a Scottish family (St. Clair et al. 1990; Blackwood et al. 2001), although this occurs at 1q42, ∼70 Mb away from our signal. Although there have been no BP findings in this region of chromosome 1, the overlap of a SZ signal highlights the growing evidence of shared susceptibility between BP and SZ, which has been noted for several other chromosomes (Berrettini 2000). In particular, association evidence for KCNN3 has focused on dimensions of SZ (Ritsner et al. 2003), and it is possible that susceptibility to a dimension that is shared between patients with BPI and those with SZ is controlled by the same gene.
The small size of our data set has limited our ability to detect linkage signals. To pursue our findings by use of linkage, we would need to gather further data from the current families, add additional families by use of the same ascertainment methods, and/or increase the density of markers. We have shown the improvement resulting from increased density and will continue in this vein. However, recruitment of additional family members in the study pedigrees and the ascertainment of new multiplex pedigrees has proven extremely difficult. Another strategy that we propose is to focus linkage efforts on BP-related clinical phenotypes that are themselves familial and are also relevant to aspects of the SZ phenotype, such as mania or psychosis. This would provide a larger set of families for linkage analysis, with potentially more homogeneous genetic etiologies, and this strategy fits well with the growing evidence of overlapping linkage results in SZ and BP.
Finally, given the evidence that LD studies can have greater power and resolution than linkage (Risch and Merikangas 1996) and given the emerging high-throughput genotyping technology for SNP analyses, we also recommend shifting strategies for BPI gene discovery toward designs optimized for association strategies. For example, although we recruited ARPs with BPI from only 41 families of Ashkenazi descent, we have collected over 340 Ashkenazi parent/child trios with a BPI proband. Therefore, collection of relatively homogeneous groups in appropriately powered sizes is feasible for LD and direct association analyses. From a pragmatic as well as a scientific perspective, association designs and analytic techniques may be the most fruitful avenue of discovery.
Acknowledgments
We thank the families for their participation in this study. We are also grateful to Peter Holmans, for sharing his P-O-O allele-sharing software, and to Kristin Nicodemus and Weimin Chen, for computational help. This project was funded by NIMH grant R01MH58153. Some of the results of this study were obtained by use of the program package SAGE, which is supported by a U.S. Public Health Service Resource Grant (RR03655) from the National Center for Research Resources.
Electronic-Database Information
The URLs for data presented herein are as follows:
- Australian Genome Research Facility (AGRF), http://www.agrf.org.au/
- Johns Hopkins Epidemiology/Genetics Program in Psychiatry, http://www.hopkinsmedicine.org/epigen/
- Marshfield Center for Medical Genetics, http://research.marshfieldclinic.org/genetics/
- NIMH Center for Genetic Studies, http://www.nimhgenetics.org/ (for DIGS, version 2.0 [click on “Interviews”])
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for BP and SZ) [PubMed]
- University of California–Santa Cruz (UCSC) Genome Bioinformatics, http://genome.ucsc.edu/cgi-bin/hgGateway
References
- Abecasis GR, Cherny SS, Cookson WO, Cardon LR (2002) Merlin—rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet 30:97–101 10.1038/ng786 [DOI] [PubMed] [Google Scholar]
- Akiskal HS (2002) The bipolar spectrum: the shaping of a new paradigm in psychiatry. Curr Psychiatry Rep 4:1–3 [DOI] [PubMed] [Google Scholar]
- American Psychiatric Association (1994) Diagnostic and statistical manual of mental disorders, fourth edition. American Psychiatric Association, Washington, DC [Google Scholar]
- Andreasen NC, Grove WM, Shapiro RW, Keller MB, Hirschfeld RM, McDonald-Scott P (1981) Reliability of lifetime diagnosis: a multicenter collaborative perspective. Arch Gen Psychiatry 38:400–405 [DOI] [PubMed] [Google Scholar]
- Badner JA, Gershon ES (2002) Meta-analysis of whole-genome linkage scans of bipolar disorder and schizophrenia. Mol Psychiatry 7:405–411 10.1038/sj.mp.4001012 [DOI] [PubMed] [Google Scholar]
- Bassett SS, Avramopoulos D, Fallin D (2002) Evidence for parent of origin effect in late-onset Alzheimer disease. Am J Med Genet 114:679–686 10.1002/ajmg.10648 [DOI] [PubMed] [Google Scholar]
- Benazzi F (2001) Sensitivity and specificity of clinical markers for the diagnosis of bipolar II disorder. Compr Psychiatry 42:461–465 10.1053/comp.2001.27901 [DOI] [PubMed] [Google Scholar]
- Berrettini WH (2000) Are schizophrenic and bipolar disorders related? A review of family and molecular studies. Biol Psychiatry 48:531–538 10.1016/S0006-3223(00)00883-0 [DOI] [PubMed] [Google Scholar]
- Berrettini WH, Ferraro TN, Goldin LR, Weeks DE, Detera-Wadleigh S, Nurnberger JI Jr, Gershon ES (1994) Chromosome 18 DNA markers and manic-depressive illness: evidence for a susceptibility gene. Proc Natl Acad Sci USA 91:5918–5921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackwood DH, Fordyce A, Walker MT, St. Clair DM, Porteous DJ, Muir WJ (2001) Schizophrenia and affective disorders: cosegregation with a translocation at chromosome 1q42 that directly disrupts brain-expressed genes: clinical and P300 findings in a family. Am J Hum Genet 69:428–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackwood DH, He L, Morris SW, McLean A, Whitton C, Thomson M, Walker MT, Woodburn K, Sharp CM, Wright AF, Shibasaki Y, St. Clair DM, Porteous DJ, Muir WJ (1996) A locus for bipolar affective disorder on chromosome 4p. Nat Genet 12:427–430 [DOI] [PubMed] [Google Scholar]
- Brzustowicz LM, Hayter JE, Hodgkinson KA, Chow EW, Bassett AS (2002) Fine mapping of the schizophrenia susceptibility locus on chromosome 1q22. Hum Hered 54:199–209 10.1159/000070665 [DOI] [PubMed] [Google Scholar]
- Brzustowicz LM, Hodgkinson KA, Chow EW, Honer WG, Bassett AS (2000) Location of a major susceptibility locus for familial schizophrenia on chromosome 1q21-q22. Science 288:678–682 10.1126/science.288.5466.678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassano GB, Dell’Osso L, Frank E, Miniati M, Fagiolini A, Shear K, Pini S, Maser J (1999) The bipolar spectrum: a clinical reality in search of diagnostic criteria and an assessment methodology. J Affect Disord 54:319–328 10.1016/S0165-0327(98)00158-X [DOI] [PubMed] [Google Scholar]
- Chowdari KV, Mirnics K, Semwal P, Wood J, Lawrence E, Bhatia T, Deshpande SN, B KT, Ferrell RE, Middleton FA, Devlin B, Levitt P, Lewis DA, Nimgaonkar VL (2002) Association and linkage analyses of RGS4 polymorphisms in schizophrenia. Hum Mol Genet 11:1373–1380 10.1093/hmg/11.12.1373 [DOI] [PubMed] [Google Scholar]
- Coryell W, Leon AC, Turvey C, Akiskal HS, Mueller T, Endicott J (2001) The significance of psychotic features in manic episodes: a report from the NIMH collaborative study. J Affect Disord 67:79–88 10.1016/S0165-0327(99)00024-5 [DOI] [PubMed] [Google Scholar]
- Craddock N, Jones I (1999) Genetics of bipolar disorder. J Med Genet 36:585–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Detera-Wadleigh SD, Badner JA, Berrettini WH, Yoshikawa T, Goldin LR, Turner G, Rollins DY, Moses T, Sanders AR, Karkera JD, Esterling LE, Zeng J, Ferraro TN, Guroff JJ, Kazuba D, Maxwell ME, Nurnberger JI Jr, Gershon ES (1999) A high-density genome scan detects evidence for a bipolar-disorder susceptibility locus on 13q32 and other potential loci on 1q32 and 18p11.2. Proc Natl Acad Sci USA 96:5604–5609 10.1073/pnas.96.10.5604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick DM, Foroud T, Flury L, Bowman ES, Miller MJ, Rau NL, Moe PR, et al (2003) Genomewide linkage analyses of bipolar disorder: a new sample of 250 pedigrees from the National Institute of Mental Health Genetics Initiative. Am J Hum Genet 73:107–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egeland JA, Gerhard DS, Pauls DL, Sussex JN, Kidd KK, Allen CR, Hostetter AM, Housman DE (1987) Bipolar affective disorders linked to DNA markers on chromosome 11. Nature 325:783–787 10.1038/325783a0 [DOI] [PubMed] [Google Scholar]
- Ekelund J, Lichtermann D, Hovatta I, Ellonen P, Suvisaari J, Terwilliger JD, Juvonen H, Varilo T, Arajarvi R, Kokko-Sahin ML, Lonnqvist J, Peltonen L (2000) Genome-wide scan for schizophrenia in the Finnish population: evidence for a locus on chromosome 7q22. Hum Mol Genet 9:1049–1057 10.1093/hmg/9.7.1049 [DOI] [PubMed] [Google Scholar]
- Ekholm JM, Kieseppa T, Hiekkalinna T, Partonen T, Paunio T, Perola M, Ekelund J, Lonnqvist J, Pekkarinen-Ijas P, Peltonen L (2003) Evidence of susceptibility loci on 4q32 and 16p12 for bipolar disorder. Hum Mol Genet 12:1907–1915 10.1093/hmg/ddg199 [DOI] [PubMed] [Google Scholar]
- Escamilla MA (2001) Population isolates: their special value for locating genes for bipolar disorder. Bipolar Disord 3:299–317 10.1034/j.1399-5618.2001.30605.x [DOI] [PubMed] [Google Scholar]
- Ewald H, Mors O, Koed K, Eiberg H, Kruse TA (1997) Susceptibility loci for bipolar affective disorder on chromosome 18? A review and a study of Danish families. Psychiatr Genet 7:1–12 [DOI] [PubMed] [Google Scholar]
- Ewen KR, Bahlo M, Treloar SA, Levinson DF, Mowry B, Barlow JW, Foote SJ (2000) Identification and analysis of error types in high-throughput genotyping. Am J Hum Genet 67:727–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallin MD, Lasseter VK, Wolyniec PS, McGrath JA, Nestadt G, Valle D, Liang KY, Pulver AE (2003) Genomewide linkage scan for schizophrenia susceptibility loci among Ashkenazi Jewish families shows evidence of linkage on chromosome 10q22. Am J Hum Genet 73:601–611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanous AH, Neale MC, Straub RE, Webb BT, O’Neill AF, Walsh D, Kendler KS (2004) Clinical features of psychotic disorders and polymorphisms in HT2A, DRD2, DRD4, SLC6A3 (DAT1), and BDNF: a family based association study. Am J Med Genet 125B:69–78 [DOI] [PubMed] [Google Scholar]
- Foulkes WD, Thiffault I, Gruber SB, Horwitz M, Hamel N, Lee C, Shia J, et al (2002) The founder mutation MSH2*1906G→C is an important cause of hereditary nonpolyposis colorectal cancer in the Ashkenazi Jewish population. Am J Hum Genet 71:1395–1412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freimer NB, Reus VI, Escamilla MA, McInnes LA, Spesny M, Leon P, Service SK, Smith LB, Silva S, Rojas E, Gallegos A, Meza L, Fournier E, Baharloo S, Blankenship K, Tyler DJ, Batki S, Vinogradov S, Weissenbach J, Barondes SH, Sandkuijl LA (1996) Genetic mapping using haplotype, association and linkage methods suggests a locus for severe bipolar disorder (BPI) at 18q22-q23. Nat Genet 12:436–441 [DOI] [PubMed] [Google Scholar]
- Garner C, McInnes LA, Service SK, Spesny M, Fournier E, Leon P, Freimer NB (2001) Linkage analysis of a complex pedigree with severe bipolar disorder, using a Markov chain Monte Carlo method. Am J Hum Genet 68:1061–1064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodwin FK, Jamison KR (1990) Manic-depressive illness. Oxford University Press, New York [Google Scholar]
- Green E, Craddock N (2003) Brain-derived neurotrophic factor as a potential risk locus for bipolar disorder: evidence, limitations, and implications. Curr Psychiatry Rep 5:469–476 [DOI] [PubMed] [Google Scholar]
- Grigoroiu-Serbanescu M, Martinez M, Nothen MM, Grinberg M, Sima D, Propping P, Marinescu E, Hrestic M (2001) Different familial transmission patterns in bipolar I disorder with onset before and after age 25. Am J Med Genet 105:765–773 10.1002/ajmg.10047 [DOI] [PubMed] [Google Scholar]
- Gurling HM, Kalsi G, Brynjolfson J, Sigmundsson T, Sherrington R, Mankoo BS, Read T, Murphy P, Blaveri E, McQuillin A, Petursson H, Curtis D (2001) Genomewide genetic linkage analysis confirms the presence of susceptibility loci for schizophrenia, on chromosomes 1q32.2, 5q33.2, and 8p21–22 and provides support for linkage to schizophrenia, on chromosomes 11q23.3–24 and 20q12.1–11.23. Am J Hum Genet 68:661–673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammer MF, Redd AJ, Wood ET, Bonner MR, Jarjanazi H, Karafet T, Santachiara-Benerecetti S, Oppenheim A, Jobling MA, Jenkins T, Ostrer H, Bonne-Tamir B (2000) Jewish and Middle Eastern non-Jewish populations share a common pool of Y-chromosome biallelic haplotypes. Proc Natl Acad Sci USA 97:6769–6774 10.1073/pnas.100115997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmans P (2003) The effects of sex-specific recombination on tests for imprinting. Paper presented at Annual Meeting of the International Genetic Epidemiology Society. Redondo Beach, CA, November 2–4 [Google Scholar]
- Hong CJ, Huo SJ, Yen FC, Tung CL, Pan GM, Tsai SJ (2003) Association study of a brain-derived neurotrophic-factor genetic polymorphism and mood disorders, age of onset and suicidal behavior. Neuropsychobiology 48:186–189 10.1159/000074636 [DOI] [PubMed] [Google Scholar]
- Huxtable SJ, Saker PJ, Haddad L, Walker M, Frayling TM, Levy JC, Hitman GA, O’Rahilly S, Hattersley AT, McCarthy MI (2000) Analysis of parent-offspring trios provides evidence for linkage and association between the insulin gene and type 2 diabetes mediated exclusively through paternally transmitted class III variable number tandem repeat alleles. Diabetes 49:126–130 [DOI] [PubMed] [Google Scholar]
- Hwu HG, Liu CM, Fann CS, Ou-Yang WC, Lee SF (2003) Linkage of schizophrenia with chromosome 1q loci in Taiwanese families. Mol Psychiatry 8:445–452 10.1038/sj.mp.4001235 [DOI] [PubMed] [Google Scholar]
- Kelsoe JR, Spence MA, Loetscher E, Foguet M, Sadovnick AD, Remick RA, Flodman P, Khristich J, Mroczkowski-Parker Z, Brown JL, Masser D, Ungerleider S, Rapaport MH, Wishart WL, Luebbert H (2001) A genome survey indicates a possible susceptibility locus for bipolar disorder on chromosome 22. Proc Natl Acad Sci USA 98:585–590 10.1073/pnas.011358498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruglyak L, Daly MJ, Reeve-Daly MP, Lander ES (1996) Parametric and nonparametric linkage analysis: a unified multipoint approach. Am J Hum Genet 58:1347–1363 [PMC free article] [PubMed] [Google Scholar]
- Lander E, Kruglyak L (1995) Genetic dissection of complex traits: guidelines for interpreting and reporting linkage results. Nat Genet 11:241–247 [DOI] [PubMed] [Google Scholar]
- Li F, Haghighi W (1999) Perl as a tool for linkage analysis. Am J Hum Genet Suppl 65:A260 [Google Scholar]
- Liang KY, Chiu YF, Beaty TH (2001) A robust identy-by-descent procedure using affected sib pairs: multipoint mapping for complex diseases. Hum Hered 51:64–78 10.1159/000022961 [DOI] [PubMed] [Google Scholar]
- Liu J, Juo SH, Dewan A, Grunn A, Tong X, Brito M, Park N, Loth JE, Kanyas K, Lerer B, Endicott J, Penchaszadeh G, Knowles JA, Ott J, Gilliam TC, Baron M (2003) Evidence for a putative bipolar disorder locus on 2p13-16 and other potential loci on 4q31, 7q34, 8q13, 9q31, 10q21-24, 13q32, 14q21 and 17q11-12. Mol Psychiatry 8:333–342 10.1038/sj.mp.4001254 [DOI] [PubMed] [Google Scholar]
- Lynch HT, Coronel SM, Okimoto R, Hampel H, Sweet K, Lynch JF, Barrows A, Wijnen J, van der Klift H, Franken P, Wagner A, Fodde R, de la Chapelle A (2004) A founder mutation of the MSH2 gene and hereditary nonpolyposis colorectal cancer in the United States. JAMA 291:718–724 [DOI] [PubMed] [Google Scholar]
- Maziade M, Raymond V, Cliche D, Fournier JP, Caron C, Garneau Y, Nicole L, Marcotte P, Couture C, Simard C, Boivin R, Rodrigue C, Boutin P, De Braekeleer M, Martinez M, Mérette C (1995) Linkage results on 11Q21-22 in Eastern Quebec pedigrees densely affected by schizophrenia. Am J Med Genet 60:522–528 [DOI] [PubMed] [Google Scholar]
- Maziade M, Roy MA, Rouillard E, Bissonnette L, Fournier JP, Roy A, Garneau Y, Montgrain N, Potvin A, Cliche D, Dion C, Wallot H, Fournier A, Nicole L, Lavallee JC, Merette C (2001) A search for specific and common susceptibility loci for schizophrenia and bipolar disorder: a linkage study in 13 target chromosomes. Mol Psychiatry 6:684–693 10.1038/sj.mp.4000915 [DOI] [PubMed] [Google Scholar]
- McGinnis R, Shifman S, Darvasi A (2002) Power and efficiency of the TDT and case-control design for association scans. Behav Genet 32:135–144 10.1023/A:1015205924326 [DOI] [PubMed] [Google Scholar]
- McInnes LA, Service SK, Reus VI, Barnes G, Charlat O, Jawahar S, Lewitzky S, Yang Q, Duong Q, Spesny M, Araya C, Araya X, Gallegos A, Meza L, Molina J, Ramirez R, Mendez R, Silva S, Fournier E, Batki SL, Mathews CA, Neylan T, Glatt CE, Escamilla MA, Luo D, Gajiwala P, Song T, Crook S, Nguyen JB, Roche E, Meyer JM, Leon P, Sandkuijl LA, Freimer NB, Chen H (2001) Fine-scale mapping of a locus for severe bipolar mood disorder on chromosome 18p11.3 in the Costa Rican population. Proc Natl Acad Sci USA 98:11485–11490 10.1073/pnas.191519098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McInnis MG, Dick DM, Willour VL, Avramopoulos D, MacKinnon DF, Simpson SG, Potash JB, et al (2003a) Genome-wide scan and conditional analysis in bipolar disorder: evidence for genomic interaction in the National Institute of Mental Health genetics initiative bipolar pedigrees. Biol Psychiatry 54:1265–1273 10.1016/j.biopsych.2003.08.001 [DOI] [PubMed] [Google Scholar]
- McInnis MG, Lan TH, Willour VL, McMahon FJ, Simpson SG, Addington AM, MacKinnon DF, Potash JB, Mahoney AT, Chellis J, Huo Y, Swift-Scanlan T, Chen H, Koskela R, Colin Stine O, Jamison KR, Holmans P, Folstein SE, Ranade K, Friddle C, Botstein D, Marr T, Beaty TH, Zandi P, DePaulo JR (2003b) Genome-wide scan of bipolar disorder in 65 pedigrees: supportive evidence for linkage at 8q24, 18q22, 4q32, 2p12, and 13q12. Mol Psychiatry 8:288–298 10.1038/sj.mp.4001277 [DOI] [PubMed] [Google Scholar]
- McInnis MG, Lan TH, Willour VL, McMahon FJ, Simpson SG, Addington AM, MacKinnon DF, Potash JB, Mahoney AT, Chellis J, Huo Y, Swift-Scanlan T, Chen H, Koskela R, Stine OC, Jamison KR, Holmans P, Folstein SE, Ranade K, Friddle C, Botstein D, Marr T, Beaty TH, Zandi P, DePaulo JR (2003c) Genome-wide scan of bipolar disorder in 65 pedigrees: supportive evidence for linkage at 8q24, 18q22, 4q32, 2p12, and 13q12. Mol Psychiatry 8:288–298 10.1038/sj.mp.4001277 [DOI] [PubMed] [Google Scholar]
- McKusick VA (1973) Genetic studies in American inbred populations with particular reference to the Old Order Amish. Isr J Med Sci 9:1276–1284 [PubMed] [Google Scholar]
- McMahon FJ, Hopkins PJ, Xu J, McInnis MG, Shaw S, Cardon L, Simpson SG, MacKinnon DF, Stine OC, Sherrington R, Meyers DA, DePaulo JR (1997) Linkage of bipolar affective disorder to chromosome 18 markers in a new pedigree series. Am J Hum Genet 61:1397–1404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon FJ, Simpson SG, McInnis MG, Badner JA, MacKinnon DF, DePaulo JR (2001) Linkage of bipolar disorder to chromosome 18q and the validity of bipolar II disorder. Arch Gen Psychiatry 58:1025–1031 10.1001/archpsyc.58.11.1025 [DOI] [PubMed] [Google Scholar]
- McMahon FJ, Stine OC, Meyers DA, Simpson SG, DePaulo JR (1995) Patterns of maternal transmission in bipolar affective disorder. Am J Hum Genet 56:1277–1286 [PMC free article] [PubMed] [Google Scholar]
- Middleton FA, Pato MT, Gentile KL, Morley CP, Zhao X, Eisener AF, Brown A, Petryshen TL, Kirby AN, Medeiros H, Carvalho C, Macedo A, Dourado A, Coelho I, Valente J, Soares MJ, Ferreira CP, Lei M, Azevedo MH, Kennedy JL, Daly MJ, Sklar P, Pato CN (2004) Genomewide linkage analysis of bipolar disorder by use of a high-density single-nucleotide-polymorphism (SNP) genotyping assay: a comparison with microsatellite marker assays and finding of significant linkage to chromosome 6q22. Am J Hum Genet 74:886–897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirnics K, Middleton FA, Stanwood GD, Lewis DA, Levitt P (2001) Disease-specific changes in regulator of G-protein signaling 4 (RGS4) expression in schizophrenia. Mol Psychiatry 6:293–301 10.1038/sj.mp.4000866 [DOI] [PubMed] [Google Scholar]
- Morissette J, Villeneuve A, Bordeleau L, Rochette D, Laberge C, Gagn B, Laprise C, Bouchard G, Plante M, Gobeil L, Shink E, Weissenbach J, Barden N (1999) Genome-wide search for linkage of bipolar affective disorders in a very large pedigree derived from a homogeneous population in Quebec points to a locus of major effect on chromosome 12q23-q24. Am J Med Genet 88:567–587 [DOI] [PubMed] [Google Scholar]
- Nanko S, Gill M, Owen M, Takazawa N, Moridaira J, Kazamatsuri H (1992) Linkage study of schizophrenia with markers on chromosome 11 in two Japanese pedigrees. Jpn J Psychiatry Neurol 46:155–159 [DOI] [PubMed] [Google Scholar]
- Nebel A, Filon D, Brinkmann B, Majumder PP, Faerman M, Oppenheim A (2001) The Y chromosome pool of Jews as part of the genetic landscape of the Middle East. Am J Hum Genet 69:1095–1112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nebel A, Filon D, Weiss DA, Weale M, Faerman M, Oppenheim A, Thomas MG (2000) High-resolution Y chromosome haplotypes of Israeli and Palestinian Arabs reveal geographic substructure and substantial overlap with haplotypes of Jews. Hum Genet 107:630–641 10.1007/s004390000426 [DOI] [PubMed] [Google Scholar]
- Neuhausen SL, Godwin AK, Gershoni-Baruch R, Schubert E, Garber J, Stoppa-Lyonnet D, Olah E, et al (1998) Haplotype and phenotype analysis of nine recurrent BRCA2 mutations in 111 families: results of an international study. Am J Hum Genet 62:1381–1388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niell BL, Long JC, Rennert G, Gruber SB (2003) Genetic anthropology of the colorectal cancer-susceptibility allele APC I1307K: evidence of genetic drift within the Ashkenazim. Am J Hum Genet 73:1250–1260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nothen MM, Cichon S, Rohleder H, Hemmer S, Franzek E, Fritze J, Albus M, Borrmann-Hassenbach M, Kreiner R, Weigelt B, Minges J, Lichtermann D, Maier W, Craddock N, Fimmers R, Holler T, Baur MP, Rietschel M, Propping P (1999) Evaluation of linkage of bipolar affective disorder to chromosome 18 in a sample of 57 German families. Mol Psychiatry 4:76–84 10.1038/sj.mp.4000454 [DOI] [PubMed] [Google Scholar]
- Ostrer H (2001) A genetic profile of contemporary Jewish populations. Nat Rev Genet 2:891–898 10.1038/35098506 [DOI] [PubMed] [Google Scholar]
- Potash JB, Chiu YF, MacKinnon DF, Miller EB, Simpson SG, McMahon FJ, McInnis MG, DePaulo JR Jr (2003) Familial aggregation of psychotic symptoms in a replication set of 69 bipolar disorder pedigrees. Am J Med Genet 116B:90–97 [DOI] [PubMed] [Google Scholar]
- Potash JB, Willour VL, Chiu YF, Simpson SG, MacKinnon DF, Pearlson GD, DePaulo JR Jr, McInnis MG (2001) The familial aggregation of psychotic symptoms in bipolar disorder pedigrees. Am J Psychiatry 158:1258–1264 10.1176/appi.ajp.158.8.1258 [DOI] [PubMed] [Google Scholar]
- Risch N, Merikangas K (1996) The future of genetic studies of complex human diseases. Science 273:1516–1517 [DOI] [PubMed] [Google Scholar]
- Ritsner M, Amir S, Koronyo-Hamaoui M, Gak E, Ziv H, Halperin T, Kitain L, Navon R (2003) Association study of CAG repeats in the KCNN3 gene in Israeli patients with major psychosis. Psychiatr Genet 13:143–150 10.1097/00041444-200309000-00002 [DOI] [PubMed] [Google Scholar]
- SAGE (2003) Statistical analysis for genetic epidemiology. Statistical Solutions, Cork, Ireland; http://darwin.cwru.edu/sage/index.php (accessed May 24, 2004)
- Santachiara Benerecetti AS, Semino O, Passarino G, Torroni A, Brdicka R, Fellous M, Modiano G (1993) The common, Near-Eastern origin of Ashkenazi and Sephardi Jews supported by Y-chromosome similarity. Ann Hum Genet 57:55–64 [DOI] [PubMed] [Google Scholar]
- Schulze TG, Chen YS, Badner JA, McInnis MG, DePaulo JR, McMahon FJ (2003) Additional, physically ordered markers increase linkage signal for bipolar disorder on chromosome 18q22. Biol Psychiatry 53:239–243 10.1016/S0006-3223(02)01492-0 [DOI] [PubMed] [Google Scholar]
- Segurado R, Detera-Wadleigh SD, Levinson DF, Lewis CM, Gill M, Nurnberger JI Jr, Craddock N, et al (2003) Genome scan meta-analysis of schizophrenia and bipolar disorder. III. Bipolar disorder. Am J Hum Genet 73:49–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serretti A, Macciardi F, Cusin C, Lattuada E, Souery D, Lipp O, Mahieu B, Van Broeckhoven C, Blackwood D, Muir W, Aschauer HN, Heiden AM, Ackenheil M, Fuchshuber S, Raeymaekers P, Verheyen G, Kaneva R, Jablensky A, Papadimitriou GN, Dikeos DG, Stefanis CN, Smeraldi E, Mendlewicz J (2000) Linkage of mood disorders with D2, D3 and TH genes: a multicenter study. J Affect Disord 58:51–61 10.1016/S0165-0327(99)00112-3 [DOI] [PubMed] [Google Scholar]
- Shaw SH, Kelly M, Smith AB, Shields G, Hopkins PJ, Loftus J, Laval SH, Vita A, De Hert M, Cardon LR, Crow TJ, Sherrington R, DeLisi LE (1998) A genome-wide search for schizophrenia susceptibility genes. Am J Med Genet 81:364–376 [DOI] [PubMed] [Google Scholar]
- Sklar P, Gabriel SB, McInnis MG, Bennett P, Lim YM, Tsan G, Schaffner S, Kirov G, Jones I, Owen M, Craddock N, DePaulo JR, Lander ES (2002) Family-based association study of 76 candidate genes in bipolar disorder: BDNF is a potential risk locus. Brain-derived neutrophic factor. Mol Psychiatry 7:579–593 10.1038/sj.mp.4001058 [DOI] [PubMed] [Google Scholar]
- Sklar P, Pato MT, Kirby A, Petryshen TL, Medeiros H, Carvalho C, Macedo A, Dourado A, Coelho I, Valente J, Soares MJ, Ferreira CP, Lei M, Verner A, Hudson TJ, Morley CP, Kennedy JL, Azevedo MH, Lander E, Daly MJ, Pato CN (2003) Genome-wide scan in Portuguese Island families identifies 5q31-5q35 as a susceptibility locus for schizophrenia and psychosis. Mol Psychiatry 9:213–218 10.1038/sj.mp.4001418 [DOI] [PubMed] [Google Scholar]
- Smyth C, Kalsi G, Brynjolfsson J, O’Neill J, Curtis D, Rifkin L, Moloney E, Murphy P, Sherrington R, Petursson H, Gurling H (1996) Further tests for linkage of bipolar affective disorder to the tyrosine hydroxylase gene locus on chromosome 11p15 in a new series of multiplex British affective disorder pedigrees. Am J Psychiatry 153:271–274 [DOI] [PubMed] [Google Scholar]
- St. Clair D, Blackwood D, Muir W, Carothers A, Walker M, Spowart G, Gosden C, Evans HJ (1990) Association within a family of a balanced autosomal translocation with major mental illness. Lancet 336:13–16 10.1016/0140-6736(90)91520-K [DOI] [PubMed] [Google Scholar]
- Stine OC, Xu J, Koskela R, McMahon FJ, Gschwend M, Friddle C, Clark CD, McInnis MG, Simpson SG, Breschel TS, Vishio E, Riskin K, Feilotter H, Chen E, Shen S, Folstein S, Meyers DA, Botstein D, Marr TG, De Paulo JR (1995) Evidence for linkage of bipolar disorder to chromosome 18 with a parent-of-origin effect. Am J Hum Genet 57:1384–1394 [PMC free article] [PubMed] [Google Scholar]
- Stone K (1989) Mania in the elderly. Br J Psychiatry 155:220–224 [DOI] [PubMed] [Google Scholar]
- Strauch K, Fimmers R, Kurz T, Deichmann KA, Wienker TF, Baur MP (2000) Parametric and nonparametric multipoint linkage analysis with imprinting and two-locus-trait models: application to mite sensitization. Am J Hum Genet 66:1945–1957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struewing JP, Hartge P, Wacholder S, Baker SM, Berlin M, McAdams M, Timmerman MM, Brody LC, Tucker MA (1997) The risk of cancer associated with specific mutations of BRCA1 and BRCA2 among Ashkenazi Jews. N Engl J Med 336:1401–1408 10.1056/NEJM199705153362001 [DOI] [PubMed] [Google Scholar]
- Tikochinski Y, Ritte U, Gross SR, Prager EM, Wilson AC (1991) mtDNA polymorphism in two communities of Jews. Am J Hum Genet 48:129–136 [PMC free article] [PubMed] [Google Scholar]
- Todd RD (2002) Genetics of early onset bipolar affective disorder: are we making progress? Curr Psychiatry Rep 4:141–145 [DOI] [PubMed] [Google Scholar]
- Tsuang MT, Faraone SV (1990) The genetics of mood disorders. The Johns Hopkins Press, Baltimore [Google Scholar]
- Weissman MM, Prusoff BA, Merikangas KR (1984) Is delusional depression related to bipolar disorder? Am J Psychiatry 141:892–893 [DOI] [PubMed] [Google Scholar]
- Zandi PP, Willour VL, Huo Y, Chellis J, Potash JB, MacKinnon DF, Simpson SG, McMahon FJ, Gershon E, Reich T, Foroud T, Nurnberger J Jr, DePaulo JR Jr, McInnis MG (2003) Genome scan of a second wave of NIMH genetics initiative bipolar pedigrees: chromosomes 2, 11, 13, 14, and X. Am J Med Genet 119B:69–76 [DOI] [PubMed] [Google Scholar]