

Abstract
Microcephalin (MCPH1) is a gene mutated in primary microcephaly, an autosomal recessive neurodevelopmental disorder in which there is a marked reduction in brain size. PCC syndrome is a recently described disorder of microcephaly, short stature, and misregulated chromosome condensation. Here, we report the finding that MCPH1 primary microcephaly and PCC syndrome are allelic disorders, both having mutations in the MCPH1 gene. The two conditions share a common cellular phenotype of premature chromosome condensation in the early G2 phase of the cell cycle, which, therefore, appears to be a useful diagnostic marker for individuals with MCPH1 gene mutations. We demonstrate that an siRNA-mediated depletion of MCPH1 is sufficient to reproduce this phenotype and also show that MCPH1-deficient cells exhibit delayed decondensation postmitosis. These findings implicate microcephalin as a novel regulator of chromosome condensation and link the apparently disparate fields of neurogenesis and chromosome biology. Further characterization of MCPH1 is thus likely to lead to fundamental insights into both the regulation of chromosome condensation and neurodevelopment.
Introduction
Primary microcephaly (MIM 251200) is a neurodevelopmental disorder characterized by a marked reduction in brain size in the absence of other malformations or significant neurological deficits. Etiologically, the reduction in brain size is likely to reflect a reduction in neural cell number, as a consequence of either reduced proliferation or increased cell death (Mochida and Walsh 2001). Primary microcephaly is inherited as an autosomal recessive trait and displays genetic heterogeneity, with six loci mapped to date: MCPH1–MCPH6 (Jackson et al. 1998; Jamieson et al. 1999, 2000; Roberts et al. 1999; Moynihan et al. 2000; Pattison et al. 2000; Leal et al. 2003). Two genes have been shown to be mutated in this disorder, microcephalin and ASPM, for the MCPH1 and MCPH5 loci, respectively (Bond et al. 2002; Jackson et al. 2002). It remains to be determined how the loss of function of these proteins causes microcephaly. MCPH1 contains several BRCT domains (Huyton et al. 2000) and, therefore, has been hypothesized to play a role in cell-cycle regulation or in DNA repair (Jackson et al. 2002).
Recently, another syndrome of microcephaly, postnatal short stature and premature chromosome condensation syndrome (PCC syndrome [MIM 606858]), has been described in two children born to consanguineous parents (Neitzel et al. 2002). PCC syndrome has been unique in being the only human disorder in which chromosome condensation is abnormal. Patients with this condition have a high number (10%–15%) of prophase-like cells in routine cytogenetic preparations and have repeatedly poor-quality metaphase G-banding, even on high-resolution chromosome preparations. It has been established that the prophase-like cells result from the early onset of chromosome condensation (hence, “premature chromosome condensation”), in an otherwise apparently unperturbed cell cycle (Neitzel et al. 2002).
Chromosome condensation is a cellular process that is essential for the integrity of mitosis and meiosis, requiring both compaction of chromatin and resolution of intertwined sister chromatids (Swedlow and Hirano 2003). Chromosome compaction requires chromatin reorganization to achieve a 10,000-fold compression in the DNA length prior to chromosome segregation. This is brought about principally by two ATP-hydrolyzing enzymes—topoisomerase II and the condensin complex. Resolution also must occur, so that sister chromatids are separated from each other, and requires the release of the cohesin protein complex from chromosomes during prophase (Losada et al. 2002). The process of chromosome condensation needs to be regulated appropriately, in conjunction with other components of the cell cycle. Precisely how this is achieved has not been established yet, although several regulators have been identified (Kimura et al. 1998; Losada et al. 2002; Watrin and Legagneux 2003).
We show here that MCPH1 primary microcephaly and PCC syndrome are allelic conditions. Both are caused by mutations in the MCPH1 gene and have in common the cellular phenotype of aberrantly regulated chromosome condensation.
Subjects and Methods
Subjects
The two families with MCPH1 primary microcephaly described here have been reported elsewhere (Jackson et al. 1998, 2002), as has the family with PCC syndrome (Neitzel et al. 2002). The study was approved by the Leeds Health Authority/United Leeds Teaching Hospitals National Health Service Trust research ethics committee and the ethics committee of the Charité Universitary Medicine Berlin, Campus Virchow Hospital.
Cytogenetics
Cytogenetic preparations were performed on peripheral blood samples with the use of standard clinical laboratory procedures to prepare G-banded metaphase spreads (Rooney 2001).
Genetic Linkage Analysis
Microsatellite analysis was performed by standard semiautomated methods with the use of an ABI PRISM 377 (Applied Biosystems). Markers were selected from the Généthon final linkage and Marshfield maps. Two-point LOD scores were calculated with LINKAGE v5.2 (Lathrop and Lalouel 1984).
Mutation Detection
The entire coding region of the MCPH1 gene was screened by direct sequencing, as described elsewhere (Jackson et al. 2002). Specifically, exon 5 was amplified by PCR and was sequenced with primers ex5F, dTGCCTTAAGCAGTTGCAGGAC, and ex5R, dACCCCACTAGTCATCATGCAAA. Sequencing was performed using Big Dye Terminator cycle sequencing kit (Applied Biosystems), in accordance with the manufacturer’s protocol, and electrophoresis was done with an ABI 377 gene sequencer.
siRNA-Mediated Depletion of MCPH1
Cells were transfected with 110 nM of RNA duplexes (Qiagen-Xeragon) with the use of Oligofectamine (Invitrogen), in accordance with the manufacturer’s instructions. The siRNA duplex sense-strand sequences were the MCPH1-derived sequence, 5′-CUCUCUGUGUGAAGCACCA-3′, and the control sequence, 5′-UUCUCCGAACGUGUCACGU-3′. Chromosome preparation was performed 48 h after transfection, with the use of standard methods, and >1,000 nuclei per preparation were scored. Transfection efficiency was estimated at 70%–80% by counting the proportion of fluorescent cells after treatment with the fluorescein-conjugated control duplex.
Cytochalasin-B Block to Demonstrate Decondensation Delay
Cytokinesis was blocked with 6 μg/ml of cytochalasin, 6 h before harvesting. Cells were subjected to hypotonic treatment with 0.075 M of KCl for 2 min and were fixed two times with a 3:1 mix of methanol and acetic acid, and the last fixation was in a 9:1 mix of methanol and glacial acetic acid. Nuclei were stained with Giemsa.
Results
Genomewide homozygosity mapping of the family with PCC syndrome had identified a single large candidate region of 64 cM on chromosome 8 (markers D8S264–D8S532), which included the MCPH1 locus (fig. 1). This finding and an overlapping clinical phenotype (fig. 2) led us to consider the possibility that these two disorders might be allelic. Severe microcephaly is present in both MCPH1 primary microcephaly (−5 to −10 SD) and PCC syndrome (−8 to −10 SD) (fig. 2a). A variable short stature is also evident in both. Magnetic resonance imaging (MRI) demonstrates a marked reduction in the size of the cerebral cortex, as well as some reduction in cerebellar and brain-stem volume (fig. 2b–2d). Overall, the neuroimaging is consistent with the classification “microcephaly with simplified gyri” type I (Dobyns 2002). However, in patients with PCC syndrome, periventricular neuronal heterotopias were also observed (fig. 2e), as well as a marked gyral simplification giving the appearance of pachygyria (fig. 2d).
Figure 1.

Depiction of the 64-cM homozygous region, identified by a genomewide search, in consanguineous siblings with PCC syndrome. The region of homozygous markers is boxed. Maximum two-point LOD scores of 1.37 (θ=0) were obtained for markers D8S264, D8S277, D8S550, and D8S560.
Figure 2.

Findings indicating that MCPH1 primary microcephaly and PCC syndrome are allelic disorders. Patients with MCPH1 primary microcephaly (black diamonds) and patients with PCC syndrome (unblackened squares) have a similar degree of microcephaly, and both groups also exhibit short stature of varying severity. a, Head circumference (OFC), height, and weight, plotted as SDs relative to the appropriate age- and sex-related population mean. b and c, Neuroimaging in MCPH1 primary microcephaly, demonstrating a small but structurally normal brain. In PCC syndrome, however, there is more marked gyral simplification resembling pachygyria (d), and periventricular neuronal heterotopias are also present (e). Subjects include an adult with MCPH1 primary microcephaly (b, T1W sagittal; c, axial T2W) and an 18-mo–old child with PCC syndrome (d, axial T2W; e, axial T1W and T2W images of lateral ventricles). f–h, MRI scans of normal subjects age matched to those of images b–d, respectively. The scale bar is 1 cm.
To investigate whether individuals with MCPH1 primary microcephaly showed evidence of the PCC cellular phenotype, archival G-banded cytogenetic preparations of peripheral blood lymphocytes were reviewed. Previous laboratory reports from these patients, indicating low-quality metaphases with poor banding resolution, were confirmed. However, multiple prophase-like cells were also identified, representing 7%–17% of the total number of cells. These prophase-like cells were identical in morphology to those seen previously in PCC syndrome (fig. 3).
Figure 3.
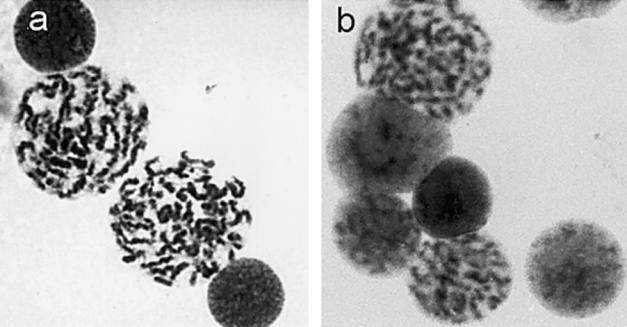
Premature chromosome condensation, present in both MCPH1 primary microcephaly and PCC syndrome. Shown are prophase-like cells in peripheral blood lymphocytes of patients with MCPH1 primary microcephaly (a) and patients with PCC syndrome (b).
Sequencing the entire coding sequence of the MCPH1 gene in patients with PCC syndrome revealed a single-nucleotide insertion in exon 5 within an A6 run (coding nt 427) (fig. 4). The resulting frameshift is predicted to generate a termination codon in exon 6 and, thus, a markedly truncated protein of 146 aa, encoding only the N-terminal BRCT domain of the MCPH1 protein. This sequence change was homozygous in both affected children, heterozygous in both parents, and not present in 220 control alleles.
Figure 4.

Homozygous ins427A mutation (arrow) of the MCPH1 gene, which is present in patients with PCC syndrome.
Next, RNAi depletion of MCPH1 was performed to confirm that loss of microcephalin function is sufficient to induce the cellular phenotype of PCC. Both HeLa and SV40-transformed fibroblast (LN9SV) cell lines, transfected with an siRNA duplex specific for MCPH1, manifested a cellular phenotype identical to that seen in affected individuals (fig. 5). Cell counts demonstrated that significantly more prophase-like cells (P≪.001, χ2) were present in both cell lines treated with the MCPH1 siRNA than in cells treated with the control duplex (fig. 5b and 5c). Thus, we infer that the cellular phenotype observed in patients is due to functional loss of the microcephalin protein.
Figure 5.

The siRNA-mediated depletion of MCPH1 is sufficient to cause premature chromosome condensation. a, Prophase-like phenotype observed in HeLa cells after siRNA-mediated depletion of MCPH1. A marked increase in prophase cells is seen after MCPH1 siRNA transfection in HeLa and LN9SV fibroblasts (b), whereas there is no significant change in mitotic index (% metaphases) (c).
Reviewing diagnostic cytogenetic slides, we observed smaller nuclei with a PCC phenotype, which raised the possibility that such cells might represent postmitotic cells that have not yet decondensed their chromatin. To address this hypothesis, lymphoblastoid cell lines from both patients with MCPH1 primary microcephaly and those with PCC syndrome were treated with cytochalasin B. By preventing cytokinesis, this treatment enables postmitotic cells to be distinguished by their binucleate nature. Many early-G1 binucleate cells from patients with MCPH1 primary microcephaly and from those with PCC syndrome had condensed chromosomes 6 h after treatment, a finding not present in controls (P≪.001, χ2) (fig. 6). From this, we infer that there is also a considerable delay in chromosome decondensation after mitosis in early G1.
Figure 6.

Delayed decondensation observed in MCPH1-deficient cells. a, Binucleate MCPH1 mutant cell with condensed chromosomes indicating a delay in decondensation. b, Binucleate control lymphoblastoid cell with decondensed chromatin. c, Proportion of cells with condensed chromosomes postmitosis in patient and control cell lines. Many cytochalasin-treated binucleate G1-phase PCC (29%) and MCPH1 (36%) mutant cells show condensed chromatin, compared with control lymphoblastoid cells (4%).
Discussion
We establish here that mutations in the MCPH1 gene are the cause of both PCC syndrome and MCPH1 primary microcephaly. Diagnostically, the PCC condensation phenotype may prove to be a cytogenetic hallmark for these disorders, and the presence of prophase cells should have considerable diagnostic utility in the identification of individuals with MCPH1 mutations. Mutation screening of future patients with “PCC” cellular phenotype will be required to establish whether this phenotype is always the result of MCPH1 mutations or whether other genes also might be implicated in this process.
Our results implicate microcephalin as a regulatorof chromosome condensation in both the G2 and G1 phases of the cell cycle. This finding reinforces the hypothesis that MCPH1 primary microcephaly is due to perturbation of the cell cycle during neurogenesis, resulting in either reduced proliferative capacity or increased cell death. Surprisingly, however, we find that neuronal heterotopias sometimes can be found also in individuals with MCPH1 mutations, suggesting that migration of neurons and glia during formation of the cerebral cortex (Barkovich et al. 2001) might be perturbed. This observation raises interesting questions regarding the possible extension of the MCPH1 primary microcephaly phenotype into neuronal heterotopia and other neuronal migration disorders.
Our findings have important implications for the cell biology of chromosome condensation and its regulation. Topoisomerase II and the condensin complex are key components in compacting chromatin into metaphase chromosomes, whereas sister chromatid resolution follows the release of cohesin from chromatin (Swedlow and Hirano 2003). However, regulation of these processes is incompletely understood. Chromatin compaction is, at least in part, controlled by loading of condensin onto chromatin. In turn, condensin function may be regulated by phosphorylation involving distinct signaling elements that include Cdk1 and Aurora B (Kimura et al. 1998; Swedlow and Hirano 2003). The abnormally early compaction of chromatin in MCPH1-deficient cells could indicate that MPCH1 negatively regulates condensin loading, and it will be important to define its relationship to condensin and these regulators. However, compaction is not entirely dependent on condensin, since removing condensin subunits in several model organisms perturbs, but does not prevent, chromosome condensation (Swedlow and Hirano 2003;Watrin and Legagneux 2003). Additionally, condensin loading onto chromosomes appears to occur during late prophase, well after the visible chromosome condensation is apparent (Watrin and Legagneux 2003), which indicates that other factors, perhaps including MCPH1, also must participate in chromatin compaction and its regulation.
In summary, our earlier finding of abnormal chromosome condensation in early G2 phase (Neitzel et al. 2002) is shown now to result from the absence of functional microcephalin. This implicates MCPH1 as a negative regulator of chromosome condensation, acting very early in the condensation process, quite possibly prior to the involvement of condensin. Homology of microcephalin with cell-cycle checkpoint proteins, such as BRCA1 and TopBP1, which regulate G2/M transition, may indicate that it acts as an intermediary between the cell cycle control and chromosome condensation apparatus. Additionally, the finding of delayed chromosome decondensation in mutant cell lines implies that postmitotic decondensation is also a regulated and active process. Further biochemical characterization of microcephalin thus promises to provide important new insights into the regulation of both condensation and decondensation of mitotic chromosomes.
Acknowledgments
We wish to acknowledge Mark Houseman for technical assistance and David Bonthron, Rob Morgan, and Bill Dobyns for helpful discussions. We also acknowledge C. G. Woods, for his work in originally ascertaining the two families with MCPH1 primary microcephaly. A.P.J. is funded by a Medical Research Council Clinician Scientist Fellowship, and S.M.B. is funded by Yorkshire Cancer Research. We are grateful to Gudrun Nürnberg for assisting with calculations of the LOD scores and to Jürgen Sperner for the MRI scans of the patients with PCC syndrome. We are indebted to all the patients, their parents, and the clinicians for their cooperation. This article is dedicated to Robert T. Johnson, on the occasion of his 60th birthday.
Electronic-Database Information
URL for data presented herein is as follows:
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for primary microcephaly and PCC syndrome)
References
- Barkovich AJ, Kuzniecky RI, Jackson GD, Guerrini R, Dobyns WB (2001) Classification system for malformations of cortical development: update 2001. Neurology 57:2168–2178 [DOI] [PubMed] [Google Scholar]
- Bond J, Roberts E, Mochida GH, Hampshire DJ, Scott S,Askham JM, Springell K, Mahadevan M, Crow YJ, Markham AF, Walsh CA, Woods CG (2002) ASPM is a major determinant of cerebral cortical size. Nat Genet 32:316–320 10.1038/ng995 [DOI] [PubMed] [Google Scholar]
- Dobyns WB (2002) Primary microcephaly: new approaches for an old disorder. Am J Med Genet 112:315–317 10.1002/ajmg.10580 [DOI] [PubMed] [Google Scholar]
- Huyton T, Bates PA, Zhang X, Sternberg MJ, Freemont PS (2000) The BRCA1 C-terminal domain: structure and function. Mutat Res 460:319–332 [DOI] [PubMed] [Google Scholar]
- Jackson AP, Eastwood H, Bell SM, Adu J, Toomes C, Carr IM, Roberts E, Hampshire DJ, Crow YJ, Mighell AJ, Karbani G, Jafri H, Rashid Y, Mueller RF, Markham AF, Woods CG (2002) Identification of microcephalin, a protein implicated in determining the size of the human brain. Am J Hum Genet 71:136–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson AP, McHale DP, Campbell DA, Jafri H, Rashid Y, Mannan J, Karbani G, Corry P, Levene MI, Mueller RF, Markham AF, Lench NJ, Woods CG (1998) Primary autosomal recessive microcephaly (MCPH1) maps to chromosome 8p22-pter. Am J Hum Genet 63:541–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamieson CR, Fryns JP, Jacobs J, Matthijs G, Abramowicz MJ (2000) Primary autosomal recessive microcephaly: MCPH5 maps to 1q25–q32. Am J Hum Genet 67:1575–1577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamieson CR, Govaerts C, Abramowicz MJ (1999) Primary autosomal recessive microcephaly: homozygosity mapping of MCPH4 to chromosome 15. Am J Hum Genet 65:1465–1469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura K, Hirano M, Kobayashi R, Hirano T (1998) Phosphorylation and activation of 13S condensin by Cdc2 in vitro. Science 282:487–490 10.1126/science.282.5388.487 [DOI] [PubMed] [Google Scholar]
- Lathrop GM, Lalouel JM (1984) Easy calculations of LOD scores and genetic risks on small computers. Am J Hum Genet 36:460–465 [PMC free article] [PubMed] [Google Scholar]
- Leal GF, Roberts E, Silva EO, Costa SM, Hampshire DJ, Woods CG (2003) A novel locus for autosomal recessive primary microcephaly (MCPH6) maps to 13q12.2. J Med Genet 40:540–542 10.1136/jmg.40.7.540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losada A, Hirano M, Hirano T (2002) Cohesin release is required for sister chromatid resolution, but not for condensin-mediated compaction, at the onset of mitosis. Genes Dev 16:3004–3016 10.1101/gad.249202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochida GH, Walsh CA (2001) Molecular genetics of human microcephaly. Curr Opin Neurol 14:151–156 10.1097/00019052-200104000-00003 [DOI] [PubMed] [Google Scholar]
- Moynihan L, Jackson AP, Roberts E, Karbani G, Lewis I, Corry P, Turner G, Mueller RF, Lench NJ, Woods CG (2000) A third novel locus for primary autosomal recessive microcephaly maps to chromosome 9q34. Am J Hum Genet 66:724–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neitzel H, Neumann LM, Schindler D, Wirges A, Tonnies H, Trimborn M, Krebsova A, Richter R, Sperling K (2002) Premature chromosome condensation in humans associated with microcephaly and mental retardation: a novel autosomal recessive condition. Am J Hum Genet 70:1015–1022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattison L, Crow YJ, Deeble VJ, Jackson AP, Jafri H, Rashid Y, Roberts E, Woods CG (2000) A fifth locus for primary autosomal recessive microcephaly maps to chromosome 1q31. Am J Hum Genet 67:1578–1580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts E, Jackson AP, Carradice AC, Deeble VJ, Mannan J, Rashid Y, Jafri H, McHale DP, Markham AF, Lench NJ, Woods CG (1999) The second locus for autosomal recessive primary microcephaly (MCPH2) maps to chromosome 19q13.1–13.2. Eur J Hum Genet 7:815–820 10.1038/sj.ejhg.5200385 [DOI] [PubMed] [Google Scholar]
- Rooney DE (2001) Human cytogenetics: a practical approach. Oxford University Press, Oxford [Google Scholar]
- Swedlow JR, Hirano T (2003) The making of the mitotic chromosome: modern insights into classical questions. Mol Cell 11:557–569 10.1016/S1097-2765(03)00103-5 [DOI] [PubMed] [Google Scholar]
- Watrin E, Legagneux V (2003) Introduction to chromosome dynamics in mitosis. Biol Cell 95:507–513 10.1016/j.biolcel.2003.08.003 [DOI] [PubMed] [Google Scholar]